

Summary
We have recently developed a sensitive and specific urine‐based antigen detection ELISA for the diagnosis of visceral leishmaniasis (VL). This assay used rabbit IgG and chicken IgY polyclonal antibodies specific for the Leishmania infantum proteins iron superoxide dismutase 1 (Li‐isd1), tryparedoxin1 (Li‐txn1) and nuclear transport factor 2 (Li‐ntf2). However, polyclonal antibodies have limitations for upscaling and continuous supply. To circumvent these hurdles, we began to develop immortalized monoclonal antibodies. We opted for recombinant camelid VHHs because the technology for their production is well established and they do not have Fc, hence providing less ELISA background noise. We report here an assay development using VHHs specific for Li‐isd1 and Li‐ntf2. This new assay was specific and had analytical sensitivity of 15‐45 pg/mL of urine. The clinical sensitivity was comparable to that obtained with the ELISA assembled with conventional rabbit and chicken antibodies to detect these two antigens. Therefore, similar to our former studies with conventional antibodies, the future inclusion of VHH specific for Li‐txn1 and/or other antigens should further increase the sensitivity of the assay. These results confirm that immortalized VHHs can replace conventional antibodies for the development of an accurate and reproducible antigen detection diagnostic test for VL.
1. INTRODUCTION
Accurate diagnosis of active visceral leishmaniasis (VL) both in humans and dogs is still a difficult task. Because VL is an insidious and slow progressing pathology, clinical diagnosis is mostly achieved at the later stages of the disease when marked signs of anaemia and hepato/splenomegaly become present. During this phase, the parasites (or their molecules) can be detected in biopsies of liver, spleen and bone marrow. Unfortunately, these tests are at best 60% sensitive.1, 2, 3 Due to the limitations of these methods (aggressive risky sample collection and low sensitivity), the presence of antiparasite antibodies is routinely used as a marker of infection.3, 4, 5, 6, 7, 8 Overall, the reported performances of these tests indicate that they have good sensitivity for the diagnosis of VL in both symptomatic and asymptomatic humans and dogs. However, due to cross‐reactions with other infectious agents (eg, Leishmania agents of cutaneous/mucosal leishmaniasis, Trypanosoma cruzi and Trypanosoma brucei), and the difficult choice of a suitable cut‐off, these conventional serological tests are less specific than PCR assessment of parasites in biopsy or blood specimens.5 Moreover, these antibody tests do not discriminate active disease from either subclinical infection or cured individuals because the antibodies can circulate for years after cure. In addition, these tests have several other limitations, including inability to predict response to treatment and very low sensitivity (57.4%) in patients co‐infected with HIV.9, 10, 11
An antigen detection test for VL was developed approximately 10 years ago as an interesting alternative for the diagnosis of active VL. This assay is a latex agglutination test (KAtex) based on leishmanial antigen detection in urine of patients with VL. This test detects a mixture of leishmanial carbohydrate complex antigens using latex beads adsorbed with specific polyclonal rabbit antibody.12, 13, 14, 15, 16 Preliminary studies using urine of VL patients demonstrated the presence of leishmanial antigen in active disease followed by its disappearance after successful treatment.17 However, recent studies involving VL patients co‐infected with HIV have shown that the sensitivity of KAtex varied from 47.7% to 85.7% and specificity from 96% to 98.7%.18 These conflicting results can perhaps be explained on the grounds of uncontrolled specificity and sensitivity (affinity/avidity) of the anticarbohydrate antibodies used in the test. The carbohydrate preparation involves the growth of leishmania promastigotes followed by several biochemical purification steps.17 These procedures usually generate a mixture of related and unrelated products. Consequently, this antigenic preparation, when used to produce a key reagent (antibody) for the test development, stimulates a mixture of a variety of antibody specificities. Therefore, the production of a test assembled using a heterogeneous antibody population is, by definition, difficult to standardize under standard operating procedures (SOP).
An alternative to native carbohydrates, microbial protein antigens are interesting molecules to circumvent the above restrictions. Protein epitopes are better defined, and recombinant proteins or synthetic peptides are easily obtainable thus facilitating production and upscaling of reliable and reproducible specific antibodies. Using this premise, we have recently identified the Leishmania infantum proteins iron superoxide dismutase 1 (Li‐isd1), tryparedoxin1 (Li‐txn1) and nuclear transport factor 2 (Li‐ntf2) in the urine of VL patients, presumably shed from the parasite during active VL.19, 20, 21 These antigens or biomarkers represented unique and promising candidate molecules for the development of an antigen detection test for the diagnosis of active VL. In our preliminary pilot studies using a capture ELISA assembled to detect these three antigens in urine samples of confirmed VL patients from Brazil, the assay was >95% sensitive and 100% specific. Unfortunately, the polyclonal antibodies that were produced in rabbits and chicken to develop the test are not adequate for mass scale production of the final test. Ideally, the antibodies should be immortalized reagents.
Camelid single‐domain recombinant antibodies (sdAbs) offer a new approach to generating such renewable diagnostic reagents. With a molecular weight of approximately 13 kDa, these high‐affinity, single‐domain variable fragments of heavy‐chain antibodies (VHHs) have the ability to refold and retain binding activity after denaturation.22 Moreover, large amounts of VHHs can be produced by standard recombinant protein expression in Escherichia coli. Therefore, VHHs are very desirable immunoreagents.22, 23, 24
Here, we report the production, characterization and initial clinical validation of immortalized camelid VHHs specific for the L. infantum biomarkers iron superoxide dismutase1 (Li‐isd1) and nuclear transport factor 2 (Li‐ntf2). The results confirm that these antibodies are promising and useful reagents for development and mass scale production of a feasible antigen detection assay for the accurate diagnosis of VL.
2. MATERIAL AND METHODS
2.1. Clinical specimens
A total of 24 urine samples from patients with New World VL were evaluated in this study. These samples were collected from patients (aged 2‐65 years old) diagnosed with VL based on the following criteria: a clinical course consistent with VL (eg, fever, anaemia and hepatosplenomegaly), and confirmatory laboratory findings (identification of Leishmania in bone marrow aspirates) and serological test (rapid immunochromatographic test, IT‐Leish®; Bio‐Rad, São Paulo, Brazil). All samples were obtained from the University Hospital Clemente of Farias (Montes Claros, Minas Gerais State, Brazil). In addition, 12 urine samples were obtained from healthy control subjects living in the same geographical area where the VL patients lived. Approval to use the samples was obtained from the Human Research Ethics Committee—COEP (CAAE‐00842112.2.0000.5149) of the Federal University of Minas Gerais. The informed consent for research involving humans was also submitted and approved by COEP/UFMG. All samples were anonymized.
2.2. Immunization of alpacas
Alpacas were obtained locally and maintained on pasture. Immunizations and bleeding procedures followed National Institute of Health guidelines and were approved by the Institutional Animal Care and Use Committee at Tufts University. Three purified recombinant L. infantum proteins were used to immunize two alpacas (Sophia and Sadie). The antigens (iron superoxide dismutase 1 [Li‐isd1], tryparedoxin1 [Li‐txn1] and nuclear transport factor 2 [Li‐ntf2]) were produced and purified as we have previously described.20 The alpacas were immunized with a pool of the three antigens (100 μg each) by five successive multisite subcutaneous (SC) injections at 3‐week intervals. For the first immunization, the antigens were in alum/CpG adjuvant, and subsequent immunizations contained only alum as an adjuvant. Fifteen days after the 4th immunization, alpacas were bled for serology.
2.3. VHH display library preparation from lymphocytes of immunized alpacas
Blood was obtained from the two alpacas 7 days after the fifth immunization followed by lymphocyte purification and RNA preparation, which was performed using the RNeasy kit (Qiagen, Valencia, CA, USA). A VHH display phage library was prepared as described previously,25 yielding a library (JRB‐2) having a complexity of ~1 × 107 independent clones with >95% containing VHH inserts.
2.4. Identification and purification of VHHs
Phage library panning, phage recovery and clone fingerprinting were performed as previously described.26, 27, 28 The JRB‐2 VHH display library was separately panned on Li‐isd1, Li‐txn1 and Li‐ntf2 coated onto Nunc Immunotubes (Thermo Fisher Scientific, Waltham, MA, USA). Initial panning was performed on plastic coated with 10 μg/mL of target, followed by a second round of panning at high stringency coated at 1 μg/mL and employing a 10‐fold lower titre of input phage, shorter binding times and longer washes. Ninety‐five random clones from the selected population were screened for the expression of VHHs that bound to each leishmanial target protein. Approximately 80% of the random clones secreted VHHs to the periplasm demonstrated significant binding to their panning target protein (>2× background) producing the strongest “bug supernatant” ELISA29 signals on plates coated with target. About 45 clones producing the strongest positive signals from both Li‐isd1 and Li‐ntf2 panning were characterized by DNA fingerprinting,29 and the sequence of the coding DNA for the best (highest ELISA signal) clone possessing each unique fingerprint was obtained (13 sequences for Li‐isd1 and 18 for Li‐ntf2). From this initial screen, we identified five unique, unrelated VHHs (ie, no evidence of a common B‐cell clonal origin) that recognized Li‐isd1 and seven unique VHHs that recognized Li‐ntf2.
2.5. Expression and purification of VHHs in Escherichia coli
Expression and purification of recombinant thioredoxin/VHH fusion proteins containing hexahistidine were performed as previously described.28 All recombinant VHHs were expressed with a carboxyl‐terminal E‐tag epitope. VHH heterodimers were engineered in which two selected VHHs were separated by a 15‐amino acid flexible spacer [(GGGGS)3], often without the thioredoxin partner and with different epitope tags. Purified VHHs were finally characterized by competition analysis to identify and deselect those that recognized overlapping epitopes on their target. Competition ELISA analysis was performed as previously described.27
2.6. Serological and antigen detection assays
Immuno Plates (96 wells, flat bottom), Maxi Sorp certified (Thermo Scientific) were used throughout. For conventional serological ELISA, plates were coated with 100 μL of 1 μg/mL recombinant antigens in PBS overnight at 4°C. Plates were blocked for 2 hours with 5% BSA in PBS. Alpaca serum or selected VHHs, in serial dilutions, were added to the wells and incubated for 1 hour at room temperature. The development was performed with goat anti‐llama HRP (Abcam, Cambridge, MA, USA) at 1/10 000 or rabbit anti‐E‐tag HRP antibody (Abcam) at 1/10 000. Plates were then washed six times with PBS‐0.1% Tween 20, and reactions were developed with TMB substrate and read at 450 nm. For the capture ELISA antigen detection tests, the plates were coated with selected VHHs or specific chicken IgY overnight at 4°C. Next, the antigens or human urine samples (100 μL) were added to the wells followed by overnight incubation at 4°C. After washing six times with PBS‐0.1% Tween 20, wells corresponding to VHH reactions were incubated for 1 hour at room temperature with rabbit anti‐E‐tag HRP antibody (Abcam) at 1/10 000. Wells corresponding to IgY as capture antibody were incubated with biotinylated specific rabbit IgG (1 μg/mL) for 1 hour at room temperature followed by incubation with streptavidin‐peroxidase (Sigma‐Aldrich, St. Louis, MO, USA) at 1/15 000 for 1 hour also at room temperature. In between these steps, plates were washed six times with PBS‐Tween 20. Reactions (VHH and IgY coated plates) were developed with TMB substrate and read at 450 nm.
2.7. Statistical analysis
Statistical significance was determined by unpaired t test for comparisons between capture ELISA results obtained for VL patients and healthy subject control groups. Comparisons were performed using the Mann‐Whitney rank‐sum test (P values <0.05 were considered statistically significant).
3. RESULTS
3.1. Generation and purification of VHHs specific for Li‐isd1, Li‐txn1 and Li‐ntf2
Subsequent to the immunization of the alpacas used to generate the VHHs, the antibody immune response was monitored by direct ELISA. Both alpacas responded with high antibody titres to Li‐isd1, Li‐txn1 and Li‐ntf2 (>1/62 500). Based on our prior experience22, 25 these are fully acceptable responses for the generation of recombinant VHHs. Initially, a VHH phage display library was prepared by RT‐PCR of the VHH coding sequences from a pool of the two alpacas B‐cell RNA and demonstrated to be of high quality. The library was separately panned for VHHs binding to each of the three leishmanial antigens coated onto plastic surfaces of microtiter plates. Following two rounds of panning, 95 random clones were selected from each library, cultured and then tested for binding to Li‐isd1, Li‐txn1 and Li‐ntf2. Numerous positive clones were identified. A summary of the results obtained for each marker is as follows:
3.1.1. Li‐isd1
About 80% of the clones displayed significant binding (>2× background) for Li‐isd1 by ELISA, and about 50% of the clones displayed strong binding (>10‐30× background), demonstrating that the antigen had elicited a robust VHH response. Forty‐five‐positive clones were picked for DNA fingerprinting and 13 showed unique fingerprints. These clones were selected for DNA sequencing. Sequencing showed that the 13 VHHs clustered into five groups that clearly derive from B cells having a common origin. The VHH producing the strongest apparent target recognition was selected from each of the five homology groups. These five VHHs had unique complementary‐determining regions (CDRs), suggesting they do not have common B‐cell origins.
3.1.2. Li‐txn1
The panning and selection process proceeded poorly despite the strong antibody titres observed in the sera of the immunized alpacas. Only one VHH could be expressed that showed binding to Li‐txn1, although with poor affinity. Moreover, this clone was not specific for Li‐txn1. Therefore, this marker was deprioritized for further evaluation.
3.1.3. Li‐ntf2
About 25% of the affinity‐selected clones displayed significant binding (>2× background) for Li‐ntf2 by ELISA. Only two clones displayed strong binding >10× background) for Li‐ntf2 by ELISA. Therefore, we repanned at a lower stringency, and 95 additional clones were picked at random. The results were quite similar to the first panning, suggesting that selection stringency was not a limiting factor. A total of 45 positive clones were picked for DNA fingerprinting. Of these, 18 clones had apparently unique fingerprints and were selected for DNA sequencing. Seven homology groups were identified that each appears to have arisen from common B‐cell origins and one VHH from each homology group was selected for expression in E. coli host cells.
After induction, VHHs were purified from the soluble phase of the E. coli cell lysates. Three anti‐Li‐isd1 and two anti‐Li‐ntf2 clones that had acceptable expression were assessed by ELISA for their binding properties to their respective specific antigens. In addition, we performed competition analysis, which showed that none of them compete with each other in their binding to Li‐isd1 or Li‐ntf2 (not shown). The three anti‐Li‐isd1 VHH/clones were named as JRD‐C1, JRD‐E5 and JRD‐E9. The two anti‐Li‐ntf2 VHH/clones were named JRF‐E3 and JRO‐H8. The binding of each of the purified VHHs is illustrated in Figure 1.
Figure 1.
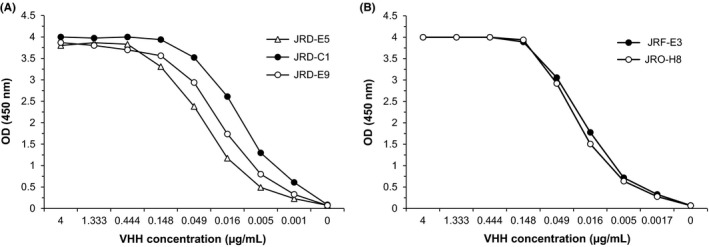
Reactivity of selected VHHs with recombinant proteins Li‐isd1 or Li‐ntf2. Microtitre plates were coated with Li‐isd1 (A) or Li‐ntf2 (B), and reactivity of the selected VHHs was tested by conventional ELISA
3.2. Production of high‐affinity homodimer and heterodimer of selected VHHs for assay development
Next, we produced a series of homodimers and heterodimers of the selected VHHs. VHH homo and heterodimers are known to increase the affinity of their interaction with the target molecules. In addition, the heterodimer recognizes two epitopes in the native target molecules thus increasing the overall ability of the reagent to detect the biomarker. The homodimers and heterodimers were obtained using synthetic genes encoding either a duplicate VHH (homodimer) or two VHHs recognizing different epitopes of the target biomarker (heterodimer). After expression and purification, using a conventional capture ELISA, these reagents were tested in varied combinations, either as capture or detection antibodies. The VHH dimers that provided the best signal to noise OD for each of the biomarkers were selected for further clinical assay development. For Li‐isd1‐specific clones, we selected JRD‐C1/JRD‐C1, a homodimer expressed as JRD‐C1/JRD‐C1/myc, and JRD‐E5/JRD‐E9, a heterodimer expressed as JRD‐E5/JRD‐E9/E‐tag. VHHs recognizing only two nonoverlapping Li‐ntf2‐specific epitopes were identified, so heterodimers could not be used as components of a Li‐ntf2 capture ELISA. Therefore, we constructed homodimers only of the two anti‐Li‐ntf2 VHH clones.
3.3. Assemble of capture ELISA with selected homo and heterodimers and determination of assay sensitivity to detect the leishmanial antigens in spiked urine
To standardize the Li‐isd1 capture ELISA, the homodimer JRD‐C1/JRD‐C1/myc was used as the capture reagent and the E‐tag heterodimer JRD‐E5/JRD‐E9/E‐tag as developing reagent. For Li‐ntf2 capture ELISA, we selected the homodimer JRO‐H8/JRO‐H8/myc as capture reagent and the homodimer JRF‐E3/JRF‐E3/E‐tag as developing reagent (Figure 1). Next, various concentrations of the VHH reagents and recombinant leishmanial antigens were tested in a checkerboard fashion. The highest signal to noise concentrations of the reagents that provided the lowest background (assay in the absence of either Li‐isd1 or Li‐ntf2) were 2 μg/mL of homodimers and 1 μg/mL of heterodimer. To investigate the analytical performance of the capture ELISA and possible interference of urine in the test sensitivity, we next diluted the antigens at decreasing concentrations (1215, 405, 135, 45, 15 and 5 pg/mL) into either PBS‐1% BSA buffer or urine from healthy donors. Results were read at OD450 nm and are illustrated in Figure 2. The limit of detection of this assay (arbitrarily defined as an OD 50% above the OD given by the background reading) for the antigens spiked into either urine or buffer was ~45‐15 pg/mL. The background OD readings for the assay performed with buffer vs urine in the absence of antigens did not differ. Importantly, normal human urine did not interfere with the sensitivity of the antigen detection capture ELISA. These results, as we have previously shown with an assay assembled with rabbit and chicken polyclonal antibodies,20 indicate that urine should not interfere with the VHH VL test using this human specimen.
Figure 2.
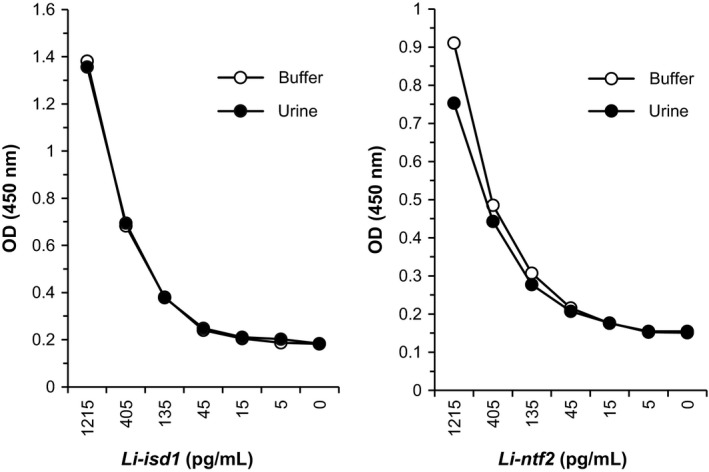
Sensitivity of capture ELISAs assembled with VHH for detection of the proteins Li‐isd1 and Li‐ntf2 spiked in buffer as well as in urine samples of normal healthy subjects. Capture VHH JRD‐C1/JRD‐C1‐C1/myc (anti‐Li‐isd1) or JRO‐H8/JRO‐H8/myc (anti‐Li‐ntf2) at 2 μg/mL was used to coat the ELISA plates. Wells were then incubated with various concentrations of Li‐isd1 or Li‐ntf2 diluted either in buffer plus 1% BSA or in urine from normal healthy subjects followed by incubation with E‐tag‐labelled developing VHH JRD‐E5/JRD‐E9 (anti‐Li‐isd1) or JRF‐E3/JRF‐E3 (anti‐Li‐ntf2). Reactions were developed after addition of peroxidase‐labelled anti‐E‐tag plus the substrate H2O2 and the chromophore TMB. Results are expressed as OD read at 450 nm. Same results were obtained using ELISA assembled with conventional rabbit IgG and chicken IgY antibodies (not shown)
3.4. Clinical validation of the assay assembled with VHHs
In order to begin the validation of the VHHs specific for Li‐isd1 and Li‐ntf2 as immortalized reagents for the development of a VL diagnostic test, we performed a capture ELISA assembled with these reagents and used urine samples from 24 patients with confirmed VL from Minas Gerais State, Brazil, and from 12 healthy control subjects from this endemic area for the disease. Urine samples from VL patients were collected prior to initiating VL therapy. In addition, the samples were also tested comparatively using the Li‐isd1 and Li‐ntf2 assays that we have previously developed.19, 20, 30 For this assay, the capture ELISA is assembled with conventional rabbit and chicken antibodies. For the VHH‐based assays, we used the reagents described in Figure 2; that is for the assay to detect Li‐isd1, we used the homodimer JRD‐C1/JRD‐C1‐C1/myc as capture and the heterodimer JRD‐E5/JRD‐E9/E‐tag as the developing reagent. For the assay to detect Li‐ntf2, we used the homodimer JRO‐H8/JRO‐H8/myc as capture and the homodimer JRF‐E3/JRF‐E3/E‐tag as the developing reagent. Using a standard cut‐off value calculated as the average of the results for the samples from the healthy control subjects plus 3 SD, 10 (41%) VL samples tested positive using the assay assembled with the anti‐Li‐isd1VHH reagents (Figure 3). In contrast 8 (33%) VL samples tested positive using the assay assembled with conventional antibodies (affinity‐purified IgY as capture and biotin‐labelled rabbit IgG as developing reagent). Although the sensitivity of either assay is not high enough to warrant a final test for the diagnosis of VL, it indeed confirms our previous results using conventional anti‐Li‐isd1 antibodies,19, 20 which was approximately 30%. A highly sensitive test (>98%) was only achieved when we used a multiplexed assay assembled to detect, in addition to Li‐isd1 the two other L. infantum‐specific biomarkers. Importantly, the results illustrated in Figure 3 show that most samples that were positive using the assay assembled with the VHHs were also positive with the assay assembled with conventional antibodies. However, a few samples among the disease group were positive only for one of the two tests. Samples 11, 16 and 20 were positive only for the test assembled with the VHHs, and sample 15 was positive only for the test assembled with conventional antibodies. Therefore, the two assays can complement each other, importantly increasing the sensitivity of tests designed to detect Li‐isd1 in the urine of VL patients. No false‐positive samples were observed for either assay.
Figure 3.
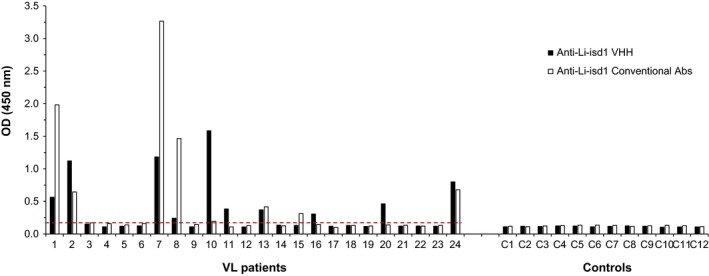
Antigen detection capture ELISA assembled with VHHs for the identification of Li‐isd1 in urine of VL patients and controls. ELISA plates were coated with either VHH JRD‐C1/JRD‐C1‐C1/myc (2 μg/mL) or antigen affinity‐purified IgY anti‐Li‐isd1 antibody (1 μg/mL). After overnight incubation with patients or control, urine samples plates were washed and wells were incubated with E‐tag‐labelled developing VHH JRD‐E5/JRD‐E9 or biotin‐labelled rabbit IgG anti‐Li‐isd1 antibody. Wells corresponding to reactions with VHHs were next incubated with peroxidase‐labelled rabbit anti‐E‐tag antibody. Wells incubated with biotinylated IgG were next incubated with streptavidin‐peroxidase. The substrate H2O2 and the chromophore TMB were then added followed by OD reading at 450 nm. Samples from VL patients and from controls were from Belo Horizonte, MG, Brazil. Dashed lines represent the cut‐off values calculated as described in the text. These are representative results of at least three experiments performed at different times with the same urine samples and same capture ELISA. VL, visceral leishmaniasis
Visceral leishmaniasis samples tested using the assays assembled with the anti‐Li‐ntf2 VHH reagents, and conventional antibodies showed similar overall results to those observed for Li‐isd1 (Figure 4). Thus, five (20.8%) VL samples tested positive using the assay assembled with the anti‐Li‐ntf2 VHH reagents. In contrast 11 (45.8%) VL samples tested positive using the assay assembled with conventional antibodies. However, one of the positive samples that was negative with the test assembled with conventional antibodies was strongly positive with the test assembled with the VHH reagents, again illustrating the possibility of increasing the antigen detection by using the combination of the assay assembled with conventional antibodies with an assay assembled with VHH reagents.
Figure 4.
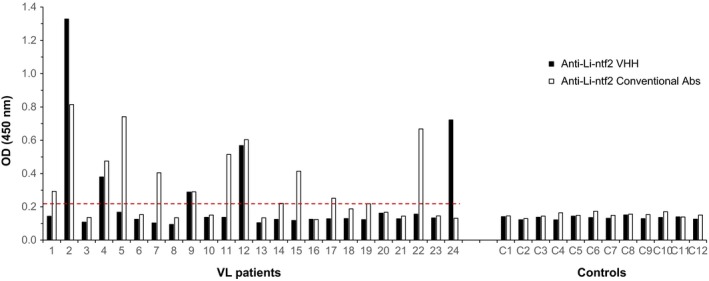
Antigen detection capture ELISA assembled with VHHs for the identification of Li‐ntf2 in urine of VL patients and controls. ELISA plates were coated with either JRO‐H8/JRO‐H8/myc (2 μg/mL) or purified IgY anti‐Li‐ntf2 antibody (1 μg/mL). After overnight incubation with patients or control urine samples, plates were washed and wells were incubated with E‐tag‐labelled developing JRF‐E3/JRF‐E3 or biotin‐labelled rabbit IgG anti‐Li‐isd1 antibody. Wells corresponding to reactions with VHHs were next incubated with peroxidase‐labelled rabbit anti‐E‐tag antibody. Wells incubated with biotinylated IgG were next incubated with streptavidin‐peroxidase. The substrate H2O2 and the chromophore TMB were then added followed by OD reading at 450 nm. Samples from VL patients and from controls were from Belo Horizonte, MG, Brazil. Dashed lines represent the cut‐off values calculated as described in the text. These are representative results of at least three experiments performed at different times with the same urine samples and same capture ELISA. VL, visceral leishmaniasis
The complementation of the results obtained with the assays assembled with VHH only and conventional Abs only is illustrated in Figure 5. Eight samples were positive only with the assay performed with anti‐Li‐isd1 VHH reagents, whereas three samples were positive only with the assay performed with anti‐Li‐ntf2 VHH reagents. Therefore, a combination of double‐positive plus single‐positive results leads to a sensitivity of 54.2%. Similarly, the combined sensitivity of the assay performed with conventional Abs only was 58.3%. Moreover, three samples (10, 16 and 20) that were negative with the assay performed with the conventional Abs were positive with the combined VHH assay thus suggesting a second level of assay complementarity.
Figure 5.
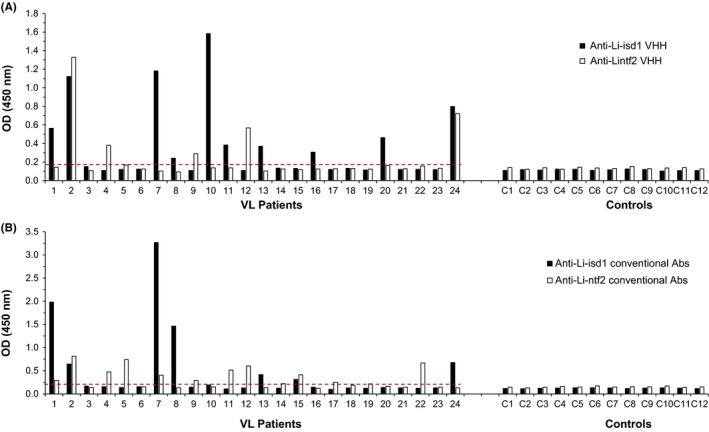
Compiled results of capture ELISA assembled with VHHs or conventional Abs for the identification of the proteins Li‐isd1 and Li‐ntf2 in urine of VL patients and controls. Note that several samples were positive only with either one of the VHH (A) or conventional Abs (B) assays, hence supporting the development of a multiplexed assay to increase the clinical sensitivity of the test. Also, the overall sensitivity of the compiled assays was highly comparable between VHH and conventional Abs assays (54.2% and 58.3% for VHH and conventional Abs, respectively). VL, visceral leishmaniasis
To experimentally confirm the complementation, we next assembled a “duplexed” assay coating the plates with both JRD‐C1/JRD‐C1‐C1/myc and JRO‐H8/JRO‐H8/myc homodimers and developing the reaction with a pool of JRD‐E5/JRD‐E9/E‐tag plus JRF‐E3/JRF‐E3/E‐tag. This duplexed assay was then used to test the urine samples from VL patients, from both healthy control subjects and from non‐VL patients who have other infectious diseases (cutaneous leishmaniasis, Chagas’ disease, schistosomiasis and tuberculosis). The results are illustrated in Figure 6 and confirm the predicted complementary illustrated in Figure 5. In fact, the overall sensitivity of the duplexed assay was slightly superior to that of the predicted sensitivity (66.6% × 54.2%), which could be due to incremental or summative OD signals in borderline results of urine samples performed with the individual VHHs. In addition, the results obtained with the duplexed assay further confirms the specificity of the test in that no positive samples were seen with urine from healthy control subjects as well as from patients having several infectious diseases other than VL.
Figure 6.
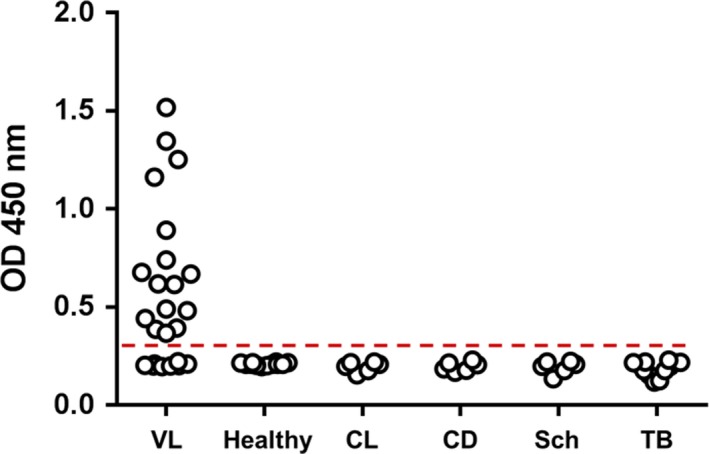
Clinical sensitivity and specificity of a capture duplexed ELISA assembled with both Li‐isd1 and Li‐ntf2 VHHs. ELISA plate wells were coated with a pool of VHH JRD‐C1/JRD‐C1‐C1/myc (anti‐Li‐isd1) and JRO‐H8/JRO‐H8/myc (anti‐Li‐ntf2) VHHs following by blocking and overnight incubation with patients or control urine samples. Plates were washed, and wells were incubated with a pool of the E‐tag‐labelled developing VHHs JRD‐E5/JRD‐E9 (anti‐Li‐isd1) plus JRF‐E3/JRF‐E3 (anti‐Li‐ntf2). Wells were next incubated with peroxidase‐labelled rabbit anti‐E‐tag antibody. The substrate H2O2 and the chromophore TMB were then added followed by OD reading at 450 nm. Samples from VL patients, n = 24; CL (cutaneous leishmaniasis) n = 6; CD (Chagas disease), n = 6; Sch (schistosomiasis), n = 6; and TB (tuberculosis), n = 12 were from Belo Horizonte, MG, Brazil. Healthy controls, n = 6 were from local donors. Dashed line represents the cut‐off values calculated as described in the text. These are representative results of at least three experiments performed at different times with the same urine samples. VL, visceral leishmaniasis
Together, these findings agree with our previous observations that showed that a multiplexed assay will have excellent sensitivity and specificity for the diagnosis of VL.19, 20, 25
4. DISCUSSION
Over the past years, using mass spectroscopy, we have shown that protein antigens from pathogens such as Mycobacterium tuberculosis and L. infantum can be detected in urine of patients with pulmonary tuberculosis and VL, respectively. The discovery of these pathogen proteins led us to the development of urine‐based antigen detection diagnostic tests for these diseases.19, 20, 30, 31, 32, 33 Capture ELISAs assembled with polyclonal antibodies raised in rabbits, and chicken have been used as proof of concept for the development of a urine‐based diagnostic test. This overall approach has proven to be a reliable discovery of pathogen biomarkers strategy, particularly for VL. A multiplexed assay based on three of the discovered biomarkers could be developed and shown to be highly sensitive and specific for the diagnosis of VL.19, 20, 30 However, a possible hindrance for upscaling and commercial production of this test exists as it relies on polyclonal antibodies. Conventional monoclonal antibodies (mAb) or recombinant single‐domain antibodies (sdAbs) are existing and proven alternatives to circumvent this limitation.
Here, we report our preliminary results on the use of sdAbs produced to the L. infantum antigens Li‐isd1 and Li‐ntf2 that we previously described as biomarkers of VL. We opted for the production of the immortalized recombinant camelid VHH because the technology for the production of such unique monoclonal antibodies is well established22 and because VHHs do not have the Fc portion of conventional immunoglobulins. Therefore, they provide less background noise due to reduced nonspecific binding between immunoglobulins (rheumatoid factor‐like) that can be seen in capture ELISA performed with conventional immunoglobulin antibody molecules.34
The initial proposal was to develop VHHs to the three L. infantum biomarkers that we have previously described (Li‐isd1, Li‐txn1 and Li‐ntf2). The immunization of two alpacas with a pool of the three antigens resulted in high serum antibody titres to the three antigens, followed by successful screening of a VHH library and generation of several VHH clones specific Li‐isd1 and Li‐ntf2. We arbitrarily selected clones that showed binding signals that were >10 times the background. We chose to use this criterion because we wanted to select VHHs that strongly bind to the target biomarkers, thus favouring the development of a sensitive capture ELISA. We are aware that such criterion reduces the number of possible VHH clones that could also be used in the assay development. However, for the present proof of principle study, we opted to concentrate only in the validation of strong binding VHHs as possible reagents for assay development, including comparison with the assay that we have previously developed using conventional chicken and rabbit antibodies. For this reason, we did not include clones that provided binding signals that were <10 times the background. Nonetheless, we were able to select five unique clones specific for Li‐isd1 and eight unique clones specific for Li‐ntf2. Out of these, we could express and purified soluble recombinant proteins from three different clones specific for Li‐isd1 and from two clones specific for Li‐ntf2. In addition, to increase the binding properties of these VHH for the assembly and development of a sensitive capture ELISA, we constructed one homodimer and one heterodimer for the anti‐Li‐isd1 and two homodimers for the clones specific for Li‐ntf1. Logistically, because we had only two clones specific for Li‐ntf2, the construction of heterodimer would not be useful for the assembly of the capture ELISA.
Unfortunately, and for reasons not clear, only one VHH clone reactive to Li‐txn1 was obtained. In addition, this VHH showed poor specificity for the antigen. It is very unusual for an antigen that induces strong antibody titres, such as Li‐txn1 yield such poor numbers of reactive VHH clones. However, it is theoretically possible that this antigen‐stimulated primary conventional antibody response in the alpacas and little or no heavy‐chain‐only antibodies, the source of VHHs. Another possibility is that Li‐txn1 undergoes conformational changes when coated onto plastic, and conformation is very important for VHH binding,35 which could also explain the lack of detection of possible Li‐txn1‐specific clones. Several approaches, including capturing the antigen on a plate coated with purified rabbit anti‐Li‐txn1 antibody followed by repanning (not shown), did not improve the detection of Li‐txn1 by VHH clones. An alternative possibility that could address this issue would be homogeneous time‐resolved fluorescence assays (HTRF). Unfortunately, the use of this approach would require a great deal of standardization and adaptation for the selection of clones producing VHH antibodies specific to Li‐tnx1 present in the soluble phase of the assay (in contrast to immobilized on the plastic surface of the ELISA plate). Therefore, because we interpreted that further clarification of whether the lack of clone detection was indeed due to low frequency of B cells or was due to conformational changes was beyond the scope of the manuscript, we deprioritized the generation of anti‐Li‐txn1 VHH in the present pilot study.
The capture ELISAs assembled with either anti‐Li‐isd1 or anti‐Li‐ntf2 VHHs showed similar biochemical sensitivity to that of our former ELISA assembled with chicken and rabbit conventional antibodies. This is a critical step for the development of a urine‐based antigen detection assay because the concentration of the pathogen's biomarker in this clinical sample is very low. Our former studies indicated that an assay sensitivity equal or higher than 100 pg/ml of the analyte in the urine sample is not sensitive enough to be used in clinical settings. We are confident that because the sensitivity of both capture ELISAs assembled with the VHHs produced in the present study was ~15‐45 pg/mL, these reagents are compatible with the development of a sensitive clinical diagnostic test for VL. Moreover, and importantly, the binding of the VHHs to their specific targets was not inhibited by human urine.
The initial clinical validation of the ELISAs assembled with VHHs clearly showed that the selected and engineered reagents have a clinical sensitivity and specificity for the diagnosis of VL that is similar to that of the assay developed with conventional antibodies. At this point, the overall sensitivity of the duplexed VHH assay (66.6%) is not sufficient for the development of a final test for the diagnosis of VL. However, these results are consistent with our former studies that indicated that a highly sensitive test requires the simultaneous detection of at least three VL biomarkers, either separately or in a multiplexed formatted test.19 Therefore, the inclusion of VHHs specific for other leishmanial markers, for example Li‐tnx1, should increase the sensitivity of the assay to accepted clinical levels. Moreover, we have recently discovered and characterized the clinical utility of four new Leishmania donovani markers (manuscript in preparation). Hence, we are confident that the addition of new VHHs specific for these additional markers will result in a development of a highly sensitive assay for the diagnosis of VL.
Finally, we believe that this assay, once optimized for higher sensitivity, will be of great utility not only for the diagnosis of active VL but also as an important tool to monitor the treatment efficacy of this disease. In fact, we have preliminary evidence of this utility using an ELISA assembled with conventional rabbit and chicken antibodies.30 This observation supports the suggestion that the assay sensitivity or antigen concentration in the urine is correlated with the pathogen's burden in the target organs. Therefore, in theory, it is possible that because urine from infected individuals contain variable amounts of pathogen antigens that a quantitative assay may be used to correlate different clinical forms and/or severity of the disease, for example asymptomatic and active VL.
In conclusion, altogether, the results confirm that VHH can replace conventional antibodies for the development of an antigen detection diagnostic test for VL. Importantly, because VHHs are immortalized reagents, they circumvent a practical and major limitation of conventional antibodies, that is the upscaling and continuous commercial supply of these reagents.
CONFLICT OF INTEREST
CA is an employee of DetectoGen Inc., has ownership option in the company and has received funding for research carried out in this work. JD is an employee of Detectogen Inc. and has no ownership or ownership option in the company. AC‐N is a co‐founder/director of DetectoGen Inc. and has ownership of the company. In addition, he has received funding for research carried out in this work.
Abeijon C, Dilo J, Tremblay JM, et al. Use of VHH antibodies for the development of antigen detection test for visceral leishmaniasis. Parasite Immunol. 2018;40:12584 10.1111/pim.12584
Funding information
This work was supported by a Universal Partnership (UP) Grant from the Massachusetts Life Sciences Center to AC‐N, and by a grant from the National Institutes of Health to CA (R44‐AI113992).
REFERENCES
- 1. Melby PC. Recent developments in leishmaniasis. Curr Opin Infect Dis. 2002;15(5):485‐490. [DOI] [PubMed] [Google Scholar]
- 2. Srivastava P, Dayama A, Mehrotra S, Sundar S. Diagnosis of visceral leishmaniasis. Trans R Soc Trop Med Hyg. 2011;105(1):1‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sundar S, Rai M. Laboratory diagnosis of visceral leishmaniasis. Clin Diagn Lab Immunol. 2002;9(5):951‐958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chappuis F, Sundar S, Hailu A, et al. Visceral leishmaniasis: what are the needs for diagnosis, treatment and control? Nat Rev Microbiol. 2007;5(11):873‐882. [DOI] [PubMed] [Google Scholar]
- 5. Dye C, Vidor E, Dereure J. Serological diagnosis of leishmaniasis: on detecting infection as well as disease. Epidemiol Infect. 1993;110(3):647‐656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Guerin PJ, Olliaro P, Sundar S, et al. Visceral leishmaniasis: current status of control, diagnosis, and treatment, and a proposed research and development agenda. Lancet Infect Dis. 2002;2(8):494‐501. [DOI] [PubMed] [Google Scholar]
- 7. Quinnell RJ, Courtenay O, Garcez L, Dye C. The epidemiology of canine leishmaniasis: transmission rates estimated from a cohort study in Amazonian Brazil. Parasitology. 1997;115(Pt 2):143‐156. [DOI] [PubMed] [Google Scholar]
- 8. Sinha PK, Pandey K, Bhattacharya SK. Diagnosis & management of leishmania/HIV co‐infection. Indian J Med Res. 2005;121(4):407‐414. [PubMed] [Google Scholar]
- 9. Houghton RL, Petrescu M, Benson DR, et al. A cloned antigen (recombinant K39) of Leishmania chagasi diagnostic for visceral leishmaniasis in human immunodeficiency virus type 1 patients and a prognostic indicator for monitoring patients undergoing drug therapy. J Infect Dis. 1998;177(5):1339‐1344. [DOI] [PubMed] [Google Scholar]
- 10. Nigro L, Vinci C, Romano F, Russo R. Comparison of the indirect immunofluorescent antibody test and the direct agglutination test for serodiagnosis of visceral leishmaniasis in HIV‐infected subjects. Eur J Clin Microbiol Infect Dis. 1996;15(10):832‐835. [DOI] [PubMed] [Google Scholar]
- 11. Santos‐Gomes G, Gomes‐Pereira S, Campino L, Araujo MD, Abranches P. Performance of immunoblotting in diagnosis of visceral Leishmaniasis in human immunodeficiency virus‐Leishmania sp.‐coinfected patients. J Clin Microbiol. 2000;38(1):175‐178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Alam M, Shamsuzzaman A, Musa A, et al. Antigen detection from urine of Kala‐azar cases by latex agglutination tes. Mymensingh Med J. 2008;17(1):22‐27. [PubMed] [Google Scholar]
- 13. Attar ZJ, Chance ML, El‐Safi S, et al. Latex agglutination test for the detection of urinary antigens in visceral leishmaniasis. Acta Trop. 2001;78(1):11‐16. [DOI] [PubMed] [Google Scholar]
- 14. Boelaert M, El‐Safi S, Hailu A, et al. Diagnostic tests for kala‐azar: a multi‐centre study of the freeze‐dried DAT, rK39 strip test and KAtex in East Africa and the Indian subcontinent. Trans R Soc Trop Med Hyg. 2008;102(1):32‐40. [DOI] [PubMed] [Google Scholar]
- 15. Diro E, Techane Y, Tefera T, et al. Field evaluation of FD‐DAT, rK39 dipstick and KATEX (urine latex agglutination) for diagnosis of visceral leishmaniasis in northwest Ethiopia. Trans R Soc Trop Med Hyg. 2007;101(9):908‐914. [DOI] [PubMed] [Google Scholar]
- 16. Sundar S, Agrawal S, Pai K, Chance M, Hommel M. Detection of leishmanial antigen in the urine of patients with visceral leishmaniasis by a latex agglutination test. Am J Trop Med Hyg. 2005;73(2):269‐271. [PubMed] [Google Scholar]
- 17. Sarkari B, Chance M, Hommel M. Antigenuria in visceral leishmaniasis: detection and partial characterisation of a carbohydrate antigen. Acta Trop. 2002;82(3):339‐348. [DOI] [PubMed] [Google Scholar]
- 18. Riera C, Fisa R, Lopez P, et al. Evaluation of a latex agglutination test (KAtex) for detection of Leishmania antigen in urine of patients with HIV‐Leishmania coinfection: value in diagnosis and post‐treatment follow‐up. Eur J Clin Microbiol Infect Dis. 2004;23(12):899‐904. [DOI] [PubMed] [Google Scholar]
- 19. Abeijon C, Campos‐Neto A. Potential non‐invasive urine‐based antigen (protein) detection assay to diagnose active visceral leishmaniasis. PLoS Negl Trop Dis. 2013;7(5):e2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Abeijon C, Kashino SS, Silva FO, et al. Identification and diagnostic utility of Leishmania infantum proteins found in urine samples from patients with visceral leishmaniasis. Clin Vaccine Immunol. 2012;19(6):935‐943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kashino SS, Abeijon C, Qin L, et al. Identification of Leishmania infantum chagasi proteins in urine of patients with visceral leishmaniasis: a promising antigen discovery approach of vaccine candidates. Parasite Immunol. 2012;34(7):360‐371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Maass DR, Sepulveda J, Pernthaner A, Shoemaker CB. Alpaca (Lama pacos) as a convenient source of recombinant camelid heavy chain antibodies (VHHs). J Immunol Methods. 2007;324(1–2):13‐25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Arbabi GM, Desmyter A, Wyns L, Hamers R, Muyldermans S. Selection and identification of single domain antibody fragments from camel heavy‐chain antibodies. FEBS Lett. 1997;414(3):521‐526. [DOI] [PubMed] [Google Scholar]
- 24. Wesolowski J, Alzogaray V, Reyelt J, et al. Single domain antibodies: promising experimental and therapeutic tools in infection and immunity. Med Microbiol Immunol. 2009;198(3):157‐174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Moayeri M, Leysath CE, Tremblay JM, et al. A heterodimer of a VHH (variable domains of camelid heavy chain‐only) antibody that inhibits anthrax toxin cell binding linked to a VHH antibody that blocks oligomer formation is highly protective in an anthrax spore challenge model. J Biol Chem. 2015;290(10):6584‐6595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Maass DR, Harrison GB, Grant WN, Shoemaker CB. Three surface antigens dominate the mucosal antibody response to gastrointestinal L3‐stage strongylid nematodes in field immune sheep. Int J Parasitol. 2007;37(8–9):953‐962. [DOI] [PubMed] [Google Scholar]
- 27. Mukherjee J, Tremblay JM, Leysath CE, et al. A novel strategy for development of recombinant antitoxin therapeutics tested in a mouse botulism model. PLoS One. 2012;7(1):e29941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tremblay JM, Kuo CL, Abeijon C, et al. Camelid single domain antibodies (VHHs) as neuronal cell intrabody binding agents and inhibitors of Clostridium botulinum neurotoxin (BoNT) proteases. Toxicon. 2010;56(6):990‐998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tremblay JM, Mukherjee J, Leysath CE, et al. A single VHH‐based toxin‐neutralizing agent and an effector antibody protect mice against challenge with Shiga toxins 1 and 2. Infect Immun. 2013;81(12):4592‐4603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Abeijon C, Singh OP, Chakravarty J, Sundar S, Campos‐Neto A. Novel antigen detection assay to monitor therapeutic efficacy of visceral leishmaniasis. Am J Trop Med Hyg. 2016;95(4):800‐802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kashino SS, Pollock N, Napolitano DR, Rodrigues V Jr, Campos‐Neto A. Identification and characterization of Mycobacterium tuberculosis antigens in urine of patients with active pulmonary tuberculosis: an innovative and alternative approach of antigen discovery of useful microbial molecules. Clin Exp Immunol. 2008;153(1):56‐62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Napolitano DR, Pollock N, Kashino SS, Rodrigues V Jr, Campos‐Neto A. Identification of Mycobacterium tuberculosis ornithine carboamyltransferase in urine as a possible molecular marker of active pulmonary tuberculosis. Clin Vaccine Immunol. 2008;15(4):638‐643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pollock NR, Macovei L, Kanunfre K, et al. Validation of Mycobacterium tuberculosis Rv1681 protein as a diagnostic marker of active pulmonary tuberculosis. J Clin Microbiol. 2013;51(5):1367‐1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Larsson A, Karlsson‐Parra A, Sjoquist J. Use of chicken antibodies in enzyme immunoassays to avoid interference by rheumatoid factors. Clin Chem. 1991;37(3):411‐414. [PubMed] [Google Scholar]
- 35. Vrentas CE, Moayeri M, Keefer AB, et al. A diverse set of single‐domain antibodies (VHHs) against the anthrax toxin lethal and edema factors provides a basis for construction of a bispecific agent that protects against anthrax infection. J Biol Chem. 2016;291(41):21596‐21606. [DOI] [PMC free article] [PubMed] [Google Scholar]