

Abstract
Following up on scattered reports on interactions of conventional chaotropic ions (for example, I−, SCN−, ClO4 −) with macrocyclic host molecules, biomolecules, and hydrophobic neutral surfaces in aqueous solution, the chaotropic effect has recently emerged as a generic driving force for supramolecular assembly, orthogonal to the hydrophobic effect. The chaotropic effect becomes most effective for very large ions that extend beyond the classical Hofmeister scale and that can be referred to as superchaotropic ions (for example, borate clusters and polyoxometalates). In this Minireview, we present a continuous scale of water–solute interactions that includes the solvation of kosmotropic, chaotropic, and hydrophobic solutes, as well as the creation of void space (cavitation). Recent examples for the association of chaotropic anions to hydrophobic synthetic and biological binding sites, lipid bilayers, and surfaces are discussed.
Keywords: aqueous solvation, cavitation, large anions, superchaotropes, superchaotropic ions
1. Introduction
Hydrogen bonding and the hydrophobic effect are the two overarching concepts in the understanding of associative and dissociative processes in water, including aqueous solvation, protein folding, and molecular self‐assembly.1, 2, 3, 4, 5, 6 Specific‐ion solvation has additionally been related, since Hofmeister, to the kosmotropic (water‐structure‐forming) and chaotropic (water‐structure‐breaking) properties of ions.7, 8, 9 Following up on scattered reports on the unusual behavior of large anions,10, 11, 12, 13, 14, 15, 16, 17, 18 the propensity of these anions to associate with hydrophobic and neutral polar phases has recently been generalized and named the chaotropic effect, contrasting the hydrophobic effect as a distinct assembly motif in chemistry.3, 19, 20 The effect becomes particularly pronounced for superchaotropic ions that reach beyond the classical Hofmeister scale.10, 18 The chaotropic effect extends to molecular recognition events with macrocyclic hosts,10, 12, 21, 22, 23, 24, 25, 26, 27 biologically relevant interactions with proteins and peptides,13, 28, 29, 30, 31, 32 association to small organic molecules,10, 33 binding to membranes,17, 34, 35, 36, 37, 38 polymers,39 as well as colloids,15, 18, 40 and it manifests itself in solid‐state structures between superchaotropic ions and organic components.10, 12, 26, 41, 42 The terms “chaotropic effect” and “superchaotropic ions” have been readily absorbed by the chemical literature in different contexts.11, 12, 13, 15, 16, 17, 18, 22, 24, 28, 40, 41, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54 This relevance in several areas of chemistry reveals the chaotropic effect as a generic driving force, orthogonal to the hydrophobic effect. Its operation and impact have been rapidly unfolding during the last three years and are therefore reviewed herein.
We will first propose a conceptual rationalization of different modes of aqueous solvation that seamlessly merge the three principle solvation patterns, which span from hydrogen bonding to the hydrophobic effect and position the chaotropic effect in the transition region. The discrete solvation modes have thermochemical fingerprints, which can be used to pinpoint the dominant driving force of supramolecular assembly processes in water. The individual phenomena, for which the chaotropic effect plays a key role, will be subsequently discussed.
2. Principle Modes of Solvation in Water
The effect of different solutes on the water structure can be understood and visualized by the way that water molecules arrange themselves around a solute (Figure 1 a). Keeping in mind the tetrahedral coordination pattern of each water molecule, both the relative orientation of the tetrahedron to the convex solute surface (apical, lateral, and basal) as well as the involved interaction sites at the apex, edge, or base (hydrogen atoms or electron lone pairs) need to be specified. Such a pictorial view of water–solute interactions derives from the previously introduced views by Herzfeld55 and Collins,56 among others. We stress that even a well‐established phenomenon such as the hydrophobic effect turns out to be extremely intricate at the level of accurate molecular understanding and quantitative predictions remain subject to debate.57, 58, 59, 60, 61, 62 The same applies for the chaotropic effect addressed herein, such that the presently proposed qualitative arguments and quantitative explanations present our own current stage of rationalization.
Figure 1.

Principal aqueous solvation patterns for anions. a) Presumed favorable orientations of water molecules around a cavity and at the surface of different solutes: kosmotropic and chaotropic ions, hydrophobic molecules, and void space. Electron lone pairs are visualized in yellow. b) Extended Hofmeister scale with specification of the superchaotropic, hydrophobic ionic, and superhydrophobic regions; see Figure S1 in the Supporting Information for a two‐dimensional illustration of the solvation pattern dependence on charge density and polarizability.
Any solvation of a solute requires the creation of a cavity (Figure 1 a, right) around which water molecules arrange themselves with the sole objective of retaining most of the stability of the original hydrogen‐bonding network. In the absence of intermolecular interactions with the solute, no preferential orientation of the individual water molecules towards the surface of the cavity results, leading to both hydrogen atoms and lone pairs pointing to the convex surface. Common kosmotropic ions are characterized by a small size and large charge density. The positioning of a kosmotropic anion in the cavity results in highly directional hydrogen‐bonding or coordinative interaction, which leads to a strong apical orientation of the water molecules (Figure 1 a, left). Chaotropic ions are typically large and charge‐delocalized. The positioning of a chaotropic ion in the cavity causes less directional ion–dipole interactions to predominate, which results in a lateral orientation of the water molecules under alignment of their dipole moment (Figure 1 a, second from the left). The positioning of a hydrophobic solute in the cavity activates predominantly distance‐dependent dispersion interactions, which lead to a preferred basal orientation of the water molecules, maximizing the proximity to bonding electrons, lone pairs, and the oxygen atom (Figure 1 a, second from the right). For cations instead of anions, the principle orientations of the water molecules (apical versus lateral) remain the same except that the interactions occur through the lone pairs instead of the hydrogens, with only the latter variant shown in Figure 1 a.
The pictorial representations of the different aqueous solvation modes allow us to advance a continuum model for solvation, which not only spans from kosmotropes to chaotropes but extends further to hydrophobes and, ultimately, an empty cavity (Figure 1 b).63 This continuum view of solvation corresponds to a gradual change of the preferred orientation of the inner‐sphere water molecules from apical (left) to lateral to basal. The continuum includes as prominent cases the established effects of hydrogen bonding (coordinative bonding) and the hydrophobic effect. As an additional effect, the chaotropic effect emerges in the transition region beyond the classical Hofmeister scale (kosmotrope–chaotrope) and the recognized hydrophobic domain, which spans from hydrophobic ions to neutral species. Others and we have recently introduced the term “superchaotropic”, akin to superhydrophobic, for those ions that exceed common chaotropic properties.10, 15, 16, 18, 23, 41, 64 Superchaotropic ions do not display hydrophobic properties by the common definitions but instead give rise to peculiar effects that are summarized in this review and can be understood as being a consequence of the chaotropic effect. The continuum scale for aqueous solvation can be further extended to superhydrophobic species (negligible dispersion) up to vacuum (no interaction); the latter describes the solvation around a formed void in water (cavitation). It is consequential that the water orientations induced by the different types of dominant intermolecular interactions in the first hydration shell (Figure 1 a) have characteristic consequences for the hydrogen‐bonding network in the entire solvation shell. This accounts for many of the physicochemical and thermochemical properties of the associated solvation processes, including the entropic signature of the hydrophobic effect,3, 20, 65, 66 the hydrogen‐bond‐forming or disrupting effect of kosmotropes and chaotropes,67 as well as the associated enthalpic signatures.68
3. Thermochemical Hydration Characteristics
The immersion of a hydrophobic species such as an alkane or noble gas in water is classically thought to be accompanied by a structuring of the solvation shell around the solute and a negative entropic contribution of hydration (Frank–Evans model).69 Accordingly, the association of such a hydrophobic species with an organic binding site, such as a macrocyclic cavity, should be characterized by an entropic driving force, which can be traced to the destruction of the structured water shell around the solvated hydrophobic solute. This presents the signature of the classical hydrophobic effect.3 For example, the aqueous association of the highly hydrophobic adamantane and triamantane cores to γ‐cyclodextrin (γ‐CD), a prototypal macrocyclic host, shows this thermochemical behavior (Figure 2).70, 71, 72, 73 Although the iceberg model suggested by Frank and Evans and the related explanation for the hydrophobic effect have been much debated, it remains appealing today because of the constant and recent experimental support in its favor. For example, the water molecules around noble gases have recently been spectroscopically demonstrated to resemble the structure of ice74, 75 and the rotational dynamics of water molecules in the solvation shell around hydrophobes is reduced.76, 77
Figure 2.

Enthalpy–entropy compensation plot for γ‐CD complexes with dodecaborate anions and previously reported γ‐CD complexes with diverse organic guests.10
Recently, we have observed that the unexpectedly strong binding of large dodecaborate anions (B12X12 2−) to the same macrocycle, γ‐CD, displays a diametric thermochemical signature; that is, it is enthalpically driven with a negative entropic component (Figure 2).10, 23 Since the driving force for this aqueous binding phenomenon could not be related to the hydrophobic effect (the dianions themselves are not hydrophobic species, see below), we considered kosmotropic and chaotropic ion properties as spanned by the Hofmeister series (SO4 2−<F−<HPO4 2−<CH3COO−<Cl−<Br−<NO3 −<I−<ClO4 −<SCN−), a scale originally defined on the basis of salting‐out and salting‐in effects of ions on egg whites and serum proteins.8 Indeed, it turned out that the dodecaborate clusters behave as superchaotropic anions; that is, their chaotropic properties such as salting‐in effects exceed those of accepted chaotropes (ClO4 −, SCN−, PF6 −≪large anions). For example, they significantly increase the solubility of adenine and riboflavin, two established salting‐in standards.10 Accordingly, we proposed a new effect, the chaotropic effect, as an alternative driving force, which drives the association of (super)chaotropic ions with hydrophobic and neutral matter. The chaotropic effect needs to be differentiated from unspecific Hofmeister effects, in that it accounts for specific chaotrope–solute interactions.
The thermochemical fingerprint for the binding of chaotropic anions can be contrasted to that for the binding of hydrophobic species. Even though both effects relate to desolvation, the hydration shell around a hydrophobe is highly structured while that around a chaotrope is less structured and less hydrogen bonded, relative to bulk water. The desolvation process is therefore classically entropically driven in the case of hydrophobes,3 while for chaotropes a dominant negative enthalpic component and an unfavorable entropic term applies. Upon desolvation, the hydration shell around the chaotrope is stripped off and allowed to regain the hydrogen bonds of the aqueous bulk.10 This rationalization is fully in line with the original classification of chaotropic ions as being “water‐structure breakers”,78 such that their desolvation (due to binding to other species) leads formally to a “water structure recovery”.10 As is the case with the debate on the origin of the hydrophobic effect, the interpretation of Hofmeister effects has been similarly controversial, with undeniable pieces of evidence in favor of water‐structure breaking effects of chaotropic ions in their solvation shell being known, for example, trends of viscosity B‐coefficients.79
Marcus has devised, based on experimental ion properties, an empirical scale for water‐structural entropies for ionic hydration (ΔS struct, listed in Table 1 for selected anions), which allows also an assessment of the (kosmotropic and chaotropic) properties of ions and of the thermochemical trends related to their (de)hydration.68, 80 In a chemometric sense, Table 1 can be viewed as a chaotropicity scale, in this case for anions. If the ΔS struct values are positive, the water structure in their surrounding decreases, which can be (again empirically) converted into an effective loss of hydrogen bonds in the entire solvation shell around the ion (negative values of ΔHB).68 Ions unambiguously qualify as chaotropes if ΔHB<−1 (or TΔS struct>5 kcal mol−1); superchaotropic anions, such as borate clusters, reach values lower than circa −2 (or TΔS struct>9 kcal mol−1).
Table 1.
Chaotropicity scale based on the ion hydration parameters proposed by Marcus (Refs. 68, 80): Water‐structural entropies of different anions (TΔS struct), their size (radius, r), and net effects on the number of surrounding hydrogen bonds (ΔHB).
| anion | r [pm][a] | TΔS struct [kcal mol−1][b] | ΔHB [c] |
|---|---|---|---|
| kosmotropes | |||
| SO4 2− | 230 | −6.7 | 0.78 |
| HPO4 2− | 238 | −4.4 | 0.46 |
| CO3 2− | 178 | −4.0 | 0.40 |
| F− | 133 | −2.1 | 0.12 |
| chaotropes | |||
| Br− | 196 | 5.8 | −1.01 |
| SCN− | 213 | 5.9 | −1.03 |
| BF4 − | 230 | 6.6 | −1.12 |
| ClO4 − | 240 | 7.6 | −1.27 |
| I− | 220 | 8.3 | −1.37 |
| PF6 − | 295[d] | 8.7 | −1.43 (1.93)[e] |
| superchaotropes | |||
| Fe(CN)6 3− | 373 | 12.8 | −2.01[e] |
| S2O8 2− | 300 | 13.0 | −2.04[e] |
| S4O6 2− | 310 | 13.2 | −2.06[e] |
| B10H10 2− | 393[f] | 11.5 | −1.83 |
| B12H12 2− | 400[f] | 14.8 | −2.31 |
| B12F12 2− | 430[f] | 12.6 | −1.98 |
| B12Cl12 2− | 525[f] | 15.3 | −2.37 |
| B12Br12 2− | 560[f] | 16.3 | −2.51 |
| B12I12 2− | 590[f] | 17.1 | −2.63 |
| PMo12O40 3− | 629[f] | 18.2 | −2.79 |
| PW12O40 3−[g] | 654[f] | 18.9 | −2.89 |
| P2W18O62 6−[h] | 701[f] | 20.3 | −3.08 |
[a] As tabulated in Refs. 80, 124. [b] Calculated according to Marcus, see Refs. 80, 124, values for ions for which no hydration enthalpies are known were interpolated (for PF6 −) or extrapolated (for borate clusters and POMs) from a correlation of ΔS struct versus r for chaotropic anions listed in this table as well as the data for SiF6 −, PdCl6 2−, PtCl6 2−, Co(CN)6 3−, and Fe(CN)6 4− (ΔS struct=4.48+0.399r, n=14, r=0.77). [c] From eq. 25 in Ref. 68, there abbreviated as ΔG HB. [d] From Ref. 125. [e] From Ref. 124, multiplied with the correction factor of 1.25 subsequently introduced in Ref. 68. [f] Calculated from the diameter (the distance between the outer atoms including the vdW radii) obtained from their geometry‐optimized structures (for borate clusters) or XRD structures (for POMs). [g] Keggin‐type POM. [h] Dawson‐type POM.
We further concluded that the chaotropic effect presents a generic driving force that accounts for the intrinsic affinity of chaotropic ions, prominently anions, for neutral organic or inorganic matter, including concave macrocyclic binding sites, lipid bilayers, membranes, and polymers.11, 12, 13, 16, 17, 18, 22, 24, 28, 40, 43, 44, 45, 46, 47, 48 It should be noted that indications for the affinity of anions to hydrophobic binding sites existed early in the literature81, 82, 83, 84, 85, 86, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96, 97, 98, 99, 100, 101, 102, 103 but were in part attributed to a putative hydrophobic (or lipophilic) property of large anions, implying the hydrophobic effect as driving force. The hydrophobic and chaotropic effect can and need to be conceptually distinguished for the reason stated above and also because the hydrophobic nature of a solute is subject to a conventional definition, which chaotropic ions do not fulfill. Specifically, as shown in Table 2, hydrophobic species display positive free energies of hydration, small enthalpies of hydration, and large positive heat capacities of hydration,104, 105, 106 prerequisites that are not fulfilled by chaotropic ions. Rather, chaotropic anions can be classified as hydrophilic on ion‐solvation scales.107, 108 Classifications of chaotropic ions as hydrophobic, occasionally found in the literature,109, 110, 111, 112, 113, 114, 115, 116, 117, 118, 119 are unfortunate because they may lead to incorrect assignments in terms of the underlying driving force for associative processes. Vice versa, hydrophobic ions do actually exist, such as BPh4 −, (C2F5)3PF3 −, and AsPh4 +,80, 120, 121, 122 and it is also important not to label these as being superchaotropic47, 51, 52, 123 when sequences of ionic properties, scales for ionic solvation, or reasons for aqueous assembly processes are being developed. Superchaotropic and hydrophobic ions are neighbors on the continuum solvation scale in Figure 1 b (because they are both large and highly polarizable), but they would differ in terms of their surface charge density and dominant water‐solvation pattern (Figure 1 a). This is reflected, among other things, in hydration thermochemical data, which in the case of hydrophobic ions are diagnostic for hydrophobic species (Table 2). Note that the primary thermochemical data in Table 2 do not allow a ready distinction between kosmotropic and chaotropic character, which led Marcus to introduce the water‐structural entropies as diagnostic measure for this ion property (Table 1), with water‐structural entropy (ΔS struct) being a function of hydration entropy of the ion (ΔS hyd) and its radius (r).68
Table 2.
Thermodynamic hydration data for selected solutes.
| kosmotropic anion |
chaotropic anion |
superchaotropic anion |
hydrophobic ion |
hydrophobic neutral species |
|||
|---|---|---|---|---|---|---|---|
| F− | SCN− | ClO4 − | B12H12 2− | BPh4 − | AsPh4 + | CH4 | |
| ΔG hyd [a] | −112.1[b] | −69.6[b] | −54.7[b] | −140[c] | 10.8[b] | 10.3[b] | 2.0[d] |
| ΔH hyd [a] | −121.9[b] | −74.3[b] | −58.8[b] | −145[e] | −11.2[b] | −11.2[b] | −2.6[d] |
| TΔS hyd [a] | −9.8[b] | −4.7[b] | −4.1[b] | −5[f] | −22.0[b] | −21.5[b] | −4.6[d] |
| ΔhydcP [g] | −14.1[h] | 1.4[h] | −2.2[h] | – | 186.7[h] | 192.0[h] | 52.0[d] |
It follows that the chaotropic and hydrophobic effect as driving forces in aqueous solution can be empirically and experimentally distinguished by inspection of the hydration characteristics of the involved species (Tables 1 and 2) and also by measurement of the thermodynamics (Figure 2) of the corresponding supramolecular assembly processes.
4. Examples
The chaotropic effect accounts for the association of chaotropic anions to neutral binding sites, surfaces, and phases. Following its nature as a generic driving force in aqueous solution, the chaotropic effect has recently been shown to have an impact on diverse areas of chemistry, ranging from molecular recognition and supramolecular chemistry to biologically relevant interactions with membranes, lipids, and proteins, to polymer and colloid chemistry. While scattered indications for such interactions of chaotropic ions, predominantly anions, were known, the conceptualization and generalization of the chaotropic effect has only emerged very recently, made possible by a sequence of striking observations for very large anions such as borate clusters and polyoxometalates (POMs) for which the effect is particularly pronounced. Accordingly, the unfolding of the chaotropic effect is tied to the appreciation of the superchaotropic character of these large anions.
4.1. Supramolecular Chemistry
Macrocyclic hosts are popular in supramolecular chemistry because they possess hydrophobic cavities that mimic biological binding sites, and which can complex hydrophobic guest molecules. Despite the hydrophobic nature of these cavities, early indications for the competitive binding of ions exist.96, 99, 130 Taraszewska and Wójcik were the first to correlate the small but significant binding of simple anions to α‐CD and β‐CD with the Hofmeister character of the anions.99 Recently, while investigating interactions of large dodecaborate cluster dianions of the type B12X12 2− (X=H, Cl, Br, and I) with γ‐CD, we observed exceedingly high binding constants,10 which for the first time rivaled the binding of highly hydrophobic guest molecules such as adamantane and triamantane.70, 71 This study has led to the introduction of the terms chaotropic effect and superchaotropic ions.10 We contrasted the distinct thermodynamic fingerprints of host–guest complexation driven by the chaotropic versus the hydrophobic effect and traced the negative enthalpies for binding of the large cluster anions back to the desolvation of these superchaotropic ions. We rationalized the affinity trends of different chaotropic and superchaotropic anions in terms of their water‐structuring properties as empirically quantified by Marcus (see Table 1). Host–guest dispersion interactions may contribute to the chaotropic effect, while for the smaller macrocyclic cavities, the release of high‐energy water may also play a supportive role (see the Supporting Information for interplay and differentiation of the chaotropic effect from the non‐classical hydrophobic effect and dispersion).
Understanding the driving forces between ions and concave binding sites is therefore of prime importance for the supramolecular design of synthetic receptors with defined affinity and selectivity. Accordingly, the unexpectedly high binding affinities of superchaotropic anions and the chaotropic effect as a generic driving force have recently found their entry into the important area of anion recognition.44, 65, 117, 131, 132, 133, 134, 135, 136, 137, 138, 139, 140, 141, 142, 143
In follow‐up studies with inclusion complexes, the affinity of dodecaborate clusters to cyclodextrins (CDs) has been extended to the larger CD homologues (Figure 3)23 as well as other borate clusters,14, 144 both in solution and in the gas phase.14, 23, 24 The binding propensity of borate clusters has also been demonstrated for calixarenes21 and a tetrathiafulvalene host24 as an alternative macrocyclic host (Figure 3). Independent evidence for an intrinsic affinity of other large anions, namely the classical Keggin POM ion (PMo12O40 3−), to γ‐CD had been obtained by Stoddart and co‐workers,26 and we concluded that the arguments in terms of a chaotropic effect being operative are also transferable to this popular class of large anions.10 The actual affinities of POMs with CDs have been studied in detail by Cadot and co‐workers12, 22 as well as others.25 The chaotropic effect also accounts for the recently reported binding of chaotropic anions to biotinuril,98 bambusuril,45, 46, 97, 145 hemicucurbituril,146 a tricarbazolo triazolophane (not studied in water),147 and the so‐called octa‐acid (see Figure 3 for structures).82, 83, 148, 149, 150, 151 The studied chaotropic anions included not only those in Table 1 but also N3 −, CNO−, SeCN−, IO4 −, ReO4 −, and SbF6 −. Noteworthy, the thermodynamic fingerprint (enthalpically driven processes with a negative entropic contribution) is identical in all cases, regardless of which type of superchaotropic anion and which macrocycle are being used.10, 12, 22, 23, 26, 97, 98 Finally, the inclusion preference of metallo‐macrocyclic cage compounds towards chaotropic anions stands out as well but has not been mechanistically rationalized in comparable detail.142, 143, 152
Figure 3.
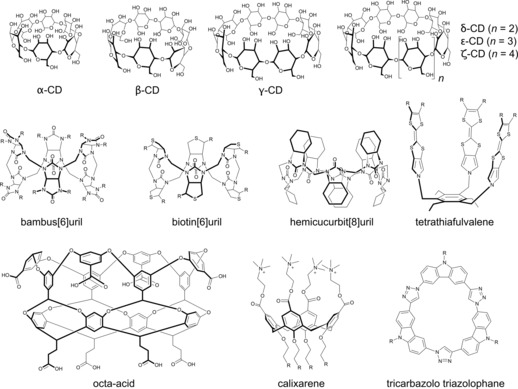
Macrocycles with intrinsic affinity for chaotropic and superchaotropic anions.
In many cases, the aqueous solution interactions of large anions with macrocyclic hosts are retained in their solid‐state structures. Inclusion complexes are obtained (Figure 4), mostly with partial inclusion owing to the large anion sizes and frequently host–guest complexes of the 2:1 type. This applies to γ‐CD/B12Br12 2− (Figure 4 a),10 γ‐CD/PMo12O40 3− (Figure 4 b),26 and β‐CD/PMo12O40 3− (Figure 4 c).26 The chaotropic effect can also be exploited for the construction of more complex hybrid and hierarchical assemblies, for example, when superchaotropic Dawson‐type anions ([P2W18O62]6−) and cations ([Ta6Br12(H2O)6]2+) interact with γ‐CD (Figure 4 d).12 Moussawi et al. extended the design to giant Mo‐blue ring‐shaped anions, [Mo154O462H14(H2O)70]14− (Mo154), which themselves are sufficiently large to encapsulate an entire γ‐CD/[P2W18O62]6− inclusion complex (Figure 4 e); the CDs were also found to associate to the outer surface of the large ring‐cluster.22 Most recently, Ivanov et al. reported strong binding between γ‐CD and chalcogenide cluster anions of the type [Re6Q8(CN)6]4− with Q=S, Se, and Te; their affinities increased in the order S<Se≪Te and the entropy–enthalpy correlation resembled that of other superchaotropic anions, namely B12X12 2−.27
Figure 4.
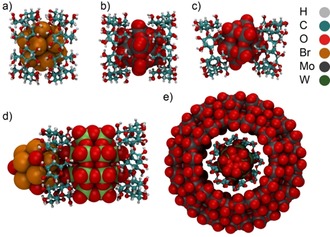
XRD structures for superchaotrope/CD complexes a) γ‐CD/B12Br12 2−, b) γ‐CD/PMo12O40 3−, c) β‐CD/PMo12O40 3−, d) γ‐CD/[P2W18O62]6−/[Ta6Br12(H2O)6]2+, and e) [Mo154]/γ‐CD/[P2W18O62]6−.
The B12H11SH2− ion has recently shown potential as an inorganic capping ligand to stabilize gold nanoparticles.21, 153 This allows for the creation of hierarchical supramolecular architectures on the surface of gold nanoparticles through strong host–guest complexation between the dodecaborate anions and an amphiphilic calixarene (Figure 5 a).21
Figure 5.
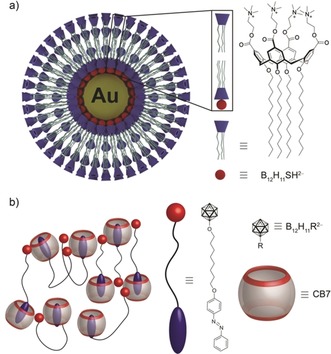
Examples of hierarchical and orthogonal solution‐phase architectures exploiting the chaotropic effect as an assembly motif: a) assembly of an amphiphilic calixarene on dodecaborate‐stabilized gold nanoparticles and b) assembly of CB7 in the presence of an amphiphilic dodecaborate‐functionalized azobenzene. The red residue illustrates a superchaotropic unit as recognition site to the exterior surface of CB7 and the blue unit represents an auxiliary hydrophobic unit that shows a preferential affinity for inclusion complexation with CB7.
The chaotropic effect accounts for the high affinity of superchaotropic anions to hydrophobic surfaces in general. It is not limited to the interaction with macrocyclic cavities (Figure 3) and the formation of inclusion complexes (Figure 4) but also applies similarly to the interactions with convex surfaces (Figure 6), which accounts for the intrinsic propensity of these large anions to form also exclusion complexes. Indeed, many studies have described their binding to the exterior of macrocycles,41, 64, 84, 86, 87, 88, 90, 91, 92, 93, 94, 154 where the chaotropic effect applies as well. Most of these cases involve macrocyclic hosts with relatively small cavity size, such that inclusion‐type complexes are not possible.41, 64, 94, 154 For example, superchaotropic anions, such as POMs and dodecaborate clusters, associate to the hydrophobic exterior of cucurbit[n]uril (CBn) macrocycles (Figure 6).41, 64, 94, 154 The large size of superchaotropic anions allows them to interact with several macrocycles in a multidentate manner, which can efficiently induce their precipitation, for example, of CBn. This is counterintuitive because conventional chaotropes are otherwise known to increase the solubility of organic solutes (salting‐in Hofmeister effect).156 The combination of the chaotropic and hydrophobic effect as assembly motifs has provided a new strategy for engineering multi‐responsive supramolecular networks in water (see Figure 5 b and Supporting Information for details).41, 64
Figure 6.
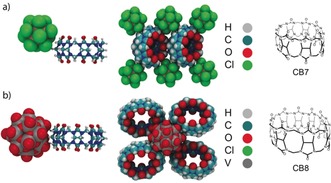
XRD structures of CBn/superchaotropic anion exclusion complexes a) CB7/B12Cl12 2− and b) CB8/[H2O⊂VIV 18O42]12−.
4.2. Biologically Relevant Interactions with Proteins, Peptides, and Membranes
The effect of salts on the water‐solubility and stability of proteins dates back to the original work by Hofmeister.8 Since then, it has become obvious that these salting‐in and salting‐out effects are unlikely to be primarily related to changes in the bulk water structure; instead, at least in the case of chaotropic anions, direct interactions between the ions and the protein or peptide play also an important role.9, 157, 158 These interactions, which have been demonstrated computationally31, 32, 159 and experimentally,7, 31, 32, 56, 160 are fully in line with the chaotropic effect, which predicts an intrinsic affinity between chaotropic ions and hydrophobic residues, in this case of biopolymers. These biologically relevant interactions bear similarities to the host–guest complexation processes discussed above because binding of chaotropic anions to specific binding sites in the protein is also observed. For example, Whitesides and co‐workers have shown that weakly hydrated anions (chaotropes) bind to cavities on the surfaces of proteins (the binding pocket of human carbonic anhydrase II) with higher affinity than strongly hydrated anions (kosmotropes).161 The thermochemical behavior (Figure 7) for the association process, namely large favorable enthalpic and unfavorable entropic contributions, is in line with the chaotropic effect as a driving force. The authors have also pointed out how the intrinsic affinity of chaotropic anions to proteins relates to other prevalent empirical concepts, such as the law of matching water affinities.161 It is worth noting that the large favorable enthalpic contributions for the interactions of large anions with their binding sites are frequently compensated by unfavorable entropic terms resulting from the formation of structured complexes or aggregates.10, 162
Figure 7.

Thermodynamics of anion binding to human carbonic anhydrase II: a) Plot showing the thermodynamic parameters for the association of different anions (298.15 K, pH 7.6, 10 mm sodium phosphate buffer). b) Plot of the free energies of binding (ΔG°bind,anion) versus the free energies of hydration (ΔG°hydration). Reprinted with permission from Ref. 161.
Indications of interactions of large anions with proteins were known even before their superchaotropic character had been introduced.29, 163, 164, 165, 166 The classification of dodecaborate clusters in 201510 sparked several lines of follow‐up work directed towards their direct binding to proteins and led to an interpretation in terms of the chaotropic effect.13, 28 For example, Kuperman et al. have recently investigated the binding of perhalogenated closo‐borates (B10X10 2− and B12X12 2−; X: Cl, Br, and I) to bovine and human serum albumins.28 The measured binding constants were higher than the values obtained with the parent hydrogen cluster165 and varied from 104 to 106 m −1. Goszczyński et al. have expanded the interactions with serum albumin from dodecaborates to other hydrophobic or superchaotropic boron cluster types, namely carboranes and metallacarboranes;13 they established the affinity order, dodecaborate anion (B12H12 2−, superchaotropic)≪carborane (C2B10H12, hydrophobic)<metallacarborane ([M(C2B9H11)2 −], superchaotropic).13 The interactions of POMs with peptides and proteins, which have recently been reviewed,167 are also a consequence of the superchaotropic character and the associated chaotropic effect, as evidenced by the thermodynamic fingerprint (enthalpically driven binding processes with large entropic penalty).164 In contrast, some of the interactions of organic ions (AsPh4 + and BPh4 −) with proteins are presumably accounted for in terms of their hydrophobic character.47, 51
Numerous examples for unspecific interactions of chaotropic anions with biological membranes are also known;7, 162, 168, 169, 170, 171, 172, 173, 174 in particular, their membrane‐disrupting properties are noteworthy.171, 172 Gabel and co‐workers investigated the interaction between dodecaborate cluster dianions and lipid bilayers;34, 35, 36, 175 at high salt concentrations they observed changes in the morphology of liposomes as well as bilayer leakage (see Figure 8 for an example). The release of the liposomal contents was attributed to interactions between the dodecaborate clusters and lipids, which leads to either the formation of pores or the destruction of the liposomes.36 In hindsight, the reported interactions of chaotropic anions with membranes are a prototypal example of the chaotropic effect, which predicts an intrinsic affinity of these anions to associate with hydrophobic phases, such as the interior of bilayer membranes.
Figure 8.
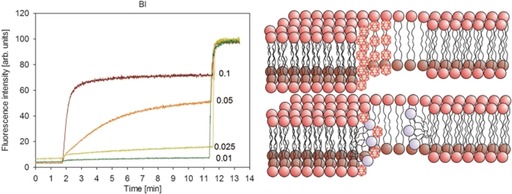
Leakage of carboxyfluorescein (CF) induced by B12I12 2− in DSPC liposomes at 37 °C. Concentrations of clusters for the individual traces in the graph are given on the left (mm). Suggested mechanism of the interaction between dodecaborate clusters and liposomes (right). Reprinted with permission from Ref. 36.
Interactions between POM anions and membranes have also been reported.37, 162, 176, 177, 178 Jing et al. reported destabilizing effects of a POM macro‐ion on the phase and morphology of lipid bilayers.162 Nabika et al. reported leakage of egg‐phosphatidylcholine vesicles caused by the Keggin‐type POM SiW12O40 4− and PW12O40 3−;176 the former anion was also found to interact with a model cell membrane.177 Most recently, Nabika and co‐workers have traced these interactions, experimentally corroborated through surface–pressure isotherms of ionic and zwitterionic lipids, to the superchaotropic nature of these anions.17
4.3. Interactions with Interfaces and Surfaces
There have been multiple indications of interactions of chaotropic anions with neutral interfaces and surfaces, involving both hydrophobic and polar interfaces as well as hard and soft surfaces. The examples extend from interactions of conventional chaotropic anions with colloids121 to their accumulation at the liquid–air interface.179, 180, 181 Recently, evidence for such interactions has been extended to large anions, including dodecaborate clusters (B12X12 2− and B12X11Y2−; X=H, Cl, Br, I and Y=SH, OH, NR3 +) interacting with hydrophilic column materials (Superdex 200, Sepharose 4B, Sephadex G‐50, Sephadex G‐100, alumina, silica gel, and anion exchange resin)182 and carborane clusters (mainly metallacarboranes) accumulating at water–lipid and water–air interfaces.183, 184 Moreover, akin to the tendency of hydrophobic and classical amphiphilic molecules to form self‐assemblies in water, carborane clusters have been found to form micelle‐type self‐assemblies in water and to show indications of surfactant behavior.183, 185, 186, 187, 188 Halogenated borate clusters accumulate on surfaces by simultaneous adsorption of small neutral molecules.189
With the description of the chaotropic effect as a generic driving force for the assembly of large anions,10 these processes can now be described within a uniform mechanistic framework. Indeed, the previously determined thermochemical signature in the solid‐phase binding of dodecaborate cluster anions (enthalpically driven binding with large entropic penalty)182 fully agrees with this new interpretation. Moreover, in two independent lines of investigation, Matějíček and co‐workers have established the operation of the chaotropic effect for boron cluster anions,11, 40, 53, 190, 191 while the group of Bauduin and Diat have expanded the observed self‐assembly and surface‐assembly tendencies to POMs as superchaotropic anions.15, 16, 18, 192
In detail, POMs adsorb at micellar surfaces and on monolayers of nonionic surfactants, as shown by using small‐angle X‐ray scattering (SAXS) and ion flotation techniques.16 Cloud point measurements (as a measure of the adsorption strength/association constant of additives with surfactants)160 of a triethylene glycol monomethyl ether solution in the presence and absence of different anions allowed the classification of POMs (SiW12O40 4− and PW12O40 3−) as superchaotropic anions, as their effect on the cloud point exceeded those of classical chaotropic anions, indicating strong adsorption of the anionic POMs onto the micellar surfaces (Figure 9).16 Most recently, other POMs have also been tested and sorted according to their superchaotropic character, PW12O40 3−>PMO12O40 3−>SiW12O40 4−>P2W18O62 6−>P2W17VO62 7−≫SCN−.18 The superchaotropic nature of the POM anions was attributed to the efficient charge delocalization over their large surface area (volume), typically at least in the nanometer range. This allows the release of hydration water molecules around the POMs upon adsorption or association, which provides the corresponding driving force,18 in line with the entropic component of the water‐structure recovery expected for the chaotropic effect.10 It should be noted that this alternative experimental scale for (super)chaotropicity is complementary to the empirical scale based on Marcus theory (see Table 1).
Figure 9.

Left: Cloud points of a 60 mm triethylene glycol monomethyl ether solution in the presence of POMs: SiW4− (SiW12O40 4−) and PW3− (PW12O40 3−) and sodium salts with various representative anions of the Hofmeister series: SCN− (chaotropic), WO4 − (kosmotropic), and Cl− (intermediate between salting‐in and salting‐out). Inset: Ion‐containing surfactant solution at temperatures below (left) and above (right) the cloud point. Right: Schematic of the adsorption of a POM anion on a hydrophilic interface covered by a polyethoxylated surfactant (right). Reprinted with permission from Ref. 16.
The tendency of different boron clusters (B10H10 2−, B12H12 2−, B12H11SH2−, 1‐carbadodecaborate, and cobalt bis(1,2‐dicarbollide)) to self‐assemble into aggregates and colloids in water has been detailed by Matějíček and co‐workers,11, 188, 190, 193 while the group of Bauduin and Diat have described the formation of charged nano‐colloids from POMs.15 The unusual behavior of anionic boron clusters can again be attributed to the chaotropic effect, which complements the classical hydrophobic effect as a supramolecular assembly and self‐assembly motif in water (Figure 10).40 The behavior of POMs has also been related to their superchaotropic character.16, 18 The intrinsic self‐assembly tendency of both superchaotropic anions (borate clusters and POMs) accounts also for their high propensity to accumulate at the water–air surface; this leads to surfactant‐like behavior, such as a reduction of the surface tension.11, 40, 53, 190, 191 Accordingly, because they lack the polar head group/hydrophobic tail design of classical amphiphiles, they have also been classified as non‐classical surfactants or “intrinsic amphiphiles”.11, 40, 53, 190, 191
Figure 10.
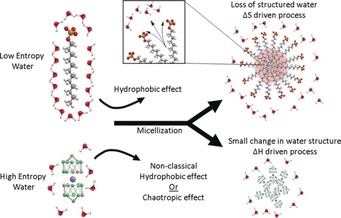
Micellization can be driven by the classical hydrophobic effect (top) or the chaotropic effect (or a combination of the chaotropic and non‐classical hydrophobic effect, bottom) with emphasized changes of the hydration shells for the classical surfactant and the large anion cobalt bis(1,2‐dicarbollide), COSAN. For a specification of the non‐classical hydrophobic effect, see the Supporting Information. Reprinted with permission from Ref. 40.
5. Conclusions and Outlook
Within less than three years, the chaotropic effect has entered the chemical literature as a generic driving force for diverse assembly processes involving superchaotropic ions. The chaotropic effect can be differentiated from the hydrophobic effect in terms of its characteristic hydration pattern and the thermochemical fingerprint of its associated binding phenomena. Examples include host–guest inclusion complexation, anion recognition, exclusion complex formation and associated solid‐state structures, direct interactions with proteins and membranes, interactions with other interfaces and surfaces, as well as self‐assembly. Future research on the chaotropic effect and superchaotropic anions will entail the dissection of dispersion interactions, the design of supramolecular architectures exploiting the hydrophobic and chaotropic effects as orthogonal assembly motifs, and their interactions with inorganic materials.
Conflict of interest
The authors declare no conflict of interest.
Biographical Information
Khaleel I. Assaf was born in Amman, Jordan, in 1985. He graduated with a MSc degree from the Hashemite University, Jordan. In 2010, he was awarded a scholarship from the German Academic Exchange Service (DAAD) to start his PhD studies at Jacobs University Bremen, Germany, under the supervision of Prof. Werner Nau. He subsequently continued as a postdoctoral fellow at the same university. His research interests lie in the areas of supramolecular chemistry, in particular molecular recognition and the design of advanced functional materials.

Biographical Information
Werner M. Nau was born in Fulda, Germany, in 1968. He started his chemistry studies in 1987 at the University of Würzburg, Germany, obtained his MSc in 1991 at St. Francis Xavier University, Canada, and his PhD at the University of Würzburg in 1994 with Waldemar Adam in organic chemistry. Following his postdoc at the University of Ottawa, Canada, in 1994/95 with J.C. “Tito” Scaiano in photochemistry, he completed his habilitation in physical chemistry with J. Wirz at the University of Basel, Switzerland, where he became Assistant Professor in 2000. Since 2002, he is Professor of Chemistry at Jacobs University Bremen, Germany, and, since 2012, Dean of the faculty. His research interests include supramolecular chemistry, photochemistry, and physical organic chemistry, focusing on water as solvent.

Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
W.M.N. and K.I.A. are grateful to the DFG for grant NA‐686/8 within the priority program SPP 1807 “Control of London Dispersion Interactions in Molecular Chemistry”. The authors would like to thank Detlef Gabel for stimulating the work with borate clusters.
K. I. Assaf, W. M. Nau, Angew. Chem. Int. Ed. 2018, 57, 13968.
References
- 1. Sherrington D. C., Taskinen K. A., Chem. Soc. Rev. 2001, 30, 83–93. [Google Scholar]
- 2. MacGillivray L. R., Atwood J. L., Nature 1997, 389, 469. [Google Scholar]
- 3. Biedermann F., Nau W. M., Schneider H.-J., Angew. Chem. Int. Ed. 2014, 53, 11158–11171; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 11338–11352. [Google Scholar]
- 4. Schneider H.-J., Acc. Chem. Res. 2015, 48, 1815–1822. [DOI] [PubMed] [Google Scholar]
- 5. Blokzijl W., Engberts J. B. F. N., Angew. Chem. Int. Ed. Engl. 1993, 32, 1545–1579; [Google Scholar]; Angew. Chem. 1993, 105, 1610–1650. [Google Scholar]
- 6. Snyder P. W., Lockett M. R., Moustakas D. T., Whitesides G. M., Eur. Phys. J. Spec. Top. 2014, 223, 853–891. [Google Scholar]
- 7. Zhang Y., Cremer P. S., Curr. Opin. Chem. Biol. 2006, 10, 658–663. [DOI] [PubMed] [Google Scholar]
- 8. Hofmeister F., Arch. Exp. Pathol. Pharmakol. 1888, 24, 247–260. [Google Scholar]
- 9. Kunz W., Lo Nostro P., Ninham B. W., Curr. Opin. Colloid Interface Sci. 2004, 9, 1–18. [Google Scholar]
- 10. Assaf K. I., Ural M. S., Pan F., Georgiev T., Simova S., Rissanen K., Gabel D., Nau W. M., Angew. Chem. Int. Ed. 2015, 54, 6852–6856; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 6956–6960. [Google Scholar]
- 11. Ďorďovič V., Tošner Z., Uchman M., Zhigunov A., Reza M., Ruokolainen J., Pramanik G., Cígler P., Kalíková K., Gradzielski M., Matějíček P., Langmuir 2016, 32, 6713–6722. [DOI] [PubMed] [Google Scholar]
- 12. Moussawi M. A., Leclerc-Laronze N., Floquet S., Abramov P. A., Sokolov M. N., Cordier S., Ponchel A., Monflier E., Bricout H., Landy D., Haouas M., Marrot J., Cadot E., J. Am. Chem. Soc. 2017, 139, 12793–12803. [DOI] [PubMed] [Google Scholar]
- 13. Goszczyński T. M., Fink K., Kowalski K., Leśnikowski Z. J., Boratyński J., Sci. Rep. 2017, 7, 9800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Eyrilmez S. M., Bernhardt E., Dávalos J. Z., Lepšík M., Hobza P., Assaf K. I., Nau W. M., Holub J., Oliva-Enrich J. M., Fanfrlík J., Hnyk D., Phys. Chem. Chem. Phys. 2017, 19, 11748–11752. [DOI] [PubMed] [Google Scholar]
- 15. Malinenko A., Jonchère A., Girard L., Parrès-Maynadié S., Diat O., Bauduin P., Langmuir 2018, 34, 2026–2038. [DOI] [PubMed] [Google Scholar]
- 16. Naskar B., Diat O., Nardello-Rataj V., Bauduin P., J. Phys. Chem. C 2015, 119, 20985–20992. [Google Scholar]
- 17. Kobayashi D., Nakahara H., Shibata O., Unoura K., Nabika H., J. Phys. Chem. C 2017, 121, 12895–12902. [Google Scholar]
- 18. Buchecker T., Schmid P., Renaudineau S., Diat O., Proust A., Pfitzner A., Bauduin P., Chem. Commun. 2018, 54, 1833–1836. [DOI] [PubMed] [Google Scholar]
- 19. Schneider H.-J., Angew. Chem. Int. Ed. 2009, 48, 3924–3977; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 3982–4036. [Google Scholar]
- 20. Chandler D., Nature 2005, 437, 640–647. [DOI] [PubMed] [Google Scholar]
- 21. Assaf K. I., Hennig A., Peng S., Guo D.-S., Gabel D., Nau W. M., Chem. Commun. 2017, 53, 4616–4619. [DOI] [PubMed] [Google Scholar]
- 22. Moussawi M. A., Haouas M., Floquet S., Shepard W. E., Abramov P. A., Sokolov M. N., Fedin V. P., Cordier S., Ponchel A., Monflier E., Marrot J., Cadot E., J. Am. Chem. Soc. 2017, 139, 14376–14379. [DOI] [PubMed] [Google Scholar]
- 23. Assaf K. I., Gabel D., Zimmermann W., Nau W. M., Org. Biomol. Chem. 2016, 14, 7702–7706. [DOI] [PubMed] [Google Scholar]
- 24. Warneke J., Jenne C., Bernarding J., Azov V. A., Plaumann M., Chem. Commun. 2016, 52, 6300–6303. [DOI] [PubMed] [Google Scholar]
- 25. Zhang B., Guan W., Yin F., Wang J., Li B., Wu L., Dalton Trans. 2018, 47, 1388–1392. [DOI] [PubMed] [Google Scholar]
- 26. Wu Y., Shi R., Wu Y.-L., Holcroft J. M., Liu Z., Frasconi M., Wasielewski M. R., Li H., Stoddart J. F., J. Am. Chem. Soc. 2015, 137, 4111–4118. [DOI] [PubMed] [Google Scholar]
- 27. Ivanov A. A., Falaise C., Abramov P. A., Shestopalov M. A., Kirakci K., Lang K., Moussawi M. A., Sokolov M. N., Naumov N. G., Floquet S., Landy D., Haouas M., Brylev K. A., Mironov Y. V., Molard Y., Cordier S., Cadot E., Chem. Eur. J. 2018, 24, 13467–13478. [DOI] [PubMed] [Google Scholar]
- 28. Kuperman M. V., Losytskyy M. Y., Bykov A. Y., Yarmoluk S. M., Zhizhin K. Y., Kuznetsov N. T., Varzatskii O. A., Gumienna-Kontecka E., Kovalska V. B., J. Mol. Struct. 2017, 1141, 75–80. [Google Scholar]
- 29. Zhang G., Keita B., Craescu C. T., Miron S., de Oliveira P., Nadjo L., Biomacromolecules 2008, 9, 812–817. [DOI] [PubMed] [Google Scholar]
- 30. Geng J., Li M., Ren J., Wang E., Qu X., Angew. Chem. Int. Ed. 2011, 50, 4184–4188; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 4270–4274. [Google Scholar]
- 31. Paterová J., Rembert K. B., Heyda J., Kurra Y., Okur H. I., Liu W. R., Hilty C., Cremer P. S., Jungwirth P., J. Phys. Chem. B 2013, 117, 8150–8158. [DOI] [PubMed] [Google Scholar]
- 32. Okur H. I., Hladílková J., Rembert K. B., Cho Y., Heyda J., Dzubiella J., Cremer P. S., Jungwirth P., J. Phys. Chem. B 2017, 121, 1997–2014. [DOI] [PubMed] [Google Scholar]
- 33. Von Hippel P. H., Schleich T., Acc. Chem. Res. 1969, 2, 257–265. [Google Scholar]
- 34. Gabel D., Awad D., Schaffran T., Radovan D., Dărăban D., Damian L., Winterhalter M., Karlsson G., Edwards K., ChemMedChem 2007, 2, 51–53. [DOI] [PubMed] [Google Scholar]
- 35. Awad D., Damian L., Winterhalter M., Karlsson G., Edwards K., Gabel D., Chem. Phys. Lipids 2009, 157, 78–85. [DOI] [PubMed] [Google Scholar]
- 36. Awad D., Bartok M., Mostaghimi F., Schrader I., Sudumbrekar N., Schaffran T., Jenne C., Eriksson J., Winterhalter M., Fritz J., Edwards K., Gabel D., ChemPlusChem 2015, 80, 656–664. [DOI] [PubMed] [Google Scholar]
- 37. Kobayashi D., Ouchi Y., Sadakane M., Unoura K., Nabika H., Chem. Lett. 2017, 46, 533–535. [Google Scholar]
- 38. Sakamoto A., Unoura K., Nabika H., J. Phys. Chem. C 2018, 122, 1404–1411. [Google Scholar]
- 39. Brus J., Zhigunov A., Czernek J., Kobera L., Uchman M., Matějíček P., Macromolecules 2014, 47, 6343–6354. [Google Scholar]
- 40. Fernandez-Alvarez R., Ďorďovič V., Uchman M., Matějíček P., Langmuir 2018, 34, 3541–3554. [DOI] [PubMed] [Google Scholar]
- 41. Wang W., Wang X., Xiang C., Zhou X., Gabel D., Nau W. M., Assaf K. I., Zhang H., ChemNanoMat 2018, 10.1002/cnma.201800377. [DOI] [Google Scholar]
- 42. Shubina E. S., Tikhonova I. A., Bakhmutova E. V., Dolgushin F. M., Antipin M. Y., Bakhmutov V. I., Sivaev I. B., Teplitskaya L. N., Chizhevsky I. T., Pisareva I. V., Bregadze V. I., Epstein L., Shur V. B., Chem. Eur. J. 2001, 7, 3783–3790. [DOI] [PubMed] [Google Scholar]
- 43. Havel V., Sindelar V., ChemPlusChem 2015, 80, 1601–1606. [DOI] [PubMed] [Google Scholar]
- 44. Gale P. A., Howe E. N. W., Wu X., Chem 2016, 1, 351–422. [Google Scholar]
- 45. Havel V., Babiak M., Sindelar V., Chem. Eur. J. 2017, 23, 8963–8968. [DOI] [PubMed] [Google Scholar]
- 46. Cova T. F. G. G., Nunes S. C. C., Valente A. J. M., Pinho e Melo T. M. V. D., Pais A. A. C. C., J. Mol. Liq. 2017, 242, 640–652. [Google Scholar]
- 47. Pérez-Fuentes L., Drummond C., Faraudo J., Bastos-González D., Soft Matter 2017, 13, 1120–1131. [DOI] [PubMed] [Google Scholar]
- 48. Qi B., Wu C., Li X., Wang D., Sun L., Chen B., Liu W., Zhang H., Zhou X., ChemCatChem 2018, 10, 2285–2290. [Google Scholar]
- 49. El Anwar S., Holub J., Tok O., Jelínek T., Růžičková Z., Fojt L., Šolínová V., Kašička V., Grüner B., J. Organomet. Chem. 2018, 865, 189–199. [Google Scholar]
- 50.V. Ďorďovič, Doctoral thesis, Self-assembly of Boron Cluster Compounds and their Co-assembly with Polymers, Charles University, 2017.
- 51.L. P. Fuentes, Doctoral thesis, Specific Effects of Organic and Inorganic Ions in Soft Interfaces, Universidad de Granada, 2016.
- 52.S. Braun, Doctoral thesis, Biophysikalische Untersuchungen zur Wechselwirkung von grenzflächenaktiven Substanzen mit Liposomen, Albert-Ludwigs-Universität Freiburg, 2016.
- 53.P. Matějíček, Habilitation thesis, Klastrové sloučeniny bóru jako nový typ amfifilů: Roztokové chování a interakce s polymery, Univerzita Karlova v Praze, 2016.
- 54.M. M. A. Moussawi, Doctoral thesis, Assemblages à base de polyoxométallates : des interactions fondamentales aux matériaux hybrides supramoléculaires, Université de Versailles-Sanit-Quentin-en-Yvelines, 2017.
- 55.J. Herzfeld, D. J. Olbris, in eLS, Wiley, Hoboken, 2001, 10.1038/npg.els.0002974. [DOI]
- 56. Collins K. D., Methods 2004, 34, 300–311. [DOI] [PubMed] [Google Scholar]
- 57. Yang L., Adam C., Cockroft S. L., J. Am. Chem. Soc. 2015, 137, 10084–10087. [DOI] [PubMed] [Google Scholar]
- 58. Marmur A., J. Am. Chem. Soc. 2000, 122, 2120–2121. [Google Scholar]
- 59. Abraham M. H., Blandamer M. J., J. Am. Chem. Soc. 2002, 124, 7853–7856. [DOI] [PubMed] [Google Scholar]
- 60. Shimizu S., Chem. Phys. Lett. 2004, 392, 456–459. [Google Scholar]
- 61. Abraham M. H., Grellier P. L., McGill R. A., J. Chem. Soc. Perkin Trans. 2 1988, 339–345. [Google Scholar]
- 62. Cremer P. S., Flood A. H., Gibb B. C., Mobley D. L., Nat. Chem. 2017, 10, 8–16. [DOI] [PubMed] [Google Scholar]
- 63.The required extension of the Hofmeister scale is implicitly contained in the work by Marcus (see Table 1). Leontidis and co-workers also proposed this extension of the Hofmeister scale towards the chaotropic end (beyond PF6 −); they termed this area, which would correspond to the area of hydrophobic ions in Figure 1, initially as “disruptors of soft matter”, and included later so-called hydrotropes and surfactants towards the right side, see Leontidis E., Christoforou M., Georgiou C., Delclos T., Curr. Opin. Colloid Interface Sci. 2014, 19, 2–8; [Google Scholar]; Leontidis E., Curr. Opin. Colloid Interface Sci. 2016, 23, 100–109. On our scale (Figure 1 b), classical amphiphiles that consist of a kosmotropic head with a hydrophobic tail would be best placed near hydrophobic ions. [Google Scholar]
- 64. Wang W., Wang X., Cao J., Liu J., Qi B., Zhou X., Zhang S., Gabel D., Nau W. M., Assaf K. I., Zhang H., Chem. Commun. 2018, 54, 2098–2101. [DOI] [PubMed] [Google Scholar]
- 65. Biedermann F., Schneider H.-J., Chem. Rev. 2016, 116, 5216–5300. [DOI] [PubMed] [Google Scholar]
- 66. Rekharsky M. V., Inoue Y., Chem. Rev. 1998, 98, 1875–1918. [DOI] [PubMed] [Google Scholar]
- 67. Saha D., Mukherjee A., J. Phys. Chem. B 2016, 120, 7471–7479. [DOI] [PubMed] [Google Scholar]
- 68. Marcus Y., Chem. Rev. 2009, 109, 1346–1370. [DOI] [PubMed] [Google Scholar]
- 69. Frank H. S., Evans M. W., J. Chem. Phys. 1945, 13, 507–532. [Google Scholar]
- 70. Voskuhl J., Waller M., Bandaru S., Tkachenko B. A., Fregonese C., Wibbeling B., Schreiner P. R., Ravoo B. J., Org. Biomol. Chem. 2012, 10, 4524–4530. [DOI] [PubMed] [Google Scholar]
- 71. Schibilla F., Voskuhl J., Fokina N. A., Dahl J. E. P., Schreiner P. R., Ravoo B. J., Chem. Eur. J. 2017, 23, 16059–16065. [DOI] [PubMed] [Google Scholar]
- 72. Cromwell W. C., Bystrom K., Eftink M. R., J. Phys. Chem. 1985, 89, 326–332. [Google Scholar]
- 73. Nekvinda J., Grüner B., Gabel D., Nau W. M., Assaf K. I., Chem. Eur. J. 2018, 24, 12970–12975. [DOI] [PubMed] [Google Scholar]
- 74. Pinto de Magalhães H., Brennwald M. S., Kipfer R., Environ. Sci. Process. Impact. 2017, 19, 405–413. [DOI] [PubMed] [Google Scholar]
- 75. Grdadolnik J., Merzel F., Avbelj F., Proc. Natl. Acad. Sci. USA 2017, 114, 322–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Bakulin A. A., Pshenichnikov M. S., Bakker H. J., Petersen C., J. Phys. Chem. A 2011, 115, 1821–1829. [DOI] [PubMed] [Google Scholar]
- 77. Laage D., Stirnemann G., Hynes J. T., J. Phys. Chem. B 2009, 113, 2428–2435. [DOI] [PubMed] [Google Scholar]
- 78. Leberman R., Soper A. K., Nature 1995, 378, 364–366. [DOI] [PubMed] [Google Scholar]
- 79. Jenkins H. D. B., Marcus Y., Chem. Rev. 1995, 95, 2695–2724. [Google Scholar]
- 80. Marcus Y., J. Solution Chem. 1994, 23, 831–848. [Google Scholar]
- 81. Cramer F., Saenger W., Spatz H. C., J. Am. Chem. Soc. 1967, 89, 14–20. [Google Scholar]
- 82. Sokkalingam P., Shraberg J., Rick S. W., Gibb B. C., J. Am. Chem. Soc. 2016, 138, 48–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Gibb C. L. D., Gibb B. C., J. Am. Chem. Soc. 2011, 133, 7344–7347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Zhao Y., Liang L.-L., Chen K., Zhang T., Xiao X., Zhang Y.-Q., Tao Z., Xue S.-F., Zhu Q.-J., CrystEngComm 2013, 15, 7987–7998. [Google Scholar]
- 85. Li Q., Qiu S.-C., Zhang Y.-Q., Xue S.-F., Tao Z., Prior T. J., Redshaw C., Zhu Q.-J., Xiao X., RSC Adv. 2016, 6, 77805–77810. [Google Scholar]
- 86. Ni X.-L., Xiao X., Cong H., Liang L.-L., Cheng K., Cheng X.-J., Ji N.-N., Zhu Q.-J., Xue S.-F., Tao Z., Chem. Soc. Rev. 2013, 42, 9480–9508. [DOI] [PubMed] [Google Scholar]
- 87. Han B.-X., Wang C.-Z., Chen K., Xiao X., Tao Z., Xue S.-F., Zhang Y.-Q., Zhu Q.-J., CrystEngComm 2014, 16, 1615–1619. [Google Scholar]
- 88. Zhao Y., Liang L.-L., Chen K., Ji N.-N., Cheng X.-J., Xiao X., Zhang Y.-Q., Xue S.-F., Zhu Q.-J., Dong N., Tao Z., Dalton Trans. 2014, 43, 929–932. [DOI] [PubMed] [Google Scholar]
- 89. Zhao G., Wang Z., Wang R., Li J., Zou D., Wu Y., Tetrahedron Lett. 2014, 55, 5319–5322. [Google Scholar]
- 90. Qiu S.-C., Li Q., Chen K., Zhang Y.-Q., Zhu Q.-J., Tao Z., Inorg. Chem. Commun. 2016, 72, 50–53. [Google Scholar]
- 91. Cheng X.-J., Ji N.-N., Zhao Y., Liang L.-L., Xiao X., Zhang Y.-Q., Xue S.-F., Zhu Q.-J., Tao Z., CrystEngComm 2014, 16, 144–147. [Google Scholar]
- 92. Cui X.-W., Chen S.-Y., Wang C.-Z., Zhao W.-X., Sun T., Ni X.-L., Zhang Y.-Q., Tao Z., Chin. Chem. Lett. 2016, 27, 173–177. [Google Scholar]
- 93. Han B.-X., Wang C.-Z., Zhao Y., Chen K., Xiao X., Zhu Q.-J., Xue S.-F., Zhang Y.-Q., Tao Z., Eur. J. Inorg. Chem. 2014, 831–835. [Google Scholar]
- 94. Fang X., Kögerler P., Isaacs L., Uchida S., Mizuno N., J. Am. Chem. Soc. 2009, 131, 432–433. [DOI] [PubMed] [Google Scholar]
- 95. Cao M., Lin J., Lü J., You Y., Liu T., Cao R., J. Hazard. Mater. 2011, 186, 948–951. [DOI] [PubMed] [Google Scholar]
- 96. Pursell J. L., Pursell C. J., J. Phys. Chem. A 2016, 120, 2144–2149. [DOI] [PubMed] [Google Scholar]
- 97. Yawer M. A., Havel V., Sindelar V., Angew. Chem. Int. Ed. 2015, 54, 276–279; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 278–281. [Google Scholar]
- 98. Lisbjerg M., Nielsen B. E., Milhoj B. O., Sauer S. P. A., Pittelkow M., Org. Biomol. Chem. 2015, 13, 369–373. [DOI] [PubMed] [Google Scholar]
- 99. Taraszewska J., Wójcik J., Supramol. Chem. 1993, 2, 337–343. [Google Scholar]
- 100. Flinn A., Hough G. C., Stoddart J. F., Williams D. J., Angew. Chem. Int. Ed. Engl. 1992, 31, 1475–1477; [Google Scholar]; Angew. Chem. 1992, 104, 1550–1551. [Google Scholar]
- 101. Buvári A., Barcza L., Inorg. Chim. Acta 1979, 33, L179–L180. [Google Scholar]
- 102. Wojcik J. F., Rohrbach R. P., J. Phys. Chem. 1975, 79, 2251–2253. [Google Scholar]
- 103. Thoma J. A., French D., J. Phys. Chem. 1961, 65, 1825–1828. [Google Scholar]
- 104. Schmid R., Monatsh. Chem. 2001, 132, 1295–1326. [Google Scholar]
- 105. Graziano G., J. Chem. Soc. Faraday Trans. 1998, 94, 3345–3352. [Google Scholar]
- 106. Ben-Naim A., Marcus Y., J. Chem. Phys. 1984, 81, 2016–2027. [Google Scholar]
- 107. Kato H., Nishikawa K., Koga Y., J. Phys. Chem. B 2008, 112, 2655–2660. [DOI] [PubMed] [Google Scholar]
- 108. Morita T., Westh P., Nishikawa K., Koga Y., J. Phys. Chem. B 2014, 118, 8744–8749. [DOI] [PubMed] [Google Scholar]
- 109. Schwierz N., Horinek D., Netz R. R., Langmuir 2010, 26, 7370–7379. [DOI] [PubMed] [Google Scholar]
- 110. Schwierz N., Horinek D., Netz R. R., Langmuir 2013, 29, 2602–2614. [DOI] [PubMed] [Google Scholar]
- 111. Davion van Mau N., Isaaurat B., Amblard G., J. Colloid Interface Sci. 1984, 101, 1–9. [Google Scholar]
- 112. Sisson A. L., Clare J. P., Davis A. P., Chem. Commun. 2005, 5263–5265. [DOI] [PubMed] [Google Scholar]
- 113. Rokitskaya T. I., Kosenko I. D., Sivaev I. B., Antonenko Y. N., Bregadze V. I., Phys. Chem. Chem. Phys. 2017, 19, 25122–25128. [DOI] [PubMed] [Google Scholar]
- 114. Jordan J. H., Gibb B. C., Chem. Soc. Rev. 2015, 44, 547–585. [DOI] [PubMed] [Google Scholar]
- 115. Kavallieratos K., Moyer B. A., Chem. Commun. 2001, 1620–1621. [DOI] [PubMed] [Google Scholar]
- 116. Beer P. D., Gale P. A., Angew. Chem. Int. Ed. 2001, 40, 486–516; [PubMed] [Google Scholar]; Angew. Chem. 2001, 113, 502–532. [Google Scholar]
- 117. Langton M. J., Marques I., Robinson S. W., Félix V., Beer P. D., Chem. Eur. J. 2016, 22, 185–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Baer M. D., Mundy C. J., Faraday Discuss. 2013, 160, 89–101. [DOI] [PubMed] [Google Scholar]
- 119. Gale P. A., Dehaen W., Anion Recognition in Supramolecular Chemistry, Springer Science & Business Media, Berlin, 2010. [Google Scholar]
- 120. Flewelling R. F., Hubbell W. L., Biophys. J. 1986, 49, 531–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Calero C., Faraudo J., Bastos-González D., J. Am. Chem. Soc. 2011, 133, 15025–15035. [DOI] [PubMed] [Google Scholar]
- 122. Ranke J., Othman A., Fan P., Müller A., Int. J. Mol. Sci. 2009, 10, 1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Bastos-González D., Pérez-Fuentes L., Drummond C., Faraudo J., Curr. Opin. Colloid Interface Sci. 2016, 23, 19–28. [Google Scholar]
- 124. Marcus Y., Ion Properties, Vol. 1, Marcel Dekker, New York, 1997. [Google Scholar]
- 125. Marcus Y., J. Phys. Chem. B 2014, 118, 2172–2175. [DOI] [PubMed] [Google Scholar]
- 126. Marcus Y., Ions in Solution and Their Solvation, Wiley, Hoboken, 2016. [Google Scholar]
- 127. Lee T. B., McKee M. L., Inorg. Chem. 2011, 50, 11412–11422. [DOI] [PubMed] [Google Scholar]
- 128. Kaczmarczyk A., Nichols W. C., Stockmayer W. H., Eames T. B., Inorg. Chem. 1968, 7, 1057–1061. [Google Scholar]
- 129. Marcus Y., Biophys. Chem. 1994, 51, 111–127. [Google Scholar]
- 130. Diard J. P., Saint-Aman E., Serve D., J. Electroanal. Chem. Interfacial Electrochem. 1985, 189, 113–120. [Google Scholar]
- 131. Liu Y., Sengupta A., Raghavachari K., Flood A. H., Chem 2017, 3, 411–427. [Google Scholar]
- 132. Langton M. J., Serpell C. J., Beer P. D., Angew. Chem. Int. Ed. 2016, 55, 1974–1987; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 2012–2026. [Google Scholar]
- 133. Liu Y., Singharoy A., Mayne C. G., Sengupta A., Raghavachari K., Schulten K., Flood A. H., J. Am. Chem. Soc. 2016, 138, 4843–4851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Qiao B., Anderson J. R., Pink M., Flood A. H., Chem. Commun. 2016, 52, 8683–8686. [DOI] [PubMed] [Google Scholar]
- 135. Kubik S., Acc. Chem. Res. 2017, 50, 2870–2878. [DOI] [PubMed] [Google Scholar]
- 136. Lim J. Y. C., Beer P. D., Chem 2018, 4, 731–783. [Google Scholar]
- 137. Sommer F., Marcus Y., Kubik S., ACS Omega 2017, 2, 3669–3680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Prochowicz D., Kornowicz A., Lewiński J., Chem. Rev. 2017, 117, 13461–13501. [DOI] [PubMed] [Google Scholar]
- 139. Brown A., Beer P. D., Chem. Commun. 2016, 52, 8645–8658. [DOI] [PubMed] [Google Scholar]
- 140. Liu Y., Singharoy A., Mayne C. G., Sengupta A., Raghavachari K., Schulten K., Flood A. H., J. Am. Chem. Soc. 2016, 138, 4843–4851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Ramsay W. J., Rizzuto F. J., Ronson T. K., Caprice K., Nitschke J. R., J. Am. Chem. Soc. 2016, 138, 7264–7267. [DOI] [PubMed] [Google Scholar]
- 142. Rizzuto F. J., Wu W. Y., Ronson T. K., Nitschke J. R., Angew. Chem. Int. Ed. 2016, 55, 7958–7962; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 8090–8094. [Google Scholar]
- 143. Zhang D., Ronson T. K., Mosquera J., Martinez A., Nitschke J. R., Angew. Chem. Int. Ed. 2018, 57, 3717–3721; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 3779–3783. [Google Scholar]
- 144. Assaf K. I., Suckova O., Al Danaf N., von Glasenapp V., Gabel D., Nau W. M., Org. Lett. 2016, 18, 932–935. [DOI] [PubMed] [Google Scholar]
- 145. Fiala T., Sleziakova K., Marsalek K., Salvadori K., Sindelar V., J. Org. Chem. 2018, 83, 1903–1912. [DOI] [PubMed] [Google Scholar]
- 146. Kaabel S., Adamson J., Topic F., Kiesila A., Kalenius E., Oeren M., Reimund M., Prigorchenko E., Lookene A., Reich H. J., Rissanen K., Aav R., Chem. Sci. 2017, 8, 2184–2190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Lee S., Hirsch B. E., Liu Y., Dobscha J. R., Burke D. W., Tait S. L., Flood A. H., Chem. Eur. J. 2016, 22, 560–569. [DOI] [PubMed] [Google Scholar]
- 148. Gibb C. L. D., Oertling E. E., Velaga S., Gibb B. C., J. Phys. Chem. B 2015, 119, 5624–5638. [DOI] [PubMed] [Google Scholar]
- 149. Carnegie R. S., Gibb C. L. D., Gibb B. C., Angew. Chem. Int. Ed. 2014, 53, 11498–11500; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 11682–11684. [Google Scholar]
- 150. Sullivan M. R., Yao W., Tang D., Ashbaugh H. S., Gibb B. C., J. Phys. Chem. B 2018, 122, 1702–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Jordan J. H., Gibb C. L. D., Wishard A., Pham T., Gibb B. C., J. Am. Chem. Soc. 2018, 140, 4092–4099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Paul R. L., Argent S. P., Jeffery J. C., Harding L. P., Lynam J. M., Ward M. D., Dalton Trans. 2004, 3453–3458. [DOI] [PubMed] [Google Scholar]
- 153. Qi B., Wu C., Xu L., Wang W., Cao J., Liu J., Zhang S., Gabel D., Zhang H., Zhou X., Chem. Commun. 2017, 53, 11790–11793. [DOI] [PubMed] [Google Scholar]
- 154. Goel T., Barooah N., Mallia M. B., Bhasikuttan A. C., Mohanty J., Chem. Commun. 2016, 52, 7306–7309. [DOI] [PubMed] [Google Scholar]
- 155. Fang X., Kögerler P., Isaacs L., Uchida S., Mizuno N., J. Am. Chem. Soc. 2009, 131, 432–433. [DOI] [PubMed] [Google Scholar]
- 156. Hatefi Y., Hanstein W. G., Proc. Natl. Acad. Sci. USA 1969, 62, 1129–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Jungwirth P., Cremer P. S., Nat. Chem. 2014, 6, 261–263. [DOI] [PubMed] [Google Scholar]
- 158. Schwierz N., Horinek D., Sivan U., Netz R. R., Curr. Opin. Colloid Interface Sci. 2016, 23, 10–18. [Google Scholar]
- 159. Mikael L., Pavel J., J. Phys. Condens. Matter 2008, 20, 494218. [Google Scholar]
- 160. Zhang Y., Furyk S., Bergbreiter D. E., Cremer P. S., J. Am. Chem. Soc. 2005, 127, 14505–14510. [DOI] [PubMed] [Google Scholar]
- 161. Fox J. M., Kang K., Sherman W., Héroux A., Sastry G. M., Baghbanzadeh M., Lockett M. R., Whitesides G. M., J. Am. Chem. Soc. 2015, 137, 3859–3866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Jing B., Hutin M., Connor E., Cronin L., Zhu Y., Chem. Sci. 2013, 4, 3818–3826. [Google Scholar]
- 163. Schaffran T., Justus E., Elfert M., Chen T., Gabel D., Green Chem. 2009, 11, 1458–1464. [Google Scholar]
- 164. Zhang G., Keita B., Craescu C. T., Miron S., de Oliveira P., Nadjo L., J. Phys. Chem. B 2007, 111, 11253–11259. [DOI] [PubMed] [Google Scholar]
- 165. Losytskyy M. Y., Kovalska V. B., Varzatskii O. A., Kuperman M. V., Potocki S., Gumienna-Kontecka E., Zhdanov A. P., Yarmoluk S. M., Voloshin Y. Z., Zhizhin K. Y., Kuznetsov N. T., Elskaya A. V., J. Lumin. 2016, 169, 51–60. [Google Scholar]
- 166. Bijelic A., Molitor C., Mauracher S. G., Al-Oweini R., Kortz U., Rompel A., ChemBioChem 2015, 16, 233–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Arefian M., Mirzaei M., Eshtiagh-Hosseini H., Frontera A., Dalton Trans. 2017, 46, 6812–6829. [DOI] [PubMed] [Google Scholar]
- 168. McLaughlin S., Bruder A., Chen S., Moser C., Biochim. Biophys. Acta Biomembr. 1975, 394, 304–313. [DOI] [PubMed] [Google Scholar]
- 169. Sachs J. N., Woolf T. B., J. Am. Chem. Soc. 2003, 125, 8742–8743. [DOI] [PubMed] [Google Scholar]
- 170. Sanderson P. W., Lis L. J., Quinn P. J., Williams W. P., Biochim. Biophys. Acta Biomembr. 1991, 1067, 43–50. [DOI] [PubMed] [Google Scholar]
- 171. Christoforou M., Leontidis E., Brezesinski G., J. Phys. Chem. B 2012, 116, 14602–14612. [DOI] [PubMed] [Google Scholar]
- 172. Leontidis E., Aroti A., Belloni L., J. Phys. Chem. B 2009, 113, 1447–1459. [DOI] [PubMed] [Google Scholar]
- 173. Macdonald P. M., Seelig J., Biochemistry 1988, 27, 6769–6775. [DOI] [PubMed] [Google Scholar]
- 174.Charge-delocalized organic ions (AsPh4 + and BPh4 −) are also known to interact (dissolve in) membranes, but this tendency is due to their well-defined hydrophobic (or even lipophilic) character, see Refs. [120] and [121].
- 175. Schaffran T., Li J., Karlsson G., Edwards K., Winterhalter M., Gabel D., Chem. Phys. Lipids 2010, 163, 64–73. [DOI] [PubMed] [Google Scholar]
- 176. Nabika H., Inomata Y., Itoh E., Unoura K., RSC Adv. 2013, 3, 21271–21274. [Google Scholar]
- 177. Nabika H., Sakamoto A., Motegi T., Tero R., Yamaguchi D., Unoura K., J. Phys. Chem. C 2016, 120, 15640–15647. [Google Scholar]
- 178. Nabika H., Unoura K., in Surface Chemistry of Nanobiomaterials, Elsevier, Amsterdam, 2016, pp. 231–263. [Google Scholar]
- 179. Levin Y., dos Santos A. P., Diehl A., Phys. Rev. Lett. 2009, 103, 257802. [DOI] [PubMed] [Google Scholar]
- 180. Levin Y., Phys. Rev. Lett. 2009, 102, 147803. [DOI] [PubMed] [Google Scholar]
- 181. Petersen P. B., Saykally R. J., Mucha M., Jungwirth P., J. Phys. Chem. B 2005, 109, 10915–10921. [DOI] [PubMed] [Google Scholar]
- 182. Fan P., Neumann J., Stolte S., Arning J., Ferreira D., Edwards K., Gabel D., J. Chromatogr. A 2012, 1256, 98–104. [DOI] [PubMed] [Google Scholar]
- 183. Bauduin P., Prevost S., Farràs P., Teixidor F., Diat O., Zemb T., Angew. Chem. Int. Ed. 2011, 50, 5298–5300; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 5410–5412. [Google Scholar]
- 184. Brusselle D., Bauduin P., Girard L., Zaulet A., Viñas C., Teixidor F., Ly I., Diat O., Angew. Chem. Int. Ed. 2013, 52, 12114–12118; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 12336–12340. [Google Scholar]
- 185. Viñas C., Tarrés M., González-Cardoso P., Farrás P., Bauduin P., Teixidor F., Dalton Trans. 2014, 43, 5062–5068. [DOI] [PubMed] [Google Scholar]
- 186. Popov A., Borisova T., J. Colloid Interface Sci. 2001, 236, 20–27. [DOI] [PubMed] [Google Scholar]
- 187. Chevrot G., Schurhammer R., Wipff G., J. Phys. Chem. B 2006, 110, 9488–9498. [DOI] [PubMed] [Google Scholar]
- 188. Uchman M., Ďordovič V., Tosner Z., Matějíček P., Angew. Chem. Int. Ed. 2015, 54, 14113–14117; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 14319–14323. [Google Scholar]
- 189. Warneke J., McBriarty M. E., Riechers S. L., China S., Engelhard M. H., Aprà E., Young R. P., Washton N. M., Jenne C., Johnson G. E., Laskin J., Nat. Commun. 2018, 9, 1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Uchman M., Abrikosov A. I., Lepšík M., Lund M., Matějíček P., Adv. Theor. Simul. 2018, 1, 1700002. [Google Scholar]
- 191. Ďordovič V., Verbraeken B., Hogenboom R., Kereïche S., Matějíček P., Uchman M., Chem. Asian J. 2018, 13, 838–845. [DOI] [PubMed] [Google Scholar]
- 192. Buchecker T., Goff X. L., Naskar B., Pfitzner A., Diat O., Bauduin P., Chem. Eur. J. 2017, 23, 8434–8442. [DOI] [PubMed] [Google Scholar]
- 193. Matějíček P., Cígler P., Procházka K., Král V., Langmuir 2006, 22, 575–581. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary