

Abstract
Over the past 15 years, three new classes of drugs, glucagon‐like peptide‐1 (GLP‐1) receptor agonists, dipeptidyl peptidase 4 (DPP‐4) inhibitors and sodium glucose cotransporter‐2 (SGLT‐2) inhibitors have been approved to treat type 2 diabetes based on effects on glycemic control. Although large randomized controlled trials have played an important role in characterizing the efficacy and safety of these agents on a population level, questions remain about how best to individualize therapy and target the “right” medicine to the “right” patient. In contrast, few medicines have been approved to treat diabetic kidney disease and initiatives have been launched on both sides of the Atlantic to facilitate the development of effective personalized medicines for the treatment of diabetic kidney disease. Increasingly, “omics,” imaging and other biomarkers will be used to match patients with therapies to which they are likely to respond best. This review addresses regulatory considerations related to precision medicine, draws lessons learned from other therapeutic areas and discusses efforts undertaken by the European (EMA) and United States (FDA) to facilitate the development of such therapies. Moving forward, an integrated approach that makes use of predictive preclinical models, innovative trial designs, observational “real‐world” data and novel statistical methodologies will likely be needed to complement inherently smaller RCTs conducted in more selected populations. Patient involvement will also be critical. Regulatory agencies are ready to engage in such approaches.
Keywords: precision medicine, regulatory science
1. INTRODUCTION
In the past 15 years, three new classes of drugs, glucagon‐like peptide‐1 (GLP‐1) receptor agonists, dipeptidyl peptidase 4 (DPP‐4) inhibitors and sodium glucose cotransporter‐2 (SGLT‐2) inhibitors have been approved for the treatment of diabetes based on their ability to lower glucose levels in patients with type 2 diabetes.1 Over this time period, another drug, rosiglitazone, which had been approved based on effects on glycemic control, was ultimately withdrawn from the European market and its use restricted in the United States because of concerns over its cardiovascular (CV) safety.2 Thus, with uncertainties remaining over the macrovascular benefits—and even potential harms—of glucose‐lowering drugs, the US Food and Drug Administration (FDA) issued guidance in 2008 on how sponsors should demonstrate that new antidiabetic agents for type 2 diabetes do not increase cardiovascular risk to an unacceptable extent.3 It should be noted that in Europe, cardiovascular outcome trials (CVOT) are not required for new antidiabetic agents, although at the time of approval, CV (and other) harms should nevertheless be excluded.4, 5 Since the introduction of the FDA guidance in 2008, a large number of CVOTs have been conducted to assess the cardiovascular risk of antidiabetic agents.1 These trials enrolled large populations and included, in total, over 100 000 patients with type 2 diabetes often at high CV risk to allow more efficient evaluation of CV safety. They demonstrated the CV safety of DPP4 inhibitors, GLP‐1 agonists and SGLT‐2 inhibitors and even important benefits for some GLP‐1 agonists and SGLT‐2 inhibitors. Although these mega‐trials have played an important role in characterizing the efficacy and safety of these new interventions on a population level, a marked heterogeneity in response at the individual level was observed.6, 7, 8 Questions thus remain about how to individualize therapy and select the “right” drug for the “right” patient.
In contrast to this experience, no medicine has been approved by the FDA or European Medicines Agency (EMA) to treat diabetic kidney disease within over 15 years. Although recent CVOTs raise hope that some of the new antidiabetic medicines may also provide important renal benefits, there is widespread recognition that better tools and a better understanding of diabetic kidney disease is needed to facilitate drug development for diabetic kidney disease. This includes the need to identify different subgroups of patients with the disease who may be more likely to progress and/or who are more likely to respond beneficially to certain treatments. Indeed, projects, like the Innovative Medicines Initiative Biomarker Enterprise to Attack Diabetic Kidney Disease (http://www.beat-dkd.eu) and the Kidney Precision Medicine Project (http://www.kpmp.org) have been initiated in Europe and the United States, respectively, to facilitate the development of effective personalized medicines for the treatment of diabetic kidney disease.
This landscape, which arguably includes both extremes—a relative abundance of medicines approved to improve glycemic control in type 2 diabetes and questions related to how to choose from among them, as well as a paucity of medicines approved to slow the progression of diabetic kidney disease‐highlights the need for more targeted or personalized treatment approaches to maximize the benefit‐to‐harm ratio of approved therapies and to facilitate the development of new ones. Such an approach reflects an approach that is increasing being adopted in other areas of medicine, in particular oncology. This review addresses regulatory considerations related to precision medicine, draws lessons learned from other therapeutic areas, and discusses efforts undertaken by the EMA and FDA to facilitate the development of such therapies.
2. PRECISION MEDICINE AND THE REGULATORY SYSTEM
Today's regulatory systems aim to ensure the quality and effectiveness/efficacy of medicines and a positive benefit‐risk profile. While the quality of the medicine is essentially the summed assessment of critical product features (eg, potency, product stability, purity), the benefit‐risk is a summed function of what the medicine brings to the patient (eg, pharmacodynamics) and what the patient brings to the medicine (eg, renal function, genotype, patient adherence). There is growing evidence that what the patient brings to the medicine is critical to patient outcomes, both in terms of efficacy/effectiveness and safety.
Finding the best patient‐therapy pair, or precision medicine, is not a new concept. Physicians have always endeavored to match available remedies to patients, considering the characteristics of the patient, the characteristics of the drug, and ideally the patient's values and priorities. But advances in pharmacogenomic science, biomarker discovery, as well as the identification of other signatures of therapy response have paved the way for greater sophistication in finding the best patient‐therapy pair. Moreover, increasingly caregivers and clinicians involve willing patients in shared decision‐making, choosing a therapy based on (expected) outcomes that are most relevant to these patients. Patients and regulators may have mostly similar values with regards to major drug effects of antidiabetic drugs, but do differ in the value they attach to minor short‐term (adverse) drug effects.9 Whereas patients are already involved in a range of activities at the EMA and FDA, a European public private partnership project, IMI‐PREFER (https://www.imi-prefer.eu/) investigates how and when to include patient‐preference in decision‐making during the drug life cycle. Moving forward, regulatory systems should help ensure that these best patient‐therapy pairs can be identified and brought together. Some products are relatively forgiving, (eg, there is a large range between doses/exposures that cause benefit and those that cause harm) and can be used in a broad swath of patients with a disease. Other products, however, need careful targeting, monitoring and follow‐up, because a “mismatch” may result in a harm that outweighs the benefit. Examples include metformin (diabetes) in patients with severe renal impairment or abacavir (HIV) in patients known to carry the HLA‐B*5701 allele. The drug label, also known as the Summary of Product Characteristics (SmPC) in Europe, is meant to help prescribers identify the best patient‐therapy match. For example, the SmPC for rosuvastatin indicates that for patients with SLCO1B1 (OATP1B1) and/or ABCG2 (BCRP) genetic polymorphisms in these transporter proteins there is a risk of increased rosuvastatin exposure and a lower daily dose is recommended.10 The SmPC for clopidogrel describes that in patients who are poor CYP2C19 metabolisers, at recommended doses clopidogrel forms less of its active metabolite and has a smaller effect on platelet function.11
There are, however, many methodological challenges associated with determining the best patient‐therapy match; we discuss some of these challenges in the next section.
3. NEW CLINICAL TRIAL DESIGNS FOR PRECISION MEDICINE IN REGULATORY CONTEXT
Regulators are well‐versed in evaluating drug effects at a population level based on randomized (double‐blind) controlled trials (RCT). Robust findings in well‐conducted RCTs allow regulators to draw conclusions about a drug's benefit‐to‐harm ratio with reasonable certainty for a population similar to the one studied in the trial. Typically, a drug is approved based on a finding in the overall population, for example, by demonstrating that in the trial population the mean effect in the test arm of the trial is larger than in the placebo arm or at least non‐inferior to an active control arm, and if adverse effects do not off‐set the observed benefit.
In contrast to this population‐based approach, precision medicine makes use of an increased understanding of disease biology to a priori define patient groups that are more likely to respond positively or negatively to a treatment; that is, improve the patient‐therapy match. Predictive biomarkers are used to individualize treatment to patients with a specific disease expression or initial treatment response.12 To date, this approach has been most widely used in oncology with therapies targeted to, and defined by genetic mutations, for example, Human Epithelial Growth Factor Receptor 1 (HER1) mutations (trastuzumab),13 programmed death ligand‐1 (PDL‐1) expression (pembrolizumab)14 and BRAF mutations (vemurafenib).15 In the majority of these oncology development programs, translational research and exploratory clinical trials informed the pivotal registration studies. In other fields, dynamic or response biomarkers thought to predict the long‐term benefit or harms of treatment are being explored to target medicines to populations more likely to derive benefit. For example, in the SONAR trial, patients with diabetic kidney disease were randomized to continued active therapy or placebo if they showed a response in albuminuria (>30% decrease) to an initial 6 weeks of atrasentan treatment; some patients who were biomarker “negative” were also enrolled for reasons discussed below.16 Such dynamic or response biomarkers are needed to individualize treatment.17
While focusing clinical trial enrollment on patients more likely to respond positively to treatment has obvious advantages, often it is unclear how well the predictive or response biomarker performs in identifying this population, and hence, regulators are often interested in treatment effects in patients who are biomarker negative. Do patients without a certain biomarker response truly not benefit in terms of (long‐term) clinical endpoints? Is there really no treatment response in the biomarker negative population? Moreover, rarely does a biomarker fully discriminate between responder and non‐responder patients. In other words, the sensitivity and specificity of the biomarker is seldom 100%. Hence, unless there are compelling data or strong scientific rationale for concluding that biomarker negative patients are unlikely to respond to a treatment, regulators encourage the inclusion of marker‐negative patients in the trial; the size/proportion of the population included in the trial depends on the level of certainty in the performance of the biomarker or proposed cut‐off in identifying responders. In the SONAR trial in patients with diabetic kidney disease, this issue was addressed by including more than 1000 patients with less than a 30% reduction in albuminuria; that is, hypothesized non‐responders. Randomization was also stratified according to prespecified albuminuria response strata, with the objective of identifying a minimum albuminuria response threshold associated with a beneficial effect of treatment.16
Indeed, the experience in oncology teaches us that even when patients with various biomarker expression levels are included in a trial, a clear cut‐off may not be easy to establish. For example, the level of tumour programmed death‐1 (PD‐1) expression impacted the effect of nivolumab + ipilimumab in the setting of unresectable metastatic melanoma, but the impact was inconsistent across various cut‐offs. Given these findings, the indication in the SmPC indicates that an increase in progression free survival and overall survival is established only in patients with low (<1%) tumour PD‐1 expression.18
Not surprisingly, the EMA guideline for anticancer European regulatory guidelines, reflects on the approach to developing targeted therapies, with preferably prospective validation of biomarkers identified in explorative studies or post hoc analyses.19 A thorough understanding of the biology is a given. Enrichment designs (only biomarker positive patients), stratified design (randomization based on biomarker expression, like in the SONAR trial) and adaptive enrichment designs (where after an interim analysis randomization can be restricted to a biomarker‐positive population only) may be efficient ways for determining the benefit‐risk in the biomarker‐positive population. There is a caveat though, that is that these enrichment designs may be non‐informative on the biomarker‐negative population.20 As described elsewhere in this issue of the Journal, platform, basket and umbrella trials may offer additional innovative study designs for evidence generation in the setting of precision‐medicine.21 In addition, observational “real‐world” data may be increasingly considered to provide further evidence postapproval.22 Finally, multiple smaller trials with clearly defined patient populations could be used to test specific treatments or combinations at various dose levels more appropriately than performing subgroup analyses in large clinical trials. Ideally, these trials would use a standardized data collection that allows future meta‐analytical approaches to answer questions that are relevant to a more general population, provided a willingness to share data among different parties.23 Regulatory experience remains, however, limited and there are many methodological challenges for which regulators, industry, and academic methodologists will need to collaborate in finding solutions. Clearly, as Eichler and Sweeney write “…the randomised controlled trial must adapt and evolve to respond to a changing environment….”23
4. EUROPEAN AND USA REGULATORY TOOLS TO SUPPORT USE OF NOVEL METHODOLOGIES IN DRUG DEVELOPMENT
In Europe, biomarkers are qualified for a certain context of use, that is, to support drug development or regulatory decision‐making. Interested parties, often consortia or companies and academia can apply at the Scientific Advice Working Party for “qualification of novel methodologies.”24 This procedure can result in “qualification advice” or a “qualification opinion.” The latter is an endorsement of the Committee of Human Medicinal Products that the novel methodology, that is, biomarker, may be used in a certain clearly defined context. For example, to enrich a study population based on a biomarker or use a patient reported outcome as a clinical endpoint (see Figure 1). A large proportion of the qualification procedures focus on development of different type of biomarkers; mostly protein and imaging biomarkers that are developed primarily for use of evaluating clinical safety, as tool for enrichment or to be used as endpoints. Another major set of procedures focuses on development of patient reported outcomes and clinical scores again in the context of use as endpoint or for enrichment purposes to detect more responsive/at risk study populations. Finally, a group of diverse procedures ranges from systems biology approaches, big data to product‐device methods that could be applied throughout the drug development context. The data needed to qualify the biomarker is highly dependent on the intended (context of) use. A qualification opinion is published on the website of the European Medicines Agency and subject to a 2‐month consultation procedure, where interested parties may submit their views on a qualification. It is expected that in the setting of precision medicine this procedure may be sought more frequently by interested parties to improve the pairing of patient and therapy.
Figure 1.
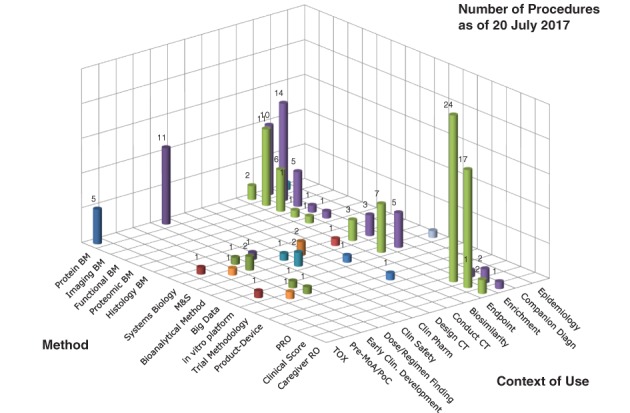
Scope of qualification of novel methodologies applications at the European medicines agency. The novel methodology (method) and their intended use (Context of Use) that were submitted to the Scientific Advice Working Party of the European Medicines Agency up to July 20, 2017 are presented in this figure. Figure courtesy of E. Manolis, EMA, London
The US FDA also has a Drug Development Tool (DDT) Qualification Program that qualifies biomarkers, clinical outcome assessments (COA) and animal models for a specific context of use in drug development.25 As in Europe, qualification of the drug development tool enables sponsors to use the tool in the qualified context of use during drug development without requesting that the regulatory body reconfirm its suitability for the qualified context of use. The process for qualification of drug development tools is changing in the United States because of recent legislation titled the “21st Century Cures Act.” This legislation formally establishes a multi‐step step process for qualification, consisting of three submissions to the qualification program: a Letter of Intent, a Qualification Plan and a Full Qualification Package. The new legislation also includes transparency provisions that apply to both requestors' submissions and FDA's formal written determinations on these submissions.25
Mechanisms also exist to enable better collaboration between FDA and EMA on products in development or under review in both regulatory regions. The EMA and FDA hold regular teleconferences focusing on special topics and therapeutic areas. Existing workgroups (referred to as “clusters”) focus on rare diseases, pediatric drug development and pharmacogenomics, among other areas. Sponsors can also seek parallel scientific advice from FDA and EMA on issues related to the development phase of a new product. These platforms, as well as others allow regulators to exchange information and discuss issues affecting a drug development program and/or more general issues affecting the development of medicines. Precision medicine and the ability to identify, select and test drugs in the most responsive patient populations are frequent topics during these discussions. Disease‐specific meetings and research consortia also provide an opportunity for regulators from around the world to meet and interact with the larger community on issues related to the development of more targeted therapies.26
5. SUMMARY AND CONCLUSION
Precision medicine presents both opportunities and challenges. Programs, such as the “qualification of novel methodologies” procedure at the EMA and the Drug Development Tools Qualification Program at the FDA are intended to support the development of tools that can be used to expedite the development of new therapies and potentially facilitate the development of much needed precision medicines. Moving forward, an integrated approach that makes use of predictive preclinical models, innovative trial designs, observational “real‐world” data and novel statistical methodologies will likely be needed to complement inherently smaller RCTs conducted in more selected populations. The speed at which we develop these medicines will depend in large part on the willingness of various stakeholders to share individual patient data, be it from trials or observational data sets. Patient involvement, and understanding of patient preferences towards different drug effects, will also be critical.
ACKNOWLEDGEMENTS
The views expressed in this article are the personal views of the authors and may not be understood or quoted as being made on behalf of or reflecting the position of the Food and Drug Administration, The Dutch Medicines Evaluation Board or the European Medicines Agency or one of its committees or working parties. This paper has been written as a reflection on the goals of the IMI BEAt‐DKD project and the Precision Medicine Symposium that took place in December 2017 in Groningen, The Netherlands. The BEAt‐DKD project has received funding from the Innovative Medicines Initiative 2 Joint Undertaking under grant agreement No 115974. This Joint Undertaking receives support from the European Union's Horizon 2020 research and innovation programme and EFPIA.
Mol PGM, Thompson A, Heerspink HJL, Leufkens HGM. Precision medicine in diabetes and diabetic kidney disease: Regulatory considerations. Diabetes Obes Metab. 2018;20(Suppl. 3):19–23. 10.1111/dom.13453
REFERENCES
- 1. Blind E, Janssen H, Dunder K, de Graeff PA. The European medicines agency's approval of new medicines for type 2 diabetes. Diabetes Obes Metab. 2018; 1‐5. 10.1111/dom.13349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Blind E, Dunder K, de Graeff PA, Abadie E. Rosiglitazone: a European regulatory perspective. Diabetologia. 2011;54:213‐218. [DOI] [PubMed] [Google Scholar]
- 3.FDA. Guidance for Industry. Diabetes mellitus: evaluating cardiovascular risk in new antidiabetic therapies to treat type 2 diabetes. https://www.fda.gov/downloads/Drugs/Guidances/ucm071627.pdf. 2008. Accessed May 11, 2018
- 4.EMA. Guideline on clinical investigation of medicinal products in the treatment or prevention of diabetes mellitus. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/06/WC500129256.pdf. 2012. Accessed May 11, 2018
- 5.EMA. Reflection paper on assessment of cardiovascular safety profile of medicinal products. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2016/03/WC500203804.pdf. 2016. Accessed May 11, 2018
- 6. Petrykiv SI, Laverman GD, de Zeeuw D, Heerspink HJL. The albuminuria‐lowering response to dapagliflozin is variable and reproducible among individual patients. Diabetes Obes Metab. 2017;19(10):1363‐1370. 10.1111/dom.12936. [DOI] [PubMed] [Google Scholar]
- 7. Zobel EH, von Scholten BJ, Lindhardt M, Persson F, Hansen TW, Rossing P. Pleiotropic effects of liraglutide treatment on renal risk factors in type 2 diabetes: Individual effects of treatment. J Diabetes Complicat. 2017;31(1):162‐168. 10.1016/j.jdiacomp.2016.09.016. [DOI] [PubMed] [Google Scholar]
- 8. Mosenzon O, Leibowitz G, Bhatt DL, et al. Effect of Saxagliptin on Renal Outcomes in the SAVOR‐TIMI 53 Trial. Diabetes Care. 2017;40(1):69‐76. 10.2337/dc16-0621. [DOI] [PubMed] [Google Scholar]
- 9. Mol PG, Arnardottir AH, Straus SM, et al. Understanding drug preferences, different perspectives. Br J Clin Pharmacol. 2015;79(6):978‐987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rosuvastatin summary of product characteristics. https://www.medicines.org.uk/emc/product/7555/smpc. Accessed June 18, 2018.
- 11.EMA. European Public Assessment Report clopidogrel http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/000174/WC500042189.pdf, Accessed June 18, 2018.
- 12.BEST (Biomarkers, EndpointS, and other Tools) resource [Internet]. Understanding prognostic versus predictive biomarkers. https://www.ncbi.nlm.nih.gov/books/NBK402284/. Accessed May 11, 2018
- 13.EMA. European public assessment report trastuzumab. http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/000278/human_med_000818.jsp&mid=WC0b01ac058001d124. Accessed May 11, 2018
- 14.EMA. European public assessment report pembrolizumab. http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/003820/human_med_001886.jsp&mid=WC0b01ac058001d124. Accessed May 11, 2018
- 15.EMA. European public assessment report vemurafenib. http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/002409/human_med_001544.jsp&mid=WC0b01ac058001d124. Accessed May 11, 2018
- 16. Heerspink HJL, Andress DL, Bakris G, et al. Rationale and protocol of the study of diabetic nephropathy with AtRasentan (SONAR) trial: a clinical trial design novel to diabetic nephropathy. Diabetes Obes Metab. 2018;20:1369‐1376. 10.1111/dom.13245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Senderowicz AM, Pfaff O. Similarities and differences in the oncology drug approval process between FDA and European Union with emphasis on in vitro companion diagnostics. Clin Cancer Res. 2014;20(6):1445‐1452. 10.1158/1078-0432.CCR-13-1761. [DOI] [PubMed] [Google Scholar]
- 18.EMA. European public assessment report nivolumab. http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/003985/human_med_001876.jsp&mid=WC0b01ac058001d124. Accessed May 30, 2018
- 19.EMA. Guideline on the evaluation of anticancer medicinal products in man. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2017/11/WC500238764.pdf. Accessed May 11, 2018
- 20. Pignatti F, Ehmann F, Hemmings R, et al. Cancer drug development and the evolving regulatory framework for companion diagnostics in the European union. Clin Cancer Res. 2014;20(6):1458‐1468. 10.1158/1078-0432.CCR-13-1571. [DOI] [PubMed] [Google Scholar]
- 21. Heerspink HJL, List J, Perkovic V. New clinical trial for establishing drug efficacy and safety in a precision medicine era. Diabetes Obes Metab. 2018; in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Breckenridge A, Eichler HG, Jarow JP. Precision medicine and the changing role of regulatory authorities. Nat Drug Discov. 2016;15:805‐806. 10.1038/nrd.2016.06. [DOI] [PubMed] [Google Scholar]
- 23. Eichler HG, Sweeney F. The evolution of clinical trials: can we address the challenges of the future? Clin Trials. 2018;15(S1):27‐32. [DOI] [PubMed] [Google Scholar]
- 24.EMA. Qualification of novel methodologies for drug development: guidance to applicants (EMA/CHMP/SAWP/72894/2008). http://www.ema.europa.eu/docs/en_GB/document_library/Regulatory_and_procedural_guideline/2009/10/WC500004201.pdf. Accessed May 11, 2018. US_FDA.
- 25. Food and Drug Administration . 21st Century Cures Act: Qualification of Drug Development Tools. https://www.fda.gov/Drugs/DevelopmentApprovalProcess/DrugDevelopmentToolsQualificationProgram/ucm561587.htm. Accessed June 15, 2018
- 26. Salgado R, Moore H, Martens JWM, et al. Societal challenges of precision medicine: bringing order to chaos. Eur J Cancer. 2017;84:325‐334. 10.1016/j.ejca.2017.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]