

Abstract
Proton detection and fast magic‐angle spinning have advanced biological solid‐state NMR, allowing for the backbone assignment of complex protein assemblies with high sensitivity and resolution. However, so far no method has been proposed to detect intermolecular interfaces in these assemblies by proton detection. Herein, we introduce a concept based on methyl labeling that allows for the assignment of these moieties and for the study of protein−protein interfaces at atomic resolution.
Keywords: methyl labeling, interfaces, proteins, protein structures, solid-state NMR
Solid‐state NMR (ssNMR) spectroscopy is a powerful technique to study the structure and dynamics of macromolecular protein assemblies, such as bacterial cytoskeletal filaments,1 phage capsids2 and amyloid fibrils.3, 4, 5, 6, 7, 8 Although ssNMR linewidths, in contrast to solution NMR, are not limited by the molecular weight of the protein complex, many protein samples remain beyond the scope of this method. This is due to heterogeneous line‐broadening, a sample‐dependent property, and the spectral crowding caused by a large number of residues. These effects lead to peak overlap, rendering spectral data ambiguous, and ssNMR as a tool ineffective for many biological systems. Recently, protein deuteration and magic angle spinning (MAS) at frequencies at or above 40 kHz have enabled proton‐detected experiments that offer a higher sensitivity and dimensionality than their traditional carbon‐detected counterparts.9, 10, 11, 12 While these methodologies allow for unambiguous spectral assignments (via a “backbone walk”) of large proteins,13, 14, 15 structural data such as long‐distance restraints, which are required for 3D structure determination, are still collected in a highly ambiguous and/or insensitive manner. To this date, most restraints are extracted from carbon‐detected experiments (e. g. 2D carbon‐carbon correlations with homonuclear mixing through space)16,17 or proton‐detected 4D HNhhNH experiments with radio frequency driven recoupling mixing (RFDR mixing)18 between protons (correlating amide groups through space).19,20 The latter experiment offers excellent resolution due to its four‐dimensionality but lacks sensitivity compared to 3D proton‐detected experiments (since increasing dimensionality decreases signal‐to‐noise).21 However, its 3D version, the 3D HNhH, yields highly ambiguous data for uniformly labeled proteins due to the ubiquity of protons in proteins. If unambiguous data are to be obtained in 2D or 3D proton‐detected experiments, this methodology clearly needs to be combined with sophisticated labeling strategies and experimental designs.22
An exceptional advantage of ssNMR as a structural biology method is that it allows for the study of protein‐protein interfaces by heterogeneous isotope labeling.23,24 Traditionally, these structural features have been analyzed by carbon‐detected experiments such as spin dilution experiments4 and mixed labeling schemes with 50 % 13C labeled protein species and 50 % 15N labeled protein species (e. g. through NhhC experiments25). Due to their carbon‐detected nature, these experiments suffer from low sensitivity and dimensionality, requiring many weeks of data acquisition, and often resulting in highly ambiguous data.
In this work, we use methyl labeling26—which has proven itself as an indispensable tool in solution NMR of large proteins27,28—to simplify spectral data and facilitate proton‐detection.29,30 The methodology presented here allows for the collection of long‐distance restraints with 3D proton‐detected ssNMR experiments between the following: 1.) methyl and amide groups globally; 2.) methyl and amide groups at protein‐protein interfaces.
Additionally, we present an assignment method for isoleucine Cδ1 methyl groups by their correlation with backbone resonances (which ought to be known from a preliminary backbone walk) using a specific isoleucine precursor (“assignment precursor”) and homonuclear transfer steps. In this manner, time‐consuming assignment by mutagenesis can be avoided, opening the way to study assemblies with very large subunits and a high number of isoleucine residues. The presented strategy is used to assign the isoleucine Cδ1 methyl groups in gp17.1, the major tail tube protein of the bacteriophage SPP1,31 and to identify a protein−protein interface within the gp17.1 polymer that is crucial for protein oligomerization.
By supplementation of the precursor 2‐Ketobutyric acid‐4‐13C‐3,3‐d2 during protein expression in deuterated media,32 isoleucine Cδ1 methyl groups are 13C and 1H labeled, and thus rendered “NMR‐visible” (Figure 1A and Figure S3 in the Supporting Information). Isoleucine residues are well distributed over the primary structure of gp17.1 (Figure 1B) and are, due to their limited number, fully resolved by their Hδ1 chemical shift as shown in the 2D hCH correlation spectrum in Figure 1C (each isoleucine Cδ1 methyl group in gp17.1 results in one peak in the spectrum). If all subunits in the prepared gp17.1 protein assembly are 15N and isoleucine Cδ1 methyl labeled, proton−proton mixing from amide to methyl protons can occur within and in between gp17.1 subunits (left panel of Figure 1D). If half of the subunits are 15N labeled and the other half are isoleucine Cδ1 methyl labeled, this mixing can only occur between two different subunits (right panel of Figure 1D). Figure 1E shows 2D planes of two 3D HNhH experiments with RFDR mixing superimposed with the 2D hNH fingerprint spectrum of gp17.1 (grey spectrum in the background, Figure S1). The 2D plane corresponding to the Hδ1 methyl frequency of Ile18 (as represented by the dashed blue line) shows magnetization transfer from nearby amide protons to the Hδ1 protons of Ile18. In the left spectrum (homogeneous labeling) all nearby amide groups are visible. However, in the right spectrum (mixed labeling) only intermolecular contacts are visible, limiting the number of peaks, and revealing a protein‐protein interface between Ile18 and the C‐terminus of gp17.1.
Figure 1.
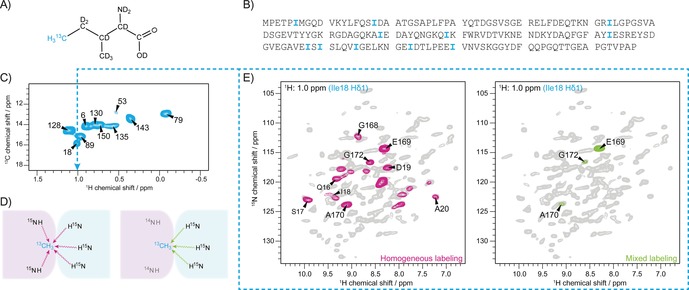
Isoleucine‐methyl labeling as a tool to map protein interfaces. A) 2‐Ketobutyric acid‐4‐13C‐3,3‐d2 as a precursor leads to labeling of isoleucines in the protein as indicated. Only the Cδ1 methyl group carries a 13C isotope as well as protons and is thus, by the choice of suitable experiments, “NMR visible”. B) Isoleucine residues are well distributed over the primary structure of gp17.1 C) 2D hCH correlation spectrum of isoleucine‐Cδ1 labeled gp17.1 at 40 kHz MAS and 900 MHz external magnetic field. The limited number of isoleucines in gp17.1 results in fully resolved unique resonances in the proton dimension. D) Schematic representation of the labeling strategy to identify intermolecular contacts. In the first sample (left), all subunits are 15N and Ile‐Cδ1 labeled. Magnetization transfer through space occurs between and within subunits. In the second sample (right), half of the subunits are 15N and the other half Ile‐Cδ1 labeled. Magnetization transfer through space can only occur between different subunits. E) 2D planes of 3D HNhH spectra with RFDR mixing superimposed with a 2D hNH correlation spectrum. The 2D planes correspond to the 1H methyl frequency of Ile18 (as represented by the blue dashed line) showing magnetization transfer from amide protons to the protons attached to Ile18 Cδ1. In the left case, all NH groups close to the methyl group of Ile18 are revealed. However, in the right case, only intermolecular contacts can be detected limiting the amount of peaks. This approach reveals a protein‐protein interface between Ile18 of one subunit and the C‐terminus (E169, A170 and G172) of another subunit.
For the isoleucine Cδ1 methyl assignment, the precursor 2‐Ketobutyric acid‐13C4‐3,3‐d2 (“assignment precursor”, Figure S3) is supplemented during protein expression, which in addition to the NMR‐visible isoleucine Cδ1 methyl group leads to the incorporation of further 13C species at the Cγ1, Cα and CO sites of all isoleucine residues, without concomitant introduction of additional protons. Using a long cross polarization (CP) step of 6 ms, magnetization can be transferred from Hδ1 to the labeled carbons in the isoleucine side chain and back to the methyl protons resulting in a 2D hCH correlation spectrum as shown in Figure 2A. This procedure enables the correlation of all labeled carbon atoms within the isoleucine side chain and Cα as part of the backbone. Additionally, in a second experiment, magnetization is transferred from Hδ1 to Cγ1, and subsequently to Cα by homonuclear dipolar recoupling enhanced by amplitude modulation (DREAM) transfer33 traversing the unlabeled Cβ through space ‐ a transfer which would be impossible in solution NMR by through‐bond mixing. By cαN and nH CP steps, magnetization is finally transferred to the backbone amide protons for detection, resulting in a 2D HccanH correlation spectrum (Figures 2B and S2). These two experiments allow for a straightforward assignment of the isoleucine Cδ1 methyl groups by their correlation with backbone resonances. If these spectra are not sufficient for assignment, the 2D HccanH spectrum can be expanded by further indirect dimensions on either Cα or amide N.
Figure 2.
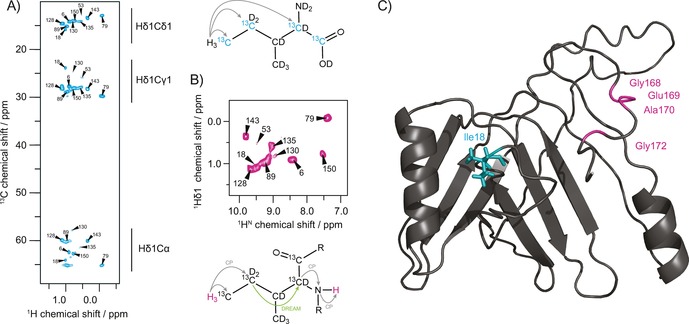
Assignment of isoleucine Cδ1 methyl groups and location of the protein−protein interface. A) 2D hCH correlation spectrum of isoleucine Cδ1 methyl labeled gp17.1 at 40 kHz MAS and 900 MHz external magnetic field. 2‐Ketobutyric acid‐13C4‐3,3‐d2 was used as a precursor resulting in the labeling of isoleucine residues as shown. A 6 ms hC cross‐polarization time enabled magnetization transfer from the Hδ1 methyl protons throughout the side‐chain and back. B) 2D HccanH spectrum to correlate methyl Hδ1 protons with their backbone amide protons for assignment. Magnetization is initially transferred from Hδ1 to Cδ1 and Cγ1 via CP, and subsequently to Cα via DREAM. Finally, magnetization transfer to HN is achieved by cαN and nH CP. C) Mapping of the determined tail tube interface onto the homology model of a polymerized gp17.1 subunit (taken from Langlois et al.31). The location of the Ile18 residue in the middle of the subunit suggests that the C‐terminus forms a molecular bridge that embraces the next monomer within the tail tube quaternary structure.
The described methodology allowed us to assign all visible isoleucine Cδ1 methyl groups, including multiple conformations, of gp17.1 (cf. the Supporting Information Table S4) and, on that basis, to identify 105 unambiguous long‐range restraints between amides and isoleucine Cδ1 methyl groups, 7 of which are unambiguously intermolecular (Figure S4, including chemical shift‐ambiguous restraints: 179/12). The determined intermolecular contacts between Ile18 and Glu169, Ala170 and Gly172 (and Gly168 which is below the contour levels in Figure 1E) are intriguing since the C‐terminus is predicted to be disordered and lacks homology to any other structurally‐described bacteriophage tail protein. Also, Ile18 is not located in the outer β‐strands of the homology model of polymerized gp17.1, which would resemble a “classic” protein interface in such an assembly, but in the middle of the subunit (Figure 2C). To check the significance of this newly discovered interface, we removed the last 7 and 14 C‐terminal residues of gp17.1 (1–170 and 1–163) and observed that both mutants are incapable of polymerization, but remain monomeric (see size‐exclusion chromatography data in Figure S5 and solution NMR data in Figure S6 in the Supporting Information).34 This emphasizes the proposition that the C‐terminus forms a molecular bridge that embraces the next monomer within the tube,35 similar to what was described for the N‐terminus of the T4 phage tail tube protein gp19.36
We expect that the methodology introduced here will turn out useful in a variety of solid‐state NMR investigations of supramolecular assemblies or membrane‐integrated proteins or protein complexes. We note that our straightforward approach to assign isoleucines can be modified to assign the methyl groups of leucine and valine (LV), threonine, alanine or methionine. This makes tedious mutation studies for methyl group assignment redundant, provided that the backbone assignment is available. Furthermore, we think that methyl groups as tools for long‐distance restraints will become indispensable for the analysis of similar or even larger protein systems. The mixed‐labeling strategy for the identification of protein−protein interfaces may also be extended to include differently methyl‐labeled subunits, e.g 50 % isoleucine Cδ1 methyl labeled; 50 % LV methyl labeled.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank Dr. Peter Schmieder for help with the solution NMR measurements and Dr. Veniamin Chevelkov for helpful discussions. This work was supported by the Leibniz‐Forschungsinstitut für Molekulare Pharmakologie (FMP) and the European Research Council (ERC Starting Grant to A.L.; Grant agreement no.: 337490).
M. Zinke, P. Fricke, S. Lange, S. Zinn-Justin, A. Lange, ChemPhysChem 2018, 19, 2457.
References
- 1. Shi C., Fricke P., Lin L., Chevelkov V., Wegstroth M., Giller K., Becker S., Thanbichler M., Lange A., Sci. Adv. 2015, 1, e1501087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Aharoni T., Goldbourt A., Chem. – A Eur. J. 2018, DOI 10.1002/chem.201800532. [Google Scholar]
- 3. Quinn C. M., Polenova T., Q. Rev. Biophys. 2017, 50, 1–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Schütz A. K., Vagt T., Huber M., Ovchinnikova O. Y., Cadalbert R., Wall J., Güntert P., Böckmann A., Glockshuber R., Meier B. H., Angew. Chem. Int. Ed. 2015, 54, 331–335; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 337–342. [Google Scholar]
- 5. Colvin M. T., Silvers R., Frohm B., Su Y., Linse S., Griffin R. G., J. Am. Chem. Soc. 2015, 137, 7509–7518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lewandowski J. R., Halse M. E., Blackledge M., Emsley L., Science. 2015, 348, 578–581. [DOI] [PubMed] [Google Scholar]
- 7. Theint T., Nadaud P. S., Aucoin D., Helmus J. J., Pondaven S. P., Surewicz K., Surewicz W. K., Jaroniec C. P., Nat. Commun. 2017, 8, DOI 101038/s41467-017-00794-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.D. T. Murray, M. Kato, Y. Lin, K. R. Thurber, I. Hung, S. L. Mcknight, R. Tycko, D. T. Murray, M. Kato, Y. Lin, Cell 2017, 171, 615–627. [DOI] [PMC free article] [PubMed]
- 9. Barbet-Massin E., Pell A. J., Retel J. S., Andreas L. B., Jaudzems K., Franks W. T., Nieuwkoop A. J., Hiller M., Higman V., Guerry P., J. Am. Chem. Soc. 2014, 136, 12489–12497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhou D. H., Nieuwkoop A. J., Berthold D. A., Comellas G., Sperling L. J., Tang M., Shah G. J., Brea E. J., Lemkau L. R., Rienstra C. M., J. Biomol. NMR 2012, 54, 291–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chevelkov V., Habenstein B., Loquet A., Giller K., Becker S., Lange A., J. Magn. Reson. 2014, 242, 180–188. [DOI] [PubMed] [Google Scholar]
- 12. Penzel S., Smith A. A., Agarwal V., Hunkeler A., Org M. L., Samoson A., Böckmann A., Ernst M., Meier B. H., J. Biomol. NMR 2015, 63, 165–186. [DOI] [PubMed] [Google Scholar]
- 13. Fraga H., Arnaud C. A., Gauto D. F., Audin M., Kurauskas V., Macek P., Krichel C., Guan J. Y., Boisbouvier J., Sprangers R., ChemPhysChem 2017, 18, 2697–2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Vasa S. K., Singh H., Rovó P., Linser R., J. Phys. Chem. Lett. 2018, 9, 1307–1311. [DOI] [PubMed] [Google Scholar]
- 15. Fricke P., Chevelkov V., Zinke M., Giller K., Becker S., Lange A., Nat. Protoc. 2017, 12, 764–782. [DOI] [PubMed] [Google Scholar]
- 16. Loquet A., Sgourakis N. G., Gupta R., Giller K., Riedel D., Goosmann C., Griesinger C., Kolbe M., Baker D., Becker S., Nature 2012, 486, 276–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Castellani F., Van Rossum B., Diehl A., Schubert M., Rehbein K., Oschkinat H., Nature 2002, 420, 23–26. [DOI] [PubMed] [Google Scholar]
- 18. Bennett A. E., Rienstra C. M., Griffiths J. M., Zhen W., Lansbury P. T., Griffin R. G., J. Chem. Phys. 1998, 108, 9463–9479. [Google Scholar]
- 19. Huber M., Hiller S., Schanda P., Ernst M., Böckmann A., Verel R., Meier B. H., ChemPhysChem 2011, 12, 915–918. [DOI] [PubMed] [Google Scholar]
- 20. Huber M., Böckmann A., Hiller S., Meier B. H., Phys. Chem. Chem. Phys. 2012, 14, 5239. [DOI] [PubMed] [Google Scholar]
- 21. Linser R., Bardiaux B., Higman V., Fink U., Reif B., J. Am. Chem. Soc. 2011, 133, 5905–5912. [DOI] [PubMed] [Google Scholar]
- 22. Jain M., Lalli D., Stanek J., Gowda C., Prakash S., Tom S., J. Phys. Chem. Lett. 2017, 8, 2399–2405. [DOI] [PubMed] [Google Scholar]
- 23. Etzkorn M., Böckmann A., Lange A., Baldus M., J. Am. Chem. Soc. 2004, 136, 14746–14751. [DOI] [PubMed] [Google Scholar]
- 24. Nieuwkoop A. J., Rienstra C. M., J. Am. Chem. Soc. 2010, 132, 7570–7571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lange A., Luca S., Baldus M., J. Am. Chem. Soc. 2002, 124, 9704–9705. [DOI] [PubMed] [Google Scholar]
- 26. Fasshuber H. K., Demers J.-P., Chevelkov V., Giller K., Becker S., Lange A., J. Magn. Reson. 2015, 252, 10–19. [DOI] [PubMed] [Google Scholar]
- 27. Sprangers R., Velyvis A., Kay L. E., Nat. Methods 2007, 4, 697–703. [DOI] [PubMed] [Google Scholar]
- 28. Kerfah R., Plevin M. J., Sounier R., Gans P., Boisbouvier J., Curr. Opin. Struct. Biol. 2015, 32, 113–122. [DOI] [PubMed] [Google Scholar]
- 29. Kurauskas V., Crublet E., Macek P., Kerfah R., Gauto D. F., Boisbouvier J., Schanda P., Chem. Commun. 2016, 52, 9558–9561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wang S., Parthasarathy S., Nishiyama Y., Endo Y., Nemoto T., Yamauchi K., Asakura T., Takeda M., Terauchi T., Kainosho M., PLoS One 2015, 10, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Langlois C., Ramboarina S., Cukkemane A., Auzat I., Chagot B., Gilquin B., Ignatiou A., Petitpas I., Kasotakis E., Paternostre M., J. Biol. Chem. 2014, 290, jbc.M114.613166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gardner K. H., Kay L. E., J. Am. Chem. Soc. 1997, 119, 7599–7600. [Google Scholar]
- 33. Verel R., Ernst M., Meier B. H., J. Magn. Reson. 2001, 150, 81–99. [DOI] [PubMed] [Google Scholar]
- 34. Anglister J., Grzesiek S., Ren H., Klee C. B., Bax A., J. Biomol. NMR 1993, 3, 121–126. [DOI] [PubMed] [Google Scholar]
- 35. Zinke M., Fricke P., Samson C., Hwang S., Wall J. S., Lange S., Zinn-Justin S., Lange A., Angew. Chem. Int. Ed. 2017, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Taylor N. M. I., Prokhorov N. S., Guerrero-Ferreira R. C., Shneider M. M., Browning C., Goldie K. N., Stahlberg H., Leiman P. G., Nature 2016, 533, 346–352. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary