

Abstract
Background & Aims
A simultaneous decline in pro‐ and anticoagulant drivers in patients with liver diseases results in a “rebalanced” haemostatic system, even in acutely ill patients. Nevertheless, both bleeding and thrombotic events are common. Here, we explored efficacy of pro‐ and antihaemostatic strategies in compensated and acutely ill cirrhotics which may be unpredictable given the profound haemostatic changes.
Methods
We tested the effects in vitro of the addition of clinically relevant doses of commonly used pro‐ and antihaemostatic strategies in plasma from healthy individuals (n = 30) and patients with compensated (n = 18) and acutely decompensated cirrhosis (n = 18), and acute‐on‐chronic liver failure (n = 10). We used thrombin generation tests and fibrin clot permeability assays to assess potency of various approaches.
Results
Fresh frozen plasma and recombinant factor VIIa modestly increased thrombin generation (10%‐20%). Prothrombin complex concentrate increased thrombin generation two‐fold in controls and 2‐4‐fold in patients. Clot permeability decreased after addition of fibrinogen concentrate by 51% in controls and by 50%‐60% in patients. Low molecular weight heparin decreased thrombin generation by 18% in controls and by 23%‐54% in patients. Similarly, dabigatran decreased thrombin generation by 33% in controls and by 47%‐100% in patients. In contrast, rivaroxaban decreased thrombin generation by 55% in controls, but only by 11%‐38% in patients.
Conclusions
These in vitro data suggest little prohaemostatic effect of fresh frozen plasma and recombinant factor VIIa in acutely ill cirrhotics, whereas prothrombin complex concentrate and fibrinogen concentrate clearly improved haemostasis. Furthermore, our data suggest the requirement for dose adjustments of commonly used anticoagulants in these patients.
Keywords: bleeding, cirrhosis, haemostasis, thrombosis
Abbreviations
- ACLF
acute of chronic liver failure
- AD
acute decompensation
- ETP
endogenous thrombin potential
- FFP
fresh frozen plasma
- LMWH
low molecular weight hepari
- PCC
prothrombin complex concentrate
- TM
thrombomodulin
Key points.
Bleeding and thrombotic events are common in patients with liver disease, but there is uncertainty on optimal management strategies
Efficacy of pro‐ and anticoagulant agents are likely altered in patients with liver disease as a result of profound changes in their haemostatic system
Using plasma samples from patients with compensated cirrhosis, acute decompensation of cirrhosis or acute‐on‐chronic liver failure we demonstrate little prohaemostatic effect of fresh frozen plasma and recombinant factor VIIa, whereas prothrombin complex concentrate and fibrinogen concentrate clearly improved haemostasis
Commonly used anticoagulants have profoundly altered anticoagulant potency in critically ill cirrhotics
1. INTRODUCTION
Patients with liver diseases frequently acquire substantial alterations in their haemostatic system.1 A simultaneous decline in pro‐ and anticoagulant drivers results in a “rebalanced” haemostatic system.2, 3, 4 External factors may tip the balance towards hypo‐ or hypercoagulability,5 and distinct hypo‐6, 7, 8, 9, 10 and hypercoagulable11, 12, 13, 14 features in patients with liver disease which may predispose them to bleeding or thrombotic complications.
Bleeding and thrombosis are not uncommon in patients with liver disease, in particular in those with advancing and decompensated illness.15 However, little evidence‐based treatment strategies for prevention or treatment of bleeding or thrombosis are available. Current expert recommendations propose a very restrictive prophylactic prohaemostatic management, and a more active anticoagulant approach.5, 16, 17 Importantly, anticoagulant therapy in patients with cirrhosis is challenging,18 and new generation anticoagulant drugs have not been extensively studied in patients with cirrhosis, although the clinical use in these patients is increasing.19, 20
Decompensation may tip the haemostatic balance of patients with liver disease towards a bleeding phenotype. A recent study has identified thrombocytopenia (with a platelet count <30 000 μL) and hypofibrinogenemia (<0.6 g/L) as independent risk factors for bleeding in acutely ill patients with cirrhosis.21 It is, however, unknown whether there is a causal link between bleeding risk and these laboratory abnormalities, and studies assessing whether reversal of thrombocytopenia and/or hypofibrinogenemia decrease the bleeding risk in these patients will be required to ascertain this. It is unknown whether acutely ill patients with cirrhosis are at risk for development of venous thrombosis similar to the increased risk in well compensated patients.22 Nevertheless, prophylactic or therapeutic antithrombotic strategies may be required in acutely ill patients with cirrhosis, in the context of venous thrombosis, portal vein thrombosis, and thrombosis of extracorporeal assist devices.15, 23
We have recently studied the haemostatic status of patients with acutely decompensated or acute‐on‐chronic liver failure and found a remarkably preserved haemostatic system.24 The relatively well‐preserved haemostatic balance therefore suggests that a defensive prohaemostatic and a proactive antihaemostatic approach may be warranted in these patients.
Given the major alterations in the haemostatic system of patients with cirrhosis, the efficacy of pro‐ and antihaemostatic strategies may be unpredictable. We have previously demonstrated that in patients with compensated cirrhosis the in vitro anticoagulant effects of some of the commonly used drugs was decreased, whereas the anticoagulant effects of others were increased as compared to anticoagulant effects in healthy individuals.25, 26 Another study has shown a lack of prohaemostatic effect of in vitro addition of fresh frozen plasma to plasma from patients with compensated cirrhosis, despite improvements in plasma levels of coagulation factors.27 Similarly, transfusion of platelets to patients with cirrhosis did increase the platelet count, but did not improve global haemostasis.28
With the aim to provide a more rational approach to pro‐ and antihaemostatic treatment of acutely ill patients with cirrhosis, and to facilitate design of future clinical studies, we tested the in vitro effects of commonly used pro‐ and antihaemostatic strategies in plasma from patients with acutely decompensated cirrhosis and acute‐on‐chronic liver failure.
2. MATERIALS AND METHODS
2.1. Patients
The study was performed at King's College Hospital, a 950‐bed tertiary hospital in London, United Kingdom, between August 2013 and August 2015. The study was approved by NRES Committee London‐Westminster, Study Number 12/LO/1417. Informed consent or assent was obtained from participants or their personal consultees. Details on patient recruitment and blood sampling have been published previously.24 In short, patients were sampled on admission and patients were only excluded when currently using antihaemostatic agents. From the published cohort we studied 30 healthy volunteers, 18 patients with acute decompensation (AD) of cirrhosis and 10 patients with acute‐on‐chronic liver failure (ACLF). Eighteen patients with well compensated cirrhosis were newly recruited in the outpatient clinic, and were not using antihaemostatic drugs at the time of sampling. Acute decompensation of chronic liver disease and ACLF were defined and graded according to number of organ failures in concordance with criteria reported in the CANONIC study.29 From the 10 patients with ACLF, 1 was classified as grade 1 and 9 were grade 3.
2.2. Routine laboratory tests
Haemoglobin, white blood cell count, albumin, creatinine, bilirubin, aspartate transaminase, alanine transaminase and gamma‐glutamyl transpeptidase were measured in the diagnostic laboratory of King's College Hospital for routine clinical care. International normalized ratios, and plasma levels of fibrinogen, antithrombin, factor II, factor VIII and factor X were measured in stored frozen samples on an automated coagulation analyzer (ACL 300 TOP) with reagents and protocols from the manufacturer (Werfen, Breda, The Netherlands).
2.3. In vitro addition of pro‐ and anticoagulants
We added the following agents to plasma samples of each patient and control:
Recombinant factor VIIa (Novo Nordisk, Bagsvaerd, Denmark)‐final concentration 50 nmol/L
Cofact (a 4‐factor prothrombin complex concentrate (PCC), Sanquin, Amsterdam, Netherlands)‐final concentration 0.5 U/mL
Pooled normal plasma (to mimick fresh frozen plasma [FFP] transfusion‐obtained by combining plasma from >200 healthy volunteers, a generous gift from Dr. J.C. Meijers, Academic Medical Center Amsterdam, the Netherlands)‐final concentration 20% (v/v)
Fibrinogen concentrate (CSL Behring, Marburg, Germany)‐final concentration 1 g/L
Rivaroxaban, a direct factor Xa inhibtor (Alsachim, Illkirch Graffenstaden, France)‐final concentration 25 ng/mL
Dabigatran, a direct thrombin inhibitor (Alsachim, Illkirch Graffenstaden, France)‐final concentration 300 ng/mL
The low molecular weight heparin (LMWH) Clexane (Sanofi‐Aventis BV, Gouda, the Netherlands)‐final concentration 0.2 U/mL
Plasma levels of procoagulant drugs were chosen to represent clinically relevant (peak) levels observed in clinical use in the general population. The final concentrations of the anticoagulant drugs were also chosen to represent clinically relevant plasmas levels and were identical to levels used in previously published experiments.25 Importantly, drug concentrations which gave appreciable (but not maximal) inhibition of thrombin generation in pooled normal plasma were selected so it would be possible to detect both increased and decreased drug effects in patients compared to controls.
2.4. Thrombin generation
The thrombin generation test was performed using platelet‐poor plasma with the fluorimetric method described by Hemker,30 Calibrated Automated Thrombography® in absence or presence of the above‐mentioned agents, except for fibrinogen concentrate. Coagulation was activated using commercially available reagents containing recombinant tissue factor (final concentration 5 pmol/L), phospholipids (final concentration 4 μmol/L), in the presence of soluble thrombomodulin (TM, the concentration of which is not revealed by the manufacturer). These reagents were purchased from Thrombinoscope BV, Maastricht, the Netherlands. Thrombin Calibrator (Thrombinoscope BV) was added to calibrate the thrombin generation curves. A fluorogenic substrate with CaCl2 (FluCa‐kit, Thrombinoscope BV, Maastricht, the Netherlands) was dispensed in each well to allow a continuous registration of thrombin generation. Fluorescence was read in time by a fluorometer, Fluoroskan Ascent® (ThermoFisher Scientific, Helsinki, Finland). All procedures were undertaken according to the protocol suggested by Thrombinoscope B.V.
The pro‐ or anticoagulant potency of the different agents was expressed as the percentual change in endogenous thrombin potential (ETP), lag time, peak or velocity index after addition of the study agent. These percentages were compared between patients and controls.
2.5. Fibrin concentration and fibrin permeability
Fibrinogen levels in plasma of patients and healthy volunteers were determined on an ACL TOP 300 analyzer using reagents from Instrumentation Laboratory (Breda, the Netherlands) according to the manufacturer's instructions.
The average pore size of the fibrin clot (expressed as the Darcy constant, Ks) was determined in permeation studies as previously described.11 In short, plasma samples (100 μL) were incubated with 10 μL of activation buffer (final concentration of 1 IU/mL thrombin, 20 mmol/L CaCl2, in tris‐buffered saline, pH 7.5) to generate clots. After mixing, 100 μL was immediately transferred to a 4.5‐cm plastic tip with a roughened interior surface, which was cut off from a 1 mL serological pipette (Corning Costar Stripette; Sigma‐Aldrich, St Louis, MO, USA), and left for 2 hours in a moist chamber at room temperature to consolidate. The plastic tip was then connected through a flexible silicon tube to a syringe containing TBS with a 4‐cm pressure drop. After a washout period of 90 minutes with tris‐buffered saline, preweighed tubes were attached to the clotting tip, and tris‐buffered saline drops passing through the clot were collected every 30 minutes for 2 hours. The total volume of liquid passing through the clot was weighed after collection. Permeation of tris‐buffered saline through the clot was quantified according to the flow rate and the following equation, Ks = (Q × L × η)/T × A × P, where Ks = Darcy's constant, Q = volume of liquid (mL), L = clot length (cm), η = viscosity (poise), T = time (s), A = cross‐sectional area of the clot (cm2), and P = pressure drop (dyne/cm).
2.6. Statistical analyses
Data are expressed as means (with standard deviations (SDs)), medians (with interquartile ranges), or numbers (with percentages) as appropriate. Multiple groups were compared using One‐way ANOVA (with the Tukey's post‐test) or Kruskal‐Wallis H test (with Dunn's post‐test) as appropriate. P values of .05 or less were considered statistically significant. GraphPad Prism (San Diego, USA) and GraphPad Instat (San Diego, USA) were used for analyses.
3. RESULTS
We studied the effects of ex vivo addition of commonly used pro‐ and anticoagulant drugs in patients with compensated and decompensated cirrhosis, and in patients with acute‐on‐chronic liver failure and compared results with those obtained in healthy controls. Table 1 summarises baseline characteristics of patients and controls.
Table 1.
Patient characteristics
| Controls (n = 30) | Compensated (n = 18) | AD (n = 18) | ACLF (n = 10) | |
|---|---|---|---|---|
| Age | 37 ± 7 | 60 ± 13 | 54 ± 14 | 56 ± 10 |
| Gender (male, %) | 15 (50%) | 12 (67%) | 5 (50%) | 13 (72%) |
| CLIF‐SOFA | n/a | n/a | 4 ± 2 | 10 ± 4 |
| MELD | n/a | 9 ± 2 | 16 ± 9 | 31 ± 9 |
| Reason for decompensation | n/a | n/a | Ascites n = 4Variceal bleeding n = 4Encephalopathy n = 10 | Sepsis n = 4Variceal bleeding n = 4SBP n = 2 |
| Haemoglobin (g/L) | n/d | 145 (113‐145) | 111 (93‐128) | 95 (77‐116) |
| WBC (× 109/L) | n/d | 5 (4‐6) | 6 (3‐9) | 10 (8‐19) |
| Platelets (× 109/L) | n/d | 119 (87‐150) | 95 (65‐132) | 89 (65‐111) |
| Albumin (g/L) | n/d | 41 (39‐45) | 33 (30‐37) | 28 (26‐33) |
| Creatinin (μmol/L) | n/d | 82 (70‐89) | 67 (56‐93) | 135 (94‐224) |
| Bilirubin (μmol/L) | n/d | 19 (11‐25) | 44 (30‐71) | 362 (116‐493) |
| AST (U/L) | n/d | 42 (37‐45) | 47 (38‐56) | 79 (50‐86) |
| ALT (U/L) | n/d | 36 (24‐43) | 25 (18‐30) | 46 (24‐61) |
| GGT (U/L) | n/d | 60 (33‐143) | 77 (47‐135) | 37 (30‐91) |
| INR | 1.0 ± 0.1 | 1.1 ± 0.1 | 1.6 ± 0.4**,*** | 2.1 ± 0.4**,**** |
| Fibrinogen (g/L) | 2.8 ± 0.4 | 3.3 ± 0.7 | 2.2 ± 0.9*,*** | 1.3 ± 0.3**,**** |
| Antithrombin (%) | 107 ± 11 | 88 ± 26* | 46 ± 18**,*** | 28 ± 14**,**** |
| FII (%) | 101 ± 18 | 78 ± 17** | 48 ± 16**,*** | 31 ± 10** |
| FVIII (%) | 99 ± 36 | 157 ± 42** | 146 ± 27* | 212 ± 93**,**** |
| FX (%) | 105 ± 23 | 85 ± 21* | 57 ± 20**,*** | 41 ± 15** |
ALT, alanine transaminase; AST, aspartate transaminase; CLIF‐SOFA, Chronic Liver Failure‐Sequential Organ Failure Assessment; F, factor; GGT, gamma‐glutamyl transpeptidase; INR, international normalised ratio; MELD, model for end‐stage liver disease; n/a, not applicable; n/d, not determined; SBP, spontaneous bacterial peritonitis; WBC, white blood cell. Shown are means ± standard deviation or medians (interquartile range). *P < .05 vs control, **P < .01 vs control, ***P < .01 vs compensated, ****P < .01 vs AD.
Without the addition of pro‐ or anticoagulant agents, patients generated more thrombin as compared to controls, using TM‐modified thrombin generation tests, which is in line with our previously published data (Table 2).
Table 2.
Thrombin generation parameters of thrombomodulin‐modified thrombin generation testing in controls and patients with compensated (CC) or acutely decompensated cirrhosis (AD), or acute‐on‐chronic liver failure (ACLF)
| ETP (nmol/L IIa × min) | Peak (nmol/L IIa) | Lag time (min) | Vel index (nmol/L IIa × min) | |
|---|---|---|---|---|
| Controls | 440.0 (343.4‐639.8) | 122.8 (93.9‐180.3) | 1.67 (1.46‐1.98) | 61.9 (46.3‐96.5) |
| CC | 699.5 (538.5‐819.3) | 184.5 (152.0‐211.3) | 1.67 (1.33‐1.67) | 99.0 (75.8‐111.0) |
| AD | 736.3 (696.2‐882.3)** | 182.3 (173.2‐215.6)** | 1.33 (1.33‐1.63)* | 102.4 (90.3‐112.1)** |
| ACLF | 648.1 (473.3‐743.7) | 124.5 (86.5‐144.6) | 1.92 (1.67‐2.00) | 60.3 (43.4‐75.0) |
Shown are medians (interquartile ranges). *P < .05, **P < .01 vs controls.
When recombinant factor VIIa (50 nmol/L) was added to plasma of healthy individuals, an increase of ~20% in ETP, peak, and velocity index was observed, with an ~20% decrease in lag time. In patients, however, addition of recombinant factor VIIa did not appreciably change ETP, peak, and velocity index, whereas the lag time was shortened to a similar extent as in controls (Table 3, Figure 1).
Table 3.
Percentual changes in thrombin generation parameters of thrombomodulin‐modified thrombin generation testing in controls and patients with compensated (CC) or acutely decompensated cirrhosis (AD), or acute‐on‐chronic liver failure (ACLF) after in vitro addition of prohaemostatic agents
| ETP | Peak | Lag time | Vel index | |
|---|---|---|---|---|
| rFVIIa | ||||
| Controls | 21 (8‐32) | 20 (8‐32) | 28 (20‐34) | 20 (11‐42) |
| CC | 3 (0‐16)* | 2 (−3 to 9)** | 21 (20‐30) | 1 (−6 to 20)* |
| AD | 2 (−1 to 5)*** | −1 (−3 to 3)*** | 24 (17‐37) | 0 (−10 to 5)*** |
| ACLF | 3 (−3 to 27)* | −1 (−4 to 19)* | 20 (15‐25)* | 3 (−6 to 25) |
| Cofact | ||||
| Controls | 92 (788‐101) | 72 (46‐89) | 0 (0‐17) | 72 (38‐93) |
| CC | 109 (82‐149) | 89 (55‐102) | 0 (0‐0) | 78 (55‐104) |
| AD | 151 (135‐197)*** | 109 (87‐150)** | 0 (−13‐6) | 97 (75‐136)* |
| ACLF | 273 (212‐393)*** | 176 (141‐219)*** | 10 (0‐17) | 178 (156‐265)*** |
| Pooled normal plasma | ||||
| Controls | 16 (2‐21) | 15 (−3‐19) | 0 (−10‐0) | 15 (−4‐19) |
| CC | 7 (−1‐21) | 7 (0‐18) | 20 (0‐26) | 6 (−4‐18) |
| AD | 19 (8‐29) | 23 (10‐33) | −11 (−25‐0) | 22 (12‐31) |
| ACLF | 38 (27‐69) *** | 56 (47‐124) *** | 0 (−9‐9) | 59 (45‐117) *** |
Shown are the percentual increase of the ETP, peak, or velocity index, and the percentual decrease in the lag time. Data are expressed as median percentages with interquartile range.*P < .05, **P < .01, ***P < .001 vs controls.
Figure 1.
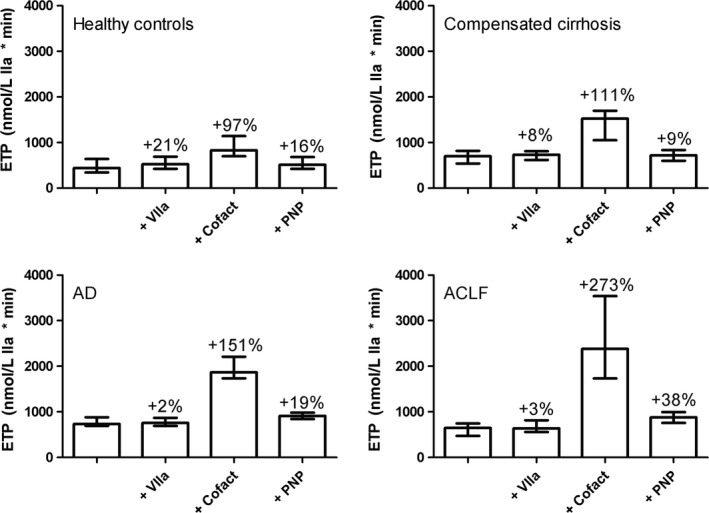
Absolute ETP values from thrombomodulin‐modified thrombin generation testing in controls and patients with compensated or acutely decompensated cirrhosis (AD), or acute‐on‐chronic liver failure (ACLF) prior to and after in vitro addition of prohaemostatic agents. Indicated are percentual changes in the ETP after addition of procoagulant agents. Shown are medians with interquartile ranges . PNP, pooled normal plasma
Addition of the PCC Cofact (0.5 U/mL) to plasma of healthy individuals resulted in an approximate doubling of total thrombin generation (to 829 [697‐1141] nmol/L IIa × min). An exaggerated response was observed in plasma from patients with a 110% increase in the compensated cirrhosis group (to 1525 [1052‐1697] nmol/L IIa × min), a 150% increase in thrombin generation in the AD group (to 1869 [1734‐2210] nmol/L IIa × min) and a 270% increase in the ACLF group (to 2383 [1731‐3538] nmol/L IIa × min). Similarly, the peak and velocity index increased substantially more in patients compared to controls. The lag time did not appreciably change in either controls or patients (Table 3, Figure 1).
Addition of pooled normal plasma (20% v/v) increased total thrombin generation in controls by 16%, with very similar changes in compensated and AD patients (9%‐19% increase). Addition of pooled normal plasma to samples of patients with ACLF led to a more profound increase in thrombin generation (38% increase), but given the lower baseline thrombin generation levels in the ACLF group, total thrombin generation after addition of pooled normal plasma was comparable between AD and ACLF patients (913 [841‐980] vs 883 [757‐993] nmol/L IIa × min) ‐ Table 3, Figure 1.
The permeability of clots generated from plasma from healthy controls was remarkably similar to that of the permeability of clots from patients, despite the lower fibrinogen levels in patient plasma. When fibrinogen concentrate (1 g/L) was added to control samples, a 51% reduction in permeability was observed. A similar effect of fibrinogen concentrate was observed in patients compensated cirrhosis, whereas a slightly more robust effect was observed in plasma from AD and ACLF patients with a 61% reduction in permeability in the AD group and a 63% reduction in the ACLF group (Table 4).
Table 4.
Fibrinogen levels and fibrinogen permeability in controls and patients with compensated (CC) or acutely decompensated cirrhosis (AD), or acute‐on‐chronic liver failure (ACLF) in the absence and presence of fibrinogen concentrate
| Fibrinogen (g/L) | Permeability (Ks) | Permeability + 1 g/L fibrinogen concentrate (Ks) | Percentual decrease in permeability | |
|---|---|---|---|---|
| Controls | 2.8 ± 0.4 | 9.2 × 10−9 ± 3.2 × 10−9 | 4.2 × 10−9 ± 1.0 × 10−9 | 51 ± 11 |
| CC | 3.3 ± 0.7 | 9.0 × 10−9 ± 4.0 × 10−9 | 4.1 × 10−9 ± 2.0 × 10−9 | 52 ± 11 |
| AD | 2.2 ± 0.9* | 8.8 × 10−9 ± 3.6 × 10−9 | 3.7 × 10−9 ± 1.1 × 10−9 | 61 ± 15 * |
| ACLF | 1.3 ± 0.3** | 10.9 × 10−9 ± 5.1 × 10−9 | 4.0 × 10−9 ± 1.8 × 10−9 | 63 ± 17* |
Data are expressed as means ± standard deviation. *P < .05, **P < .001 vs controls.
Addition of rivaroxaban (25 ng/mL) to plasma from healthy controls resulted in a 55% decrease in the ETP (to 178 [152‐339] nmol/L IIa × min), with similar changes in peak and velocity index. The reduction in total thrombin generation was less profound in patients, particularly in the AD group in which the ETP was only reduced by 11% (to 629 [527‐692] nmol/L IIa × min) with a 25% reduction in the ACLF group (to 465 [367‐570] nmol/L IIa × min), and a 38% reduction in the compensated group (to 403 [271‐510] nmol/L IIa × min). Although the reduction in thrombin generation was substantially decreased in patients, the lag time was substantially more prolonged in patients compared to controls, particularly in the ACLF group (Table 5, Figure 2).
Table 5.
Percentual decrease in thrombin generation parameters of thrombomodulin‐modified thrombin generation testing in controls and patients with compensated (CC) or acutely decompensated cirrhosis (AD), or acute‐on‐chronic liver failure (ACLF) after addition of anticoagulants
| ETP | Peak | Lag time | Vel index | |
|---|---|---|---|---|
| Rivaroxaban | ||||
| Controls | 55 (50‐63) | 62 (56‐69) | 34 (25‐46) | 70 (62‐75) |
| CC | 42 (32‐50)* | 53 (44‐57)* | 60 (50‐100)*** | 60 (44‐68)* |
| AD | 11 (10‐30)*** | 19 (18‐35)*** | 52 (41‐63)* | 33 (27‐47)*** |
| ACLF | 25 (11‐35)** | 43 (27‐52)** | 70 (56‐87)*** | 54 (43‐64)* |
| Dabigatran | ||||
| Controls | 33 (16‐48) | 33 (20‐65) | 571 (314‐711) | 30 (5‐76) |
| CC | 50 (35‐63) | 56 (41‐73) | 455 (365‐617) | 53 (29‐75) |
| AD | 75 (93‐73)*** | 92 (84‐98)*** | 483 (298‐614) | 95 (87‐99)** |
| ACLF | No thrombin formed*** | No thrombin formed*** | No thrombin formed*** | No thrombin formed*** |
| LMWH | ||||
| Controls | 18 (9‐28) | 15 (6‐26) | 0 (0‐10) | 14 (−2‐32) |
| CC | 25 (16‐30) | 15 (5‐27) | 26 (17‐54) | 8 (−4‐20) |
| AD | 41 (26‐48)** | 26 (13‐30) | 17 (7‐29)** | 16 (1‐29) |
| ACLF | 54 (39‐61)*** | 29 (24‐42)** | 9 (0‐28) | 25 (14‐41) |
Shown are the percentual decrease of the ETP, peak, or velocity index, and the percentual increase in the lag time. Data are expressed as median percentages with interquartile range.*P < .05, **P < .01, ***P < .001 vs controls.
Figure 2.
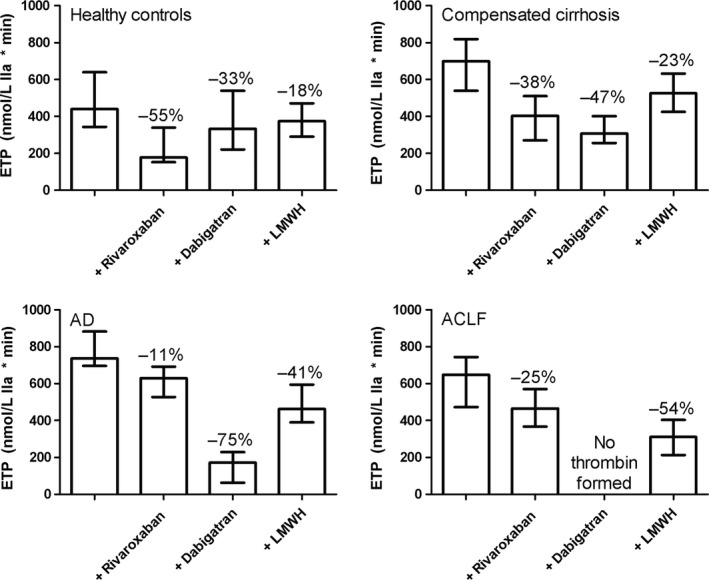
Absolute ETP values from thrombomodulin‐modified thrombin generation testing in controls and patients with compensated or acutely decompensated cirrhosis (AD), or acute‐on‐chronic liver failure (ACLF) prior to and following addition of anticoagulant agents. Indicated are percentual changes in the ETP after addition of anticoagulant agents. Shown are medians with interquartile ranges
Addition of dabigatran (300 ng/mL) decreased total thrombin generation in controls by 33% (to 333 [221‐539] nmol/L IIa × min), and prolonged the lag time more than 6‐fold. In patients, total thrombin generation was decreased by 47% in the compensated group (to 308 [256‐401] nmol/L IIa × min), by 75% in the AD group (to 171 [63‐229] nmol/L IIa × min), and none of the patients in the ACLF group generated any thrombin in the presence of dabigatran (Table 5, Figure 2).
LMWH (0.2 U/mL) decreased the ETP by 18% in controls (to 374 [291‐471] nmol/L IIa × min), with similar changes in peak and velocity index. The decrease in the ETP was more pronounced in patients with a 23% decrease in the compensated group (to 526 [425‐632]), a 41% decrease in the AD group (to 463 [390‐594] nmol/L IIa × min) and a 54% decrease (to 311 [213‐403] nmol/L IIa × min) in the ACLF group (Table 5, Figure 2). However, given the higher baseline ETP in patients, the ETP in the presence of LMWH were similar between patients and controls.
4. DISCUSSION
Here we have studied the in vitro effects of commonly used pro‐ and anticoagulant strategies in compensated and acutely ill patients with cirrhosis in comparison to healthy individuals and found remarkable differences in the potency of these strategies between patients and controls. Notably, the in vitro efficacy of the procoagulant PCC and the anticoagulants dabigatran and LMWH increased with increasing severity of disease, and the efficacy of rivaroxaban was lower in patients although not proportional to the severity of disease. The increase in efficacy of PCC, dabigatran, and LMWH with increasing severity of disease mirror the haemostatic changes that are more profound in the sicker patients. The results of this study may have clinical relevance as it suggests a requirement for dose‐adjustments for a number of agents tested, and suggests some agents to be clinically ineffective in these patients. Specifically, the lack of a procoagulant effect of recombinant factor VIIa in patients, and the minimal effect of FFP in the AD group are in line with the lack of evidence supporting the use of recombinant factor VIIa in patients with liver diseases,31, 32, 33 and with the doubts on the use of FFP in patients with liver disease in general.34 Our data suggest a procoagulant approach with PCCs and or fibrinogen concentrate to substantially improve haemostatic status in acutely ill patients with liver disease given the profound procoagulant effects of PCC in thrombin generation tests and the improvement of fibrin clot structure by fibrinogen concentrate. This strategy has been reported to be effective in managing bleeding during liver transplantation.35 However, based on our data, dosing of PCCs should be performed conservatively in acutely ill patients with cirrhosis given the profoundly exaggerated procoagulant response in patients compared to controls. Similarly, our data suggest that substantial dose‐adjustments may be required when low molecular weight heparin or direct oral anticoagulants are considered for prophylaxis or treatment of thrombotic complications in acutely ill patients with cirrhosis.
The lack of a procoagulant effect of recombinant factor VIIa in patients with cirrhosis suggests competent tissue factor‐mediated activation of coagulation in these patients, and is also in line with a very modest increase in thrombin generation following administration of recombinant factor VIIa to healthy volunteers.36 However, as recombinant factor VIIa also has important tissue factor‐independent procoagulant activity,37 it cannot be presumed that this drug does not have some prohaemostatic effect in acutely ill patients with cirrhosis in vivo. It has been previously demonstrated that FFP has no appreciable effects on in vitro thrombin generation in plasma from patients with compensated cirrhosis, despite clear changes in plasma levels of coagulation factors and a decrease in the INR.27 Here we demonstrate comparable effects of pooled normal plasma (mimicking FFP transfusion) in plasma from healthy individuals and patients with acutely decompensated cirrhosis, suggesting FFP to be ineffective in AD patients. The slight increase in thrombin generation in our control group is likely clinically insignificant and may reflect minor differences in coagulation profiles between our healthy individuals (from London, UK) and our pooled normal plasma (which was generated in Amsterdam, the Netherlands). Our data suggests FFP does have some haemostatic effect in patients with ACLF. However, the drawback of FFP administration in these acutely ill patients is the risk of volume overload and exacerbation of portal hypertension.
The substantially increased procoagulant capacity of PCCs in plasma from acutely ill patients with cirrhosis likely relates to the low baseline plasma levels of procoagulant proteins. The same dose of PCC thus results in a much larger relative increase in levels of the vitamin K‐dependent factors in patients compared to controls. Baseline thrombin generating capacity is elevated in patients compared to controls, despite the much lower levels of procoagulants, as the levels of anticoagulant proteins are also low. Therefore, the procoagulant effects of PCCs are much more prominent in patients compared to controls. The preserved fibrin clot structure in patients is in line with our previous results showing preserved fibrin clot structure in patients with compensated cirrhosis to be related to oxidative modifications to the fibrinogen molecule.11 Fibrinogen concentrate profoundly decreases fibrin clot permeability in acutely ill patients with cirrhosis, which is in line with our previous study showing that fibrinogen concentrate normalizes fibrin clot permeability in samples taken from patients during liver transplantation.38
The increased anticoagulant potency of LMWH and profoundly increased anticoagulant potency of dabigatran are in line with previous studies in patients with compensated cirrhosis,25, 39 and suggest a requirement for conservative dosing particularly of dabigatran in acutely ill patients with cirrhosis. Importantly, although it remains to be established whether dose‐adjustments and/or monitoring of drug levels improves the anticoagulant management of acutely ill patients with cirrhosis, monitoring of LMWH by anti‐Xa testing is unreliable in patients with cirrhosis.40 Thrombin generation tests are currently not ready for clinical use, but the development of whole blood generation tests may result in a point‐of‐care thrombin generation test which may be suitable for anticoagulant monitoring,41 which might be used to monitor anticoagulant treatment in these difficult patients. Rivaroxaban appears less effective in patients compared to controls which is in line with studies in patients with compensated cirrhosis,25 and dose increases may be required. However, importantly, although rivaroxaban is less effective in patients as assessed by total thrombin generation, it appears more effective compared to controls in prolonging the lag time of thrombin generation which was also observed in our previous study in compensated cirrhosis.25
Taken together, our results suggest a profound difference in clinical efficacy of some of the commonly used pro‐ and anticoagulant strategies in compensated and acutely ill patients with cirrhosis. Dose adjustments of some of these agents may be required, either to improve efficacy or to decrease the risk of side effects. Although our studies have been performed using in vitro, plasma‐based systems, which do not take the role of blood cells in haemostasis into account, these results may still be clinically relevant. We propose that our study is the starting point for future preclinical and clinical studies that will further explore the need for alternative dosing of pro‐ and anticoagulants in patients with cirrhosis. Our data may assist in development of urgently needed clinical studies to assess safety and efficacy of strategies to prevent or treat bleeding and thrombotic complications in these complex patients.
CONFLICT OF INTEREST
The authors do not have any disclosures to report.
Lisman T, Kleiss S, Patel VC, et al. In vitro efficacy of pro‐ and anticoagulant strategies in compensated and acutely ill patients with cirrhosis. Liver Int. 2018;38:1988–1996. 10.1111/liv.13882
Funding information
This study was funded in part by the Tekke Huizinga Foundation (The Netherlands).
Handling Editor: Juan Abraldes
REFERENCES
- 1. Lisman T, Leebeek FW, de Groot PG. Haemostatic abnormalities in patients with liver disease. J Hepatol. 2002;37:280‐287. [DOI] [PubMed] [Google Scholar]
- 2. Lisman T, Porte RJ. Rebalanced hemostasis in patients with liver disease: evidence and clinical consequences. Blood. 2010;116:878‐885. [DOI] [PubMed] [Google Scholar]
- 3. Lisman T, Stravitz RT. Rebalanced hemostasis in patients with acute liver failure. Semin Thromb Hemost. 2015;41:468‐473. [DOI] [PubMed] [Google Scholar]
- 4. Tripodi A, Mannucci PM. The coagulopathy of chronic liver disease. N Engl J Med. 2011;365:147‐156. [DOI] [PubMed] [Google Scholar]
- 5. Lisman T, Caldwell SH, Burroughs AK, et al. Hemostasis and thrombosis in patients with liver disease: the ups and downs. J Hepatol. 2010;53:362‐371. [DOI] [PubMed] [Google Scholar]
- 6. Bedreli S, Sowa JP, Malek S, et al. Rotational thromboelastometry can detect factor XIII deficiency and bleeding diathesis in patients with cirrhosis. Liver Int. 2017;37:562‐569. [DOI] [PubMed] [Google Scholar]
- 7. Kleinegris MC, Bos MH, Roest M, et al. Cirrhosis patients have a coagulopathy that is associated with decreased clot formation capacity. J Thromb Haemost. 2014;12:1647‐1657. [DOI] [PubMed] [Google Scholar]
- 8. Narvaiza MJ, Fernandez J, Cuesta B, Paramo JA, Rocha E. Role of sialic acid in acquired dysfibrinogenemia associated with liver cirrhosis. Ric Clin Lab. 1986;16:563‐568. [DOI] [PubMed] [Google Scholar]
- 9. Leebeek FW, Kluft C, Knot EA, de Maat MP, Wilson JH. A shift in balance between profibrinolytic and antifibrinolytic factors causes enhanced fibrinolysis in cirrhosis. Gastroenterology. 1991;101:1382‐1390. [DOI] [PubMed] [Google Scholar]
- 10. Laffi G, Marra F, Gresele P, et al. Evidence for a storage pool defect in platelets from cirrhotic patients with defective aggregation. Gastroenterology. 1992;103:641‐646. [DOI] [PubMed] [Google Scholar]
- 11. Hugenholtz GC, Mccrae FL, Adelmeijer J, et al. Procoagulant changes in fibrin clot structure in patients with cirrhosis are associated with oxidative modifications of fibrinogen. J Thromb Haemost. 2016;15:1054‐1066. [DOI] [PubMed] [Google Scholar]
- 12. Raparelli V, Basili S, Carnevale R, et al. Low‐grade endotoxemia and platelet activation in cirrhosis. Hepatology. 2017;65:571‐581. [DOI] [PubMed] [Google Scholar]
- 13. Saliola M, Lorenzet R, Ferro D, et al. Enhanced expression of monocyte tissue factor in patients with liver cirrhosis. Gut. 1998;43:428‐432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lisman T, Bongers TN, Adelmeijer J, et al. Elevated levels of von willebrand factor in cirrhosis support platelet adhesion despite reduced functional capacity. Hepatology. 2006;44:53‐61. [DOI] [PubMed] [Google Scholar]
- 15. Roberts LN, Bernal W. Management of bleeding and thrombosis in critically ill patients with liver disease. Semin Thromb Hemost. 2015;41:520‐526. [DOI] [PubMed] [Google Scholar]
- 16. Saner FH, Kirchner C. Monitoring and treatment of coagulation disorders in end‐stage liver disease. Visc Med. 2016;32:241‐248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Northup PG, Caldwell SH. Coagulation in liver disease: a guide for the clinician. Clin Gastroenterol Hepatol. 2013;11:1064‐1074. [DOI] [PubMed] [Google Scholar]
- 18. Lisman T, Kamphuisen PW, Northup PG, Porte RJ. Established and new‐generation antithrombotic drugs in patients with cirrhosis ‐ possibilities and caveats. J Hepatol. 2013;59:358‐366. [DOI] [PubMed] [Google Scholar]
- 19. De Gottardi A, Trebicka J, Klinger C, et al. Antithrombotic treatment with direct‐acting oral anticoagulants in patients with splanchnic vein thrombosis and cirrhosis. Liver Int. 2017;37:694‐699. [DOI] [PubMed] [Google Scholar]
- 20. Intagliata NM, Henry ZH, Maitland H, et al. Direct oral anticoagulants in cirrhosis patients pose similar risks of bleeding when compared to traditional anticoagulation. Dig Dis Sci. 2016;61:1721‐1727. [DOI] [PubMed] [Google Scholar]
- 21. Drolz A, Horvatits T, Roedl K, et al. Coagulation parameters and major bleeding in critically ill patients with cirrhosis. Hepatology. 2016;64:556‐568. [DOI] [PubMed] [Google Scholar]
- 22. Ambrosino P, Tarantino L, Di Minno G, et al. The risk of venous thromboembolism in patients with cirrhosis. A systematic review and meta‐analysis. Thromb Haemost. 2017;117:139‐148. [DOI] [PubMed] [Google Scholar]
- 23. Northup PG, Sundaram V, Fallon MB, et al. Hypercoagulation and thrombophilia in liver disease. J Thromb Haemost. 2008;6:2‐9. [DOI] [PubMed] [Google Scholar]
- 24. Fisher C, Patel VC, Stoy SH, et al. Balanced haemostasis with both hypo‐ and hyper‐coagulable features in critically ill patients with acute‐on‐chronic‐liver failure. J Crit Care. 2017;43:54‐60. [DOI] [PubMed] [Google Scholar]
- 25. Potze W, Arshad F, Adelmeijer J, et al. Differential in vitro inhibition of thrombin generation by anticoagulant drugs in plasma from patients with cirrhosis. PLoS ONE. 2014;9:e88390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Potze W, Adelmeijer J, Lisman T. Decreased in vitro anticoagulant potency of rivaroxaban and apixaban in plasma from patients with cirrhosis. Hepatology. 2015;61:1435‐1436. [DOI] [PubMed] [Google Scholar]
- 27. Tripodi A, Chantarangkul V, Primignani M, et al. Thrombin generation in plasma from patients with cirrhosis supplemented with normal plasma: considerations on the efficacy of treatment with fresh‐frozen plasma. Intern Emerg Med. 2012;7:139‐144. [DOI] [PubMed] [Google Scholar]
- 28. Tripodi A, Primignani M, Chantarangkul V, et al. Global hemostasis tests in patients with cirrhosis before and after prophylactic platelet transfusion. Liver Int. 2013;33:362‐367. [DOI] [PubMed] [Google Scholar]
- 29. Moreau R, Jalan R, Gines P, et al. Acute‐on‐chronic liver failure is a distinct syndrome that develops in patients with acute decompensation of cirrhosis. Gastroenterology. 2013;144:1426‐1437.e1‐9. [DOI] [PubMed] [Google Scholar]
- 30. Hemker HC, Giesen P, Al Dieri R, et al. Calibrated automated thrombin generation measurement in clotting plasma. Pathophysiol Haemost Thromb. 2003;33:4‐15. [DOI] [PubMed] [Google Scholar]
- 31. Lodge JP, Jonas S, Jones RM, et al. Efficacy and safety of repeated perioperative doses of recombinant factor VIIa in liver transplantation. Liver Transpl. 2005;11:973‐979. [DOI] [PubMed] [Google Scholar]
- 32. Planinsic RM, van der Meer J, Testa G, et al. Safety and efficacy of a single bolus administration of recombinant factor VIIa in liver transplantation due to chronic liver disease. Liver Transpl. 2005;11:895‐900. [DOI] [PubMed] [Google Scholar]
- 33. Bosch J, Thabut D, Albillos A, et al. Recombinant factor VIIa for variceal bleeding in patients with advanced cirrhosis: a randomized, controlled trial. Hepatology. 2008;47:1604‐1614. [DOI] [PubMed] [Google Scholar]
- 34. Shah NL, Intagliata NM, Northup PG, Argo CK, Caldwell SH. Procoagulant therapeutics in liver disease: a critique and clinical rationale. Nat Rev Gastroenterol Hepatol. 2014;11:675‐682. [DOI] [PubMed] [Google Scholar]
- 35. Kirchner C, Dirkmann D, Treckmann JW, et al. Coagulation management with factor concentrates in liver transplantation: a single‐center experience. Transfusion. 2014;54:2760‐2768. [DOI] [PubMed] [Google Scholar]
- 36. Bijsterveld NR, Moons AH, Boekholdt SM, et al. Ability of recombinant factor VIIa to reverse the anticoagulant effect of the pentasaccharide fondaparinux in healthy volunteers. Circulation. 2002;106:2550‐2554. [DOI] [PubMed] [Google Scholar]
- 37. Lisman T, de Groot PG. The role of cell surfaces and cellular receptors in the mode of action of recombinant factor VIIa. Blood Rev. 2015;29:223‐229. [DOI] [PubMed] [Google Scholar]
- 38. Groeneveld DJ, Adelmeijer J, Hugenholtz GC, Ariens RA, Porte RJ, Lisman T. Ex vivo addition of fibrinogen concentrate improves fibrin network structure in plasma samples taken during liver transplantation. J Thromb Haemost. 2015;13:2192‐2201. [DOI] [PubMed] [Google Scholar]
- 39. Senzolo M, Rodriguez‐Castro KI, Rossetto V, et al. Increased anticoagulant response to low‐molecular‐weight heparin in plasma from patients with advanced cirrhosis. J Thromb Haemost. 2012;10:1823‐1829. [DOI] [PubMed] [Google Scholar]
- 40. Potze W, Arshad F, Adelmeijer J, et al. Routine coagulation assays underestimate levels of antithrombin‐dependent drugs but not of direct anticoagulant drugs in plasma from patients with cirrhosis. Br J Haematol. 2013;163:666‐673. [DOI] [PubMed] [Google Scholar]
- 41. Ninivaggi M, Apitz‐Castro R, Dargaud Y, de Laat B, Hemker HC, Lindhout T. Whole‐blood thrombin generation monitored with a calibrated automated thrombogram‐based assay. Clin Chem. 2012;58:1252‐1259. [DOI] [PubMed] [Google Scholar]