

Abstract
Microtubules are cytoskeletal filaments that serve as attractive scaffolds for developing nanomaterials and nanodevices because of their unique structural properties. The functionalization of the outer surface of microtubules has been established for this purpose. However, no attempts have been made to encapsulate molecules inside microtubules with 15 nm inner diameter. The encapsulation of various molecular cargos inside microtubules constitutes a new concept for nanodevice and nanocarrier applications of microtubules. Here, we developed peptide motifs for binding to the inner surface of microtubules, based on a repeat domain of the microtubule‐associated protein Tau. One of the four Tau‐derived peptides, 2N, binds to a taxol binding pocket of β‐tubulin located inside microtubules by preincubation with tubulin dimer and subsequent polymerization of the peptide‐tubulin complex. By conjugation of 2N to gold nanoparticles, encapsulation of gold nanoparticles inside microtubules was achieved. The methodology for molecular encapsulation inside microtubules by the Tau‐derived peptide is expected to advance the development of microtubule‐based nanomaterials and nanodevices.
Keywords: gold nanoparticle, microtubules, nanotechnology, peptide, Tau
Introduction
Biosupramolecular assemblies, including cytoskeletal filaments (microtubules, F‐actin, and intermediate filaments), amyloid fibrils, and collagen fibrils, have been utilized to develop nanomaterials and nanodevices by templating the highly regulated assembled structures.1, 2 Especially, microtubules, which are hollow tubes formed by polymerization of tubulin dimers (α‐ and β‐tubulin), are attractive molecular scaffolds because of their unique structural properties.3, 4, 5, 6, 7, 8, 9, 10 Directional transport of various cargos on microtubules has been established by using motor proteins (kinesin and dynein), which move along microtubules powered by ATP.11, 12, 13, 14, 15, 16 The accumulation of metal nanoparticles on the outer surface of microtubules has been reported by Behrens et al.17, 18, 19 and other groups.20, 21, 22, 23 Dynamic self‐organization of microtubules and their associated motor proteins has been utilized to form unique nanostructures such as asters, networks, and rings.24, 25, 26, 27, 28 Although the functionalization of the outer surface of microtubules has been established for the examples mentioned above, no attempts have been made to encapsulate molecules inside microtubules. Molecular encapsulation inside microtubules with large internal space (15 nm inner diameter) is thus a new concept for the fabrication of microtubule‐based nanomaterials and nanodevices. The encapsulated cargos are transported by the kinesin‐based motor system and released due to the depolymerization capability of the microtubules. Although taxol is known to bind to a pocket of β‐tubulin located inside microtubules,29 the use of a taxol scaffold for molecular encapsulation in microtubules has not been reported, which is probably due to the complex structure and limited number of reactive groups that can be used for functionalization, restricting the conjugation of taxol with cargo molecules. Thus, a new methodology is required to encapsulate various molecular cargos in microtubules.
To design new motifs for the binding to the inner surface of microtubules, we focused on Tau, which is one of the microtubule‐associated proteins.30 It is known that Tau stabilizes microtubules and promotes their assembly by interacting with tubulin and the microtubules (Figure 1 a).31 The microtubule‐binding region of Tau consists of imperfect repeat sequences (R1, R2, R3, and R4) linked by interrepeat sequences (Figure 1 b). Various binding models have been proposed for the repeats of the full‐length Tau with tubulin and microtubules.32, 33, 34, 35, 36, 37, 38, 39 Amos et al. labeled the full‐length Tau with a gold nanoparticle (AuNP) in a position of the repeat domain of Tau to analyze the localization of the repeat domain when bound to microtubules.32 The AuNP was located close to the taxol binding pocket of β‐tubulin, suggesting the binding of the repeat domain in Tau to the inner surface of the microtubules. Based on their study, a conserved PGGG motif in the repeats is important for the binding to the pocket. An NMR study has shown that parts of the R1‐interrepeat (269–284) and R2‐interrepeat (298–312) of Tau form a hairpin conformation around the PGGG motif upon binding to the microtubules (Figure 1 b).34 Thus, the two Tau domains are attractive candidates for motifs binding to the inner surface of microtubules. Although a variety of Tau peptides were investigated to evaluate the binding of Tau to microtubules,40, 41, 42, 43, 44, 45, 46, 47, 48 these peptides have not been utilized for molecular encapsulation inside microtubules.
Figure 1.
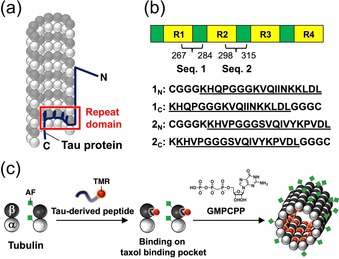
a) Part of a microtubule with exposed inner surface. b) Design of the Tau‐derived peptides 1N, 1C, 2N, and 2C based on the binding repeats of Tau. The interrepeat regions that link the repeats (R1, R2, R3, and R4) are shown in green. The sequences 1 (267–284) and 2 (298–315) used in the Tau‐derived peptides are underlined. c) Encapsulation of tetramethylrhodamine (TMR)‐labeled Tau‐derived peptide into Alexa Fluor 430 (AF)‐labeled microtubules by preincubation with tubulin and subsequent polymerization (“Before” method).
Herein, we developed four peptides based on the repeat domain of Tau for the binding to the inner surface of microtubules (Figure 1 b). One of the four Tau‐derived peptides binds to a taxol binding pocket by complexation with tubulin and subsequent polymerization (Figure 1 c). We also demonstrate that the peptide is useful for encapsulating AuNPs inside microtubules by the formation of peptide‐AuNP conjugates.
Results
Design of the microtubule‐binding peptides
The binding of parts of the Tau repeats to tubulin was modeled by molecular mechanics (MM) calculations using a MacroModel module (see Experimental Section). The two reported hairpin structures, residues 267–284 (sequence 1) and 298–315 (sequence 2), were extracted and modified from Tau (267–312)34 and put into a taxol binding pocket of β‐tubulin49 by replacing a taxol. The modeled structures show that the core hairpin motifs (PGGGKVQII for 1 and PGGGSVQIV for 2) fit well into the taxol binding pocket (Figure 2 and S1). The results agree with the previous hypothesis that the hairpin is a molecular hook anchoring Tau to microtubules by insertion into the pocket of β‐tubulin.32, 34 We designed the Tau‐derived peptides 1N, 1C, 2N, and 2C by introducing cysteines to the N‐ or C‐terminal of 1 and 2 using a flexible linker (GGG) for the conjugation of a fluorescent dye (Figure 1 b). The peptides were synthesized by standard solid‐phase 9‐fluorenylmethyloxycarbonyl (Fmoc) chemistry, purified by reverse‐phase high‐performance liquid chromatography (RP‐HPLC), and confirmed by matrix‐assisted laser desorption‐ionization time‐of‐flight mass spectrometry (MALDI‐TOF‐MS; Figures S2a–d). To monitor the binding of 1N, 1C, 2N, and 2C to tubulin and microtubules, cysteine residues of the peptides were conjugated with a red fluorescent tetramethylrhodamine (TMR)‐5‐maleimide. The TMR‐peptides (1N‐TMR, 1C‐TMR, 2N‐TMR, and 2C‐TMR) were purified by RP‐HPLC and the conjugation of TMR was confirmed by MALDI‐TOF‐MS (Figures S2e–h).
Figure 2.
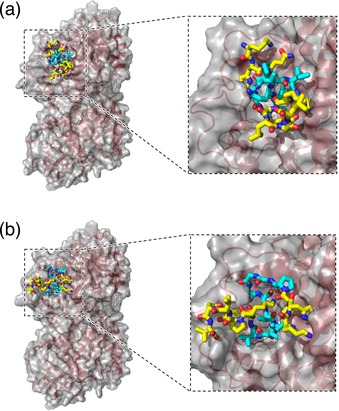
Model of the binding of sequences: a) 1, and b) 2 (stick representation) in the taxol binding pocket of β‐tubulin, obtained by molecular mechanics (MM) calculations. Cyan indicates the core hairpin motifs (PGGGKVQII for 1 and PGGGSVQIV for 2).
Binding affinity of the TMR‐labeled peptides to tubulin
We investigated the binding of TMR‐peptides to tubulin by equilibrium dialysis (see Experimental Section). TMR‐peptides were incubated with tubulin at various concentrations and dialyzed, then the fluorescence intensity of the dialyzed bulk solution was measured (Figure S3). The dissociation constant (Kd) and binding site occupancy (n) of TMR‐peptide per tubulin were estimated by fitting the data using a quadratic binding equation (Table 1). All TMR‐peptides show a higher affinity compared with the reported interrepeat‐R1 of Tau (244–277) (Kd=64.1 μm)47 and taxol derivative (N‐debenzoyl‐N‐[3‐(dimethylamino)benzoyl]taxol) (Kd=49 μm).50
Table 1.
Binding affinity of TMR‐peptides to tubulin.
| Sample | K d [μm] | Binding site occupancy n |
|---|---|---|
| 1N‐TMR | 3.4 | 0.14 |
| 1C‐TMR | 16.7 | 0.17 |
| 2N‐TMR | 6.0 | 0.12 |
| 2C‐TMR | 40.0 | 0.15 |
| Interrepeat‐R1 of Tau (244–277)[a] | 64.1 | 0.25 |
| Taxol derivative[b] | 49 | – |
Binding analysis of the TMR‐labeled peptides to microtubules by CLSM
Binding of the TMR‐peptides to microtubules was observed by confocal laser scanning microscopy (CLSM). To monitor the microtubules, a green fluorescent Alexa Fluor 430 (AF) was conjugated with tubulin (tubulin‐AF) based on a previously reported method.51 Given that it has been reported that the repeat domain of Tau binds to the taxol binding pocket when Tau and tubulin are co‐assembled before microtubule formation,32, 36 we added the TMR‐peptides before microtubule formation (“Before” method; Figure 1 c). In this method, tubulin and tubulin‐AF were preincubated with the TMR‐peptides for 30 min at 25 °C and then polymerized for 30 min at 37 °C to form microtubules by incubation with guanosine‐5′‐[(α,β)‐methyleno]triphosphate (GMPCPP), which is a slowly hydrolysable GTP analogue that is used to form stabilized microtubules.52 The binding of all of the four TMR‐peptides onto microtubules was confirmed by colocalization of red fluorescent TMR‐peptides and green fluorescent labeled microtubules (Figure 3). For comparison, the TMR‐peptides were added to GMPCPP‐stabilized microtubules (“After” method). Even using the “After” method, the four TMR‐peptides were bound to microtubules (Figure S4). Binding of 5(6)‐carboxytetramethylrhodamine (TMR‐COOH) to microtubules was not observed in the “Before” and “After” methods (Figure S5), indicating the importance of peptide moieties for the binding.
Figure 3.
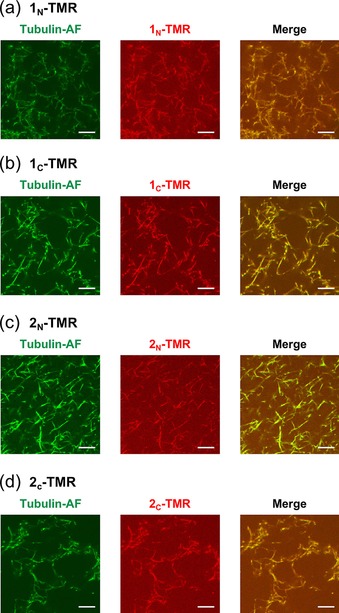
Confocal laser scanning microscopy (CLSM) images of microtubules incubated with: a) 1N‐TMR, b) 1C‐TMR, c) 2N‐TMR, and d) 2C‐TMR by the “Before” method (scale bar: 10 μm). Final concentrations: [Tubulin]=[Tubulin‐AF]=2 μm, [1N‐TMR]=[1C‐TMR]=[2N‐TMR]=[2C‐TMR]=7.5 μm, and [GMPCPP]=20 μm.
To confirm whether the TMR‐peptides were bound to the taxol binding pocket of the microtubules, competition binding experiments of taxol with TMR‐peptide‐incorporated microtubules were carried out. Considering that the binding of taxol to a β‐tubulin pocket of GMPCPP‐stabilized microtubules is strong (K d=10 nm),53 it is expected that the TMR‐peptides bound to the taxol‐binding pocket are replaced with taxol (Figure 4 a). When 2N‐TMR‐incorporated microtubules were prepared by using the “Before” method, the TMR fluorescence on microtubules was dramatically decreased by taxol (Figure 4 b), indicating the binding of 2N‐TMR to the taxol binding pocket. However, 2N‐TMR was still bound to microtubules even after taxol treatment when prepared by the “After” method (Figure 4 b). To estimate the amount of TMR‐peptides on microtubules, the ratio of the TMR and AF fluorescence intensity of each microtubule was calculated from the CLSM images. The TMR/AF ratio of 2N‐TMR‐incorporated microtubules decreases with increasing taxol concentration in the “Before” method, whereas the ratio is not affected in the “After” method (Figure 4 c). This comparison demonstrates that 2N‐TMR binds to the taxol binding pocket only when it is preincubated with tubulin. The TMR/AF ratios of microtubules incorporated with other TMR‐peptides, 1N‐TMR, 1C‐TMR, and 2C‐TMR are not significantly influenced by taxol in both the “Before” and “After” methods (Figure 4 d–f). These results indicate that the main binding sites of 1N‐TMR, 1C‐TMR, and 2C‐TMR are not the taxol binding pocket even in the “Before” method.
Figure 4.
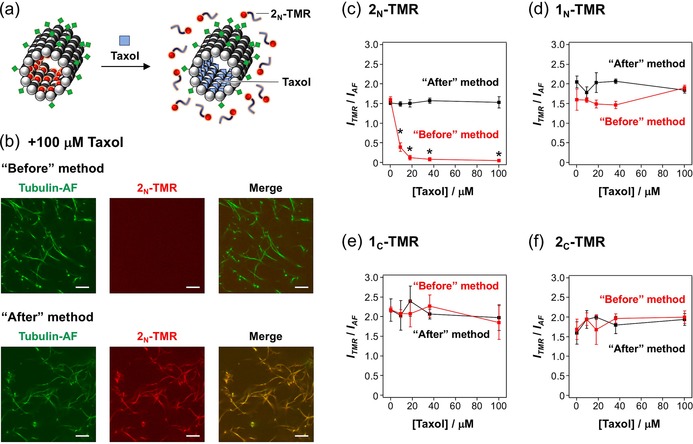
a) Substitution of 2N‐TMR in microtubules by taxol. b) CLSM images of 2N‐TMR‐incorporated microtubules prepared by the “Before” and “After” methods and further treatment with 100 μm taxol (scale bar: 10 μm). Final concentrations: [Tubulin]=[Tubulin‐AF]=2 μm, [2N‐TMR]=7.5 μm, [Taxol]=100 μm. Concentration dependence of taxol on TMR fluorescence (ITMR) per AF fluorescence (IAF) of each: c) 2N‐TMR‐, d) 1N‐TMR‐, e) 1C‐TMR‐, and f) 2C‐TMR‐incorporated microtubule. The error bars represent the SEM (N=10). *P<0.01 compared to the sample without taxol treatment, t test.
To investigate the binding site of 2N‐TMR in the “After” method and 1N‐TMR, 1C‐TMR, and 2C‐TMR in the “Before” and “After” methods, we used an anti‐tubulin antibody that binds to the C‐terminal region of tubulin on the outer surface of the microtubules. The C‐terminal region is known as one of the binding sites of Tau.54 The treatment of the antibody to GMPGPP‐stabilized microtubules and subsequent addition of 2N‐TMR (“After” method) induce a large decrease of the TMR/AF ratio (Figures 5 a and b), indicating the binding of 2N‐TMR on the outer surface of microtubules. Given that the TMR/AF ratio is not influenced by the antibody when prepared by the “Before” method (Figure 5 a and b), the main binding site of 2N‐TMR in the “Before” method is the inner surface of the microtubules, not the outer surface. These results are in good agreement with the competition binding experiments using taxol (Figure 4 c). The TMR/AF ratios of 1N‐TMR‐, 1C‐TMR‐, and 2C‐TMR‐incorporated microtubules in both the “Before” and “After” methods dramatically decrease upon antibody treatment (Figure 5 c–e), indicating that the main binding sites of 1N‐TMR, 1C‐TMR, and 2C‐TMR are on the outer surface of the microtubules. The binding models of TMR‐peptides to microtubules in the “Before” and “After” methods are summarized in Figure 6. In the “Before” method, 2N‐TMR binds to the taxol binding pocket to form the 2N‐TMR‐encapsulated microtubules, whereas other TMR‐peptides may bind to the C‐terminal region of tubulin, resulting in the accumulation of the TMR‐peptides on the outer surface (Figure 6 a). Although the sequence of 2N and 2C are similar, the binding sites are different in the “Before” method. MM calculations show that the minima conformations of 2N are close to the binding conformation in the taxol binding pocket compared with that of 2C (see Experimental Section, Figure S6). Thus, 2N may easily form the binding conformation compared with 2C, explaining why 2N can bind to the pocket but 2C cannot. In the “After” method, all of the four TMR‐peptides bind to the outer surface of microtubules (Figure 6 b). Thus, only 2N‐TMR binds to the inner surface of microtubules by preparation with the “Before” method.
Figure 5.
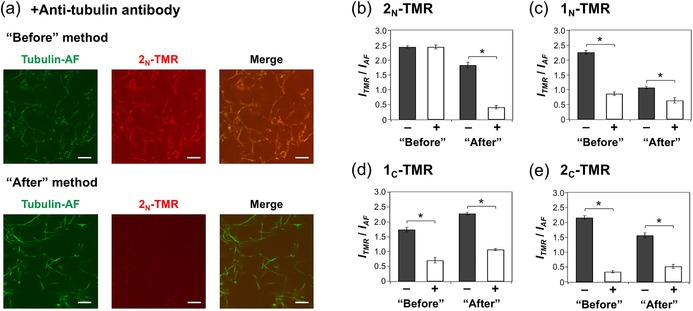
Effect of the anti‐tubulin antibody for binding of TMR‐peptides to microtubules. a) CLSM images of 2N‐TMR‐incorporated microtubules prepared by the “Before” and “After” methods with pretreatment of the anti‐tubulin antibody before addition of 2N‐TMR (scale bar: 10 μm). Final concentrations: [Tubulin]=[Tubulin‐AF]=2 μm, [2N‐TMR]=7.5 μm, and [anti‐tubulin antibody]=15 μg mL−1. ITMR/IAF of each: b) 2N‐TMR‐, c) 1N‐TMR‐, d) 1C‐TMR‐, and e) 2C‐TMR‐incorporated microtubule in the absence (black bar) and presence (white bar) of the anti‐tubulin antibody, analyzed from CLSM images. The error bars represent the SEM (N=10). *P<0.01, t test.
Figure 6.
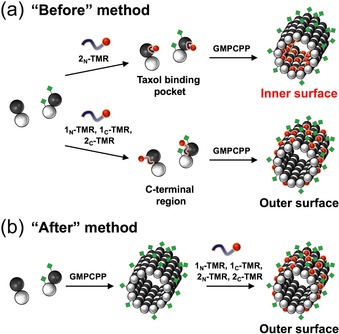
Cartoon illustrating the binding of TMR‐peptides to microtubules in the: a) “Before”, and b) “After” methods.
Effect of Tau‐derived peptides on tubulin polymerization
To evaluate the effect of the binding of Tau‐derived peptides on tubulin polymerization, we carried out turbidity measurements. The addition of 2N to tubulin in the presence of GTP at 37 °C increases the turbidity (optical density at 350 nm) due to tubulin polymerization, whereas other peptides (1N, 1C, and 2C) are less effective (Figure 7). The results indicate that the binding of 2N at the taxol binding site promotes tubulin polymerization, similar to the effect of taxol.29 The binding of the other three peptides to the outer surface of the microtubules has little influence on tubulin polymerization. Interestingly, the 2N‐treated microtubules completely depolymerized when decreasing the temperature to 4 °C, whereas taxol‐treated microtubules remained stable (Figure 7). Thus, the 2N‐treated microtubules are less stable than the taxol‐treated microtubules. The reversible polymerization/depolymerization capability is useful for the encapsulation/release of molecular cargos in microtubules, even upon binding with 2N.
Figure 7.
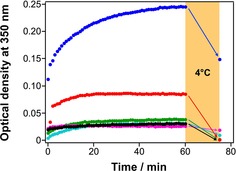
Time course of turbidity caused by tubulin polymerization. Optical density at 350 nm was measured with 4 μm tubulin and 1 mm GTP in the absence (black) or presence of 10 μm taxol (blue), 1N (cyan), 1C (green), 2N (red), 2C (magenta) at 37 °C. After 60 min measurements, the samples were cooled at 4 °C for 15 min and measured again.
Encapsulation of AuNPs inside microtubules
To evaluate the molecular encapsulation capability of 2N inside microtubules, 2N was conjugated with 5 nm AuNPs by thiol‐gold interaction. Tubulin was incubated with the 2N‐AuNP conjugates and then polymerized by GMPCPP (“Before” method; Figure 8 a). The accumulation of 2N‐AuNPs on both the inside and outside of the microtubules was observed by transmission electron microscopy (TEM; Figure 8 b (left) and S7). By treatment of the anti‐tubulin antibody, 2N‐AuNPs on the outer surface were removed (Figure 8 b (right) and S8). When 2N‐AuNPs were added to preassembled microtubules (“After” method), 2N‐AuNPs accumulated mainly on the outer surface of the microtubules (Figure 8 c (left) and S9) and were removed by the anti‐tubulin antibody (Figure 8 c (right) and S10). For analysis of the distribution of 2N‐AuNPs, the number of 2N‐AuNPs binding to the inner surface and outer surface of microtubules was counted from the TEM images (Figures 8 d). In the “Before” method, 2N‐AuNPs were efficiently bound to the inner surface of microtubules (ca. 10 AuNPs per microtubule) compared with the “After” method (Figure 8 d). By treatment of the anti‐tubulin antibody, the amount of 2N‐AuNPs binding to the outer surface of microtubules was decreased in both the “Before” and “After” methods, whereas the amount of 2N‐AuNPs inside microtubules was not affected by the treatment (Figure 8 d). The encapsulation of 2N‐AuNPs inside microtubules only by the “Before” method is in good agreement with the binding properties of 2N‐TMR. The AuNPs without conjugation of 2N did not efficiently accumulate inside microtubules, even when using the “Before” method (Figure 8 d and Figure S11). These results demonstrate that the conjugation of 2N is a useful methodology to encapsulate AuNPs in microtubules.
Figure 8.
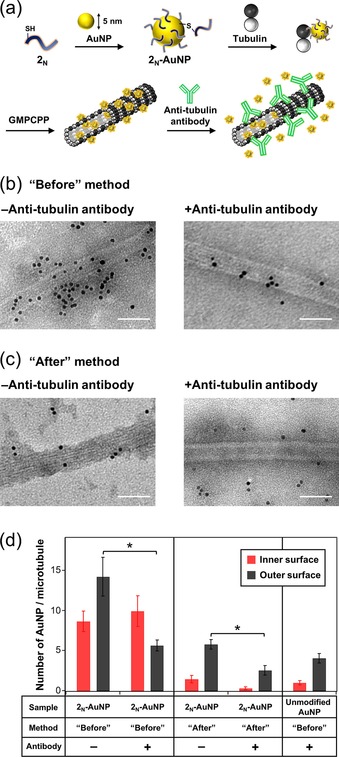
a) Encapsulation of 2N‐AuNPs inside microtubules by the “Before” method. TEM images of microtubules incubated with 2N‐AuNP by the: b) “Before”, and c) “After” methods, without (left) or with (right) treatment of the anti‐tubulin antibody (scale bar: 50 nm). The sample was stained with 2 % Gd(CH3CO2)3(H2O)n aqueous solution. d) Number of AuNPs binding to the inner surface (red) and outer surface (black) of microtubules. The error bars represent the SEM (N=15). *P<0.01, t test.
Discussion
In this study, four peptides were designed from the repeat domain of Tau for molecular encapsulation inside microtubules (Figure 1). Addition of taxol to 2N‐TMR‐incorporated microtubules in the “Before” method decreased the fluorescence of 2N‐TMR from microtubules (Figure 4 c), indicating the binding of 2N‐TMR in the taxol‐binding site. Alternatively, even if the binding site of 2N‐TMR is not the taxol‐binding site, it is possible that the binding of taxol induces a conformational change in tubulin, which affects the binding of 2N‐TMR. However, because treatment of the anti‐tubulin antibody did not affect the binding of 2N‐TMR on microtubules in the “Before” method (Figure 5 b), the binding site of 2N‐TMR is not on the outer surface. In addition, turbidity assay suggests that 2N binds to a taxol‐binding site to induce tubulin polymerization (Figure 7). Although the binding stoichiometry of 2N to tubulin is low (Table 1), it is sufficient to affect tubulin polymerization because a small number of bound taxol per tubulin can stabilize microtubules.55 As a direct evidence of molecular encapsulation inside microtubules by using 2N, we observed the encapsulation of 2N‐conjugated AuNPs inside microtubules by the “Before” method (Figure 8). In the “Before” method, the amount of 2N‐AuNPs inside microtubules was not significantly changed by treatment of the anti‐tubulin antibody, whereas 2N‐AuNPs binding to the outer surface were released by the treatment (Figure 8 d). These results indicate that 2N‐AuNPs were encapsulated inside microtubules and the counted 2N‐AuNPs inside microtubules were not due to the miscount of the 2N‐AuNPs on the outer surface. Amos et al. showed that the repeat domain of full‐length Tau conjugated with AuNP binds to the taxol‐binding site of microtubules.32 Compared to the study, we showed that a small peptide 2N from Tau is enough to bind to the inside of microtubules and encapsulate AuNPs inside microtubules. In addition, binding sites of 2N can be modulated to the inner surface or outer surface of microtubules, dependent on the “Before” or “After” methods, respectively (Figure 6). Thus, molecular cargos can be introduced to the inside or outer surface of microtubules by using 2N, according to the purpose.
Our results indicate that 2N‐TMR binds to the taxol‐binding site in the “Before” method, whereas the binding site is the outer surface in the “After” method (Figure 6). The different binding sites of 2N‐TMR in the two methods are consistent with the previous studies analyzing the binding of full‐length Tau to microtubules.32, 36, 56 Cryomicroscopy measurement showed that the repeat domain of the full‐length Tau binds to the taxol‐binding site when Tau and tubulin are co‐assembled, similar to our “Before” method.32 In contrast, when Tau is incorporated to preassembled microtubules, similar to our “After” method, Tau is solely observed on the microtubule protofilament ridges on the outer surface.56 Lew et al. revealed that there are two distinct binding sites of Tau on microtubules, dependent on the co‐assembly of Tau and tubulin (“Before” method) or binding of Tau to preassembled microtubules (“After” method).36 These studies suggest that Tau has a binding site inside microtubules, which is only found in the “Before” method, similar to 2N‐TMR. Model analysis suggested that molecular diffusion inside microtubules through the open ends of microtubules is slow when the diffusing molecules bind to the interior surface.57 Although small molecules such as taxol (854 Da) can enter the inside of microtubules by passing through pores (1.5 nm×2.0 nm) in the walls of GMPCPP‐microtubules,58, 59, 60 the pore size is not large enough for 2N‐TMR (2778 Da) to enter. Thus, 2N‐TMR cannot go through the inside of preassembled microtubules, explaining why 2N‐TMR does not bind to the taxol‐binding site inside microtubules in the “After” method.
Motility assay of microtubules with 2N‐TMR and 2N‐cargo conjugate on the kinesin‐coated plate is a next step to evaluate the effect of the encapsulation on the transport of microtubules. When 2N‐cargo and kinesin are labeled with different fluorescent molecules, which are used as a donor and acceptor in a fluorescence resonance energy transfer (FRET) measurement, it is possible to measure the FRET efficiency between 2N‐cargo and kinesin to obtain information about the distribution of 2N‐cargo on microtubules in the “Before” or “After” methods. It is also important to analyze the effect of the encapsulation of 2N‐TMR and 2N‐cargo on the assembly/disassembly process of microtubules. For instance, disassembly of microtubules can provide force for the membrane movement, as evaluated in the model system.61 Thus, evaluation of motility, dynamics, and structural properties (stability, rigidity, and length) of microtubules with encapsulated cargos will provide insight into the bioactivity of microtubules.
Conclusions
We developed Tau‐derived peptides for molecular encapsulation in microtubules. We designed four peptides with different repeat sequences of Tau and different positions of cysteine residues at the terminal. Among them, TMR‐labeled 2N was bound to the taxol binding pocket located at the inner surface of microtubules by preincubation with tubulin and subsequent polymerization of the peptide‐tubulin complex, whereas other peptides were bound on the outer surface of the microtubules. By conjugation of 2N to AuNPs, AuNP‐encapsulated microtubules were successfully constructed. These results are proof of the concept of molecular encapsulation inside microtubules based on natural microtubule‐binding proteins. Further characterization of the precise binding site of 2N‐cargo conjugate in microtubules is under investigation. We envision that 2N can be utilized for the encapsulation of various cargos, such as metal nanoparticles, quantum dots, and proteins inside microtubules, for nanodevice and nanocarrier applications by combination with the kinesin‐based transporting system and depolymerization of microtubules.
Experimental Section
Equipment and materials
High‐performance liquid chromatography (HPLC) was performed with a Shimadzu LC‐6AD liquid chromatograph with GL Science Inertsil WP300 C18 columns (4.6 mm×250 mm for analysis and 20 mm×250 mm for purification). Microwave‐assisted solid‐phase peptide synthesis was carried out with an Initiator+ (Biotage). Matrix‐assisted laser desorption ionization time‐of‐flight (MALDI‐TOF) mass spectra were taken with a Bruker Daltonics Autoflex TII with α‐cyano‐4‐hydroxycinnamic acid (α‐CHCA) as a matrix. UV/Vis spectra were obtained with a Jasco V‐630. Transmission electron microscope (TEM) was measured with a Jeol JEM 1400 Plus with a grid (C‐SMART Plus TEM grid, ALLIANCE Biosystems Inc., Osaka, Japan). Confocal laser scanning microscopy (CLSM) measurement was carried out with a FluoView FV10i (Olympus). Tubulin was purified from porcine brain by using a reported procedure.62 The reagents used were purchased from Watanabe Chemical Ind., Ltd., Tokyo Chemical Industry Co., Dojindo Laboratories Co., Ltd. and Wako Pure Chemical Industries. All the chemicals were used without further purification.
Molecular modeling
MM calculations were performed with MacroModel 10.4 (Schrödinger, Inc., New York, NY) using optimized potentials for liquid simulations (OPLS) 2005 force field with default setting. The refined structure of the α‐ and β‐tubulin dimer at 3.5 Å resolution (PDB ID: 1JFF)49 was used for molecular modeling and ligand docking. Addition of missing hydrogen atoms to the model was carried out using Maestro interface ver. 10.4 (Schrödinger) based on an explicit all atom model. As ligands, the sequences 1 (KHQPGGGKVQIINKKLDL) and 2 (KHVPGGGSVQIVYKPVDL) were extracted from residues 267–284 and 298–312 of Tau(267–312) (PDB ID: 2MZ7), respectively.34 The missing residues 313–315 (VDL) in the NMR structures were put to the residue 298–312 manually and the structure of residues 313–315 was energy‐minimized. The sequences 1 and 2 were put to the taxol‐binding pocket of β‐tubulin instead of taxol. The initial positions of 1 and 2 were determined based on the criteria that 1) the N‐ and C‐terminal of these residues are surface‐exposed and 2) the hairpin structures, PGGGKVQII for 1 and PGGGSVQIV for 2 are close to the core taxane ring of taxol. Firstly, 1 and 2 with the surrounding residues around 5.0 Å were energy‐minimized. The minimum energy conformation was then used as a starting point for a Monte Carlo conformational search with up to 1000 search steps, an energy window of 200 kJ mol−1 for saving structures, the loosened threshold for conformer redundancy: the root mean square deviation (RMSD) cutoff of 1.0 Å. In the calculations, 1 and 2 with its surrounding residues within 3.0 Å were applied for the conformational search. Finally, the obtained structures were energy‐minimized by using the same parameters above.
Comparison of 2N and 2C in MM calculations
The binding conformations of 2N (CGGGKHVPGGGSVQIVYKPVDL) and 2C (KHVPGGGSVQIVYKPVDLGGGC) in the taxol‐binding pocket of β‐tubulin were calculated by using the procedure described above. Minima conformations of 2N and 2C were searched by a Monte Carlo conformational search with up to 30 000 search steps, an energy window of 100 kJ mol−1 for saving structures, the loosened threshold for conformer redundancy: RMSD cutoff of 1.0 Å. From the minima conformations found in the step, six conformations of 2N and 7 conformations of 2C within 20 kJ mol−1 conformational gap energy from global minima were superposed to the binding conformations in the taxol‐binding pocket by alignment in the core hairpin motif (PGGGSVQIV) (Figure S6). The RMSD values of the core hairpin motif between the tubulin‐binding conformations and the minima conformations were within 3.45–3.71 Å for 2N and 4.00–5.60 Å for 2C.
Synthesis of peptides
For 1N, H‐Cys(Trt)‐Gly‐Gly‐Gly‐Lys(Boc)‐His(Trt)‐Gln(Trt)‐Pro‐Gly‐Gly‐Gly‐Lys(Boc)‐Val‐Gln(Trt)‐Ile‐Ile‐Asn(Trt)‐Lys(Boc)‐Lys(Boc)‐Leu‐Asp(OtBu)‐Leu‐Alko‐PEG resin was synthesized on Fmoc‐Leu‐Alko‐PEG resin (Watanabe Chemical Ind. Ltd) using standard Fmoc‐based solid phase chemistry (4 equiv Fmoc‐amino acids). N‐Methylpyrrolidone (NMP) solution of 1‐[(1‐(cyano‐2‐ethoxy‐2‐oxoethylideneaminooxy)‐dimethylamino‐morpholinomethylene)] methanaminium hexafluorophosphate (COMU, 4 equiv) and diisopropylethylamine (DIPEA, 4 equiv) were used as coupling reagents. Each condensation reaction was performed at RT for 2 h or by a microwave reaction with 35 W microwave power at 75 °C for 5 min. Deprotection of Fmoc groups from the resin was performed using 40 % and 20 % piperidine in N,N‐dimethylformamide (DMF). The peptidyl‐resin was washed with NMP, CH2Cl2, and CH3OH and then dried under vacuum. The peptide was deprotected and cleaved from the resin by treatment with a cleavage cocktail (trifluoroacetic acid (TFA)/thioanisole/water/ethanedithiol/triisopropylsilane=91.5:2.5:2.5:2.5:1, v/v/v/v/v). The mixture was kept at RT for 3 h. After filtration, the peptide was precipitated by adding ice‐cooled tert‐butylmethylether. After centrifugation, the peptide was washed with tert‐butylmethylether three times. The precipitated peptide was dried under vacuum. The crude product was purified by RP‐HPLC with water/acetonitrile (both containing 0.1 % TFA, 95:5 to 0:100, v/v for 100 min, linear gradient, 10 mL min−1, detected at 220 nm). The isolated yield was 74 %. MALDI‐TOF‐MS: m/z found 2248 ([M+H]+), calcd 2249 (Figure S2a). 1C (H‐Lys‐His‐Gln‐Pro‐Gly‐Gly‐Gly‐Lys‐Val‐Gln‐Ile‐Ile‐Asn‐Lys‐Lys‐Leu‐Asp‐Leu‐Gly‐Gly‐Gly‐Cys‐OH) was prepared by the procedure described above using H‐Cys(Trt)‐Trt(2‐Cl) resin. The isolated yield was 7.2 %. MALDI‐TOF‐MS: m/z found: 2250 ([M+H]+), calcd 2249 (Figure S2b). 2N (H‐Cys‐Gly‐Gly‐Gly‐Lys‐Lys‐His‐Val‐Pro‐Gly‐Gly‐Gly‐Ser‐Val‐Gln‐Ile‐Val‐Tyr‐Lys‐Pro‐Val‐Asp‐Leu‐OH) was prepared by the procedure described above using Fmoc‐Leu‐Alko‐PEG resin. The isolated yield was 12 %. MALDI‐TOF‐MS: m/z found: 2297 ([M+H]+), calcd 2297 (Figure S2c). 2C (H‐Lys‐Lys‐His‐Val‐Pro‐Gly‐Gly‐Gly‐Ser‐Val‐Gln‐Ile‐Val‐Tyr‐Lys‐Pro‐Val‐Asp‐Leu‐Gly‐Gly‐Gly‐Cys‐OH) was prepared by the procedure described above using H‐Cys(Trt)‐Trt(2‐Cl) resin. The isolated yield was 4.4 %. MALDI‐TOF‐MS: m/z found: 2297 ([M+H]+), calcd 2297 (Figure S2d).
Preparation of tetramethylrhodamine (TMR)‐labeled peptides (Scheme S1)
A DMSO solution of TMR‐5‐maleimide (5 equiv) was added to 20 μm 1N, 1C, 2N, or 2C in 200 mm sodium phosphate buffer (pH 7.0) containing 1 mm tris(2‐carboxyethyl)phosphine (TCEP) hydrochloride. The reaction mixture was stirred at 25 °C for 12 h in the dark. The mixture was dialyzed against water and purified by RP‐HPLC with water/acetonitrile (both containing 0.1 % TFA, 95:5 to 0:100, v/v for 100 min, linear gradient, 10 mL min−1, detected at 220 nm). MALDI‐TOF‐MS for 1N‐TMR: m/z found: 2731 ([M+H]+), calcd 2731 (Figure S2e), 1C‐TMR: m/z found: 2751 ([M+Na]+), calcd 2752 (Figure S2f), 2N‐TMR: m/z found: 2777 ([M+H]+), calcd 2778 (Figure S2g), 2C‐TMR: m/z found: 2777 ([M+H]+), calcd 2778 (Figure S2h).
Estimation of binding affinity by equilibrium dialysis
Aqueous solution (20 μL) containing 1 μm 1N‐TMR, 1C‐TMR, 2N‐TMR, or 2C‐TMR and 0, 2, 4, 6, 8, 10, 20, or 50 μm tubulin in BRB80 buffer was incubated at 25 °C for 30 min in the dark. The solution was dialyzed to equilibrium (24 h) against 1380 μL BRB80 buffer at 25 °C using Xpress Micro Dialyzer MD100 (6–8 kDa cut‐off, Scienova GmbH, Germany). The population of bound TMR‐peptides to tubulin was calculated as ΔI/I0, where I0 is the fluorescence intensity of the dialyzed bulk solution in the absence of tubulin, ΔI=I0−I, here I is the fluorescence intensity of the dialyzed bulk solution in the presence of each tubulin concentration. ΔI/I0 was plotted as a function of tubulin concentration, and the Kd and binding site occupancy n=ΔI max/I 0, where ΔI max is a saturated fluorescence difference, were determined by fitting to a quadratic binding function to Equation (1) using Excel and Solver.
| (1) |
where [TMR] is initial concentration of TMR‐peptides (1 μm) and [Tubulin] is initial concentration of tubulin (0–50 μm) [Eq. (1)].
Preparation of Alexa Fluor 430‐labeled tubulin (tubulin‐AF)
Tubulin‐AF was prepared using Alexa Fluor™ 430 NHS ester (Thermo Fisher Scientific) according to the standard procedure.51 The labeling ratio of tubulin‐AF was determined by measuring the absorbance of the protein and Alexa Fluor 430 at 280, and 430 nm, respectively.
Construction of TMR‐peptide‐incorporated microtubules
In the “Before” method, 1N‐TMR, 1C‐TMR, 2N‐TMR, or 2C‐TMR (375 μm, 2 μL) was added to a solution (6 μL) containing tubulin (33 μm) and tubulin‐AF (33 μm) in BRB80 buffer (80 mm PIPES pH 6.9, 1.0 mm MgCl2, 1.0 mm EGTA). The mixture (8 μL) was kept at 25 °C for 30 min in the dark. Then 2 μL of GMPCPP premix (1 mm GMPCPP, 80 mm PIPES pH 6.9, 21 mm MgCl2, 1.0 mm EGTA) was added to the mixture and kept at 37 °C for 30 min in the dark. The mixture was diluted 10‐fold with BRB80 buffer and used for CLSM imaging. For inhibition experiments with taxol, the mixture was diluted 10‐fold with various concentrations of taxol in BRB80 buffer. In the “After” method, GMPCPP premix (2 μL) was added to a solution (6 μL) containing tubulin (33 μm) and tubulin‐AF (33 μm) in BRB80 buffer. The mixture (8 μL) was kept at 37 °C for 30 min in the dark. Then 1N‐TMR, 1C‐TMR, 2N‐TMR, or 2C‐TMR (375 μm, 2 μL) was added to the mixture and kept at 25 °C for 30 min in the dark. The mixture was diluted 10‐fold with BRB80 buffer and used for CLSM imaging. For inhibition experiments with taxol, the mixture was diluted 10‐fold by various concentrations of taxol in BRB80 buffer.
CLSM measurements
The glass bottom dishes (Matsunami, Osaka, Japan) were coated with 1 mg mL−1 poly‐l‐lysine (Mw: 30000–70000, Sigma) at RT for 1 h, then removed and dried. The microtubule samples were put on the plate and kept at RT for 1 h, then observed by CLSM. Tubulin‐AF was excited with 428 nm and observed through a 536 nm emission band‐pass filter (Green). TMR‐labeled peptide was excited with 550 nm and observed through a 574 nm emission band‐pass filter (Red). AF and TMR fluorescence intensity per microtubule were measured from the fluorescence images by subtracting the background intensity using ImageJ software. The background‐subtracted TMR fluorescence intensity per AF fluorescence intensity for each microtubule (N=10) was calculated to estimate the binding of TMR‐peptides to microtubules from at least three images.
Treatment of anti‐tubulin antibody
Anti‐β‐tubulin, monoclonal antibody (Wako, Japan) in the antibody buffer (10 mm sodium phosphate buffer pH 7.4, 150 mm NaCl, 50 % (w/v) glycerol) was used. In the “Before” method, the anti‐tubulin antibody in the antibody buffer (0.5 mg mL−1, 3 μL) or the antibody buffer (3 μL) was added to a solution (3 μL) containing tubulin (67 μm) and tubulin‐AF (67 μm) in BRB80 buffer and kept at 25 °C for 60 min in the dark. 1N‐TMR, 1C‐TMR, 2N‐TMR, or 2C‐TMR (375 μm, 2 μL) was added to the mixture and kept at 25 °C for 30 min in the dark. Then 2 μL of GMPCPP premix was added to the mixture and kept at 37 °C for 30 min in the dark. The mixture was diluted 10‐fold with BRB80 buffer and used for CLSM imaging.
In the “After” method, GMPCPP premix (2 μL) was added to a solution (3 μL) containing tubulin (67 μm) and tubulin‐AF (67 μm) in BRB80 buffer. The mixture (5 μL) was kept at 37 °C for 30 min in the dark. The anti‐tubulin antibody in the antibody buffer (0.5 mg mL−1, 3 μL) or the antibody buffer (3 μL) was added to the mixture and kept at 25 °C for 60 min in the dark. 1N‐TMR, 1C‐TMR, 2N‐TMR, or 2C‐TMR (375 μm, 2 μL) was added to the mixture and kept at 25 °C for 30 min in the dark. The mixture was diluted 10‐fold with BRB80 buffer and used for CLSM imaging.
Turbidity measurement
Turbidity experiments were performed with 4 μm tubulin and 1 mm GTP in the absence or presence of 10 μm taxol, 1N, 1C, 2N, or 2C in BRB80 buffer at 37 °C. Optical density at 350 nm was monitored with a UV/Vis spectrometer for 60 min at 1 min intervals. After 60 min measurements, the samples were cooled at 4 °C for 15 min and the optical density was measured again.
Encapsulation of 2N‐AuNP conjugates in microtubules
The mixture of 50 μm 2N and 5 μm AuNP (Sigma, 5 nm, stabilized suspension in citrate buffer) was incubated at 25 °C for 60 min to construct 2N‐AuNP conjugates. The mixture (5 μL) of the 2N‐AuNP conjugates (0.2 μm AuNP, 2 μm 2N) and 2 μm tubulin in BRB80 buffer was kept at 25 °C for 30 min. GMPCPP premix (2 μL) was added to the mixture and kept at 37 °C for 30 min. The anti‐tubulin antibody in the antibody buffer (0.5 mg mL−1, 3 μL) or the antibody buffer (3 μL) was added to the mixture and kept at 25 °C for 60 min in the dark. The solution was put on a positively‐charged C‐SMART Plus TEM grid (ALLIANCE Biosystems Inc.), allowed to stand for 1 min, and then removed. Given that 2 % phosphotungstic acid (Na3(PW12O40) (H2O)n) aqueous solution was not suitable for staining of microtubules, the grid was exposed to 2 % Gd(CH3CO2)3 (H2O)n aqueous solution (5 μL) for staining, which was allowed to stand for 1 min, and then removed. The resulting grid was dried in vacuo and observed by TEM using an accelerating voltage of 80 kV.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank Professor Y. Kawata and Dr. T. Mizobata (Tottori University) for permission of usage of their ultracentrifuge. This work was supported by a Grant‐in‐Aid for Young Scientist (B) for H.I. (No. 17K14517) from Ministry of Education, Culture, Sports, Science and Technology, Japan.
H. Inaba, T. Yamamoto, A. M. R. Kabir, A. Kakugo, K. Sada, K. Matsuura, Chem. Eur. J. 2018, 24, 14958.
Contributor Information
Dr. Hiroshi Inaba, Email: hinaba@tottori-u.ac.jp.
Prof. Dr. Kazunori Matsuura, Email: ma2ra-k@tottori-u.ac.jp.
References
- 1. Webber M. J., Appel E. A., Meijer E. W., Langer R., Nat. Mater. 2016, 15, 13–26. [DOI] [PubMed] [Google Scholar]
- 2. Krieg E., Bastings M. M. C., Besenius P., Rybtchinski B., Chem. Rev. 2016, 116, 2414–2477. [DOI] [PubMed] [Google Scholar]
- 3. Hess H., Ross J. L., Chem. Soc. Rev. 2017, 46, 5570–5587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wu K. C.-W., Yang C.-Y., Cheng C.-M., Chem. Commun. 2014, 50, 4148–4157. [DOI] [PubMed] [Google Scholar]
- 5. Bachand G. D., Spoerke E. D., Stevens M. J., Biotechnol. Bioeng. 2015, 112, 1065–1073. [DOI] [PubMed] [Google Scholar]
- 6. Bachand G. D., Bouxsein N. F., VanDelinder V., Bachand M., Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2014, 6, 163–177. [DOI] [PubMed] [Google Scholar]
- 7. Malcos J. L., Hancock W. O., Appl. Microbiol. Biotechnol. 2011, 90, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Goodman B. S., Derr N. D., Reck-Peterson S. L., Trends Cell Biol. 2012, 22, 644–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. van den Heuvel M. G. L., Dekker C., Science 2007, 317, 333–336. [DOI] [PubMed] [Google Scholar]
- 10. Agarwal A., Hess H., Prog. Polym. Sci. 2010, 35, 252–277. [Google Scholar]
- 11. Diez S., Reuther C., Dinu C., Seidel R., Mertig M., Pompe W., Howard J., Nano Lett. 2003, 3, 1251–1254. [Google Scholar]
- 12. Bachand G. D., Rivera S. B., Boal A. K., Gaudioso J., Liu J., Bunker B. C., Nano Lett. 2004, 4, 817–821. [Google Scholar]
- 13. Brunner C., Wahnes C., Vogel V., Lab Chip 2007, 7, 1263–1271. [DOI] [PubMed] [Google Scholar]
- 14. Jia Y., Dong W., Feng X., Li J., Li J., Nanoscale 2015, 7, 82–85. [DOI] [PubMed] [Google Scholar]
- 15. Aoyama S., Shimoike M., Hiratsuka Y., Proc. Natl. Acad. Sci. USA 2013, 110, 16408–16413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Song W., Möhwald H., Li J., Biomaterials 2010, 31, 1287–1292. [DOI] [PubMed] [Google Scholar]
- 17. Behrens S., Rahn K., Habicht W., Bohm K. J., Adv. Mater. 2002, 14, 1621–1625. [Google Scholar]
- 18. Behrens S., Wu J., Habicht W., Unger E., Chem. Mater. 2004, 16, 3085–3090. [Google Scholar]
- 19. Behrens S., Habicht W., Wu J., Unger E., Surf. Interface Anal. 2006, 38, 1014–1018. [Google Scholar]
- 20. Achermann M., Jeong S., Balet L., Montano G. A., Hollingsworth J. A., ACS Nano 2011, 5, 1761–1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Spoerke E. D., Connor B. A., Gough D. V., McKenzie B. B., Bachand G. D., Part. Part. Syst. Charact. 2014, 31, 863–870. [Google Scholar]
- 22. Platt M., Muthukrishnan G., Hancock W. O., Williams M. E., J. Am. Chem. Soc. 2005, 127, 15686–15687. [DOI] [PubMed] [Google Scholar]
- 23. Spoerke E. D., Boal A. K., Bachand G. D., Bunker B. C., ACS Nano 2013, 7, 2012–2019. [DOI] [PubMed] [Google Scholar]
- 24. Surrey T., Nedelec F., Leibler S., Karsenti E., Science 2001, 292, 1167–1171. [DOI] [PubMed] [Google Scholar]
- 25. Lam A. T., VanDelinder V., Kabir A. M. R., Hess H., Bachand G. D., Kakugo A., Soft Matter 2016, 12, 988–997. [DOI] [PubMed] [Google Scholar]
- 26. Hess H., Clemmens J., Brunner C., Doot R., Luna S., Ernst K.-H., Vogel V., Nano Lett. 2005, 5, 629–633. [DOI] [PubMed] [Google Scholar]
- 27. Kakugo A., Kabir A. M. R., Hosoda N., Shikinaka K., Gong J. P., Biomacromolecules 2011, 12, 3394–3399. [DOI] [PubMed] [Google Scholar]
- 28. Keya J. J., Suzuki R., Kabir A. M. R., Inoue D., Asanuma H., Sada K., Hess H., Kuzuya A., Kakugo A., Nat. Commun. 2018, 9, 453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Xiao H., Verdier-Pinard P., Fernandez-Fuentes N., Burd B., Angeletti R., Fiser A., Horwitz S. B., Orr G. A., Proc. Natl. Acad. Sci. USA 2006, 103, 10166–10173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Avila J., Lucas J. J., Pérez M., Hernández F., Physiol. Rev. 2004, 84, 361–384. [DOI] [PubMed] [Google Scholar]
- 31. Amos L. A., Org. Biomol. Chem. 2004, 2, 2153–2160. [DOI] [PubMed] [Google Scholar]
- 32. Kar S., Fan J., Smith M. J., Goedert M., Amos L. A., EMBO J. 2003, 22, 70–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kar S., Florence G. J., Paterson I., Amos L. A., FEBS Lett. 2003, 539, 34–36. [DOI] [PubMed] [Google Scholar]
- 34. Kadavath H., Jaremko M., Jaremko Ł., Biernat J., Mandelkow E., Zweckstetter M., Angew. Chem. Int. Ed. 2015, 54, 10347–10351; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 10488–10492. [Google Scholar]
- 35. Li X.-H., Culver J. A., Rhoades E., J. Am. Chem. Soc. 2015, 137, 9218–9221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Makrides V., Massie M. R., Feinstein S. C., Lew J., Proc. Natl. Acad. Sci. USA 2004, 101, 6746–6751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ross J. L., Santangelo C. D., Makrides V., Fygenson D. K., Proc. Natl. Acad. Sci. USA 2004, 101, 12910–12915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Elbaum-Garfinkle S., Cobb G., Compton J. T., Li X.-H., Rhoades E., Proc. Natl. Acad. Sci. USA 2014, 111, 6311–6316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kadavath H., Hofele R. V., Biernat J., Kumar S., Tepper K., Urlaub H., Mandelkow E., Zweckstetter M., Proc. Natl. Acad. Sci. USA 2015, 112, 7501–7506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kadavath H., Cabrales Fontela Y., Jaremko M., Jaremko Ł., Overkamp K., Biernat J., Mandelkow E., Zweckstetter M., Angew. Chem. Int. Ed. 2018, 57, 3246–3250; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 3301–3305. [Google Scholar]
- 41. Aizawa H., Kawasaki H., Murofushi H., Kotani S., Suzuki K., Sakai H., J. Biol. Chem. 1989, 264, 5885–5890. [PubMed] [Google Scholar]
- 42. Butner K. A., Kirschner M. W., J. Cell Biol. 1991, 115, 717–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gustke N., Trinczek B., Biernat J., Mandelkow E. M., Mandelkow E., Biochemistry 1994, 33, 9511–9522. [DOI] [PubMed] [Google Scholar]
- 44. Goode B. L., Feinstein S. C., J. Cell Biol. 1994, 124, 769–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Panda D., Goode B. L., Feinstein S. C., Wilson L., Biochemistry 1995, 34, 11117–11127. [DOI] [PubMed] [Google Scholar]
- 46. Goode B. L., Denis P. E., Panda D., Radeke M. J., Miller H. P., Wilson L., Feinstein S. C., Mol. Biol. Cell 1997, 8, 353–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Devred F., Douillard S., Briand C., Peyrot V., FEBS Lett. 2002, 523, 247–251. [DOI] [PubMed] [Google Scholar]
- 48. Santarella R. A., Skiniotis G., Goldie K. N., Tittmann P., Gross H., Mandelkow E.-M., Mandelkow E., Hoenger A., J. Mol. Biol. 2004, 339, 539–553. [DOI] [PubMed] [Google Scholar]
- 49. Löwe J., Li H., Downing K. H., Nogales E., J. Mol. Biol. 2001, 313, 1045–1057. [DOI] [PubMed] [Google Scholar]
- 50. Sengupta S., Boge T. C., Georg G. I., Himes R. H., Biochemistry 1995, 34, 11889–11894. [DOI] [PubMed] [Google Scholar]
- 51. Peloquin J., Komarova Y., Borisy G., Nat. Methods 2005, 2, 299–303. [DOI] [PubMed] [Google Scholar]
- 52. Hyman A. A., Salser S., Drechsel D. N., Unwin N., Mitchison T. J., Mol. Biol. Cell 1992, 3, 1155–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Caplow M., Shanks J., Ruhlen R., J. Biol. Chem. 1994, 269, 23399–23402. [PubMed] [Google Scholar]
- 54. Chau M.-F., Radeke M. J., de Inés C., Barasoain I., Kohlstaedt L. A., Feinstein S. C., Biochemistry 1998, 37, 17692–17703. [DOI] [PubMed] [Google Scholar]
- 55. Jordan M. A., Wilson L., Nat. Rev. Cancer 2004, 4, 253–265. [DOI] [PubMed] [Google Scholar]
- 56. Al-Bassam J., Ozer R. S., Safer D., Halpain S., Milligan R. A., J. Cell Biol. 2002, 157, 1187–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Odde D., Eur. Biophys. J. 1998, 27, 514–520. [DOI] [PubMed] [Google Scholar]
- 58. Meurer-Grob P., Kasparian J., Wade R. H., Biochemistry 2001, 40, 8000–8008. [DOI] [PubMed] [Google Scholar]
- 59. Díaz J. F., Valpuesta J. M., Chacón P., Diakun G., Andreu J. M., J. Biol. Chem. 1998, 273, 33803–33810. [DOI] [PubMed] [Google Scholar]
- 60. Maccari G., Mori M., Rodríguez-Salarichs J., Fang W., Díaz J. F., Botta M., J. Chem. Theory Comput. 2013, 9, 698–706. [DOI] [PubMed] [Google Scholar]
- 61. Fu M., Li Q., Sun B., Yang Y., Dai L., Nylander T., Li J., ACS Nano 2017, 11, 7349–7354. [DOI] [PubMed] [Google Scholar]
- 62. Castoldi M., Popov A. V., Protein Expression Purif. 2003, 32, 83–88. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary