

Abstract
The first law of photochemistry, as described by Theodor von Grotthuß and John W. Draper, states that only the light absorbed by the irradiated matter can effect photochemical change. Consequently, the photochemical behavior of a molecule can be controlled by bringing its absorbance properties in line with the emission of the light source. A compound with a chromophore that only absorbs light at short wavelengths will not be excited by light of longer wavelengths. If one can reversibly modify the photophysical properties of a compound with a chemical activator, then it is possible to photoexcite only the activated species. For α,β‐unsaturated carbonyl compounds, the use of Lewis acids, Brønsted acids, or the formation of the respective iminium ions can bring about the desired chromophore activation to catalyze a photochemical reaction at a given wavelength. In this Minireview, the concept of chromophore activation will be illustrated, and examples of its implementation in enantioselective catalysis will be discussed.
Keywords: asymmetric catalysis, enantioselectivity, homogeneous catalysis, photocatalysis, photochemistry
1. Introduction
When identifying potential catalysts for enantioselective photochemical processes,1 it is a key question how the racemic background reaction of a molecule in its excited state can be suppressed while it is not in the chiral environment of the catalyst. A possible solution to this problem is the use of chiral triplet sensitizers.2, 3 Here, the strong distance dependence of the energy transfer ensures that only catalyst‐bound substrates are sensitized.4 Alternatively, catalysts that change the photophysical properties of the substrate, that is, the chromophore, upon substrate binding can be employed. Classically, this activation is achieved by causing a bathochromic shift in the chromophore absorption (Scheme 1; X1,X2=O for an α,β‐unsaturated carbonyl compound). The lowest‐energy transition corresponds to the excitation from the ground state S0 to the first excited state S1. If the general structure I is in equilibrium with a structurally different species II (in this case, defined by substituents Y1, Y2), then it is possible to selectively excite II. However, the absorption of II at λ′max must occur at a longer wavelength than that for I at λ max.
Scheme 1.
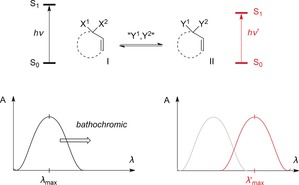
Chromophore activation by a bathochromic shift of the absorbance from I to II.
The selective irradiation of a given substrate requires emitters of near‐monochromatic light, which, owing to recent advances in light‐emitting diodes (LEDs), is possible at a large range of wavelengths.
Analogously, it is possible to selectively excite a species II formed from a species I if the triplet energy (E T) of II is lower than that of I (Scheme 2). If the triplet energy of the sensitizer E T (sens) lies between E T and E T′, then the energy transfer will occur exclusively, or at least with a considerably increased rate, from the triplet sensitizer to II. For this concept to work efficiently, it is crucial that the excitation wavelength for the triplet sensitizer is low enough in energy to preclude direct excitation of I and II.
Scheme 2.
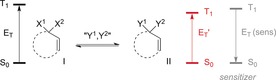
Chromophore activation by a change in the triplet energy from I to II.
This Minireview will discuss possible scenarios for the chromophore activation of α,β‐unsaturated carbonyl compounds, including the methods described in Schemes 1 and 2. Although some of the methods of activation have been known for some time, it has only been demonstrated fairly recently that these phenomena can be employed as a tool in enantioselective catalysis. This Minireview will be limited to compounds with a carbonyl group in conjugation with a single double bond. Solid‐state reactions will not be covered. Reactions where the photophysical properties of the chromophore remain unchanged and only the ground‐state properties, for example, the redox potential,5 are altered will also not be discussed. Where possible, the specific wavelengths of the light sources are given in the reaction schemes.1b, 6
2. Activation by Lewis Acids
2.1. Direct Excitation (Singlet)
Reports of Lewis acids being able to change the outcome of a photochemical reaction date back to the beginning of the 20th century. Praetorius and Korn observed as early as 1910 that irradiation of dibenzylideneacetone (1) resulted in resinification, whereas the desired reaction proceeded smoothly in the presence of uranyl chloride (Scheme 3).7 It is likely that a bathochromic shift of the absorption of 1 occurred, which contributed to improved chemoselectivity.
Scheme 3.
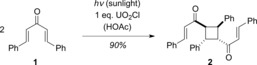
[2+2] Photodimerization of dibenzylideneacetone with sunlight:7 The presence of a Lewis acid leads to changes in the chemo‐ and regioselectivity.
The Lewis acid SnCl4 also promotes the photochemical transformation to generate 2;8 the structure of this compound was confirmed in 1975 by Alcock and co‐workers.9, 10
2.1.1. α,β‐Unsaturated Carboxylic Acid Esters (2‐Alkenoates)
The photophysical properties of 2‐alkenoates are greatly affected by Lewis acids (Scheme 4),11 resulting in a bathochromic shift of the absorption corresponding to a ππ* transition. This effect can be observed for acrylates (e.g., 3), cinnamates (e.g., 4), and 3‐aryl‐substituted acrylates (e.g., 5). Consequently, these compounds are suitable candidates for selective excitation.
Scheme 4.
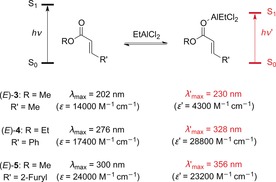
Changes in the UV/Vis properties of various α,β‐unsaturated carboxylic acid esters induced by the presence of the Lewis acid EtAlCl2.
To date, however, enantioselective photochemical transformations of these compounds under Lewis acid catalysis have not been reported. This may be due to the fact that Lewis acids are poor catalysts for the [2+2] photocycloaddition of 2‐alkenoates.12 For example, cinnamate 6 was only fully consumed after 52 hours when reacted with 1.5 equivalents of BF3⋅OEt2 and 20 equivalents of cis‐2‐butene (Scheme 5).
Scheme 5.
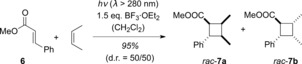
Lewis acid catalyzed [2+2] photocycloaddition of cinnamate 6 and cis‐2‐butene.
The inefficiency of the process is likely due to competing E/Z isomerization. It has been shown that the photochemical E/Z isomerization of 2‐alkenoates can be controlled under Lewis acidic conditions.11, 13 Furthermore, cycloaddition to the products rac‐7 occurred stereospecifically, which indicates that the reaction proceeds on the singlet hyperface.
2.1.2. Coumarins
The photochemical behavior of coumarin and its analogues differs significantly from that of the structurally similar cinnamates. As for cinnamates, [2+2] photodimerization and [2+2] photocycloaddition reactions of coumarins are inefficient and occur with quantum yields of approximately Φ≈10−3 . 14 In the presence of Lewis acids, however, these reactions are significantly accelerated. This effect was first reported by Lewis and co‐workers in 1983 for the [2+2] photodimerization of coumarin.15 They observed that in the presence of only 12.5 mol % BF3⋅OEt2, the quantum yield increased to Φ=0.16. Analogously, it is possible to catalyze [2+2] photocycloadditions with a Lewis acid. For example, the cycloaddition of coumarin 8 with 2,3‐dimethylbutene was complete after only five hours in the presence of substoichiometric BF3⋅OEt2, furnishing product rac‐9 in 57 % yield (Scheme 6).16
Scheme 6.

Lewis acid catalyzed [2+2] photocycloaddition of coumarin (8) with 2,3‐dimethylbutene.
The change in the reactivity of coumarin17 with respect to the presence or absence of a Lewis acid is due to a number of reasons. It is important to note that the Lewis acid stabilizes the excited state, which would otherwise rapidly undergo non‐radiative decay back to its ground state. Additionally, spin change from the singlet hyperface to the triplet hyperface (intersystem crossing, ISC) is accelerated for coumarins in the presence of Lewis acids, which is not the case for cinnamates.18 This is supported by the fact that no stereospecificity is observed in the cycloaddition with 2‐butene.16 Moreover, in analogy to the 2‐alkenoates, addition of a Lewis acid results in a bathochromic shift of the ππ* absorption. However, this effect is masked by the fact that coumarins give rise to a strong nπ* band in the UV/Vis spectrum, which is already bathochromically shifted with respect to the ππ* transition.
In 2010, the effect of a Lewis acid on the intramolecular [2+2] photocycloaddition of coumarin 10 a was investigated (Scheme 7).19 At λ=366 nm, the reaction proceeded sluggishly, whereas addition of a Lewis acid resulted in significant acceleration.20 This paved the way for the development of a chromophore‐activation‐aided enantioselective [2+2] photocycloaddition. After a screen of various chiral Lewis acids and further optimization, the AlBr3 complex 12 of a chiral oxazaborolidine21 was identified as an ideal catalyst to promote the reaction with excellent enantioselectivity. At −75 °C, cycloadduct 11 a was afforded in an enantiomeric excess (ee) of 82 %.
Scheme 7.
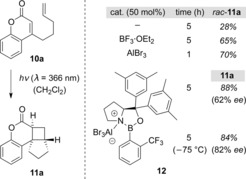
The catalytic effect of Lewis acids on the intramolecular [2+2] photocycloaddition of coumarin 10 a.
The cycloaddition did not occur stereospecifically, and in a later study, it was shown that the aforementioned reasons for the catalytic effect of a Lewis acid also applied to this reaction.22 The quantum yield Φ of the reaction 10 a→11 a amounts to 2×10−3 without a Lewis acid, whereas Φ=0.09 in the presence of chiral Lewis acid 12. The substrate scope was expanded by using a variety of 4‐substituted coumarins, which generated five‐membered rings upon intramolecular [2+2] photocycloaddition; tetracyclic products 11 were thus obtained in up to 90 % ee (Figure 1).23
Figure 1.
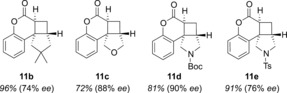
Substrate scope of the Lewis acid catalyzed intramolecular [2+2] photocycloaddition of coumarins (50 mol % 12, −75 °C, λ=366 nm).
2.1.3. 2‐Alkenones and 2‐Alkenamides
In contrast to cinnamates (Scheme 4), α,β‐unsaturated ketones give rise to an nπ* absorption that is bathochromically shifted with respect to the ππ* absorption. The nπ* transition to the excited state S1 is predominantly responsible for the photochemical behavior of 2‐alkenones. Additionally, this forbidden transition leads to a rapid population of the triplet state T1, from which [2+2] photocycloadditions of 2‐cycloalkenones III occur (Scheme 8).6 Upon coordination to a Lewis acid, for example, EtAlCl2, the ππ* transition is bathochromically shifted to overlap with the nπ* absorption of the uncomplexed species.
Scheme 8.
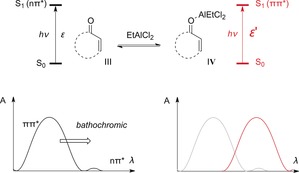
Chromophore activation by a change in the molar extinction coefficient upon formation of complex IV.
Despite having been mentioned in previous publications,24 the Lewis acid mediated bathochromic shift as a tool for enantioselective synthesis was not employed until this decade. Indeed, one would not expect a Lewis acid to exert a catalytic effect as direct excitation of III, which subsequently undergoes rapid ISC to T1, leads to highly efficient photoreactions.25 However, in 2013, our group reasoned that at a certain wavelength, the larger molar extinction coefficient ϵ′ of complex IV could lead to a higher photon absorption by IV than by complex III. Indeed, at λ=360 nm, the absorbance cross‐section of dihydropyridone 13 a was very small,[26] whereas its EtAlCl2 complex strongly absorbed at this wavelength (Scheme 9).
Scheme 9.
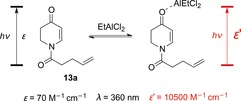
The influence of the Lewis acid EtAlCl2 on the molar extinction coefficient ϵ of dihydropyridone 13 a at λ=360 nm.
Based on this observation, chiral Lewis acid mediated enantioselective [2+2] photocycloadditions appeared to be feasible as photons would only be absorbed by the substrate when bound to the chiral Lewis acid.27 Initial experiments supported this hypothesis, and following irradiation (λ=366 nm) of dihydropyridone 13 a in the presence of Lewis acid 12, the intramolecular [2+2] photocycloaddition product 14 a was furnished in 88 % ee (Scheme 10). Analogously, a variety of products 14 were synthesized in high enantioselectivities. Moreover, product 14 g was successfully employed in total syntheses of the lupin alkaloids (+)‐lupinine and (+)‐thermopsine.
Scheme 10.
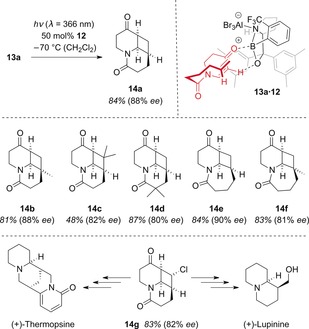
Enantioselective intramolecular [2+2] photocycloaddition of dihydropyridones to products 14 and total syntheses of lupin alkaloids starting from 14 g.
As a model for the formation of the observed absolute configuration, it is proposed that the substrate (e.g., 13 a) forms a 1:1 complex with the Lewis acid 12. Here, it is suggested that an oxazaborolidine aryl group blocks one enantiotopic face of the enone, thus forcing the cycloaddition to occur on the opposite face. Whereas the enantioface differentiation by the Lewis acid occurs in analogy to that with the coumarins, the reaction mechanisms differ significantly. In the presence of a Lewis acid, a rate enhancement was observed for the coumarins, whereas the reactions of enones were retarded.22 The quantum yields are consistent with these observations. For the racemic reaction 13 a→rac‐14 a, the quantum yield was Φ=0.23 whereas the Lewis acid mediated reaction was determined to have a lower quantum yield of Φ=4×10−3. The main reason for this effect is likely to be inefficient ISC from S1 to T1 in the presence of a Lewis acid. According to the El‐Sayed rules,25 this is probably due to the ππ* character of the S1 excited state.
In pursuit of new substrates that undergo enantioselective intramolecular [2+2] photocycloadditions, we turned our attention to 3‐alkenyloxy‐2‐cycloalkenones (15), which contain a comparatively basic oxygen atom. The donor–acceptor moiety in the cyclobutane products 16 also appeared to be a feasible handle for further derivatizations (Scheme 11).28
Scheme 11.
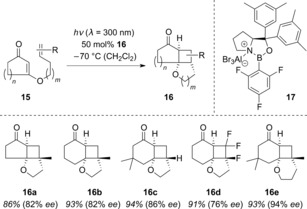
Enantioselective intramolecular [2+2] photocycloadditions of 3‐alkenyloxy‐2‐cycloalkenones 15 catalyzed by Lewis acid 17.
Owing to the short‐wavelength ππ* absorption of compounds 15, it was necessary to irradiate at a shorter wavelength (λ=300 nm). Additionally, minor modifications to the Lewis acid were required. Using Lewis acid 17, a variety of cycloalkenones 15 were converted into the corresponding cyclobutane products 16. Owing to an increased racemic background reaction, the light source had to be attenuated in some cases.
The cyclobutane products 16 were further derivatized by ring opening at the donor‐ (oxygen) and acceptor‐substituted (carbonyl) cyclobutane C−C bond, effectively generating ring‐expanded products.28
The congeners of β‐substituted enones 15, namely α‐substituted enones, were identified as promising candidates for enantioselective photoreactions. Here, however, two‐point coordination to the oxazaborolidine (see Scheme 10) is no longer possible owing to the lack of an α‐hydrogen atom. Consequently, alternative Lewis acids were investigated that could be chelated by the carbonyl and aryloxy oxygen atoms. The [6π] photocyclization of substrates 18 29, 30 was relatively unexplored at the time and was therefore chosen as a model reaction for enantioselective photochemistry starting from α‐substituted enones 18 (Scheme 12).31 As expected, a considerable bathochromic shift was observed with EtAlCl2 as the Lewis acid. The ππ* absorption of enone 18 a (R=Me, X=H) shifted from λ max=241 nm (ϵ=10 460 m −1 cm−1) to λ′max=292 nm (ϵ=10 840 m −1 cm−1), which is close to the weak nπ* absorption at λ=320 nm (ϵ=114 m −1 cm−1). Unfortunately, it was not possible to identify a Lewis acid that was strong enough to cause a similar bathochromic shift while simultaneously achieving good enantioinduction.
Scheme 12.
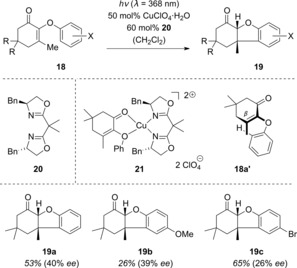
Enantioselective [6π] photocyclization of 2‐aryloxycyclohex‐2‐enones (18) mediated by a chiral copper complex.
The highest selectivities were achieved with an in situ generated copper–bis(oxazoline) complex with ligand 20. With stochiometric ligand loadings, the products 19 were obtained in up to 60 % ee. Using a substoichiometric amount of the Lewis acid catalyst, lower selectivities were observed. Enones where R=Me showed the highest enantioselectivities. The absolute configuration of the major enantiomers is consistent with the proposed model for enantioinduction where 18 a chelates to the Lewis acid, as shown in complex 21. It has previously been demonstrated that the bis(oxazoline) backbone twists the complex out of the square‐planar configuration.32 This results in 18 a adopting a helical conformation 18 a′, which itself is chiral and therefore provides the enantioface differentiation for cyclization at the β‐carbon atom. Attempts to lower the triplet energy wih a Lewis acid to accelerate the reaction according to the method described in Scheme 2 were not successful.
In 2017, Meggers and co‐workers successfully applied a chelating Lewis acid to an enantioselective [2+2] photocycloaddition.33 Here, the Lewis acid is a transition‐metal complex that is chiral at the metal center. The catalyst architecture is structurally related to known ruthenium and iridium catalysts that had previously been employed in other photochemical transformations.34 Aromatic 2‐alkenones 22 were investigated as substrates in preliminary studies, where the R substituent was either an aryl or a styryl moiety. Irradiation of 22 with a blue LED of an unspecified wavelength in the presence of 2,3‐dimethylbutadiene and complex 23 afforded the intermolecular [2+2] photocycloaddition products 24 (Scheme 13). The diastereoselectivities varied, and the other diastereomer was always observed as well (d.r.=diastereomeric ratio).
Scheme 13.
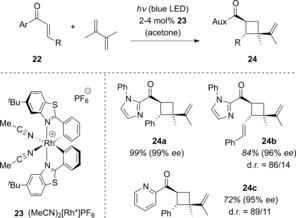
Enantioselective rhodium‐catalyzed intermolecular [2+2] photocycloaddition of 2‐alkenenones 22 to cyclobutanes 24.
Analogously, 2‐alkenylpyrazolides can be employed to access cyclobutanes 25 (Figure 2). In addition to the aforementioned 2,3‐dimethylbutadiene, enynes (product 25 a), enol ethers (product 25 b), and styrenes (product 25 c) were successfully employed in the [2+2] photocycloaddition.35 Furthermore, the reaction is stereoconvergent because the double bond configuration of the photoexcited substrate has no influence on the relative stereochemistry of the product.
Figure 2.
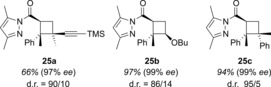
Products 25 of the enantioselective rhodium‐catalyzed intermolecular [2+2] photocycloaddition of 1‐(3′,5′‐dimethyl‐1H‐pyrazol‐1′‐yl)‐3‐phenyl‐2‐butenone.
Having demonstrated that the reaction does not occur by a single‐electron‐transfer process, the authors suggested that cycloaddition occurs from the triplet state. The chromophore activation is therefore similar to the above‐mentioned activation of enones with Lewis acids (Scheme 8). Upon association of substrate 22 a to the catalyst, a chelate complex 26 is formed, with a vastly increased molar extinction coefficient at λ=400 nm (Scheme 14). Consequently, the substrate–catalyst complex is selectively excited, with remarkably efficient enantioface differentiation being provided by the C 2‐symmetric rhodium moiety.
Scheme 14.
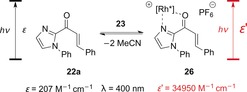
The influence of Lewis acid 23 on the molar extinction coefficient ϵ of alkenone 22 a at λ=400 nm.
A key advantage of the rhodium‐based Lewis acid 23 appears to be its ability to accelerate the ISC to the reactive triplet hyperface. The quantum yield of the reaction to 24 was determined to be Φ=0.27, which in itself underlines the extraordinary efficiency of the process. Considering that theoretical studies on the Lewis acid catalyzed [2+2] photocycloaddition to 14 proposed a considerable heavy‐atom effect on the ISC,36 this effect is expected to be several orders of magnitude higher for the rhodium‐catalyzed reactions.
The Meggers group was also able to intercept the photoexcited Lewis acid complexes of 2‐alkenylpyrayolides (27; PMP=para‐methoxyphenyl) with alkenyl azides (Scheme 15).37 Here, the authors proposed that following addition to the double bond and subsequent dinitrogen extrusion, a 1,5‐diradical is initially formed. The 1‐pyrroline products 28 were obtained with high levels of enantioselectivity.
Scheme 15.
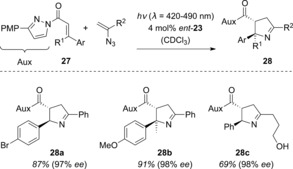
Enantioselective rhodium‐catalyzed [3+2] photocycloaddition to generate pyrrolines 28.
The intermolecular [2+2] photocycloaddition has found a myriad of applications in organic synthesis.38 Consequently, the development of an enantioselective variant with cyclic enones and simple alkenes, such as ethene and isobutene, was desirable. Unfortunately, the oxazaborolidine complexes 12 and 17 yielded poor results, with the observed chemo‐ and enantioselectivities falling far short of expectations. Surprisingly, changing the oxazaborolidine aryl group at the prolinol carbon atom resulted in a dramatic improvement of the observed chemo‐ and enantioselectivities. In a recently published study, it was demonstrated that Lewis acid 30 is able to induce high levels of enantioselectivities in the reactions of various enones 29 with simple alkenes (Scheme 16).39 The cyclobutanes 31 were obtained in 82–96 % ee (16 examples), and the observed chemo‐ and regioselectivities were far superior to those of the uncatalyzed reaction. With 1,1‐disubstituted alkenes, only one regioisomer was formed whereas the uncatalyzed reaction yielded both regioisomers, which were tedious to separate.
Scheme 16.
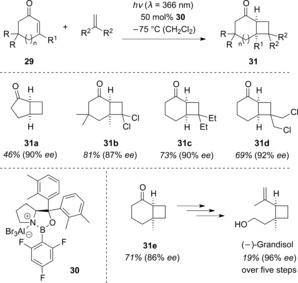
Enantioselective intermolecular [2+2] photocycloaddition of 2‐cycloalkenones (29) with simple alkenes to cyclobutanes 31.
As alluded to previously, cyclobutanes 31 are building blocks for a number of natural products. For example, the monoterpene (−)‐grandisol was accessed from 31 e in only five steps. Furthermore, the ee could be increased by simple recrystallization.
2.2. Sensitization (Triplet)
In 2016, Yoon and co‐workers disclosed an important study, describing the first example of an enantioselective reaction by triplet chromophore activation.40 The main focus of this study was the activation of 2′‐hydroxychalcones. The authors were able to demonstrate that the triplet energy of the parent compound 32 a is decreased significantly (E T′=138 kJ−1 mol−1) upon complexation to the Lewis acid Sc(OTf)3 to generate complex 33 (Scheme 17).
Scheme 17.
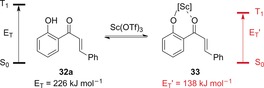
The influence of the Lewis acid Sc(OTf)3 on the triplet energy of chalcone 32 a.
According to the method outlined in Scheme 2, energy transfer to the chiral Lewis acid complex should only occur if a triplet sensitizer is employed with a triplet energy E T (sens) between E T and E T′. The well‐known ruthenium complex 35 fulfills these requirements. In combination with chiral bis(oxazoline) 34, chalcone 32 a was successfully converted with 2,3‐dimethylbutadiene into cyclobutane 36 a with high enantioselectivity (Scheme 18). In this initial study, the substrate scope was expanded by varying the chalcone and diene substituents (products 36 b–36 e).
Scheme 18.
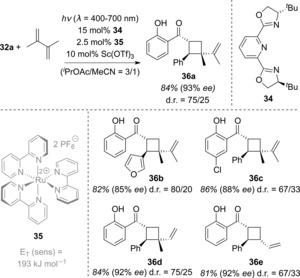
Enantioselective intermolecular [2+2] photocycloaddition of 2′‐hydroxychalcones by selective triplet energy transfer to a chiral Lewis acid complex.
In a second study,41 the substrate scope was expanded by using styrenes as the alkene partner in the cycloaddition. 1,2‐Diaryl‐substituted cyclobutanes, such as 37, were generated with high levels of enantioselectivity (Scheme 19). Cyclobutane 37 was then employed in a total synthesis of the natural product (+)‐norlignan. First, the hydroxyphenyl group was degraded in three steps, which was followed by a substitution of the aryl bromine atom.
Scheme 19.
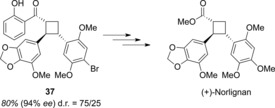
Total synthesis of (+)‐norlignan starting from cyclobutane 37, which was generated in an enantioselective [2+2] photocycloaddition.
3. Activation by Brønsted Acids
In general, the behavior of Brønsted acids towards carbonyl compounds is very similar to that of Lewis acids. With the protonation occurring at the carbonyl oxygen atom, one can expect similar photophysical effects. However, reports of chiral Brønsted acid mediated enantioselective photochemical reactions are scarce.
3.1. Direct Excitation (Singlet)
As early as the 1960s, Zalewski and Dunn investigated the UV/Vis properties of 2‐alkenals, 2‐alkenones, and 2‐alkenoates upon protonation by sulfuric acid.42 As exemplified here for crotonaldehyde (38), the ππ* absorption was shifted to longer wavelengths for all compounds (Scheme 20). Furthermore, the protonated species had larger molar extinction coefficients, as exemplified here for 39.
Scheme 20.
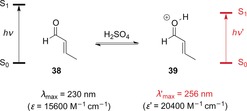
Changes in the UV/Vis properties of crotonaldehyde upon protonation by sulfuric acid.
Comprehensive studies concerning the photochemistry of quantitatively protonated compounds were carried out in the 1970s and 1980s.43 For example, Childs and co‐workers investigated the photochemical E/Z isomerization of cations of type 39, observing a preference for the Z isomer.44 As for Lewis acids (see Section 2.1), it was shown that Brønsted acids are able to catalyze the photochemical E/Z isomerization of α,β‐unsaturated carboxylic acids and ketones.45 The first photochemical rearrangement of protonated enones was reported by Cornell and Filipescu in 1977.46 Here, enone rac‐40 was irradiated in sulfuric acid, and rearranged to 42 via the protonated species 42⋅H+ (Scheme 21). From a mechanistic point of view, upon irradiation of cationic rac‐39, two consecutive 1,2‐shifts occur to complete the rearrangement. Later experiments also indicated that cations such as rac‐40⋅H+ can be stabilized by a hydride shift if the β‐position of the enone is not substituted.47 These reactions were generally associated with poor yields, and in light of the similarity to previously established rearrangements of enones,48 catalytic enantioselective variants were not pursued.
Scheme 21.
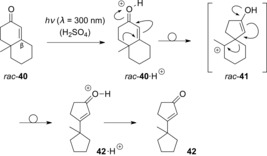
Photochemical rearrangement of 2‐cycloalkenone rac‐40 under acidic conditions.
In 2014, Sibi, Sivaguru, and co‐workers reported a catalytic enantioselective variant of the above‐mentioned coumarin photocycloaddition (see Section 2.1.2 and Scheme 7), using a chiral thiourea (Scheme 22).49 A key feature of catalyst 43 is that in addition to the two acidic thiourea protons, a further acidic site is present in the chiral binaphthyl backbone. Consequently, three acidic protons are available to aid the coordination of substrate 10 a. The authors proposed that the carbonyl oxygen atom is bound by the thiourea protons while the lactone oxygen atom is bound by the naphthol proton. This three‐point binding ensures efficient enantioface differentiation. Consequently, cyclobutane ent‐11 a and the related compounds ent‐11 f and ent‐11 g were obtained with high enantioselectivities.50
Scheme 22.
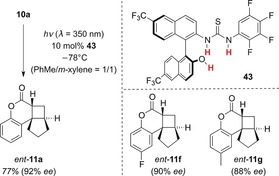
Enantioselective catalysis of the intramolecular [2+2] photocycloaddition of coumarin 10 a mediated by chiral Brønsted acid 43.
From a mechanistic point of view, it was suggested that in addition to a bathochromic shift, the lifetime of the excited state was prolonged. Furthermore, the rate of the ISC was increased. The authors concluded these three effects to be the reasons for the catalytic efficiency of the process.51
In the early 1980s, Childs and Hagar reported that upon protonation of O,O‐acetals and subsequent elimination, onium ions were formed that closely resemble the cations of protonated α,β‐unsaturated carbonyl compounds.52 In this way, O,O‐acetal 44, which does not absorb above 200 nm, was converted into cation 45, which absorbs strongly in the UV region (Scheme 23).
Scheme 23.
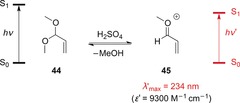
Formation of onium ion 45 in the presence of Brønsted acids.
Considering these reports, our research group hoped to selectively excite onium ions generated by Brønsted acid catalysts. However, even under anhydrous conditions, the O,O‐acetals of α,β‐unsaturated ketones were found to be in equilibrium with the free ketone species. Therefore, direct excitation of the free ketones, rather than the onium ions, could not be excluded with the wavelengths required for onium excitation. Furthermore, a catalytic effect was not observed, and attempts to carry out the reaction in an enantioselective fashion with chiral Brønsted acids proved to be unsuccessful.53 The S,S‐acetals of α,β‐unsaturated ketones, however, yielded more promising results. Here, complete dissociation of the dithiol from the ketone was not observed. Upon treatment with the Brønsted acid HNTf2 (Tf=trifluoromethylsulfonyl), a thionium ion was formed with a strongly bathochromically shifted absorption (Scheme 24). Whereas the S,S‐acetal 46 does not absorb above λ>280 nm, the thionium ion 47 exhibits a strong absorption band at λ′max=349 nm with a high molar extinction coefficient.54
Scheme 24.
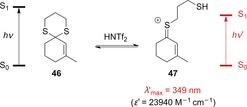
Formation of thionium ion 47 from S,S‐acetal 46 in the presence of Brønsted acids.
Based on these results, the S,S‐acetals of cyclic enones appeared to be ideal candidates for Brønsted acid catalyzed photochemical reactions mediated by visible light. Indeed, following irradiation of 48 a with visible light (λ=405 nm), product rac‐50 a was generated in high yield in the presence of imide 49 (Scheme 25).55 The cyclobutanes rac‐50 could be generated in an analogous fashion. The observed diastereoselectivity was near‐perfect for rac‐50 c (≥95:5 d.r.) and high for rac‐50 d (90:10 d.r.).
Scheme 25.
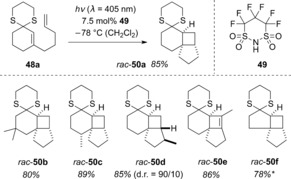
Brønsted acid catalyzed intramolecular [2+2] photocycloaddition to cyclobutanes and cyclobutenes rac‐50. *: The reaction was carried out at λ=366 nm.
The S,S‐acetals rac‐50 could subsequently be hydrolyzed to the corresponding carbonyl compounds or reductively desulfurized to hydrocarbons by a Mozingo reduction.55
3.2. Sensitization (Triplet)
To date, no Brønsted acid catalyzed photoreactions have been reported where following chromophore activation and subsequent sensitization, the triplet state is populated. The energy transfer from a protonated coumarin to EuCl3 suggests that in principle, though, this concept might be feasible.56 The phosphorescence of the coumarinium ion (excitation at λ=340 nm) was quenched efficiently by energy transfer to the europium ion, resulting in EuCl3 luminescence.
4. Activation via Iminium Ions
One of the key publications that sparked the renaissance of the field of organocatalysis was a study by MacMillan and co‐workers in 2000 on the enantioselective catalysis of Diels–Alder reactions with chiral secondary amines.57 Carbonyl compounds were activated by the in situ generation of iminium ions. This activation is akin to the activation with Lewis and Brønsted acids as the electrophilicity is increased by lowering the energy of the π* orbital, which represents the lowest unoccupied molecular orbital (LUMO). In recent times, iminium catalysis has been applied to photochemistry, and selected examples will be discussed in this Minireview.58
4.1. Direct Excitation (Singlet)
The formation of an iminium ion from a carbonyl compound leads to a bathochromic shift of the ππ* absorption. For example, upon condensation of cinnamaldehyde (51 a) with dimethylammonium perchlorate under acidic conditions, the iminium ion 52 is generated, which absorbs at a considerably longer wavelength than cinnamaldehyde (Scheme 26).59
Scheme 26.
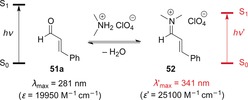
Changes in the absorbance properties upon formation of iminium ion 52 from cinnamaldehyde (51 a).
The same effect is observed for enones. A photochemical reaction under iminium catalysis, however, is challenging. As the enone nπ* transition is bathochromically shifted with respect to the iminium ππ* transition, selective excitation of the iminium ion is almost impossible.60 Nevertheless, Mariano and co‐workers demonstrated that stoichiometrically generated iminium ions of α,β‐unsaturated ketones can be directly photoexcited to undergo an intramolecular [2+2] photocycloaddition. The process takes place on the singlet hyperface and is inefficient. With a C 2‐symmetric pyrrolidine as a chiral auxiliary, the reaction could be carried out enantioselectively, with 82 % ee at 40 % conversion.61
More recently, Melchiorre and co‐workers applied the formation of iminium ions of type 52 for the selective photoexcitation of iminium ions to the S1 state. More importantly, they were able to identify reaction conditions (TFA=trifluoroacetic acid) under which the formation of a chiral iminium ion from an α,β‐unsaturated aldehyde was reversible (Scheme 27).62 Using pyrrolidine catalyst 54 (TDS=thexyldimethylsilyl) and various silanes, aldehydes 51 were enantioselectively alkylated to generate the products 55. The authors proposed that the comparatively high oxidation power of the photoexcited iminium ion 56, with a redox potential of E 1/2(56 +*/57 .)≈+2.4 V (vs. Ag/Ag+ in MeCN), enabled single electron transfer from the silane. Following cleavage of the silyl group (formally as a cation), a radical is generated, which recombines with radical 57. The recombination occurs on the top face of radical 57, resulting in the observed absolute configuration. A crucial criterion in the development of the catalyst was its high stability with respect to strongly oxidizing conditions, which in this case was achieved through installation of the fluorine substituents. In a recent related publication, Melchiorre and co‐workers employed dihydropyridines instead of silanes 53 as the alkylation reagents.63
Scheme 27.
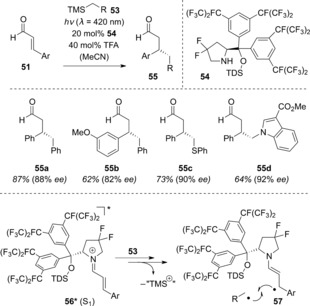
Pyrrolidine‐catalyzed enantioselective alkylation of α,β‐unsaturated aldehydes under visible‐light irradiation. TMS=trimethylsilyl.
Mariano and co‐workers demonstrated in the early 1980s that photoexcited iminium ions are able to oxidize cyclopropanols of type rac‐58, which then fragment to the corresponding β‐ketoalkyl radicals (Scheme 28).64 The Melchiorre group recently applied this method to a formal [3+2] cycloaddition of α,β‐unsaturated aldehydes with cyclopropanols. In the presence of chiral pyrrolidine 59 as the catalyst, the cyclopentanol products 60 were generated with excellent enantioselectivities.65
Scheme 28.
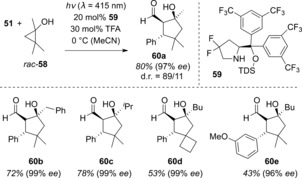
Pyrrolidine‐catalyzed enantioselective [3+2] cycloaddition of α,β‐unsaturated aldehydes under visible‐light irradiation.
4.2. Sensitization (Triplet)
To apply the full photochemical toolbox to iminium ions of α,β‐unsaturated carbonyl compounds, the population of their triplet state would be desirable. It should be noted that ISC from S1 to T1 is highly inefficient for iminium ions,25 with the majority of their photochemical reactions taking place on the singlet hyperface. Our research group investigated whether iminium ions can enable chromophore activation by lowering the triplet energy of an α,β‐unsaturated iminium ion (eniminium ion) below that of the corresponding carbonyl compound. The triplet energy of enone 61 was determined by phosphorescence spectroscopy, with its value of 289 kJ mol−1 being in agreement with those of analogous compounds.66, 67 The triplet energy of iminium ion 62 could not be determined by spectroscopic means. An intramolecular [2+2] photocycloaddition of 62 proceeded with triplet sensitizers with triplet energies E T(sens)≥253 kJ mol−1 , whereas no reaction was observed with triplet sensitizers where E T(sens)≤245 kJ mol−1. Consequently, the triplet energy E T′ of iminium ion 62 can be estimated to be approximately 40 kJ mol−1 lower than that of its corresponding carbonyl compound (Scheme 29).60
Scheme 29.
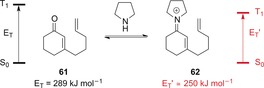
Changes in the triplet energy upon formation of iminium ion 62 from α,β‐unsaturated ketone 61.
The use of a chiral iminium ion enabled a selective [2+2] photocycloaddition via triplet sensitization that furnished the corresponding cyclobutane upon hydrolysis in 78 % yield and 88 % ee.
5. Summary and Outlook
The main conclusion that one can draw from the studies described in this Minireview is that the key principles that hold for the enantioselective catalysis of thermal reactions can also be applied to photochemistry. Upon coordination of Lewis or Brønsted acids to α,β‐unsaturated carbonyl compounds or upon iminium ion formation, the lowest unoccupied molecular orbital (LUMO) is lowered in energy, which effectively activates the substrate.57, 68 Whereas in thermal reactions, the lowering in energy of the π* orbital results in increased electrophilicity, in photochemistry, S1 and T1 states with ππ* character become closer in energy to the ground state. This enables the selective excitation of the activated substrate at longer wavelengths (for S1). As the activated substrate possesses a lower triplet energy (T1), the triplet state of the activated substrate can be selectively populated with an appropriate triplet sensitizer. In recent times, the use of chiral catalysts has led to the development of several enantioselective photochemical processes. However, it is important to note that significant challenges remain in taming reactions that do not take place on the ground‐state hyperface but several 100 kJ mol−1 higher in energy. Consequently, photochemists remain pioneers in conquering the treacherous altitudes of the excited state, with many unexplored peaks yet to be surmounted.
Conflict of interest
The authors declare no conflict of interest.
Biographical Information
Thorsten Bach carried out his undergraduate studies at the University of Heidelberg and at the University of Southern California (G. A. Olah) and was subsequently awarded his PhD in 1991 from the University of Marburg (M. T. Reetz). Following a postdoctoral fellowship at Harvard University (D. A. Evans), he completed his habilitation at the University of Münster (1992–1996). He then moved to the University of Marburg as an Associate Professor in 1997 and was appointed to the Chair of Organic Chemistry I at the Technische Universität München (TUM) in 2000. He has been an elected member of the German Academy of Sciences (Leopoldina) since 2006 and of the Bavarian Academy of Sciences since 2009.

Biographical Information
John D. Jolliffe was awarded his Master in Chemistry (MChem) from the University of Newcastle upon Tyne (M. J. Hall) in 2009. He subsequently carried out his doctoral studies at the University of Oxford (M. D. Smith), receiving his DPhil in 2017. At the end of 2017, he moved to the research group of Prof T. Bach to take up a position as a postdoctoral fellow and is currently working on the development of new catalysts for enantioselective photochemical reactions. His work has been recognized by an EPSRC Doctoral Prize in 2017 and an Alexander von Humboldt fellowship in 2018.

Biographical Information
Christoph Brenninger studied chemistry at the Technische Universität München (M.Sc. 2014). During his undergraduate studies, he completed a six‐month industrial placement with Hoffmann‐La Roche in Basel. Since 2015, he has been carrying out research as a doctoral student in the research group of Prof. T. Bach on Brønsted acid catalyzed photoreactions.

Acknowledgements
Funding of our own work in this field is provided by the European Research Council under the European Union's Horizon 2020 research and innovation programme (grant agreement no. 665951—ELICOS), the Fonds der Chemischen Industrie, and the Alexander von Humboldt foundation.
C. Brenninger, J. D. Jolliffe, T. Bach, Angew. Chem. Int. Ed. 2018, 57, 14338.
Contributor Information
M. Sc. Christoph Brenninger, http://www.oc1.ch.tum.de/
Prof. Dr. Thorsten Bach, Email: thorsten.bach@ch.tum.de.
References
- 1.For reviews, see:
- 1a. Silvi M., Melchiorre P., Nature 2018, 554, 41–49; [DOI] [PubMed] [Google Scholar]
- 1b. Brimioulle R., Lenhart D., Maturi M. M., Bach T., Angew. Chem. Int. Ed. 2015, 54, 3872–3890; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 3944–3963; [Google Scholar]
- 1c. Meggers E., Chem. Commun. 2015, 51, 3290–3301. [DOI] [PubMed] [Google Scholar]
- 2.For seminal studies, see:
- 2a. Hammond G. S., Cole R. S., J. Am. Chem. Soc. 1965, 87, 3256–3257; [Google Scholar]
- 2b. Demuth M., Raghavan P. R., Carter C., Nakano K., Schaffner K., Helv. Chim. Acta 1980, 63, 2434–2439; [Google Scholar]
- 2c. Inoue Y., Yokoyama T., Yamasaki N., Tai A., Nature 1989, 341, 225–226; [Google Scholar]
- 2d. Kim J.-I., Schuster G. B., J. Am. Chem. Soc. 1992, 114, 9309–9317. [Google Scholar]
- 3.For more recent studies, see:
- 3a. Müller C., Maturi M. M., Bauer A., Cuquerella M. C., Miranda M. A., Bach T., J. Am. Chem. Soc. 2011, 133, 16689–16697; [DOI] [PubMed] [Google Scholar]
- 3b. Maturi M. M., Wenninger M., Alonso R., Bauer A., Pöthig A., Riedle E., Bach T., Chem. Eur. J. 2013, 19, 7461–7472; [DOI] [PubMed] [Google Scholar]
- 3c. Alonso R., Bach T., Angew. Chem. Int. Ed. 2014, 53, 4368–4371; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 4457–4460; [Google Scholar]
- 3d. Maturi M. M., Bach T., Angew. Chem. Int. Ed. 2014, 53, 7661–7664; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 7793–7796; [Google Scholar]
- 3e. Tröster A., Alonso R., Bauer A., Bach T., J. Am. Chem. Soc. 2016, 138, 7808–7811; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3f. Skubi K. L., Kidd J. B., Jung H., Guzei I. A., Baik M.-H., Yoon T. P., J. Am. Chem. Soc. 2017, 139, 17186–17192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dexter D. L., J. Chem. Phys. 1953, 21, 836–850. [Google Scholar]
- 5.For reviews, see:
- 5a. Schultz D. M., Yoon T. P., Science 2014, 343, 1239176; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5b. Reckenthäler M., Griesbeck A. G., Adv. Synth. Catal. 2013, 355, 2727–2744; [Google Scholar]
- 5c. Prier C. K., Rankic D. A., MacMillan D. W. C., Chem. Rev. 2013, 113, 5322–5363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Poplata S., Tröster A., Zou Y.-Q., Bach T., Chem. Rev. 2016, 116, 9748–9815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Praetorius P., Korn F., Ber. Dtsch. Chem. Ges. 1910, 43, 2744–2746. [Google Scholar]
- 8. Stobbe H., Färber E., Ber. Dtsch. Chem. Ges. 1925, 58, 1548–1553. [Google Scholar]
- 9. Alcock N. W., Herron N., Kemp T. J., Shoppee C. W., J. Chem. Soc. Chem. Commun. 1975, 785–786. [Google Scholar]
- 10.For the uncatalyzed [2+2] photocycloaddition of dibenzylideneacetone, see: Shoppee C. W., Wang Y.-s., Sternhell S., Brophy G. C., J. Chem. Soc. Perkin Trans. 1 1976, 1880–1886, and references therein. [Google Scholar]
- 11.
- 11a. Lewis F. D., Oxman J. D., J. Am. Chem. Soc. 1981, 103, 7345–7347; [Google Scholar]
- 11b. Lewis F. D., Oxman J. D., Gibson L. L., Hampsch H. L., Quillen S. L., J. Am. Chem. Soc. 1986, 108, 3005–3015. [Google Scholar]
- 12.
- 12a. Lewis F. D., Oxman J. D., Huffman J. C., J. Am. Chem. Soc. 1984, 106, 466–468; [Google Scholar]
- 12b. Lewis F. D., Quillen S. L., Hale P. D., Oxman J. D., J. Am. Chem. Soc. 1988, 110, 1261–1267. [Google Scholar]
- 13.
- 13a. Lewis F. D., Howard D. K., Barancyk S. V., Oxman J. D., J. Am. Chem. Soc. 1986, 108, 3016–3023; [Google Scholar]
- 13b. Lewis F. D., Howard D. K., Oxman J. D., Upthagrove A. L., Quillen S. L., J. Am. Chem. Soc. 1986, 108, 5964–5968; [DOI] [PubMed] [Google Scholar]
- 13c. Lewis F. D., Elbert J. E., Upthagrove A. L., Hale P. D., J. Org. Chem. 1991, 56, 553–561. [Google Scholar]
- 14. Wells P. P., Morrison H., J. Am. Chem. Soc. 1975, 97, 154–159. [DOI] [PubMed] [Google Scholar]
- 15. Lewis F. D., Howard D. K., Oxman J. D., J. Am. Chem. Soc. 1983, 105, 3344–3345. [Google Scholar]
- 16. Lewis F. D., Barancyk S. V., J. Am. Chem. Soc. 1989, 111, 8653–8661. [Google Scholar]
- 17.For related studies on [2+2] photodimerizations, see:
- 17a. Shim S. C., Kim E. I., Lee K. T., Bull. Korean Chem. Soc. 1987, 8, 140–144; [Google Scholar]
- 17b. Shim S. C., Lee S. S., Bull. Korean Chem. Soc. 1989, 10, 324–326. [Google Scholar]
- 18. Görner H., Wolff T., Photochem. Photobiol. 2008, 84, 1224–1230. [DOI] [PubMed] [Google Scholar]
- 19. Guo H., Herdtweck E., Bach T., Angew. Chem. Int. Ed. 2010, 49, 7782–7785; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 7948–7951. [Google Scholar]
- 20.H. Guo, T. Bach, unpublished results.
- 21.For a review, see: Corey E. J., Angew. Chem. Int. Ed. 2009, 48, 2100–2117; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 2134–2151. [Google Scholar]
- 22. Brimioulle R., Bauer A., Bach T., J. Am. Chem. Soc. 2015, 137, 5170–5176. [DOI] [PubMed] [Google Scholar]
- 23. Brimioulle R., Guo H., Bach T., Chem. Eur. J. 2012, 18, 7552–7560. [DOI] [PubMed] [Google Scholar]
- 24.
- 24a. Sato T., Yoshiie S., Imamura T., Hasegawa K., Miyahara M., Yamamura S., Ito O., Bull. Chem. Soc. Jpn. 1977, 50, 2714–2730; [Google Scholar]
- 24b. Ogawa T., Masui Y., Ojima S., Suzuki H., Bull. Chem. Soc. Jpn. 1987, 60, 423–425. [Google Scholar]
- 25.
- 25a. El-Sayed M. A., Acc. Chem. Res. 1968, 1, 8–16; [Google Scholar]
- 25b. Klán P., Wirz J., Photochemistry of Organic Compounds, Wiley, Chichester, 2009, pp. 38–39. [Google Scholar]
- 26.For [2+2] photocycloadditions of dihydropyridones, see:
- 26a. Guerry P., Neier R., J. Chem. Soc. Chem. Commun. 1989, 1727–1728; [Google Scholar]
- 26b. Guerry P., Blanco P., Brodbeck H., Pasteris O., Neier R., Helv. Chim. Acta 1991, 74, 163–178. [Google Scholar]
- 27. Brimioulle R., Bach T., Science 2013, 342, 840–843. [DOI] [PubMed] [Google Scholar]
- 28. Brimioulle R., Bach T., Angew. Chem. Int. Ed. 2014, 53, 12921–12924; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 13135–13138. [Google Scholar]
- 29.
- 29a. Schultz A. G., Lucci R. D., J. Org. Chem. 1975, 40, 1371–1372; [Google Scholar]
- 29b. Schultz A. G., Lucci R. D., Fu W. Y., Berger M. H., Erhardt J., Hagmann W. K., J. Am. Chem. Soc. 1978, 100, 2150–2162. [Google Scholar]
- 30.For a recent more comprehensive study, see: Münster N., Parker N., van Dijk L., Paton R. S., Smith M. D., Angew. Chem. Int. Ed. 2017, 56, 9468–9472; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 9596–9600. [Google Scholar]
- 31. Edtmüller V., Pöthig A., Bach T., Tetrahedron 2017, 73, 5038–5047. [Google Scholar]
- 32.
- 32a. Evans D. A., Peterson G. S., Johnson J. S., Barnes D. M., Campos K. R., Woerpel K. A., J. Org. Chem. 1998, 63, 4541–4544; [Google Scholar]
- 32b. Evans D. A., Johnson J. S., Burgey C. S., Campos K. R., Tetrahedron Lett. 1999, 40, 2879–2882. [Google Scholar]
- 33. Huang X., Quinn T. R., Harms K., Webster R. D., Zhang L., Wiest O., Meggers E., J. Am. Chem. Soc. 2017, 139, 9120–9123. [DOI] [PubMed] [Google Scholar]
- 34. Ma J., Zhang X., Huang X., Luo S., Meggers E., Nat. Protocols 2018, 13, 605–632, and references therein. [DOI] [PubMed] [Google Scholar]
- 35.For a related study on the enantioselective [2+2] photocycloaddition of similar alkenes with benzofuran-2-carboxamides, see: Hu N., Jung H., Zheng Y., Lee J., Zhang L., Ullah Z., Xie X., Harms K., Baik M.-H., Meggers E., Angew. Chem. Int. Ed. 2018, 57, 6242–6246; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 6350–6354. [Google Scholar]
- 36. Wang H., Cao X., Chen X., Fang W., Dolg M., Angew. Chem. Int. Ed. 2015, 54, 14295–14298; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 14503–14506. [Google Scholar]
- 37. Huang X., Li X., Xie X., Harms K., Riedel R., Meggers E., Nat. Commun. 2017, 8, 2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.
- 38a. Kärkäs M. D., J. A. Porco, Jr. , Stephenson C. R. J., Chem. Rev. 2016, 116, 9683–9747; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38b. Bach T., Hehn J. P., Angew. Chem. Int. Ed. 2011, 50, 1000–1045; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 1032–1077; [Google Scholar]
- 38c. Hoffmann N., Chem. Rev. 2008, 108, 1052–1103. [DOI] [PubMed] [Google Scholar]
- 39. Poplata S., Bach T., J. Am. Chem. Soc. 2018, 140, 3228–3231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Blum T. R., Miller Z. D., Bates D. M., Guzei I. A., Yoon T. P., Science 2016, 354, 1391–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Miller Z. D., Lee B. J., Yoon T. P., Angew. Chem. Int. Ed. 2017, 56, 11891–11895; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 12053–12057. [Google Scholar]
- 42.
- 42a. Zalewski R. I., Dunn G. E., Can. J. Chem. 1968, 46, 2469–2470; [Google Scholar]
- 42b. Zalewski R. I., Dunn G. E., Can. J. Chem. 1969, 47, 2263–2270. [Google Scholar]
- 43.The reactions of di- and trienones, which are not covered in this Minireview, have been studied particularly by the groups of Hart and Childs; see:
- 43a. Hart H., Pure Appl. Chem. 1973, 33, 247–267; [Google Scholar]
- 43b. Childs R. F., Res. Chem. Intermed. 1980, 3, 285–314. [Google Scholar]
- 44.
- 44a. Childs R. F., Lund E. F., Marshall A. G., Morrisey W. J., Rogerson C. V., J. Am. Chem. Soc. 1976, 98, 5924–5931; [Google Scholar]
- 44b. Childs R. F., Mahendran M., Blackburn C., Antoniadis G., Can. J. Chem. 1988, 66, 1355–1358. [Google Scholar]
- 45. Childs R. F., Duffey B., Mika-Gibala A., J. Org. Chem. 1984, 49, 4352–4358. [Google Scholar]
- 46. Cornell D. G., Filipescu N., J. Org. Chem. 1977, 42, 3331–3336. [Google Scholar]
- 47.
- 47a. Lupon P., Ferrer J. C., Piniella J. F., Bonet J.-J., J. Chem. Soc. Chem. Commun. 1983, 718–719; [Google Scholar]
- 47b. Lupón P., Canals F., Iglesias A., Ferrer J. C., Palomer A., Bonet J.-J., Briansó J. L., Piniella J. F., Germain G., King G. S. D., J. Org. Chem. 1988, 53, 2193–2198. [Google Scholar]
- 48. Childs R. F., Hine K. E., Hung F. A., Can. J. Chem. 1979, 57, 1442–1445. [Google Scholar]
- 49. Vallavoju N., Selvakumar S., Jockusch S., Sibi M. P., Sivaguru J., Angew. Chem. Int. Ed. 2014, 53, 5604–5608; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 5710–5714. [Google Scholar]
- 50. Vallavoju N., Selvakumar S., Jockusch S., Prabhakaran M. T., Sibi M. P., Sivaguru J., Adv. Synth. Catal. 2014, 356, 2763–2768. [Google Scholar]
- 51.For a comprehensive analysis, see: Vallavoju N., Selvakumar S., Pemberton B. C., Jockusch S., Sibi M. P., Sivaguru J., Angew. Chem. Int. Ed. 2016, 55, 5446–5451; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 5536–5541. [Google Scholar]
- 52. Childs R. F., Hagar M. E., Can. J. Chem. 1980, 58, 1788–1794. [Google Scholar]
- 53.C. Brenninger, Y.-Q. Zou, T. Bach, unpublished results.
- 54. Brenninger C., Bach T., Top. Catal. 2018, 61, 623–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Brenninger C., Pöthig A., Bach T., Angew. Chem. Int. Ed. 2017, 56, 4337–4341; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 4401–4405. [Google Scholar]
- 56. Tarassoff P. G., Filipescu N., J. Chem. Soc. Chem. Commun. 1975, 208–209. [Google Scholar]
- 57.
- 57a. Ahrendt K. A., Borths C. J., MacMillan D. W. C., J. Am. Chem. Soc. 2000, 122, 4243–4244; [Google Scholar]
- 57b. Lelais G., MacMillan D. W. C., Aldrichimica Acta 2006, 39, 79–87. [Google Scholar]
- 58.For a review, see: Zou Y.-Q., Hörmann F. M., Bach T., Chem. Soc. Rev. 2018, 47, 278–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.
- 59a. Kuroyanagi M., Noro T., Fukushima S., Aiyama R., Ikuta A., Itokawa H., Morita M., Chem. Pharm. Bull. 1983, 31, 1544–1550; [Google Scholar]
- 59b. Childs R. F., Dickie B. D., J. Am. Chem. Soc. 1983, 105, 5041–5046. [Google Scholar]
- 60. Hörmann F. M., Chung T. S., Rodriguez E., Jakob M., Bach T., Angew. Chem. Int. Ed. 2018, 57, 827–831; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 835–839. [Google Scholar]
- 61.
- 61a. Cai X., Chang V., Chen C., Kim H.-J., Mariano P. S., Tetrahedron Lett. 2000, 41, 9445–9449; [Google Scholar]
- 61b. Chen C., Chang V., Cai X., Duesler E., Mariano P. S., J. Am. Chem. Soc. 2001, 123, 6433–6434. [DOI] [PubMed] [Google Scholar]
- 62. Silvi M., Verrier C., Rey Y. P., Buzzetti L., Melchiorre P., Nat. Chem. 2017, 9, 868–873. [DOI] [PubMed] [Google Scholar]
- 63. Verrier C., Alandini N., Pezzetta C., Moliterno M., Buzzetti L., Hepburn H. B., Vega-Peñaloza A., Silvi M., Melchiorre P., ACS Catal. 2018, 8, 1062–1066. [Google Scholar]
- 64.
- 64a. Stavinoha J., Bay E., Leone A., Mariano P. S., Tetrahedron Lett. 1980, 21, 3455–3458; [Google Scholar]
- 64b. Mariano P. S., Stavinoha J., Bay E., Tetrahedron 1981, 37, 3385–3395. [Google Scholar]
- 65. Woźniak Ł., Magagnano G., Melchiorre P., Angew. Chem. Int. Ed. 2018, 57, 1068–1072; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 1080–1084. [Google Scholar]
- 66. Schuster D. I., Dunn D. A., Heibel G. E., Brown P. B., Rao J. M., Woning J., Bonneau R., J. Am. Chem. Soc. 1991, 113, 6245–6255. [Google Scholar]
- 67.T. S. Chung, T. Bach, unpublished results.
- 68.
- 68a. Houk K. N., Strozier R. W., J. Am. Chem. Soc. 1973, 95, 4094–4096; [Google Scholar]
- 68b. Yamamoto H., Lewis Acids in Organic Synthesis, Wiley-VCH, Weinheim, 2000. [Google Scholar]