

Abstract
Objective
No evidence of progression or active disease (NEPAD) is a novel combined endpoint defined by the absence of both progression and inflammatory disease activity in primary progressive multiple sclerosis (PPMS). In the placebo‐controlled phase III ORATORIO study (NCT01194570), we investigated the effect of ocrelizumab on this comprehensive outcome and its components in a post‐hoc analysis.
Methods
The proportion of patients with NEPAD (no evidence of progression [NEP; no 12‐week confirmed progression of ≥1/≥0.5 points on the Expanded Disability Status Scale if the baseline score was ≤5.5/>5.5 points, respectively; no 12‐week confirmed progression of ≥20% on the Timed 25‐Foot Walk test and 9‐Hole Peg Test], no brain magnetic resonance imaging activity [no new/enlarging T2 lesions and no T1 gadolinium‐enhancing lesions], and no protocol‐defined relapse) from baseline to week 120 was determined in ocrelizumab‐ (600 mg; n = 465) and placebo‐treated (n = 234) patients.
Results
The majority of ORATORIO study patients with PPMS experienced clinical progression or evidence of disease activity. From baseline to week 120, 29.9% and 42.7% ocrelizumab‐treated compared to 9.4% and 29.1% placebo‐treated patients maintained NEPAD (relative risk [95% confidence interval {CI}], 3.15 [2.07–4.79]; p < 0.001) and NEP (relative risk [95% CI], 1.47 [1.17–1.84]; p < 0.001), respectively. Effects on the individual components of both measures were consistent with the compound outcomes.
Interpretation
Compared to placebo, ocrelizumab enhanced 3‐fold the proportion of PPMS patients with no evidence of either progression or inflammatory disease activity. NEPAD may represent a sensitive and meaningful comprehensive measure of disease control in patients with PPMS. Ann Neurol 2018;84:527–536
Primary progressive multiple sclerosis (PPMS) is characterized by insidiously increasing neurological disability from disease onset, with or without temporary plateaus in progression and with or without superimposed relapses.1 Preventing progression is an essential treatment goal in PPMS, a goal that drugs tested in previous clinical studies failed to achieve.2, 3, 4 Ascertaining the absence of progression in both clinical trials and clinical practice requires reliable and comprehensive measures of disease progression. Although widely used,5, 6 the Expanded Disability Status Scale (EDSS) has limited ability to reliably capture change in functioning in a range of domains, notably ambulation speed and hand/arm function, which are major contributors to the overall disability burden in patients with progressive multiple sclerosis (MS). No evidence of progression (NEP) is an endpoint that measures absence of disability progression beyond the limits of EDSS as a standalone, including upper limb function and ambulation speed. Maintaining NEP status (Fig 1) reflects stable clinically apparent disease with no worsening in three major measures of MS disability: EDSS, ambulation assessed by the Timed 25‐Foot Walk (T25FW) test, and upper limb function assessed by the 9‐Hole Peg Test (9HPT), although clinical and magnetic resonance imaging (MRI) measures of acute disease activity are not captured.
Figure 1.
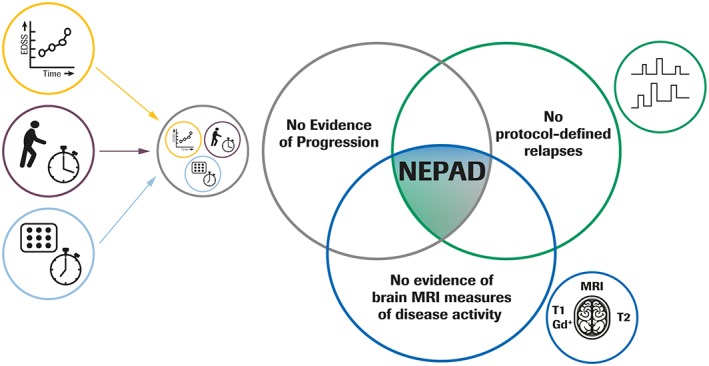
Clinical and subclinical components of NEPAD. No Evidence of Progression defined as: no 12‐week CDP, as measured by the EDSS; no evidence of 12‐week confirmed ≥ 20% progression on hand/arm function as measured by 9HPT; no evidence of 12‐week confirmed ≥ 20% progression on ambulation as measured by the T25FW test. 9HPT = 9‐Hole Peg Test; CDP = confirmed disability progression; EDSS = Expanded Disability Status Scale; Gd+ = gadolinium‐enhancing; MRI = magnetic resonance imaging; NEPAD = no evidence of progression or active disease; T25FW = Timed 25‐Foot Walk.
In PPMS, signs of acute disease activity, as defined by relapses, T1 gadolinium (Gd)‐enhancing lesions, and new or enlarging T2 hyperintense lesions on brain MRI, are present but less frequent than in relapse‐onset MS.7 With “no evidence of progression or active disease” (NEPAD), we propose a novel endpoint that assesses the combined absence of three measures of disability progression (NEP) and clinical and MRI measures of active disease (Fig 1). NEPAD may represent a more sensitive and comprehensive measure of disease control in patients with PPMS.
Ocrelizumab is a recombinant, humanized monoclonal antibody that selectively depletes CD20‐expressing B cells,8, 9 B lymphocytes being a significant contributor to the pathogenesis of MS.10, 11 The randomized, double‐blind, placebo‐controlled phase III trial, ORATORIO, evaluated the efficacy and safety of ocrelizumab in patients with PPMS and demonstrated superior efficacy of ocrelizumab on the primary endpoint, time‐to‐onset of 12‐week confirmed disability progression (CDP), as measured by the EDSS.12 Based on such results, ocrelizumab is the first disease‐modifying therapy approved for PPMS.13, 14
The objective of the current analyses was to assess the effect of ocrelizumab on NEP and NEPAD and their components, two novel, more comprehensive and sensitive outcome measures for patients with PPMS in the ORATORIO trial.
Patients and Methods
Trial Design and Patients
ORATORIO was a phase III, randomized, placebo‐controlled, double‐blind, multicenter trial (NCT01194570); details have been reported previously.12 Key eligibility criteria included age of 18 to 55 years, diagnosis of PPMS (2005 revised McDonald criteria),15 and an EDSS score of 3.0 to 6.5 at screening. All patients were required to have a documented history, or the presence at screening, of an elevated immunoglobulin G (IgG) index or at least one IgG oligoclonal band detected in the cerebrospinal fluid, and no history of previous clinical relapse. Patients (n = 732) were randomized (2:1) to receive either ocrelizumab 600mg by intravenous (IV) infusion every 24 weeks or placebo every 24 weeks for ≥ 120 weeks.12 Randomization was stratified by region (USA versus rest of world [ROW]) and age (≤45 versus > 45 years).
Clinical and MRI Endpoints
Prespecified Outcome Measures
CDP was defined as a ≥1‐point increase in EDSS score from a baseline EDSS score ≤5.5, or a 0.5‐point increase in EDSS score from a baseline EDSS score >5.5, sustained for at least 12 weeks.12 Protocol‐defined relapses were new or worsening neurological symptoms attributable to MS that lasted longer than 24 hours; were preceded by neurological stability for at least 30 days; and were accompanied by worsening on examination reflected as ≥0.5‐step increase from previous EDSS score or ≥2‐point increase in one qualifying Functional Systems Score (FSS), or 1‐point increase in two or more qualifying FSSs.12 Brain MRI was performed at baseline and weeks 24, 48, and 120; new or enlarging T2 lesions and/or T1 Gd‐enhancing lesions on any postbaseline scan were considered evidence of MRI disease activity.
NEP and NEPAD
NEP status was defined as the absence of 12‐week confirmed clinical progression, as measured by: no 12‐week CDP on EDSS, no 12‐week confirmed ≥ 20% progression on hand/arm function as measured by the 9HPT, and no 12‐week confirmed ≥ 20% progression on ambulation as measured by the T25FW test. The 20% cutoff for progression on the T25FW test and the 9HPT has previously been shown to be a clinically meaningful magnitude of disease progression.16 NEPAD was defined as: NEP, no brain MRI‐measured disease activity (no new or enlarging T2 lesions and no T1 Gd‐enhancing lesions), and no protocol‐defined relapses (NEP and NEPAD summarized in Fig 1). The schedule of MRI scans and the assessment of disability measures, including EDSS score, T25FW, and 9HPT, are shown in Figure 2.
Figure 2.
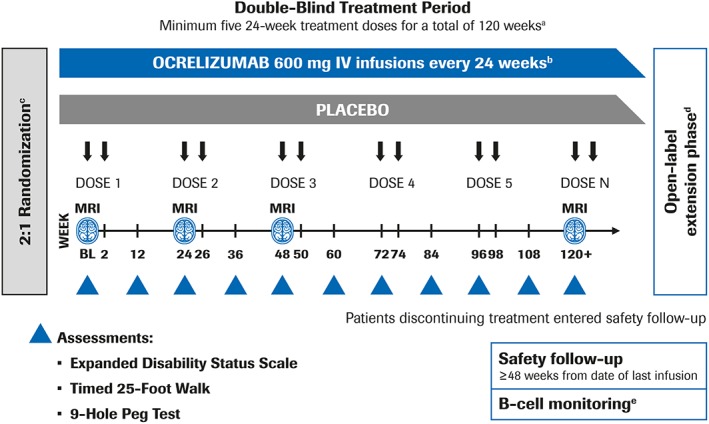
Schedule of MRI scans and assessment of disability measures in the ORATORIO trial. aThe blinded treatment period continued until the last patient completed at least 120 weeks and a target of 253 CDP events was reached. bPatients received methylprednisolone before each ocrelizumab infusion or placebo infusion. c2:1 ocrelizumab:placebo randomization stratified by age (<45 vs > 45 years) and region (USA vs rest of world). dPatients who declined to participate in the OLE entered safety follow‐up. eContinued monitoring occurred if B cells were not repleted. BL = baseline; CDP = confirmed disability progression; IV = intravenous; MRI = magnetic resonance imaging; OLE = open‐label extension.
Statistical Analyses
The exploratory post‐hoc analysis of the proportion of patients with NEP and NEPAD encompassed the period from baseline to week 120, which represents the minimum duration of the double‐blind phase for individual patients in the event‐driven ORATORIO study. The NEP analysis population (230 placebo‐ and 461 ocrelizumab‐treated patients), hereafter termed modified intention‐to‐treat (mITT)‐NEP population, was the ITT population excluding patients with missing baseline score for EDSS, T25FW, or 9HPT, or withdrawn for reasons other than lack of efficacy or death preceding the week 120 visit without evidence of progression (EP) before withdrawal (n = 41). Applying the same exclusion criteria, 234 placebo‐ and 465 ocrelizumab‐treated patients were evaluable from baseline to week 120 (excluded patients, n = 33) in the mITT‐NEPAD analysis population. Patients withdrawn from treatment preceding the week 120 visit because of lack of efficacy or death were imputed as having EP or evidence of progression or active disease (EPAD), in respective analysis of NEP and NEPAD.
Sensitivity analyses were also conducted in the ITT population where patients excluded from the mITT‐NEPAD population were imputed as having NEPAD (“best outcome scenario”) or EPAD (“worst outcome scenario”). Based on previous evidence that sex may influence the rate of progression in PPMS,2 an additional exploratory subgroup analysis of NEPAD by sex was also conducted. NEP and NEPAD data are presented using a Venn diagram surface‐proportional representation. NEP, NEPAD, and the respective components were compared in patients treated with ocrelizumab with those receiving placebo using the Cochran–Mantel–Haenszel test stratified by age (≤45 versus > 45 years) and region (USA versus ROW).
Results
Baseline Demographics and Disease Characteristics
All major baseline demographics and characteristics were balanced between treatment groups in the mITT‐NEPAD and mITT‐NEP analysis populations (Supplementary Table S1). When patients were categorized by EP/NEP or EPAD/NEPAD status over 120 weeks, baseline demographics and disease characteristics were similar. However, compared to patients with EP or EPAD, patients who maintained NEP or NEPAD over 120 weeks had lower T2 lesion volume and EDSS score at baseline and slightly better baseline functional performance as assessed by the T25FW test, 9HPT, and the Paced Auditory Serial Addition Test scores (Supplementary Tables S2 and S3). In addition, patients who maintained NEPAD also had a lower number of T1 Gd‐enhancing lesions at baseline compared to patients with EPAD (Supplementary Table S3).
Proportions of Patients With NEP
In the mITT‐NEP analysis population, ocrelizumab treatment increased the proportion of patients with PPMS maintaining NEP from baseline to week 120 by 47% compared to placebo (ocrelizumab 47.2% versus placebo 29.9%; relative risk [95% confidence interval {CI}], 1.47 [1.17–1.84]; p < 0.001; Fig 3).17 This was also reflected in superiority of ocrelizumab compared to placebo across the individual components of NEP from baseline to week 120 (no confirmed ≥ 20% progression on T25FW, p = 0.002; no confirmed ≥ 20% progression on 9HPT, p = 0.001), although significance was not reached for CDP on EDSS (p = 0.14; the lower effect size and lack of significance compared to ORATORIO primary endpoint12 may be explained by differences in the analysis population, statistical methods used, and exposure duration, ie, NEP assessment ends at week 120 whereas the primary endpoint was a time‐to‐event analysis based on the totality of the event‐driven study duration). In addition, compared to placebo, ocrelizumab increased the proportion of patients with no progression on all pairwise combinations of NEP components assessed: no 12‐week confirmed progression on EDSS and no 12‐week confirmed ≥ 20% progression on T25FW (p < 0.001); no 12‐week confirmed progression on EDSS and no 12‐week confirmed ≥ 20% progression on 9HPT (p = 0.050), and no 12‐week confirmed ≥ 20% progression on T25FW and on 9HPT (p = 0.002; Table 2).
Figure 3.
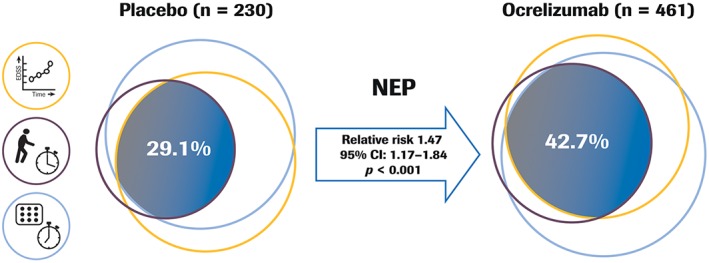
Proportion of patients with NEP from baseline to week 120 in ORATORIO. Venn diagram with approximately surface‐proportional representation. Exploratory analysis; p value from a Cochran–Mantel–Haenszel test stratified by age (≤45 vs > 45 years) and region (USA vs rest of world). NEP analysis population is ITT population excluding patients with missing baseline score for EDSS, T25FW, or 9HPT, or withdrawn for reasons other than efficacy failure or death preceding the week 120 visit, and without evidence of progression (n = 41). Imputation is used for patients withdrawn from the treatment prior to the week 120 visit and who had no event; patients withdrawn because of efficacy failure or death are considered as having an event. Relative risk for ocrelizumab vs placebo. 9HPT = 9‐Hole Peg Test; CI = confidence interval; EDSS = Expanded Disability Status Scale; ITT = intention‐to‐treat; NEP = no evidence of progression; T25FW = Timed 25‐Foot Walk.
Table 2.
Proportion of Patients With Pairwise Components of NEP and NEPAD From Baseline to Week 120 in ORATORIO
| Pairwise Components of NEP (mITT‐NEP population), % (n) | Placebo n = 230 | Ocrelizumab n = 461 | Relative Risk (95% CI) | p |
|---|---|---|---|---|
| No 12‐week confirmed progression on EDSS and no 12‐week confirmed ≥ 20% progression on T25FW |
31.3 (72) | 44.7 (206) | 1.43 (1.15–1.78) |
<0.001 |
| No 12‐week confirmed progression on EDSS and no 12‐week confirmed ≥ 20% progression on 9HPT |
53.5 (123) | 61.4 (283) | 1.15 (0.99–1.32) |
0.05 |
| No 12‐week confirmed ≥ 20% progression on T25FW and on 9HPT |
36.1 (83) | 48.4 (223) | 1.34 (1.10–1.63) |
0.002 |
| Pairwise Components of NEPAD (mITT‐NEPAD population), % (n) | Placebo n = 234 | Ocrelizumab n = 465 | Relative Risk (95% CI) | p |
|---|---|---|---|---|
| No NEP and no brain MRI‐measured disease activity | 9.4 (22) | 30.3 (141) | 3.19 (2.10–4.85) |
<0.001 |
| No NEP and no protocol‐defined relapses | 29.9 (70) | 42.2 (196) | 1.41 (1.13–1.76) |
0.002 |
| No protocol‐defined relapses and no brain MRI‐measured disease activity |
25.2 (59) | 68.0 (316) | 2.68 (2.13–3.36) |
<0.001 |
Relative risk for ocrelizumab versus placebo.
9HPT = 9‐Hole Peg Test; CI = confidence interval; EDSS = Expanded Disability Status Scale; mITT = modified intention‐to‐treat; MRI = magnetic resonance imaging; NEP = no evidence of progression; NEPAD = no evidence of progression or active disease; T25FW = Timed 25‐Foot Walk.
Proportions of Patients With NEPAD
In the mITT‐NEPAD analysis population, 91% of patients in the placebo group experienced clinical progression or active disease (EPAD) from baseline to week 120, which was principally attributed to the occurrence of MRI features characteristic of new inflammatory activity. Ocrelizumab treatment increased by 3‐fold the proportion of patients with NEPAD from baseline to week 120 compared to placebo (ocrelizumab 29.9% versus placebo 9.4%; relative risk [95% CI], 3.15 [2.07–4.79]; p < 0.001; Fig 4). This was reflected in numerical superiority across all the individual components of NEPAD with ocrelizumab compared to placebo from baseline to week 120, where ocrelizumab increased the proportion of patients with NEP, no MRI‐measured disease activity, including no new or enlarging T2 lesions and no T1 Gd‐enhancing lesions, and no protocol‐defined relapse (Table 1). Ocrelizumab also significantly increased the proportion of patients with pairwise components of NEPAD at week 120 compared to placebo: NEP and no MRI‐measured disease activity; NEP and no protocol‐defined relapse; and no protocol‐defined relapse and no MRI‐measured disease activity compared to placebo (Table 2).
Figure 4.
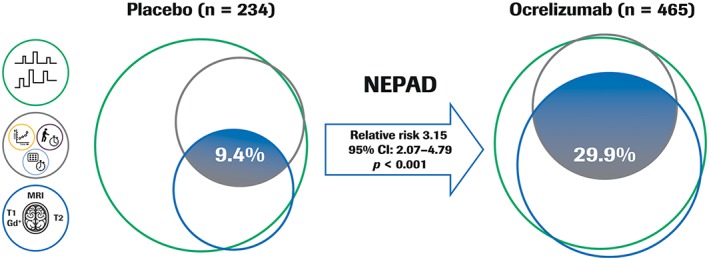
Proportion of patients with NEPAD from baseline to week 120 in ORATORIO. Exploratory analysis; the relative risk and p value are from a Cochran–Mantel–Haenszel test stratified by age (≤45 vs > 45 years) and region (USA vs rest of world). NEPAD analysis population is a modified ITT population. Patients who discontinued treatment early with at least one event before discontinuation were considered as having EPAD. Patients with no reported event before early discontinuation were considered as having EPAD if the reason for early treatment discontinuation was reported to be lack of efficacy or death; otherwise, they were excluded from the analysis. In this approximately surface‐proportional Venn diagram representation, the sector including two ocrelizumab patients with protocol‐defined relapse but with no MRI activity and NEP cannot be displayed. Relative risk for ocrelizumab vs placebo. CI = confidence interval; EPAD = evidence of progression or active disease; Gd+ = gadolinium‐enhancing; ITT = intention‐to‐treat; MRI = magnetic resonance imaging; NEP = no evidence of progression; NEPAD = no evidence of progression or active disease.
Table 1.
Proportion of Patients With NEPAD (and Its Individual Components) in the Complete mITT‐NEPAD Population, and in Male and Female Patients From Baseline to Week 120 in ORATORIO
| mITT‐NEPAD Population | Male | Female | ||||
|---|---|---|---|---|---|---|
| Placebo n = 234 | Ocrelizumab n = 465 | Placebo n = 117 | Ocrelizumab n = 241 | Placebo n = 117 | Ocrelizumab n = 224 | |
| NEPAD (mITT‐NEPAD population), % (n) | 9.4 (22) | 29.9 (139) | 7.7 (9) | 29.9 (72) | 11.1 (13) | 29.9 (67) |
| Relative risk (95% CI) p value | 3.15 (2.07–4.79) p < 0.001 | 3.73 (1.94–7.17) p < 0.001 | 2.70 (1.56–4.68) p < 0.001 | |||
| No evidence of progression (NEP)a, % (n) | 30.3 (71) | 43.2 (201) | 28.2 (33) | 44.0 (106) | 32.5 (38) | 42.4 (95) |
| Relative risk (95% CI) p value | 1.42 (1.14–1.77) p = 0.001 | 1.56 (1.13–2.16) p = 0.004 | 1.30 (0.96–1.76) p = 0.080 | |||
| No 12‐week confirmed progression on EDSS, % (n) | 63.7 (149) | 68.8 (320) | 59.0 (69) | 70.5 (170) | 68.4 (80) | 67.0 (150) |
| Relative risk (95% CI) p value | 1.08 (0.97–1.21) p = 0.16 | 1.19 (1.00–1.42) p = 0.035 | 0.98 (0.84–1.14) p = 0.79 | |||
| No 12‐week confirmed ≥ 20% progression on T25FW, % (n) | 39.7 (93) | 51.4 (239) | 36.8 (43) | 51.0 (123) | 42.7 (50) | 51.8 (116) |
| Relative risk (95% CI) p value | 1.29 (1.08–1.55) p = 0.004 | 1.38 (1.06–1.81) p = 0.012 | 1.20 (0.94–1.54) p = 0.13 | |||
| No 12‐week confirmed ≥ 20% progression on 9HPT, % (n) | 71.8 (168) | 82.4 (383) | 68.4 (80) | 80.5 (194) | 75.2 (88) | 84.4 (189) |
| Relative risk (95% CI) p value | 1.14 (1.04–1.25) p = 0.002 | 1.17 (1.02–1.34) p = 0.015 | 1.12 (1.00–1.26) p = 0.037 | |||
| No evidence of brain MRI‐measured disease activity, % (n) | 26.5 (62) | 70.3 (327) | 23.9 (28) | 68.5 (165) | 29.1 (34) | 72.3 (162) |
| Relative risk (95% CI) p value | 2.64 (2.12–3.28) p < 0.001 | 2.75 (1.98–3.83) p < 0.001 | 2.51 (1.87–3.38) p < 0.001 | |||
| No new or enlarging T2 lesions, % (n) | 26.5 (62) | 70.5 (328) | 23.9 (28) | 68.9 (166) | 29.1 (34) | 72.3 (162) |
| Relative risk (95% CI) p value | 2.64 (2.12–3.29) p < 0.001 | 2.77 (1.99–3.85) p < 0.001 | 2.51 (1.87–3.38) p < 0.001 | |||
| No T1 Gd‐enhancing lesions, % (n) | 58.1 (136) | 93.3 (434) | 57.3 (67) | 93.0 (224) | 59.0 (69) | 93.8 (210) |
| Relative risk (95% CI) p value | 1.60 (1.43–1.79) p < 0.001 | 1.60 (1.37–1.88) p < 0.001 | 1.60 (1.37–1.87) p < 0.001 | |||
| No protocol‐defined relapses, % (n) | 82.9 (194) | 92.7 (431) | 80.3 (94) | 92.1 (222) | 85.5 (100) | 93.3 (209) |
| Relative risk (95% CI) p value | 1.12 (1.05–1.19) p < 0.001 | 1.14 (1.04–1.26) p = 0.001 | 1.09 (1.01–1.19) p = 0.018 | |||
Relative risk for ocrelizumab versus placebo.
Minor differences in NEP component numbers (when NEP was herein analyzed in the mITT‐NEPAD population) compared to those presented in Figure 3 (mITT‐NEP population) originate from the differences between mITT‐NEP and mITT‐NEPAD population sample size, as outlined in Supplementary Table S1.
9HPT = 9‐Hole Peg Test; CI = confidence interval; EDSS = Expanded Disability Status Scale; Gd = gadolinium; mITT = modified intention‐to‐treat; MRI = magnetic resonance imaging; NEP = no evidence of progression; NEPAD = no evidence of progression or active disease; T25FW = Timed 25‐Foot Walk.
Sensitivity analyses performed in the ITT population (excluding 1 patient with missing baseline EDSS, following the approach originally used in the primary endpoint analysis),12 with imputation for patients who withdrew early for reasons other than “lack of efficacy” or “death” and who had NEPAD at time of study treatment discontinuation, were consistent with the main results (Supplementary Table S4). This was true both for NEPAD imputation of early discontinuers (“best outcome scenario”: ocrelizumab 33.5% versus placebo 13.1%; relative risk [95% CI]: 2.55 [1.80–3.60]; p < 0.001) and EPAD imputation of early discontinuers (“worst outcome scenario”: ocrelizumab 28.8% versus placebo 9.0%; relative risk [95% CI]: 3.17 [2.08–4.83]; p < 0.001).
Additional sensitivity analyses of NEPAD from baseline to week 120 where NEP components were based on 24‐week instead of 12‐week confirmed clinical progression events was also consistent with the main results (Supplementary Table S5) as was an analysis of NEPAD using an alternative time‐to‐event Kaplan–Meier approach over the totality of ORATORIO study duration (Fig 5; Supplementary Table S6).
Figure 5.
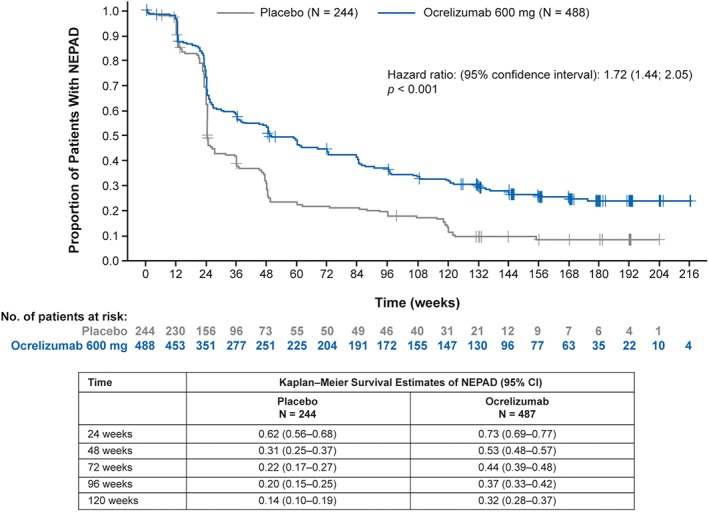
Kaplan–Meier analysis of time to evidence of progression or active disease in the ITT population during the double‐blind controlled period in ORATORIO. Patients are considered as failing NEPAD if one of the following events occurred: protocol‐defined relapse, 12‐week CDP, 12‐week confirmed progression on T25FW or 9HPT, or MRI activity. Hazard ratio estimates are obtained from a Cox model stratified by region (ROW; US) and including age, disease duration from onset, baseline EDSS score, baseline T25FW, and baseline 9HPT. p value is from a log‐rank test stratified by region (ROW; US), age (>45; ≤ 45 years), and baseline EDSS score category (<4; ≥ 4). 9HPT = 9‐Hole Peg Test; CDP = confirmed disability progression; CI = confidence internal; EDSS = Expanded Disability Status Scale; ITT = intention‐to‐treat; MRI = magnetic resonance imaging; NEPAD = no evidence of progression or active disease; ROW = rest of world; T25FW = Timed 25‐Foot Walk.
Exploratory analyses of NEPAD by sex were also consistent with the primary results. Compared to placebo, ocrelizumab significantly increased the proportion of patients with NEPAD for both male and female patients (male, p < 0.001; female, p < 0.001; Table 1). This was reflected in numerical superiority across all the individual components of NEPAD in male and female patients with ocrelizumab compared to placebo from baseline to week 120, except for no 12‐week confirmed progression on EDSS where proportions were similar in both groups for female patients (Table 1). In addition, there was no evidence for the heterogeneity of the treatment effect in relation to sex (p > 0.5 for the treatment‐by‐sex interaction in logistic regression models for NEPAD and its three components with adjustments for age, region, disease duration, baseline values of the T25FW test and 9HPT, and presence of T1 Gd‐enhancing lesions at baseline). Further exploratory subgroup analyses were performed by age at study baseline. Proportions of patients with NEPAD from baseline to week 120 were significantly higher in ocrelizumab‐treated patients compared to the placebo group, both in younger patients (age at study baseline ≤45 years): ocrelizumab 28.9% versus placebo 5.4%; relative risk [95% CI]: 5.24 [2.35–11·69]; (p < 0.001), and older patients (age at study baseline >45 years): ocrelizumab 30.8% versus placebo 13.1%; relative risk [95% CI]: 2.34 [1.42–3.84]; (p < 0.001). The numerically lower magnitude of ocrelizumab treatment effect on NEPAD in the older subgroup appeared to be, in part, driven by the differential behavior of the placebo group where twice as many of the older patients experienced NEPAD compared to the younger subgroup (Supplementary Table S7). There was no statistical evidence for a treatment effect heterogeneity on NEPAD combined outcome (p = 0.18 for the treatment‐by‐age group interaction in the model as described above).
Discussion
In this post‐hoc analysis of the phase III ORATORIO study, ocrelizumab increased the proportion of patients with PPMS maintaining NEP and NEPAD status compared to placebo, which is consistent with ocrelizumab efficacy on the primary and secondary endpoints in this trial.12
NEP may represent a more comprehensive clinical trial outcome for patients with PPMS, given that the Venn diagram representation of NEP components illustrates that the three outcome measures capture substantial nonoverlapping domains of MS disability burden. In the placebo arm of ORATORIO, more than half of patients with PPMS who had no confirmed disability progression, as measured by EDSS, experienced confirmed worsening in ambulation when measured by the T25FW test. Similarly, a sizeable proportion of patients who had no confirmed progression on EDSS, worsened on hand/arm function, as assessed by 9HPT. Treatment with ocrelizumab increased the proportion of patients with NEP at 120 weeks by 47% compared to placebo. A consolidated demonstration of absence of worsening in ambulation using the T25FW test in those patients who might have a stable EDSS may be of utmost importance, given that walking impairment negatively impacts employment status, the ability to perform activities of daily living, and health‐related quality of life of patients with MS. 18, 19 Furthermore, maintenance of walking ability has been reported by patients with MS as the most highly valued bodily function.20 In addition, given the asynchronous disease course in progressive MS,21 a comprehensive ascertainment of clinical stability should require confirmation of the absence of progression on upper limb function, as assessed by 9HPT, in patients stable on EDSS and the T25FW test. Evaluation of NEP status may therefore represent a novel, clinically meaningful endpoint and a pragmatic goal in the management of patients with PPMS in clinical practice.
NEPAD is an extension of NEP incorporating relapses, new or enlarging T2 lesions, and T1 Gd‐enhancing lesions, which are the inflammatory disease activity descriptors of progressive MS phenotypes.22 On this composite outcome that includes all descriptors of progressive disease as per the Lublin et al (2014) revised consensus on MS disease course definitions,22 the proportion of patients with NEPAD was 3‐fold higher with ocrelizumab compared to placebo. Sensitivity analyses with opposite imputation of missing data confirmed the robustness of the primary findings. NEPAD can also be viewed as an extension of NEDA (no evidence of disease activity),23 integrating aspects of disability burden (hand/arm function and ambulation impairment), which are important aspects of the clinical course of progressive MS.22 Notwithstanding this integration of two additional clinical components, the relative importance of MRI activity components remains dominant in the determination of NEPAD outcome as commonly observed for NEDA analyses24, 25, 26 in patients with relapsing multiple sclerosis (RMS). Although NEDA is increasingly seen as a treatment goal, and some long‐term data support the prognostic value of NEDA for predicting future disability progression in patients with RMS with high efficacy therapies,24, 25, 26 similar analyses are warranted to investigate the prognostic utility of NEPAD in progressive MS populations. In particular, the added value of MRI components of NEPAD for predicting long‐term outcomes remains unknown in patients with PPMS.
Similar proportions of male and female patients with PPMS maintained NEPAD status with significantly greater proportions in the ocrelizumab‐treated group compared to placebo within each sex subgroup. Except for 12‐week confirmed disability progression on EDSS, a numerical superiority favoring ocrelizumab was observed across all individual components of NEP and NEPAD to a similar extent in male and female patients. When patients were analyzed in subgroups by age at study baseline, significantly greater proportions of patients with NEPAD were observed in the ocrelizumab‐treated group compared to placebo within each age subgroup. The higher effect of ocrelizumab on NEPAD outcome noted in younger patients appeared to reflect, in part, age‐related differences in the behavior of the placebo group.
PPMS patients with NEP or NEPAD versus those with evidence of progression or active disease did not differ in age, MS disease duration, or normalized brain volume at baseline, whereas patients with NEP or NEPAD had a lower total lesion burden and disability at baseline. Whether such imbalances may reflect objective differences between those baseline covariates as potential prognostic factors for NEP/NEPAD in patients with PPMS remains to be explored.
In conclusion, ocrelizumab increased the proportion of patients with PPMS with no evidence of progression and no clinical and subclinical disease activity compared to placebo. As such, NEPAD may represent a meaningful and comprehensive disease outcome in patients with PPMS. Keeping in mind that this analysis was post hoc and not adjusted for multiplicity testing, further validation of the clinical utility of NEPAD beyond the ORATORIO results should be sought by similar analyses in other primary and secondary progressive MS study populations. In RMS, NEPAD status was recently evaluated in the pooled OPERA I and OPERA II studies, with similar findings favoring ocrelizumab compared to interferon beta‐1a.27
NEPAD represents the first incremental step of adaptation of NEDA to more comprehensively capture the different facets of progression. It is a reasonable assumption that in the specific intent of ascertaining the complete absence/remission of any measurable “signs of disease activity/progression,” the NEPAD combined endpoint may outperform any of its individual components. Subsequent evolution of the NEPAD measure may include the integration of cognitive and visual performance outcomes such as the symbol‐digit modalities test or low‐contrast letter acuity, respectively. Chronic T2 lesions with evolving T1 hypointensity may also be considered as a readout for permanent tissue damage accumulation, as well as whole‐brain volume loss, where more research is needed to discriminate pathological atrophy from normal aging or noise of the measurement, and determine personalized thresholds of annualized rates of brain volume change.28, 29, 30
Future analyses, including data from the open‐label extension of the ORATORIO study and real‐world evidence, will determine whether NEPAD maintained throughout 120 weeks will translate into sustained NEPAD and enhanced protection against accrual of disability in patients with PPMS over the long term.
Acknowledgment
This research was funded by F. Hoffmann‐La Roche Ltd (Basel, Switzerland). Editorial assistance for this article was provided by Terence Smith of Articulate Science, UK, and funded by F. Hoffmann‐La Roche Ltd. The authors had full editorial control of the manuscript and provided their final approval of all content.
We thank all patients, their families, and the investigators who participated in this trial (including the ORATORIO study Steering Committee, which provided study oversight: Douglas L. Arnold, Amit Bar‐Or, Giancarlo Comi, Gavin Giovannoni, Hans‐Peter Hartung, Stephen L. Hauser, Bernhard Hemmer, Ludwig Kappos, Fred Lublin, Xavier Montalban, Kottil Rammohan, Krzysztof Selmaj, Anthony Traboulsee, and Jerry S. Wolinsky). We also thank the independent data monitoring committee for performing data analysis and safety monitoring (Stephen C. Reingold [Chair], Magnhild Sandberg‐Wollheim [Vice‐Chair], Frederik Barkhof, Thomas Dörner, Scott Evans, Henry F. McFarland, and Israel Steiner). We are grateful to Gisèle von Büren and Fabian Model (of F. Hoffmann‐La Roche Ltd.) for additional critical review of this manuscript and technical advice
Potential Conflicts of Interest
Commercial firms whose product (ocrelizumab) was used in this study are F. Hoffmann‐La Roche Ltd. and Genentech, Inc. The following authors disclosed financial relationships with these companies. S.B. is an employee and/or shareholder of F. Hoffmann‐La Roche Ltd. C.B. is an employee and/or shareholder of F. Hoffmann‐La Roche Ltd. P.C. is an employee and/or shareholder of Genentech, Inc. G.D.‐B. is an employee and/or shareholder of F. Hoffmann‐La Roche Ltd. H.G. is an employee and/or shareholder of F. Hoffmann‐La Roche Ltd. G.G. honoraria from F. Hoffmann‐La Roche Ltd. L.K. research support for activities as principal investigator and member or chair of planning and steering committees or advisory boards for trials sponsored by F. Hoffmann‐La Roche Ltd. S.L.H. travel reimbursement from F. Hoffmann‐La Roche Ltd. X.M. speaking honoraria, travel expense, steering committee member, and advisory boards for F. Hoffmann‐La Roche Ltd. P.V. research support F. Hoffmann‐La Roche Ltd. J.S.W. advisory boards, data monitoring or steering committees, consulting agreements, and speaker honoraria from F. Hoffmann‐La Roche Ltd. and Genentech, Inc.
Data Sharing Statement
Qualified researchers may request access to individual patient‐level data through the clinical study data request platform (http://www.clinicalstudydatarequest.com). Further details on Roche's criteria for eligible studies are available here (https://clinicalstudydatarequest.com/Study-Sponsors/Study-Sponsors-Roche.aspx). For further details on Roche's Global Policy on the Sharing of Clinical Information and how to request access to related clinical study documents, see here (https://www.roche.com/research_and_development/who_we_are_how_we_work/clinical_trials/our_commitment_to_data_sharing.htm).
Supporting information
Additional supporting information can be found in the online version of this article.
Author Contributions
Study concept and design: P.C., H.G., L.K., S.L.H., X.M., and J.S.W. Data acquisition and analysis: S.B., C.B., P.C., G.D.‐B., H.G., G.G., L.K., S.L.H., X.M., P.V., and J.S.W. Drafting the manuscript and figures: S.B., C.B., P.C., G.D.‐B., H.G., G.G., L.K., S.L.H., X.M., P.V., and J.S.W. All authors edited and approved the final version of the manuscript.
References
- 1. Lublin FD. New multiple sclerosis phenotypic classification. Eur Neurol 2014;72(suppl 1):1–5. [DOI] [PubMed] [Google Scholar]
- 2. Wolinsky JS, Narayana PA, O'Connor P, et al. Glatiramer acetate in primary progressive multiple sclerosis: results of a multinational, multicenter, double‐blind, placebo‐controlled trial. Ann Neurol 2007;61:14–24. [DOI] [PubMed] [Google Scholar]
- 3. Hawker K, O'Connor P, Freedman MS, et al. Rituximab in patients with primary progressive multiple sclerosis: results of a randomized double‐blind placebo‐controlled multicenter trial. Ann Neurol 2009;66:460–471. [DOI] [PubMed] [Google Scholar]
- 4. Lublin F, Miller DH, Freedman MS, et al. Oral fingolimod in primary progressive multiple sclerosis (INFORMS): a phase 3, randomised, double‐blind, placebo‐controlled trial. Lancet 2016;387:1075–1084. [DOI] [PubMed] [Google Scholar]
- 5. Balabanov P, Haas M, Elferink A, et al. Addressing the regulatory and scientific challenges in multiple sclerosis—a statement from the EU regulators. Mult Scler 2014;20:1282–1287. [DOI] [PubMed] [Google Scholar]
- 6. Cadavid D, Cohen JA, Freedman MS, et al. The EDSS‐Plus, an improved endpoint for disability progression in secondary progressive multiple sclerosis. Mult Scler 2017;23:94–105. [DOI] [PubMed] [Google Scholar]
- 7. Miller DH, Leary SM. Primary‐progressive multiple sclerosis. Lancet Neurol 2007;6:903–912. [DOI] [PubMed] [Google Scholar]
- 8. Klein C, Lammens A, Schäfer W, et al. Response to: monoclonal antibodies targeting CD20. MAbs 2013;5:337–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Genovese MC, Kaine JL, Lowenstein MB, et al. Ocrelizumab, a humanized anti‐CD20 monoclonal antibody, in the treatment of patients with rheumatoid arthritis: a phase I/II randomized, blinded, placebo‐controlled, dose‐ranging study. Arthritis Rheum 2008;58:2652–2661. [DOI] [PubMed] [Google Scholar]
- 10. Hauser SL. The Charcot Lecture | beating MS: a story of B cells, with twists and turns. Mult Scler 2015;21:8–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kinzel S, Weber MS. B cell‐directed therapeutics in multiple sclerosis: rationale and clinical evidence. CNS Drugs 2016;30:1137–1148. [DOI] [PubMed] [Google Scholar]
- 12. Montalban X, Hauser SL, Kappos L, et al. Ocrelizumab versus placebo in primary progressive multiple sclerosis. N Engl J Med 2017;376:209–220. [DOI] [PubMed] [Google Scholar]
- 13. U.S. Food and Drug Administration News Release . FDA approves new drug to treat multiple sclerosis. First drug approved for Primary Progressive MS. 2018. https://www.fda.gov/newsevents/newsroom/pressannouncements/ucm549325.htm. Accessed September 18, 2018.
- 14. European Medicines Agency Press Release . New medicine for multiple sclerosis. 2017. http://www.ema.europa.eu/ema/index.jsp?curl=pages/news_and_events/news/2017/11/news_detail_002847.jsp&mid=WC0b01ac058004d5c1. Accessed September 18, 2018.
- 15. Polman CH, Reingold SC, Edan G, et al. Diagnostic criteria for multiple sclerosis: 2005 revisions to the “McDonald Criteria”. Ann Neurol 2005;58:840–846. [DOI] [PubMed] [Google Scholar]
- 16. Bosma LV, Kragt JJ, Brieva L, et al. Progression on the Multiple Sclerosis Functional Composite in multiple sclerosis: what is the optimal cut‐off for the three components? Mult Scler 2010;16:862–867. [DOI] [PubMed] [Google Scholar]
- 17. Montalban X, Arnold DL, Bar‐Or A, et al. Evaluation of no evidence of progression using composite disability outcome measures in patients with primary progressive multiple sclerosis in the ORATORIO trial. Presented at 32nd Congress of the European Committee for Treatment and Research in Multiple Sclerosis, 14–17 September 2016, London, United Kingdom. Oral presentation 167.
- 18. Panitch H, Applebee A. Treatment of walking impairment in multiple sclerosis: an unmet need for a disease‐specific disability. Expert Opin Pharmacother 2011;12:1511–1521. [DOI] [PubMed] [Google Scholar]
- 19. Pike J, Jones E, Rajagopalan K, et al. Social and economic burden of walking and mobility problems in multiple sclerosis. BMC Neurol 2012;12:94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Heesen C, Böhm J, Reich C, et al. Patient perception of bodily functions in multiple sclerosis: gait and visual function are the most valuable. Mult Scler 2008;14:988–991. [DOI] [PubMed] [Google Scholar]
- 21. Giovannoni G, Cutter G, Sormani MP, et al. Is multiple sclerosis a length‐dependent central axonopathy? The case for therapeutic lag and the asynchronous progressive MS hypotheses. Mult Scler Relat Disord 2017;12:70–78. [DOI] [PubMed] [Google Scholar]
- 22. Lublin FD, Reingold SC, Cohen JA, et al. Defining the clinical course of multiple sclerosis: the 2013 revisions. Neurology 2014;83:278–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Giovannoni G, Turner B, Gnanapavan S, et al. Is it time to target no evident disease activity (NEDA) in multiple sclerosis? Mult Scler Relat Disord 2015;4:329–333. [DOI] [PubMed] [Google Scholar]
- 24. Prosperini L, Fanelli F, Pozzilli C. Long‐term assessment of No Evidence of Disease Activity with natalizumab in relapsing multiple sclerosis. J Neurol Sci 2016;364:145–147. [DOI] [PubMed] [Google Scholar]
- 25. Havrdova E, Arnold DL, Cohen JA, et al. Durable efficacy of alemtuzumab on clinical outcomes over 5 years in treatment‐naive patients with active relapsing‐remitting multiple sclerosis with most patients not receiving treatment for 4 years: CARE‐MS I extension study. Presented at 31st Congress of the European Committee for Treatment and Research in Multiple Sclerosis, 7–10 October 2015, Barcelona, Spain. Oral presentation O152.
- 26. University of California, San Francisco MS‐EPIC Team , Cree BA, Gourraud PA, et al. Long‐term evolution of multiple sclerosis disability in the treatment era. Ann Neurol 2016;80:499–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Havrdova E, Arnold DL, Bar‐Or A, et al. Evaluation of no evidence of progression or active disease (NEPAD) in patients with relapsing multiple sclerosis in the OPERA I and OPERA II trials. Presented at 3rd Congress of the European Academy of Neurology, 24–27 June 2017, Amsterdam, The Netherlands. Oral presentation PR1092.
- 28. Andorra M, Nakamura K, Lampert EJ, et al. Assessing biological and methodological aspects of brain volume loss in multiple sclerosis. JAMA Neurol. 2018. Jul 2. doi: 10.1001/jamaneurol.2018.1596. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. De Stefano N, Stromillo ML, Giorgio A, et al. Establishing pathological cut‐offs of brain atrophy rates in multiple sclerosis. J Neurol Neurosurg Psychiatry 2016;87:93–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wattjes MP, Rovira À, Miller D, et al. Evidence‐based guidelines: MAGNIMS consensus guidelines on the use of MRI in multiple sclerosis—establishing disease prognosis and monitoring patients. Nat Rev Neurol 2015;11:597–606. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional supporting information can be found in the online version of this article.