

Abstract
Ubiquitin‐fold modifier 1 (UFM1) is a reversible post‐translational modifier that is covalently attached to target proteins through an enzymatic cascade and removed by designated proteases. Abnormalities in this process, referred to as Ufmylation, have been associated with a variety of human diseases. Given this, the UFM1‐specific enzymes represent potential therapeutic targets; however, understanding of their biological function has been hampered by the lack of chemical tools for activity profiling. To address this unmet need, a diversifiable platform for UFM1 activity‐based probes (ABPs) utilizing a native chemical ligation (NCL) strategy was developed, enabling the generation of a variety of tools to profile both UFM1 conjugating and deconjugating enzymes. The use of the probes is demonstrated in vitro and in vivo for monitoring UFM1 enzyme reactivity, opening new research avenues.
Keywords: activity-based probes, chemical biology, native chemical ligation, post-translational modifications, UFM1
Post‐translational modification (PTM) of proteins by chemical groups, peptides, complex molecules, or even small proteins facilitates dynamic protein diversification to modulate cellular responses. Ubiquitination is one of the most common PTMs and a number of Ub‐like proteins (Ubls) have been subsequently identified. Ubiquitin‐fold modifier 1 (UFM1) is one of the recently identified Ubls and displays a similar tertiary structure, yet it has little sequence identity to Ubiquitin (Ub).1 Analogous to ubiquitin, it is covalently attached to the lysine residues of its substrates by the sequential action of three dedicated enzymes‐E1 (UBA5), E2 (Ufc1), and E3 (Ufl1) and is cleaved by UFM1‐specific proteases (Ufsps).2 This process, referred to as Ufmylation, is initiated by the adenylation of the exposed C‐terminal glycine of mature UFM1 and subsequent nucleophilic reaction with the active‐site cysteine of UBA5. The resulting high‐energy thioester bond allows the transfer onto the catalytic site cysteine of the E2 enzyme Ufc1, in a trans‐thioesterification reaction. Lastly, the E3‐like enzyme Ufl1 mediates the transfer of activated UFM1 onto the lysine residues of the protein substrates resulting in the formation of an isopeptide linkage. In addition to releasing UFM1 from its substrates, the UFM1 specific proteases‐Ufsp1 and Ufsp2‐mediate the maturation of pro‐UFM1.3 Although Ufmylation has been connected to biological processes including ER homeostasis,4, 5, 6 vesicle trafficking,5 blood progenitor development and differentiation,7, 8 G‐coupled protein receptor (GPCR) maturation,9 transcriptional control,10 mitosis,11 and autophagy,7, 12 the underlying mechanisms remain to be studied. Furthermore, abnormalities in the UFM1 cascade are reported to be associated with a number of human diseases, including cancer,13 diabetes,14 schizophrenia,15 and ischemic heart disease6 and to play a pivotal role in embryonic development and hematopoiesis.8, 16 Notwithstanding the biochemical and structural studies of UFM1‐conjugating and deconjugating enzymes that have been undertaken,17, 18, 19, 20, 21 their biological function remains enigmatic primarily owing to the lack of activity‐based reagents. By contrast, diverse reagents and ABPs have been developed for both Ub‐conjugating and deconjugating enzymes22, 23, 24, 25, 26, 27 and have been expanded to Ubls such as SUMO28, 29, 30 and Nedd8.24, 26 This advancement of assay and activity‐based reagents has greatly propelled discoveries in the ubiquitin field, yet such a diversifiable synthetic platform for UFM1 needs to be developed.31 While UFM1 has been prepared using multiple segment ligations based on KAHA chemistry, this is a time‐consuming process requiring the incorporation of a (S)‐5‐oxaproline building block at multiple sites.32 We first attempted to generate UFM1 using linear solid‐phase peptide synthesis (SPPS) by incorporating aggregation breakers such as pseudoproline33 at permissible sites (Supporting Information, Figure S1). Although this linear synthesis approach yields full‐length UFM1, which has been utilized in a recent study,34 the synthesis wasn't efficient, presumably owing to inefficient coupling of amino acid 36 onwards (see Figure S2). To circumvent this issue, we present herein a more practical two‐segment native chemical ligation (NCL) approach35, 36 towards full‐length UFM1 and UFM1 activity‐based reagents (Figure 1). Given the increasing knowledge on the importance of UFM1, our synthetic strategy gives access to valuable tools that allow the in vitro and in vivo characterization of enzymatic activity, thereby enabling insights into the dynamics of Ufmylation.
Figure 1.
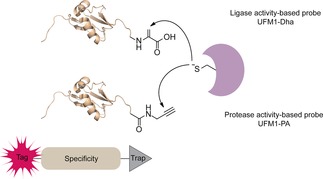
Schematic illustration of the UFM1 toolbox featuring activity‐based probes to study preference and selectivity of both proteases and ligases by covalently capturing active enzymes.
To improve the UFM1 synthesis, we devised a practical native chemical ligation (NCL) strategy to generate full‐length UFM1 based on a N‐terminal peptide thioester fragment (AA 1–44) and C‐terminal peptide fragment (AA 45–83) with alanine at position 45 replaced by a cysteine (Scheme 1). This method permitted the generation of full‐length UFM1 and a complete repertoire of probes in an efficient manner using a minimal amount of building blocks (Scheme 1).
Scheme 1.
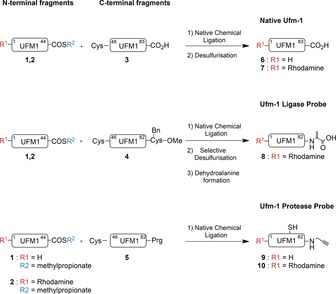
Native chemical ligation strategy towards native UFM1 and UFM1 activity‐based probes.
Using SPPS and standard coupling conditions (namely 4 equiv Fmoc‐protected amino acid, 4 equiv PyBOP, 8 equiv DIPEA, and double coupling cycles), we prepared the N‐terminal fragment (AA 1–44) on a hyper‐acid labile trityl resin to give amine 1 or functionalized its N‐termini with a Rhodamine 110 fluorophore (2).37 Then, selective cleavage from the resin with 20 % hexafluoro‐2‐propanol (HFIP) in CH2Cl2 liberated the C‐terminal carboxylate leaving all other protective groups in place. The C‐terminal carboxylate was then converted into the mercaptomethylpropionate thioester by reaction with mercaptomethylpropionate, followed by global deprotection and HPLC purification. Subsequently, the C‐terminal fragment (A45C‐83, 3) was obtained by Fmoc‐based SPPS, followed by global deprotection and HPLC purification.
Native chemical ligation of the N‐terminal thioester‐fragment (1 or 2) and C‐terminal cysteine‐fragment (3) was performed under denaturing conditions in 8 m Gdn⋅HCl containing 100 mm tris(2‐carboxyethyl)phosphine (TCEP) and 100 mm mercaptophenylacetic acid (MPAA) at pH 7.6 and 37 °C. Conversion to the full‐length UFM1 product (6 or 7) proceeded very efficiently in just 30 min. Precipitation from water and subsequent redissolution in Gdn⋅HCl followed by radical desulfurization using VA‐044, glutathione, and TCEP for 16 h at 37 °C resulted in formation of the full‐length native UFM1 as the cysteine residue at the ligation site was converted into the native alanine. Subsequent HPLC purification followed by gel‐filtration yielded UFM1 and Rho‐UFM1 in 85 % and 79 % yield, respectively.
Correct folding of purified synthetic UFM1 (Figure 2 A) was verified by circular dichroism (CD) spectroscopy (Figure 2 B). To further verify the correct folding and thus biochemical function, we compared synthetic and recombinant UFM1 in an enzymatic reaction with UBA5 (E1). Efficient formation of the UBA5∼UFM1 thioester proved that synthetic UFM1 is processed with the same efficiency as recombinant UFM1 (Figure 2 C and Figure S3).
Figure 2.
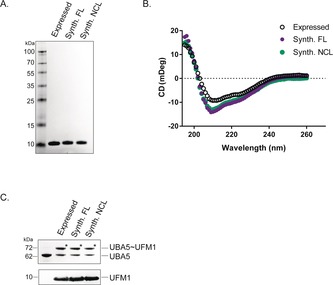
Validation of synthetic UFM1. A) SDS‐PAGE gel showing synthetic and expressed UFM1. B) CD measurements of expressed UFM1 and synthetic UFM1 made by linear synthesis or native chemical ligation. Synth. FL=UFM1 generated by linear synthesis; Synth. NCL=UFM1 generated by NCL (6). C) UBA5 reacts with both expressed and synthetic UFM1. The asterisk (*) indicates UFM1‐thioester‐UBA5.
Having developed a convenient UFM1 synthesis, we then focused on the generation of a UFM1‐based toolbox to enable the profiling and visualization of all enzymes involved in the Ufmylation cascade (Figure 1, Scheme 1). For this purpose, we synthesized UFM1‐Dha to target the conjugating class of enzymes26 and UFM1‐PA to target the proteases.24 To achieve this, we synthesized a C‐terminal fragment lacking the last amino acid (A45C‐82). Upon cleavage from the resin with HFIP, the C‐termini were equipped with either Cys(Bn)‐OMe (4) or propargylamide (5). After global deprotection and HPLC purification, the desired C‐terminal fragments were obtained and subsequently used in a NCL reaction as described above. ABP precursors for the UFM1 ligase probe (8) was successfully constructed by NCL followed by a radical desulfurization reaction to obtain UFM1‐Cys(Bn)‐OMe. This was subsequently transformed by oxidative elimination with O‐mesitylenesulfonylhydroxyl‐amine (MSH). Finally, the methyl ester was hydrolysed, to generate the UFM1‐Dha probe (8). ABPs targeting UFM1‐specific cysteine proteases (9–10) were easily constructed by native chemical ligation as well. However, one limitation we encountered was that the remaining cysteine after NCL could not be desulphurized, as radical desulfurization conditions compromise the integrity of the propargyl warhead. But since the ligation position (AA 45) is situated outside the critical C‐terminal region for UFM1 recognition by Ufsp enzymes,38 we expect this small thiol group to be tolerated. Furthermore, the routine addition of 1,4‐dithiothreitol (DTT) to the enzymatic assay reaction buffers reduces any potential disulfide bridges occurring at this cysteine that might affect enzymatic processing.
With these ABPs in hand, we tested their reactivity towards UFM1 ligases and proteases in cell lysates. Firstly, we assessed the reactivity of the E1 enzyme‐UBA5‐towards Rho‐UFM1‐Dha (8). Similar to UbDha, UFM1‐Dha forms an adenylate with the C‐terminal glycine of UFM1. The activated methylene group of this adenylate intermediate can then undergo a nucleophilic attack of the active‐site cysteine to yield either the covalent UBA5‐UFM1‐thioether adduct or the UBA5‐UFM1∼thioester26 (Figure 3 A). Upon reacting UBA5 with the ligase probe Rho‐UFM1‐Dha (8), a complex is formed that remains stable in the presence of DTT, indicative of the thioether bond formed between the active site cysteine and the dehydroalanine moiety of 8 in contrast to the thioester bond formed between UBA5 and UFM1 (7) (Figure 3 B and Figure S3). Furthermore, UFM1‐Dha does not display cross‐reactivity towards UBE1, the Ub‐activating E1 enzyme, or towards the Ufm1‐specific protease Ufsp1 underscoring the selectivity of this ABP (Figures S4 and S5).
Figure 3.
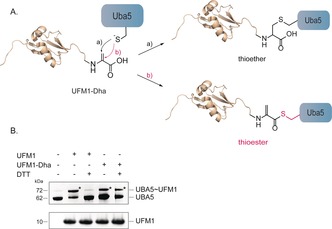
Reactivity of E1 (UBA5) towards UFM1. A) Scheme depicting the reactivity of UBA5 towards UFM1‐Dha permitting the formation of either the thioether (a) or the thioester adduct (b). B) SDS‐PAGE gel of reaction of synthetic UFM1 (6) or Rho‐UFM1‐Dha (8) and recombinant UBA5.
Next, we tested our UFM1‐PA protease probe.34 As expected, Rho‐UFM1‐PA (10) generated by NCL (containing Cys‐45) showed comparable reactivity towards Ufsp1 as the Rho‐UFM1‐PA probe generated by linear SPPS (containing the native Ala‐45), indicating that the cysteine at this position does not interfere with protease recognition, which is in agreement with literature38 (Figure S6). Given that human Ufsp1 is thought to be catalytically inactive (Figure S7), we assessed the reactivity of the active murine Ufsp1 instead. To this end, HEK293 cells transiently overexpressing Flag‐tagged murine Ufsp1 or untransfected cells were lysed and incubated with Rho‐UFM1‐PA at 37 °C (Figure 4 A). Visualization by in‐gel fluorescence scanning followed by immunoblotting revealed that Ufsp1 engages more readily than Ufsp2 with this ABP3 (Figure 4 B and Figure S8). While murine Ufsp1 completely reacts with UFM1‐PA after 45 min, endogenous Ufsp2 engages with UFM1‐PA after prolonged incubation of nearly 6 h (Figure 4 A,B). The two UFM1‐specific proteases known to date are cysteine‐proteases exhibiting a conserved papain‐like fold with the classical catalytic Cys–His–Asp triad but share no sequence homology with DUBS or Ub‐like‐protein‐specific proteases (ULPs) but rather constitute a new cysteine protease subfamily.20, 21 To assess whether deubiquitinating enzymes (DUBs) and SUMO‐specific proteases (SENPs) display cross reactivity towards UFM1‐PA, a panel of representative deubiquitinating and deSUMOylating enzymes were profiled. As expected, only UFM‐1 specific proteases recognize and bind specifically to UFM1‐PA (Figure 4 C–F and Figure S8).
Figure 4.

Assessment of UFM1‐PA reactivity and specificity against a panel of cysteine protease subfamilies. A) Immunoblot showing time‐dependent reactivity of Flag‐Ufsp1 (murine) towards Rho‐UFM1‐PA (10), B) and time‐dependent labeling of endogenous Ufsp2 (human). C,F) DUBs Flag‐UCHL‐1 and GFP‐OTUB2 do not react with UFM1‐PA. D,E) Flag‐SENP6 and Flag‐SENP1 react with S2‐PA (=SUMO2‐PA) but are unreactive towards UFM1‐PA. For fluorescence scans and actin blots see Figure S8.
Subsequently, we addressed the efficacy of UFM1‐PA by monitoring UFM1 protease activity in a cellular environment by electroporation of Rho‐UFM1‐PA (10) into HeLa cells ectopically expressing Flag‐Ufsp1 or its catalytically inactive C53A mutant as previously described.26 In‐gel fluorescence analysis followed by immunoblotting revealed labeling of endogenous Ufsp2 in cells with Rho‐UFM1‐PA, while in Ufsp2‐depleted HeLa cells reactivity was abolished (see Figure 5 A and Figures S9 and S10). As expected, only active Flag‐Ufsp1 reacted with UFM1‐PA but not its catalytically inactive version. Having demonstrated that our UFM1 ABP probe can be used to monitor enzymatic reactivity in living cells, we used the fluorescent ABP to visualize Ufsp reactivity by confocal microscopy. After introduction of Rho‐UFM1‐PA by electroporation into either unmodified or HeLa cells ectopically transfected with murine Flag‐Ufsp1, probe distribution was observed both throughout the cytoplasm and the nucleus (Figure 5 B). However, incorporation of 10 into HeLa cells ectopically expressing murine Flag‐Ufsp1 or its catalytically inactive C53A mutant, showed substantial co‐localization with wild‐type but not with the catalytically incompetent enzyme (Figure 5 C).
Figure 5.
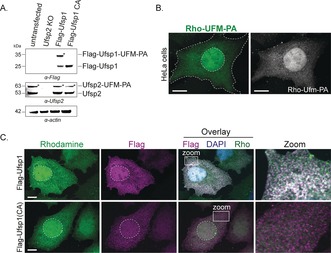
Reactivity of Hela cells electroporated with Rho‐UFM1‐PA in the absence or presence of ectopically overexpressed murine Flag‐Ufsp1. A) Immunoblots of Flag‐Ufsp1 and Flag‐Ufsp1(C53A) and endogenous Ufsp2 in untransfected cells following electroporation. B) Confocal images of untransfected HeLa cells or C) in the presence of Flag‐Ufsp1 or Flag‐Ufsp1 (C53A) after probe electroporation. Cell boundaries and nuclei are indicated by a dashed line, and insets correspond to zoom‐ins. Scale bars=10 μm. Quantification of colocalization (Mander's overlap coefficient) is shown in Figure S11.
In conclusion, we present a practical native chemical ligation‐based synthetic approach for generating UFM1 activity‐based probes. Using this facile strategy, a wide variety of UFM1 ABPs equipped with fluorescent tags, as well as diverse reactive groups targeting proteases or ligases are accessible. The strategies presented in this study highlight a variety of assays and possibilities to utilize our ABPs in the interrogation of enzymatic activities in the Ufmylation cascade. The straightforward nature of the experimental setup is expected to make them readily adoptable to comparative profiling in the presence of various perturbations, facilitating the understanding of UFM1 biology and additionally potentiate the discovery of specific UFM1 enzyme inhibitors. We anticipate that our toolset will greatly facilitate ongoing studies on the UFM1 enzyme cascade.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We would like to thank Dr. Aimee Boyle at Leiden University for assistance with the CD measurements. This work was supported by a VICI grant (NWO‐724.013.002) to H.O. from the Netherlands Organization for Scientific Research (N.W.O.).
K. F. Witting, G. J. van der Heden van Noort, C. Kofoed, C. Talavera Ormeño, D. el Atmioui, M. P. C. Mulder, H. Ovaa, Angew. Chem. Int. Ed. 2018, 57, 14164.
Contributor Information
Dr. Monique P. C. Mulder, Email: M.P.C.Mulder@lumc.nl.
Prof. Dr. Huib Ovaa, Email: H.Ovaa@lumc.nl
References
- 1. Komatsu M., Chiba T., Tatsumi K., Iemura S., Tanida I., Okazaki N., Ueno T., Kominami E., Natsume T., Tanaka K., EMBO J. 2004, 23, 1977–1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wei Y., Xu X., Genomics Proteomics Bioinf. 2016, 14, 140–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kang S. H., Kim G. R., Seong M., Baek S. H., Seol J. H., Bang O. S., Ovaa H., Tatsumi K., Komatsu M., Tanaka K., Chung C. H., J. Biol. Chem. 2007, 282, 5256–5262. [DOI] [PubMed] [Google Scholar]
- 4. Liu J., Wang Y., Song L., Zeng L., Yi W., Liu T., Chen H., Wang M., Ju Z., Cong Y. S., Nat. Commun. 2017, 8, 14186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhang Y., Zhang M., Wu J., Lei G., Li H., PLoS One 2012, 7, e48587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Azfer A., Niu J., Rogers L. M., Adamski F. M., Kolattukudy P. E., Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H1411–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhang M., Zhu X., Zhang Y., Cai Y., Chen J., Sivaprakasam S., Gurav A., Pi W., Makala L., Wu J., Pace B., Tuan-Lo D., Ganapathy V., Singh N., Li H., Cell Death Differ. 2015, 22, 1922–1934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cai Y., Pi W., Sivaprakasam S., Zhu X., Zhang M., Chen J., Makala L., Lu C., Wu J., Teng Y., Pace B., Tuan D., Singh N., Li H., PLoS Genet. 2015, 11, e1005643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chen C., Itakura E., Weber K. P., Hegde R. S., de Bono M., PLoS Genet. 2014, 10, e1004082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hu X., Pang Q., Shen Q., Liu H., He J., Wang J., Xiong J., Zhang H., Chen F., Int. J. Mol. Med. 2014, 33, 1539–1546. [DOI] [PubMed] [Google Scholar]
- 11. Merbl Y., Refour P., Patel H., Springer M., Kirschner M. W., Cell 2013, 152, 1160–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. DeJesus R., Moretti F., McAllister G., Wang Z., Bergman P., Liu S., Frias E., Alford J., Reece-Hoyes J. S., Lindeman A., Kelliher J., Russ C., Knehr J., Carbone W., Beibel M., Roma G., Ng A., Tallarico J. A., Porter J. A., Xavier R. J., Mickanin C., Murphy L. O., Hoffman G. R., Nyfeler B., eLife 2016, 5, e17290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yoo H. M., Kang S. H., Kim J. Y., Lee J. E., Seong M. W., Lee S. W., Ka S. H., Sou Y. S., Komatsu M., Tanaka K., Lee S. T., Noh D. Y., Baek S. H., Jeon Y. J., Chung C. H., Mol. Cell 2014, 56, 261–274. [DOI] [PubMed] [Google Scholar]
- 14. Lu H., Yang Y., Allister E. M., Wijesekara N., Wheeler M. B., Mol. Cell. Proteomics 2008, 7, 1434–1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rubio M. D., Wood K., Haroutunian V., Meador-Woodruff J. H., Neuropsychopharmacology 2013, 38, 1910–1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cai Y., Singh N., Li H., Exp. Hematol. 2016, 44, 442–446. [DOI] [PubMed] [Google Scholar]
- 17. Oweis W., Padala P., Hassouna F., Cohen-Kfir E., Gibbs D. R., Todd E. A., Berndsen C. E., Wiener R., Cell Rep. 2016, 16, 3113–3120. [DOI] [PubMed] [Google Scholar]
- 18. Habisov S., Huber J., Ichimura Y., Akutsu M., Rogova N., Loehr F., McEwan D. G., Johansen T., Dikic I., Doetsch V., Komatsu M., Rogov V. V., Kirkin V., J. Biol. Chem. 2016, 291, 9025–9041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mizushima T., Tatsumi K., Ozaki Y., Kawakami T., Suzuki A., Ogasahara K., Komatsu M., Kominami E., Tanaka K., Yamane T., Biochem. Biophys. Res. Commun. 2007, 362, 1079–1084. [DOI] [PubMed] [Google Scholar]
- 20. Ha B. H., Ahn H. C., Kang S. H., Tanaka K., Chung C. H., Kim E. E., J. Biol. Chem. 2008, 283, 14893–14900. [DOI] [PubMed] [Google Scholar]
- 21. Ha B. H., Jeon Y. J., Shin S. C., Tatsumi K., Komatsu M., Tanaka K., Watson C. M., Wallis G., Chung C. H., Kim E. E., J. Biol. Chem. 2011, 286, 10248–10257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mulder M. P. C., El Oualid F., ter Beek J., Ovaa H., ChemBioChem 2014, 15, 946–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. de Jong A., Merkx R., Berlin I., Rodenko B., Wijdeven R. H., El Atmioui D., Yalcin Z., Robson C. N., Neefjes J. J., Ovaa H., ChemBioChem 2012, 13, 2251–2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ekkebus R., van Kasteren S. I., Kulathu Y., Scholten A., Berlin I., Geurink P. P., de Jong A., Goerdayal S., Neefjes J., Heck A. J., Komander D., Ovaa H., J. Am. Chem. Soc. 2013, 135, 2867–2870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Flierman D., van Noort G. J. V., Ekkebus R., Geurink P. P., Mevissen T. E. T., Hospenthal M. K., Komander D., Ovaa H., Cell Chem. Biol. 2016, 23, 472–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mulder M. P. C., Witting K., Berlin I., Pruneda J. N., Wu K. P., Chang J. G., Merkx R., Bialas J., Groettrup M., Vertegaal A. C., Schulman B. A., Komander D., Neefjes J., El Oualid F., Ovaa H., Nat. Chem. Biol. 2016, 12, 523–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. de Jong A., Witting K., Kooij R., Flierman D., Ovaa H., Angew. Chem. Int. Ed. 2017, 56, 12967–12970; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 13147–13150. [Google Scholar]
- 28. Sommer S., Weikart N. D., Linne U., Mootz H. D., Bioorg. Med. Chem. 2013, 21, 2511–2517. [DOI] [PubMed] [Google Scholar]
- 29. Albrow V. E., Ponder E. L., Fasci D., Bekes M., Deu E., Salvesen G. S., Bogyo M., Chem. Biol. 2011, 18, 722–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mulder M. P. C., Merkx R., Witting K., Hameed D., El Atmioui D., Lelieveld L., Liebelt F., Neefjes J., Berlin I., Vertegaal A., Ovaa H., Angew. Chem. Int. Ed. 2018, 57, 8958–8962; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 9096–9100. [Google Scholar]
- 31. Witting K. F., Mulder M. P. C., Ovaa H., J. Mol. Biol. 2017, 429, 3388–3394. [DOI] [PubMed] [Google Scholar]
- 32. Ogunkoya A. O., Pattabiraman V. R., Bode J. W., Angew. Chem. Int. Ed. 2012, 51, 9693–9697; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 9831–9835. [Google Scholar]
- 33. Melnyk O., Vicogne J., Tetrahedron Lett. 2016, 57, 4316–4324. [Google Scholar]
- 34. Hermanns T., Pichlo C., Woiwode I., Klopffleisch K., Witting K. F., Ovaa H., Baumann U., Hofmann K., Nat. Commun. 2018, 9, 799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dawson P. E., Muir T. W., Clark-Lewis I., Kent S. B., Science 1994, 266, 776–779. [DOI] [PubMed] [Google Scholar]
- 36. Kent S., Bioorg. Med. Chem. 2017, 25, 4926–4937. [DOI] [PubMed] [Google Scholar]
- 37. Geurink P. P., van Tol B. D., van Dalen D., Brundel P. J., Mevissen T. E., Pruneda J. N., Elliott P. R., van Tilburg G. B., Komander D., Ovaa H., ChemBioChem 2016, 17, 816–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kim K. H., Ha B. H., Kim E. E., FEBS Lett. 2018, 592, 263–273. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary