

Abstract
Mucosa‐associated lymphoid tissue 1 (Malt1) regulates immune cell function by mediating the activation of nuclear factor κB (NF‐κB) signaling through both its adaptor and proteolytic function. Malt1 is also a target of its own protease activity and this self‐cleavage further contributes to NF‐κB activity. Until now, the functional distinction between Malt1 self‐cleavage and its general protease function in regulating NF‐κB signaling and immune activation remained unclear. Here we demonstrate, using a new mouse model, the importance of Malt1 self‐cleavage in regulating expression of NF‐κB target genes and subsequent T cell activation. Significantly, we further establish that Treg homeostasis is critically linked to Malt1 function via a Treg intrinsic and extrinsic mechanism. TCR‐mediated Malt1 proteolytic activity and self‐cleavage was found to drive Il2 expression in conventional CD4+ T cells, thereby regulating Il2 availability for Treg homeostasis. Remarkably, the loss of Malt1‐mediated self‐cleavage alone was sufficient to cause a significant Treg deficit resulting in increased anti‐tumor immune reactivity without associated autoimmunity complications. These results establish for the first time that inhibition of MALT1 proteolytic activity could be a viable therapeutic strategy to augment anti‐tumor immunity.
Keywords: MALT1, NF‐κB, TCR, Regulatory T cells
Introduction
Antigen receptor signaling controls lymphocyte development and is a key step regulating T cell and B cell activation. Antigen recognition by the T cell receptor (TCR) is one of the most complex pathways of the immune system, where depletion of key signaling enzymes results in severe immunodeficiency in both humans and mice 1, 2, 3, 4. Binding of the TCR to the peptide‐major histocompatibility complex (MHC) leads to the formation of the CARMA1, BCL10, and MALT1 (CBM) protein complex, resulting in NF‐κB activation 5, 6. As an adaptor, MALT1 is integral for the formation of the CBM complex by binding to BCL10 and CARMA1, which is key for phosphorylation of IκBα and NF‐κB activation 7, 8, 9, 10. The MALT1 protease function catalyzes proteolytic cleavage of multiple negative regulators of NF‐κB signaling, including RELB, CYLD and MCPIP1. As a consequence, Malt1 knock‐out (Malt1−/−) and Malt1 protease‐dead mice (Malt1PD) show defective T‐cell responses 11, 12. This finding makes Malt1 protease activity an attractive target for the treatment of auto‐inflammatory diseases, with multiple inhibitors currently in pre‐clinical development.
More recently, we and others have demonstrated that MALT1 also possesses auto‐proteolytic activity, resulting in two MALT1 fragments, p16 and p76 13, 14. The auto‐proteolytic removal of the so‐called N‐terminal death domain (p16) results in the formation of an active C‐terminal p76 fragment of MALT1 that dissociates from BCL10 and oligomerizes to promote the transcriptional activity of NF‐κB complexes in a TRAF6‐dependent manner 13, 14. In vitro data suggest that a self‐cleavage resistant MALT1 (MALT1SR) results in defective activation of NF‐κB target genes 13, thereby adding another level of complexity in how MALT1 regulates NF‐κB function.
Regulatory T cells (Treg) are a specialized subpopulation of CD4+ T cells, characterized by the expression of the transcription factor Foxp3 15. Treg cells act to suppress immune reactivity against self‐antigens, thus preventing autoimmunity. The size of the circulating Treg pool is dependent on Il2 availability that is primarily produced by CD4+ T cells 16, 17. Mice, genetically deficient in Il2, Il2ra or Il2rb, have severely reduced Treg cell numbers and develop lethal autoimmune disease 18, 19, 20, 21. Conversely, Treg enrichment within the tumor microenvironment can protect tumor cells by inhibiting anti‐tumor immunity 22.
To better understand the role of Malt1 self‐cleavage versus its general protease activity in regulating NF‐κB signaling and immune cell function in vivo, we generated a new Malt1 self‐cleavage resistant mouse model and compared it to the Malt1 protease‐dead mouse model. Our findings suggest that Malt1 self‐cleavage regulates TCR signal transduction via amplification of NF‐κB activation. This was most exemplified by the reduction of thymic Treg differentiation in Malt1SR/SR animals. Furthermore, we report that the homeostasis of Tregs was altered due to Malt1‐impairment in a cell extrinsic manner. Here, Malt1 proteolytic function and its self‐cleavage were pivotal for Il2 production by conventional CD4+ T cells. This Il2 deficiency prevented Treg expansion and reduced the levels of phospho‐Stat5 in Treg. As a consequence, we also show that the disruption of the Treg pool size in the Malt1SR/SR animals resulted in increased anti‐tumor immune reactivity.
Results
Self‐cleavage defective Malt1 does not alter IκBα phosphorylation and retains global protease activity
MALT1 protease activity is required for TCR‐mediated signaling via NF‐κB. Auto‐proteolytic MALT1 cleavage after Arginine 149 results in two protein fragments, p16 and p76 (Fig. 1A). An un‐cleavable MALT1‐R149A mutant (self‐cleavage resistant MALT1) has been shown to induce normal activation of an NF‐κB reporter gene expression, unaltered initial IκBα phosphorylation and nuclear accumulation of NF‐κB subunits 13.
Figure 1.
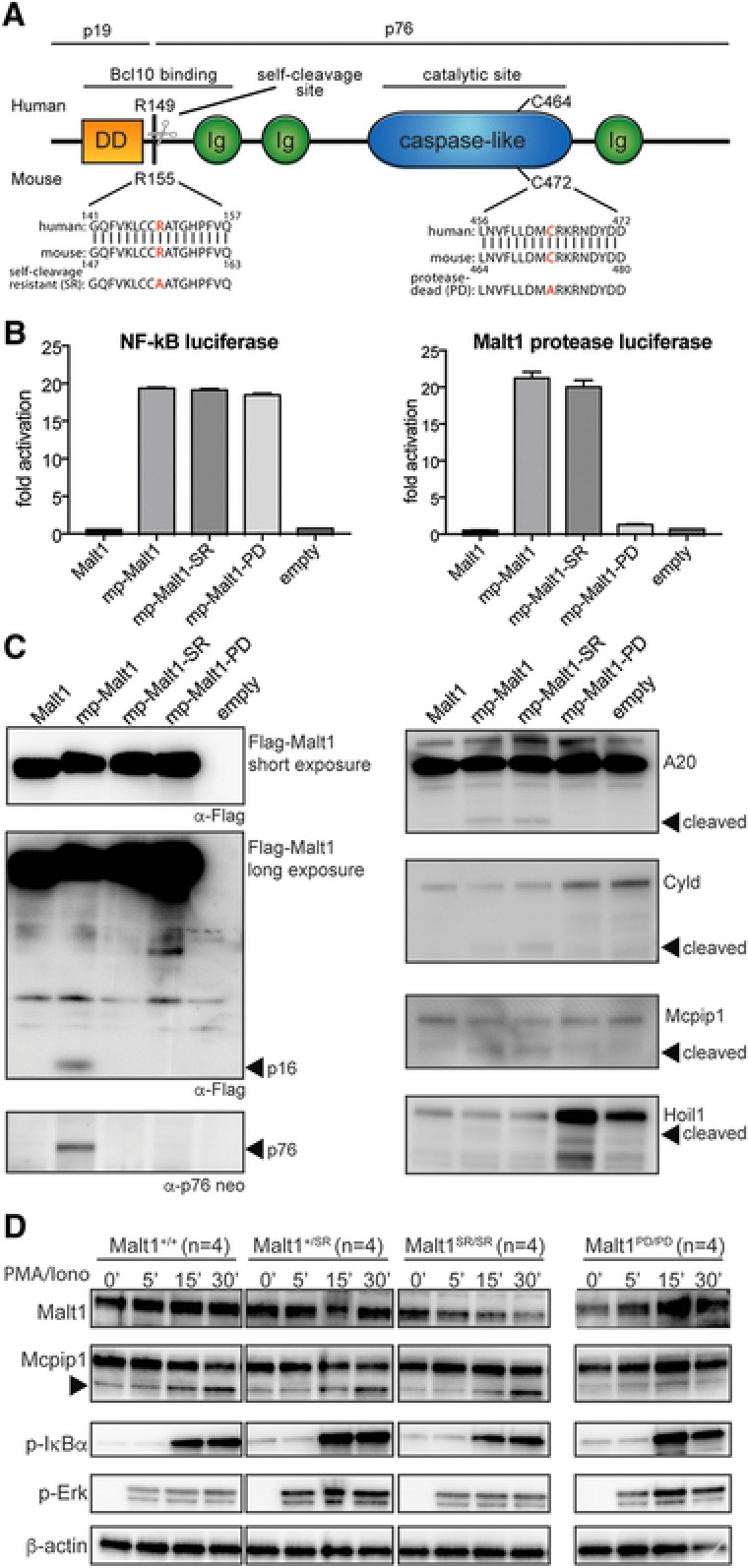
Malt1 R155A knock‐in mice express a catalytically active form of Malt1 but lack self‐cleavage activity (A) Schematic representation of Malt1 protein and its functional death domain (DD), immunoglobulin‐like domains (Ig), auto processing site and catalytic site. (B) MALT1 protease reporter assays of 293T‐BM cells transiently expressing a mouse wild‐type Malt1 (mp‐Malt1), the self‐cleavage resistant R155A mutant (mp‐Malt1‐SR), the protease‐death C472A mutant (mp‐Malt1‐PD) or empty vector (mock). Malt1 protease‐dependent luciferase activity is shown as fold induction of vector‐transfected cells. The data are shown as mean +SD of four pooled independent experiments, which were each performed in triplicates. (C) Cell lysates were immunoblotted with the indicated antibodies to detect MALT1, its N‐ and C‐terminal auto‐cleavage fragments (p16 and p76 respectively) and its proteolytic targets. Arrows indicate the cleavage fragments generated by Malt1 protease activity. (D) Immunoblot analysis of spleen of Malt1+/+, Malt1+/SR, Malt1SR/SR and Malt1PD/PD mice. Splenocytes from 4 mice were combined and then incubated with or without PMA and ionomycin for the indicated times. A non‐specific band generated with the Mcpip1 antibody was used a loading control. Data are representative of three independent experiments.
We have previously shown that targeting MALT1 to the plasma membrane via a N‐terminal myristoylation‐palmitoylation (mp‐MALT1) signal cells induces its cleavage into 16 and 76 kDa fragments 13. Mutation of the homologous mouse Malt1 auto‐proteolytic site arginine to alanine (R155A), similarly induced normal activation of an NF‐κB reporter gene but prevented self‐cleavage of membrane‐associated Malt1 (mp‐Malt1SR, Fig. 1B and C). However, the R155A mutation did not affect the protease activity of Malt1 as measured via a Malt1 bioluminescent protease assay or via the cleavage of known Malt1 substrates (Fig. 1B and C). Proteolytically inactive Malt1 (C472A) (mp‐Malt1PD), however, abolished reporter activity and substrate cleavage (Fig. 1B and C).
To selectively disrupt Malt1 auto‐proteolytic activity in vivo, we generated a Malt1 self‐cleavage resistant knock‐in mouse model by introducing the R155A mutation through targeted homologous recombination. Mice, heterozygous and homozygous for the Malt1 self‐cleavage resistant (Malt1SR) alleles were viable and born in Mendelian ratios (data not shown). Western blotting of cell lysates from the spleen of Malt1+/+, Malt1+/SR, Malt1SR/SR and Malt1PD/PD mice showed equivalent Malt1 protein expression and protein stability (data not shown) and initial IκBα and Erk phosphorylation upon PMA/ionomycin treatment, in line with our previous observations (Fig. 1D) 13. Furthermore, Malt1SR/SR cells had normal cleavage of the Malt1 proteolytic substrate Mcpip1 upon stimulation, whereas Malt1PD/PD cells showed lack of cleavage of this Malt1 target (Fig. 1D). These results show that Malt1SR/SR mice have normal Malt1 protein expression and protease activity but are unable to undergo auto‐proteolysis.
Malt1 auto‐proteolytic activity is required for optimal NF‐κB activation and T cell proliferation
Given the important role of Malt1 in mediating TCR activation and resulting T cell response, we next asked whether the lack of Malt1 auto‐proteolysis downregulated TCR signaling to NF‐κB. To this end we performed RNA sequencing analysis on unstimulated and anti‐CD3/CD28 stimulated naïve CD4+ T cells from Malt1+/+, Malt1SR/SR and Malt1PD/PD background. Analysis of the induction of known NF‐κB target genes globally in stimulated versus unstimulated CD4+ T cells showed decreased upregulation of these genes in Malt1PD/PD T cells (Supporting Information Fig. 1A). Blocking Malt1 auto‐proteolytic activity also affected the induction of NF‐κB target genes upon TCR stimulation, but to a lesser extent than observed in T cells harboring the proteolytically‐inactive Malt1 (Supporting Information Fig. 1 A). This general decrease in both Malt1PD/PD and Malt1SR/SR T cells was further corroborated using quantitative real‐time PCR analysis for key Malt1 protease‐dependent NF‐κB target genes, except for Bcl2l1 that remained unchanged in Malt1SR/SR T cells (Supporting Information Fig. 1B).
Next, we asked whether Malt1 self‐cleavage resistance also affected T cell proliferation in response to anti‐CD3/CD28 stimulation. Both Malt1SR/SR and Malt1PD/PD cells showed impaired TCR‐induced proliferation compared to Malt1+/+ CD4+ T cells, as measured by CFSE dilution (Supporting Information Fig. 1C). However, the proliferation was significantly less in the complete protease‐dead Malt1 when compared to the self‐cleavage resistant Malt1 (Supporting Information Fig. 1 C). These findings implied that the auto‐proteolytically cleaved Malt1 p76 protein product directly mediates activation of NF‐κB and the expression of NF‐κB target genes. To test this hypothesis, full‐length Malt1 or the p76 cleavage product were expressed in Jurkat cells. Here, expression of p76, but not full‐length Malt1, induced the activation of an NF‐κB luciferase‐reporter gene in the absence of TCR stimulation (Supporting Information Fig. 1 D). Taken together, these results show that the Malt1 self‐cleavage activity is not required for initial IκBα activation or NF‐κB translocation but is essential for the optimal activation of NF‐κB target genes. This is further elaborated by the observation that Malt1 p76 by itself induced transcription of common NF‐κB target genes.
Self‐cleavage resistant Malt1 mice do not develop T cell mediated autoimmunity
Malt1PD/PD animals develop fatal autoimmune disease but Malt1−/− mice do not due to their inability to mount an immune response 11, 12. Therefore, we next sought to determine whether the Malt1SR/SR mice would also develop a similar autoimmune disorder to Malt1PD/PD mice. As expected, the homozygous Malt1PD/PD mice developed symptoms of autoimmune disease, starting at 8–10 weeks of age, exemplified by the significant weight loss (Supporting Information Fig. 2 A). Autoimmune disease in Malt1PD/PD was confirmed by histological examination showing severe multifocal lymphocyte infiltration and gastritis lymphoid hyperplasia and pancreatic infiltration (Supporting Information Fig. 2 B). Strikingly, Malt1SR/SR mice showed no signs of autoimmune disease after 15 weeks and gained weight comparable to wild type Malt1+/+ animals (Supporting Information Fig. 2 C).
Self‐cleavage resistant Malt1 mice have normal levels of T‐ and B cell frequencies
The lack of autoimmune disease in the Malt1SR/SR mice prompted us to examine the peripheral lymphoid compartment in more detail. The Malt1SR/SR mice had normal frequencies of CD4+ and CD8+ T cells and no increased effector memory T cells when compared to Malt1PD/PD mice which had an increased expansion of CD8+ T cells and 2‐fold increase of effector/memory CD4+ T cells (Fig. 2A‐‐C). Interestingly, although Malt1SR/SR animals displayed a ∼25% decrease in natural killer (NK) cells, this decrease was only half that as observed for Malt1PD/PD (Fig. 2D). There was no difference in B cell frequency among the different cohorts (Fig. 2E), but marginal zone B cells were almost absent in Malt1PD/PD mice, and unchanged in Malt1SR/SR animals (Fig. 2F). Notably, the Malt1SR/SR mice did not show any signs of aberrant cytokine production or T cell polarization, in line with the absence of signs of disease development (Fig 2 G). This is in contrast to the Malt1PD/PD animals which showed a strong Th1 polarization of CD4+ T cells and increased Ifn‐γ and Il6 serum cytokine levels concordant with the signs of autoimmune disease (Fig. 2G, H).
Figure 2.
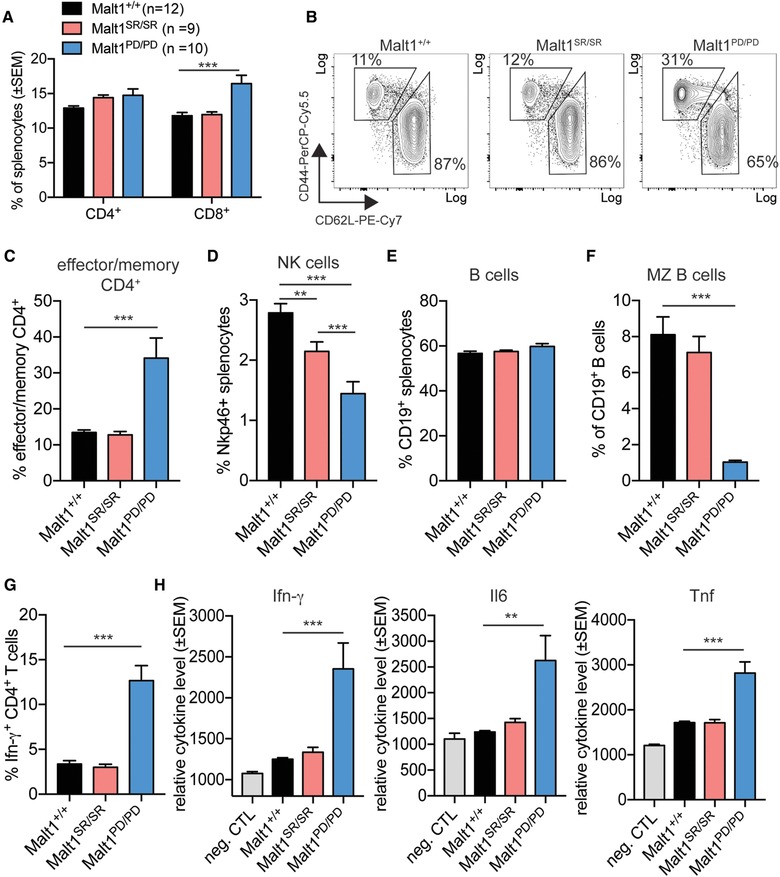
Malt1 self‐cleavage resistance rescues mice from T cell‐mediated autoimmunity (A) Frequency of CD4+ and CD8+ T cells within the spleen of Malt1+/+ (n = 12), Malt1SR/SR (n = 9) and Malt1PD/PD (n = 10) mice was analyzed by flow cytometry. (B) Representative flow cytometry data and (C) frequency of CD4+ T cells with an activated/memory (CD44hi, CD62Llo) or naïve (CD44lo, CD62Lhi) phenotype. (D) Frequency of Nkp46+ NK cells and (E) CD19 + B cells of total splenocytes. (F) Frequency of marginal zone (MZ) B cells, as identified by CD23 and CD21 expression within the total B cell population. (G) Frequency of Ifn‐γ ‐positive CD4 + T cell within the splenocyte population. (H) Ifn‐γ, Il6 and Tnf cytokine concentration was measured in the serum of mice of the different cohorts by cytometric bead array. Assay buffer was used as background fluorescence negative control to assess relative serum concentrations (neg. CTL). Data are shown as mean + SD and are representative of three independent experiments with four mice per experiment. Student's t test was used for statistical analysis. Sample means were considered significantly different at p < 0.05, with *p < 0.05, **p < 0.01 and ***p < 0.001.
Malt1 is essential for thymic development of regulatory T cells
Previously, the autoimmunity observed in Malt1PD/PD mice was linked to defective thymic Treg development 11, 12. We, therefore, sought to determine whether Treg development was also altered within the Malt1SR/SR mice. Thymic tissue from aged matched Malt1PD/PD and Malt1SR/SR mice had normal numbers of CD4+, CD8+ double‐positive, CD4SP and CD8SP cells (Fig 3 A). However, Malt1SR/SR mice, on the other hand, had a ∼44% reduction in thymic Treg (Fig. 3B). In line with this, Malt1−/− mice were reported to be almost completely depleted of thymic Treg cells 11, 23. Malt1PD/PD mice, on the other hand, had ∼81% reduction in Treg cells. Interestingly, heterozygous animals still had ∼22% reduced Treg numbers in the thymus (Fig. 3B).
Figure 3.
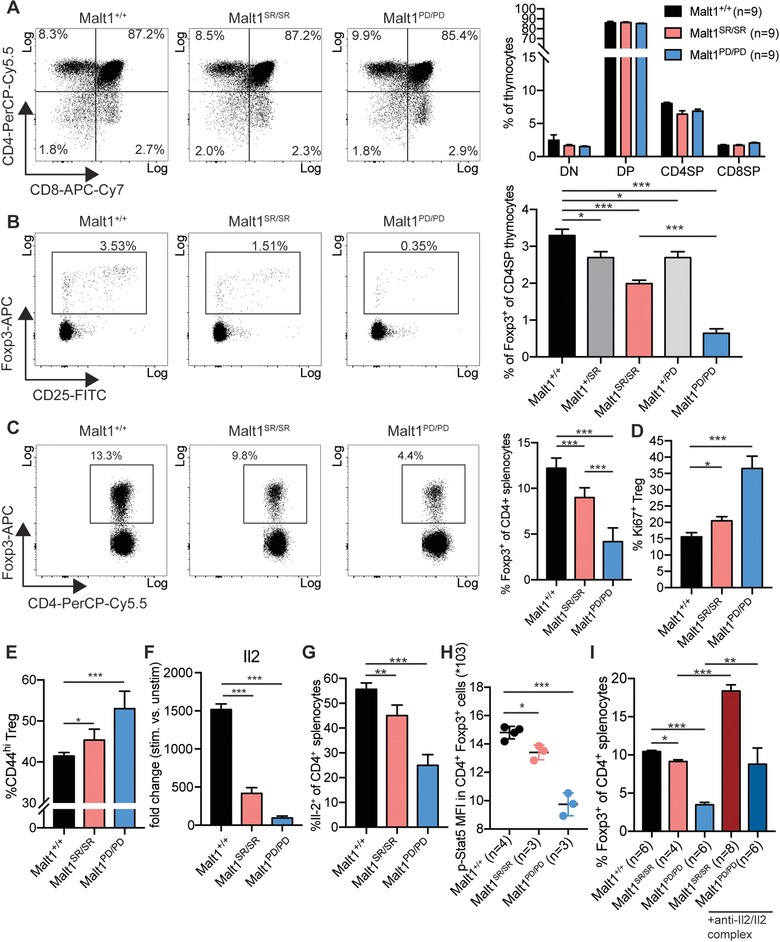
Malt1 self‐cleavage is required for thymic Treg differentiation and impairs peripheral Treg homeostasis (A) Representative flow cytometry data and frequency CD4, CD8 double negative (DN), double positive (DP), CD4 single‐positive (CD4SP) and CD8 single‐positive (CD8SP) thymocytes. The data are shown as mean + SD of three pooled independent experiments, which were each performed in triplicates with three animals per group. (B) Representative flow cytometry data and frequency of Foxp3+ regulatory T cells within CD4SP thymocytes. The data are shown as mean + SD of three pooled independent experiments, which were each performed in triplicates with three animals per group. (C) Representative flow cytometry data and frequency of Foxp3+ Treg within CD4+ splenocytes. The data are shown as mean + SD of three pooled independent experiments, which were each performed in triplicates with three animals per group. (D) Treg proliferation was measured by Ki67‐positivity within CD4+ Foxp3+ splenocytes. The data are shown as mean + SD of three pooled independent experiments, which were each performed in triplicates with three animals per group. (E) Frequency of Treg with an activated/memory phenotype, as measured by the expression of CD44 and CD62L. The data are shown as mean + SD of three pooled independent experiments, which were each performed in triplicates with three animals per group. (F) Real‐time quantitative PCR analysis Il2 induction in stimulated versus unstimulated naïve CD4+ T cells from Malt1+/+, Malt1SR/SR and Malt1PD/PD mice (n = 3 animals per group). (G) Frequency of Il2 producing cells within the population of CD4+ T cells in mice of the different cohorts. Experiments were performed with Malt1+/+ (n = 12), Malt1SR/SR (n = 9) and Malt1PD/PD (n = 10) mice in three independent experiments. Data were pooled and are shown as mean + SD. (H) Mean florescence intensity (MFI) of phospho‐Stat5 (Tyr694) in CD4+ Foxp3+ cells are shown as mean + SD for two pooled experiments with 4 mice per group. (I) Frequency of CD4+ Foxp3+ cells in mouse splenocytes of the different cohorts after treatment with the anti‐Il2/Il2 complex or control treatment. Data are shown as mean + SD with Malt1+/+ n = 6, treated Malt1SR/SR n = 8, treated Malt1PD/PD n = 6, untreated Malt1SR/SR n = 4, untreated Malt1PD/PD n = 6 animals per group.
The upregulation of Foxp3 has been shown to depend on the NF‐κB subunits RelA and c‐Rel, which are controlled by Malt1 protease activity. Consistent with this idea, we observed that anti‐CD3 stimulation mediated upregulation of Foxp3 in naïve CD4+ T cells was impaired in Malt1SR/SR and Malt1PD/PD mice (Supporting Information Fig. 3 ). Malt1SR/SR cells were still able to upregulate Foxp3, but to a lesser extent than Malt1+/+ T cells. Malt1PD/PD CD4+ T cells, however, showed almost no Foxp3 induction in response to CD3 stimulation, in line with previous observations 11.
Thus, Malt1 has an important function in regulating TCR signaling and might help to translate signaling strength to NF‐κB transcriptional activity and Foxp3 induction through its adaptor, self‐cleavage and proteolytic functions.
Malt1 mediated Il2 expression controls peripheral Treg homeostasis
Treg homeostasis is a plastic process and numbers rapidly normalize after perturbation of Treg cell population. In models of impaired thymic Treg differentiation or inducible Treg depletion, cell numbers are compensated through expansion of the remaining Treg pool in an Il2 dependent manner 24, 25, 26, 27. We, therefore, analyzed the peripheral Treg compartment in Malt1SR/SR and Malt1PD/PD animals compared to Malt1+/+ mice and found that Malt1SR/SR mice had a ∼25% reduction in Treg numbers in the spleen whilst the Malt1PD/PD mice had a ∼65% decrease (Fig. 3 C). Since Malt1 controls the downstream TCR signaling pathway, we hypothesized that Treg homeostasis was impaired due to the decreased TCR‐mediated proliferation signals. Treg from both Malt1SR/SR and Malt1PD/PD mice displayed increased proliferative activity, as measured by Ki67‐positivity (Fig. 3 D). Furthermore, acquisition of an activated/memory Treg phenotype (CD44high, CD62Llo), previously shown to be TCR‐signaling dependent 28, was not blocked in Malt1SR/SR or Malt1PD/PD animals and both subgroups even showed a subtle increase in their frequency (Fig. 3E).
Treg cell homeostasis and their survival is also critically linked to Il2 availability which is primarily produced by CD4+ T cells 17, 29, 30. However, TCR‐signaling mediated expression of the direct NF‐κB target gene Il2 was impaired in both Malt1SR/SR and Malt1PD/PD CD4+ T cells (Fig. 3F). Indeed, in vivo frequencies of Il2 producing CD4+ T cells was significantly reduced in Malt1SR/SR mice and further reduced in Malt1PD/PD animals (Fig. 3G), indicating Il2 deficiency in these animals. Therefore, we next asked if this observed Il2 deficiency directly affected Treg cells. Il2r signaling in Treg induces Stat5 phosphorylation and activation of its downstream target genes 29. We assessed the levels of phosphorylated Stat5 in Treg from the different mouse cohorts and the levels of phospho‐Stat5 correlated to the Il2 availability. The Malt1SR/SR Treg cells showed only a minor, but significant, reduction in phospho‐Stat5 levels when compared to Malt1PD/PD Tregs that had a much larger and more significant decrease in phospho‐Stat5 levels (Fig. 3H). In order to confirm that Treg frequencies were reduced due an Il2 deficiency and not due to the Treg cells being refractory to Il2, we treated mice with an Il2 complex, as described previously 31. Indeed, increasing Il2 availability expanded the Treg population in Malt1SR/SR and Malt1PD/PD animals, indicating that Malt1‐deficiency does not alter Il2 responsiveness (Fig. 3I). In conclusion, self‐cleavage and protease activity of Malt1 are critical within CD4+ T cells for Il2 production, thereby regulating Treg homeostasis in a cell extrinsic manner.
Malt1‐mediated Treg deficiency results in increased anti‐tumor immune response
The previous in vivo observations that the Malt1SR/SR and Malt1PD/PD mice have reduced Treg led us to speculate that Il2 mediated Treg de‐regulation and impaired Treg homeostasis could result in an increased anti‐tumor immune response. To this end, the MC38 colon carcinoma cell line was implanted into syngeneic Malt1+/+, Malt1SR/SR or Malt1PD/PD mice. Caliper measurements showed significantly reduced tumor growth in Malt1SR/SR and an even stronger reduction in Malt1PD/PD animals (Fig. 4A). Furthermore, Malt1 impairment resulted also in a significant reduction of the tumor weight at end stage (Fig. 4B).
Figure 4.
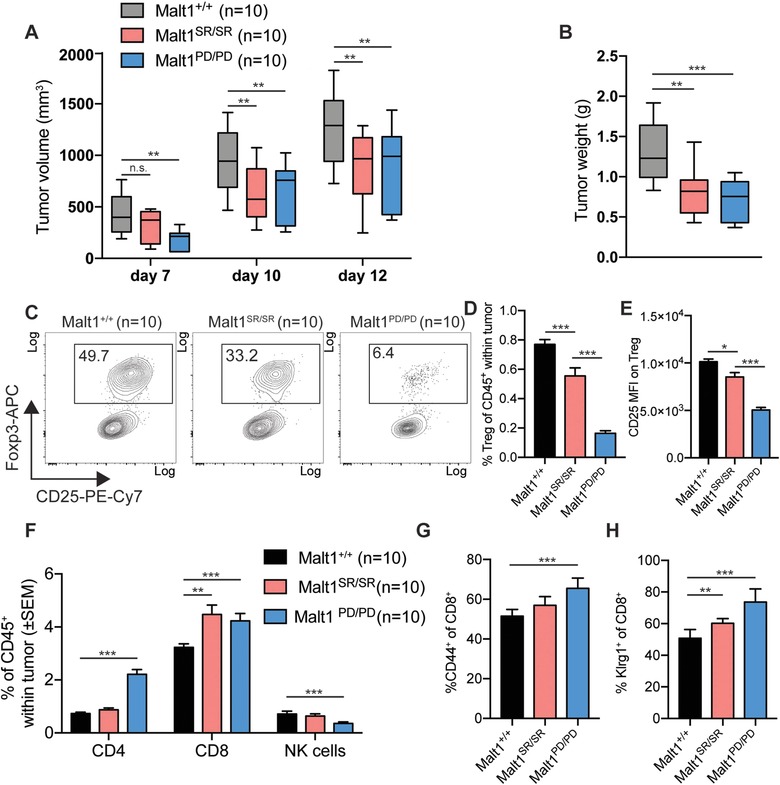
Malt1‐deficiency reduces MC38 tumor growth, by improving anti‐tumor immunity due to Treg impairment (A) Tumor volume was determined by blinded repeated caliper measurements at indicated time points in animals of the different cohorts. (B) Tumor weight at end stage. (C) Frequency of CD4+ Foxp3+ regulatory T cells in the tumor microenvironment was measured by flow cytometry (Malt1+/+ n = 10, Malt1SR/SR n = 10, Malt1PD/PD n = 10). (D) Frequency of total tumor infiltrating Treg as percentage of infiltrating CD45+ lymphocytes (Malt1+/+ n = 10, Malt1SR/SR n = 10, Malt1PD/PD n = 10). (E) CD25 MFI was measured by flow cytometry on tumor infiltrating Treg(Malt1+/+ n = 10, Malt1SR/SR n = 10, Malt1PD/PD n = 10). (F) Frequency of total tumor infiltrating CD4+ Foxp3− T cells, CD8+ T cells and Nkp46+ NK cells (Malt1+/+ n = 10, Malt1SR/SR n = 10, Malt1PD/PD n = 10). (G) Expression of the activation marker CD44 and (H) Klrg1 on CD8+ T cells within the tumor microenvironment. The data are shown as mean +SD (Malt1+/+ n = 10, Malt1SR/SR n = 10, Malt1PD/PD n = 10) and are representative of three independent experiments.
Analysis of the immune cell composition within the tumors revealed that regulatory T cell frequency in tumor infiltrating lymphocytes was significantly reduced in Malt1SR/SR tumors and even further reduced in Malt1PD/PD tumors (Fig. 4C, D). Indeed, tumor‐infiltrating Treg from Malt1SR/SR and Malt1PD/PD mice also showed significantly decreased expression levels of CD25 on their cell surface, indicating reduced Il2 availability in the microenvironment of these tumors (Fig. 4E) 32, 33. Strikingly, although the Malt1SR/SR mice only had minor decrease in Treg frequency compared to the Malt1PD/PD mice, there were equivalent frequencies of tumor infiltrating CD8+ T cells in both Malt1SR/SR and Malt1PD/PD tumors (Fig. 4F). There was also a reduced number of NK cells within the tumor of these animals, which might also be linked to the Il2 deficiency (Fig. 4F). Interestingly, increased infiltration of CD4+ effector T cells was only observed in Malt1PD/PD tumors (Fig. 4F). The increased anti‐tumor immune response was further characterized by a more activated effector phenotype of the infiltrating CD8+ T cells, as measured by the expression of the activation markers CD44 and Klrg1 (Fig. 4G).
Taken together, these data show that a minor reduction in Treg numbers within the tumor microenvironment mediated by the self‐cleavage deficient Malt1 protein is sufficient to drive an improved anti‐tumor response.
Discussion
Our recent work and that of others have established that Malt1 cleaves itself in response to TCR activation 13, 14. Here, we provide several lines of evidence for an essential role of MALT1 auto‐proteolysis in vivo in regulating NF‐κB target genes and most significantly regulatory T cell numbers. Blocking Malt1 self‐cleavage reduced NF‐κB target genes induction upon TCR stimulation, albeit to a lesser extent than observed in protease‐dead Malt1 T cells. Consequently, TCR stimulation induced proliferative response of CD4+ T cells was reduced in Malt1 self‐cleavage resistant animals, but not to the same extent than in Malt1PD/PD cells. In Jurkat cells, however, expression of self‐cleavage resistant Malt1 completely abrogated NF‐κB mediated transcription in response to TCR stimulation 13. Interestingly, we show that expression of Malt1 p76 by itself induced transcription of NF‐κB target genes. Thus, in vivo data suggest that the formation of an active C‐terminal p76 fragment of Malt1, dissociating from Bcl10 and oligomerizing to promote the transcriptional activity of NF‐κB, amplifies TCR signaling and NF‐κB activity.
In Treg cell precursors, TCR stimulation triggers the activation of NF‐κB family members, which bind to the Foxp3 promoter and enhancer regions, initiating transcription 34. Interestingly, the selective inactivation of Malt1 proteolytic activity, disrupted Foxp3 induction, but to a slightly lesser extent than complete Malt1‐inactivation 11. Here we show that self‐cleavage of Malt1 has an important function in TCR‐mediated NF‐κB activation and subsequent Foxp3 induction. Malt1 self‐cleavage resistant animals had an intermediate decrease in Treg frequency when compared to the proteolytically inactive Malt1 mice, suggesting that the level of Malt1 activity together with its ability to undergo self‐cleavage all aid to translate the TCR signaling strength to an output in terms of Foxp3 expression.
In the current study we further validated that disruption of TCR‐signaling via Malt1‐impairment in Malt1PD/PD animals resulted in severe autoimmune disease. Previous work indicated that this autoimmunity was at least in part due to the thymic deficiency in generating thymic Treg cells 11, 12. Here we show, that Malt1SR/SR mice, however, were protected from autoimmunity. Indeed, Malt1SR/SR animals only had a 40% reduction in Treg numbers, in comparison to 80% in Malt1PD/PD mice. Therefore, this reduced loss of Tregs was sufficient to prevent development of autoimmune disease and also indicates that a Treg threshold exists below which auto‐immunity results.
The most significant and novel finding was the observed defective Treg homeostasis in both Malt1PD/PD and Malt1SR/SR animals. In line with previous reports, Malt1 protease activity and self‐cleavage contributed significantly to NF‐κB‐mediated Il2 transcription 9, 11, 13. Interestingly, mice deficient in Il2, Il2ra or Il2rb have severely reduced regulatory T cell numbers and develop lethal autoimmune disease, which was also true for mice with Treg‐specific deletion of Il2ra or Il2rb 18, 19, 20, 21, 29. In this report, we show that Il2 deficiency in Malt1SR/SR and Malt1PD/PD Tregs also reduced levels of phosphorylated Stat5, the major downstream regulator of Il2 signaling. However, Tregs were still Il2 responsive and administration of anti‐Il2/Il2 complex did restore Treg numbers suggesting that the Tregs are not defective and only limited in number due to reduced Il2 availability. Similarly, previous studies showed that in c‐Rel deficient mice, Treg deficiency could also be restored by exogenous IL‐2 administration 35. In line with these observations, patients with Malt1 loss‐of‐function mutations, also show signs of autoimmune disease, IL‐2 deficiency and reduced Treg frequency 3, 36.
This imbalance between the residual partial T cell activation potential on the one hand, and the lack of regulatory T cells on the other hand, led us to ask which effect would dominate in a setting of an anti‐cancer immune response. Here, the observed Treg‐deficiency was more important as shown by reduced growth of MC38 tumors in Malt1PD/PD and Malt1SR/SR animals. Even though Malt1SR/SR animals had a more moderate reduction of Tregs than Malt1PD/PD mice, this was overcome by the increase in TCR responsiveness in these animals. Therefore, our data indicate that affecting the balance between Treg and effector cells, by disruption of Il2 driven Treg homeostasis, might be an effective strategy to augment anti‐tumor immunity without necessarily triggering intolerable systemic auto‐immunity. Indeed, clinical studies in cohorts of patients affected by multiple distinct neoplasms, also showed that increased tumor Treg cells were associated with a high rate of mortality 37, 38, 39. Therefore, strategies to impair Tregs, such as MALT1 inhibition, may be suitable to improve anti‐tumor responses seen with other immunotherapies such as checkpoint blockade.
Materials and methods
Immunoblot analysis and plasmids
Cells were lysed in ice‐cold HEPES‐NaCl lysis buffer (50 mM HEPES pH 7.4, 150 mM NaCl, 1% Triton‐X‐100) complemented with protease inhibitor cocktail (Sigma). Cell lysates were boiled with reducing SDS sample buffer, size separated on SDS‐polyacrylamide gels (Criterion XT Bis‐Tris gel 4–15%) and immunoblotted according to standard protocols. Primary antibodies specific for the Flag epitope (M2) were from Sigma‐Aldrich, MCPIP1 (MAB7875) from R&D systems, A20 (A‐12, sc‐166692) and CYLD (E10, sc‐74435) from Santa Cruz Biotechnology, and P‐IκB‐α (Ser32/36, 5A5) and P‐ERK (Thr202/Tyr204, #9101) from Cell Signaling Technology. Anti‐MALT1‐C and anti‐p76 neo were reported previously (Baens 2006 Cancer research, Baens 2014 Plos one).
Constructs for expressing proteins in eukaryotic cells were made in pcDNA3.1 (Clonetech Laboratories) encoding an N‐terminal Flag‐ epitope, with or without the myristoylation/palmitoylation (mp) motif of Lck (MGCVCSSNPEDD) inserted in front of the Flag epitope (pcD‐mp‐F‐Malt1) to direct membrane association of Malt1. For expression of native p76, its coding sequence was cloned in frame with HA‐tagged ubiquitin at the N‐terminus in pcDNA3.1.
Mice
The KU Leuven animal ethics committee approved all experiments. Malt1SR and Malt1PD mice were generated by Cyagen Biosciences using homologous recombination or Crispr/Cas9 technology respectively with C57BL/6 ES cells. Mice were housed in a specific pathogen‐free facility according to the FELASA recommendations.
Cell culture
HEK293T cells deficient for MALT1 and BCL10 (293T‐BM) were cultured in DMEM‐F12 (Life Technologies) supplemented with 10% fetal calf serum (FCS) at 37°C in 5% CO2. Jurkat T cells were grown in RPMI medium 1640 (Life technologies) supplemented with antibiotics and 10% FCS. For electroporation, Jurkat cells were washed twice with serum free medium and one million cells were electroporated with 5 μg DNA of full‐length Malt1 or Malt1 p76 expression plasmids or control DNA, using a BioRad Gene‐Pulser Xcell. Jurkat clones were incubated for 18 h prior to luciferase measurements. As positive control, cells were stimulated with 75 ng/ml PMA – 150 ng/mL ionomycin for 4 h.
Primary mouse naïve CD4+ T cells or total CD4+ T cells were isolated using negative selection kits (Thermo Fisher Scientific). Cells were cultured in RPMI medium, supplemented with 2 mM L‐Glutamine, 10 mM HEPES, antibiotics and 10% FCS. Cells were labeled with 1 μM CFSE (Thermo Fisher Scientific) and stimulated using mouse T‐activator CD3/CD28 dynabeads (Thermo Fisher Scientific).
Quantitative RT‐PCR and RNA sequencing
RNA isolation (NucleoSpin RNA Plus; Macherey‐Nagel) and cDNA synthesis (GoScript Reverse Transcription System; Promega) were performed using standard protocols. Quantitative RT‐PCR was performed with the LightCycler 480 SYBR Green I master mix (Roche Diagnostics) and analyzed using the 2(−ΔΔCt) method with Hprt1 as a reference control. For RNA sequencing, the libraries were prepared according to the standard Illumina TruSeq RNA sample preparation protocol (Illumina). The single‐end RNA‐ sequencing data was first cleaned (i.e. removal of adapters and low‐quality parts) with the fastq‐mcf software after which a quality control was performed with FastQC. The reads were then mapped to the Mus Musculus (mm10) genome with Tophat2. To identify the gene expression HTSeq‐count was used to count the number of reads per gene. These read count numbers were then normalized to the sample size. Differential gene expression analysis was performed with the R‐package DESeq2. Gene‐set for NF‐kB targets (http://www.bu.edu/nf-kb/gene-resources/targetgenes) were used for global analysis of NF‐kB target gene induction.
Malt1 bioluminescent protease assay
A bioluminescent Malt1 protease assay was developed using a circularly permuted form of firefly luciferase (Fan et al, Curr Chem Genomics 2008 2:16‐28). Malt1‐mediated cleavage of the RelB recognition sequence (LVSR) cloned in the linker region joining the native enzyme termini efficiently activates luciferase activity. HEK‐293T cells deficient for MALT1 (293T‐M) were generated using TALENs that target a BfaI site (ttctttctgttgctttcAGTTGCCTAGACCTGgagcagtgttctcttaa) in the 5′ end of exon2 of MALT1 (Cellectis). Successive disruption of BCL10 expression (293T‐BM) was obtained via genome editing with CRISPR/Cas9 technology (Bcl10 CRISPR/Cas9 KO Plasmid, sc‐400483) and confirmed via immunoblotting (Suppl 293T‐BM). 293T‐BM cells (5 × 105) were transfected with 1 μg DNA containing 100 ng Malt1‐protease luciferase reporter, 20 ng β‐galactosidase expression vector (pEL1‐β‐gal) and 50 ng of the expression constructs to test using GeneJuice Transfection Reagent (Millipore). Twenty‐four hours after transfection, cells were harvested and lysed in 30 μL Passive lysis buffer (Promega). Cell lysates (5 μL) were assayed for luciferase and β‐galactosidase activities using a FLUOstar Galaxy Plate Reader (BMG Labtechnologies). Fold Malt1 protease activation was calculated by dividing the luciferase activity, normalized to β‐galactosidase activity for each sample, by that observed in the control containing only empty expression vector.
Flow cytometry analysis
Single cell suspension was prepared from thymus, spleen and lymph nodes. Splenocytes were lysed with red blood cell lysis buffer prior to staining. For flow cytometry analysis cells were incubated with a fixable viability dye (Thermo Fisher Scientific) and stained with the following antibodies (Thermo Fisher Scientific, eBioscience): CD4 (clone GK1.5), CD8 (53‐6.7), CD25 (PC61.5), CD44 (IM‐7), CD62L (MEL‐14), Klrg1 (2F1), CD19 (eBio1D3), CD21/35 (eBio4E3), CD23 (B3B4), Nkp46 (29A1.4), CD45 (30‐F11).
For intracellular stainings cells were fixed with Transcription Factor staining Buffer set (Thermo Fisher Scientific), followed by staining with Foxp3 (FJK‐16) and/or Ki67 (SolA15).
For intracellular staining of cytokines, splenocytes were incubated for 4h with cell stimulation cocktail (Thermo Fisher Scientific), followed by cell fixation (IC fix buffer; Thermo Fisher Scientific). For intracellular staining the following antibodies were used: Ifng (clone XMG1.2), Il‐2 (JES6‐5H4).
For serum cytokine measurements, the BD Cytometric Bead Array (CBA) was used following the instruction manual.
For co‐staining of Foxp3 and p‐Stat5 splenocytes were cultured for 4h in the presence of anti‐CD3/CD28, followed by fixation with the transcription factor phospho buffer set (BD Bioscience). Subsequently, cells were permeabilized with perm Buffer III (BD Bioscience) and stained for CD4, Foxp3 and p‐Stat5 (SRBCZX).
Cells and CBA were analyzed on a FACS Verse flow cytometer (BD Biosciences). Data were analyzed with FlowJo software (Tree Star). Flow cytometry data were analyzed according to the guidelines for the use of flow cytometry and cell sorting in immunological studies 40.
Foxp3 induction
For in vitro Foxp3 induction experiments purified peripheral naïve CD4+ T cells were stimulated with indicated amounts of plate‐bound anti‐CD3 (clone 17A2, eBioscience) in the presence of TGF‐β (5 ng/mL, PeproTech) and mouse IL‐2 (100 U/mL, PeproTech).
Treatment with anti‐Il‐2–Il‐2 mAb complexes
Mice were injected i.p. at day 0, 1 and 2 with anti–IL‐2–IL‐2 immune complexes containing 1 μg recombinant murine IL‐2 (Thermo Fisher Scientific) and 5 μg anti–IL‐2 antibody (JES6‐1A12 clone; Thermo Fisher Scientific) diluted in PBS (Webster et al., JEM 206 (4): 751–760). Mice were sacrificed at day 5 and splenocytes were analyzed by flow cytometry.
MC38 cancer growth
Malt1+/+, Malt1SR/SR and Malt1PD/PD C57/Bl6 mice were subcutaneously injected with 1*106 MC38 cells. Mice were monitored daily for disease and tumors volume was determined by multiple, independent and double‐blinded caliper measurements. Tumors were weight at takedown and single cell suspension for flow cytometric analysis was prepared by using the mouse Tumor Dissociation Kit (Miltenyi Biotec).
Statistical analysis
Sample means were compared using the Student's t test using GraphPad Prism software. Sample means were considered significantly different at p < 0.05, with p < 0.05 (*), p < 0.01 (**) and p < 0.001 (***).
Conflict of interest
Authors declare no commercial or financial conflict of interest.
Abbreviations
- Malt1
Mucosa‐associated lymphoid tissue
- NF‐κB
Nuclear factor κB
- TCR
T cell receptor
- Treg
regulatory T cells
Supporting information
Supporting Information
Peer review correspondence
Acknowledgements
This work was funded by KU Leuven grant GOA/11/010. R.S., D.V. and S.B. are Aspirants of FWO‐Vlaanderen. T.B., R.S., C.e.d.B., Y.L., D.V. and S.B. performed experiments. T.B. and S.B analyzed all the data and Y.L., C.e.d.B., J.C., and P.M. provided conceptual and intellectual input. T.B., and S.B. conceived the study and wrote the manuscript.
Contributor Information
Mathijs Baens, Email: thijs.baens@cistim.be.
Simon Bornschein, Email: simon.bornschein@kuleuven.vib.be.
References
- 1. Turvey, S. E. , Durandy, A. , Fischer, A. , Fung, S. Y. , Geha, R. S. , Gewies, A. , Giese, T. et al., The CARD11‐BCL10‐MALT1 (CBM) signalosome complex: stepping into the limelight of human primary immunodeficiency. J. Allergy Clin. Immunol. 2014. 134: 276–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Thome, M. , CARMA1, BCL‐10 and MALT1 in lymphocyte development and activation. Nat. Rev. Immunol. 2004. 4: 348–359. [DOI] [PubMed] [Google Scholar]
- 3. McKinnon, M. L. , Rozmus, J. , Fung, S.‐Y. , Hirschfeld, A. F. , Del Bel, K. L. , Thomas, L. , Marr, N. et al., Combined immunodeficiency associated with homozygous MALT1 mutations. J. Allergy Clin. Immunol. 2014. 133: 1458–1462.e7. [DOI] [PubMed] [Google Scholar]
- 4. Jabara, H. H. , Ohsumi, T. , Chou, J. , Massaad, M. J. , Benson, H. , Megarbane, A. , Chouery, E. et al., A homozygous mucosa‐associated lymphoid tissue 1 (MALT1) mutation in a family with combined immunodeficiency. J. Allergy Clin. Immunol. 2013. 132: 151–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Li, Q. , Verma, I. M. , NF‐kappaB regulation in the immune system. Nat. Rev. Immunol. 2002. 2: 725–34. [DOI] [PubMed] [Google Scholar]
- 6. Thome, M. , Charton, J. E. , Pelzer, C. , Hailfinger, S. , Antigen receptor signaling to NF‐kappaB via CARMA1, BCL10, and MALT1. Cold Spring Harb. Perspect. Biol. 2010. 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ruland, J. , Duncan, G. S. , Wakeham, A. , Mak, T. W. , differential requirement for Malt1 in T and B cell antigen receptor signaling. Immunity. 2003. 19: 749–758. [DOI] [PubMed] [Google Scholar]
- 8. Ruefli‐Brasse, A. A. , French, D. M. , Dixit, V. M. , Regulation of NF‐κB‐dependent lymphocyte activation and development by paracaspase. Science. 2003. 302: 1581–1584. [DOI] [PubMed] [Google Scholar]
- 9. Coornaert, B. , Baens, M. , Heyninck, K. , Bekaert, T. , Haegman, M. , Staal, J. , Sun, L. et al., T cell antigen receptor stimulation induces MALT1 paracaspase–mediated cleavage of the NF‐κB inhibitor A20. Nat. Immunol. 2008. 9: 263–271. [DOI] [PubMed] [Google Scholar]
- 10. Rebeaud, F. , Hailfinger, S. , Posevitz‐Fejfar, A. , Tapernoux, M. , Moser, R. , Rueda, D. , Gaide, O. et al., The proteolytic activity of the paracaspase MALT1 is key in T cell activation. Nat. Immunol. 2008. 9: 272–281. [DOI] [PubMed] [Google Scholar]
- 11. Jaworski, M. , Marsland, B. J. , Gehrig, J. , Held, W. , Favre, S. , Luther, S. A. , Perroud, M. et al., Malt1 protease inactivation efficiently dampens immune responses but causes spontaneous autoimmunity. EMBO J. 2014. 33: 2765–2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gewies, A. , Gorka, O. , Bergmann, H. , Pechloff, K. , Petermann, F. , Jeltsch, K. M. , Rudelius, M. et al., Uncoupling Malt1 threshold function from paracaspase activity results in destructive autoimmune inflammation. Cell Rep. 2014. 9: 1292–1305. [DOI] [PubMed] [Google Scholar]
- 13. Baens, M. , Bonsignore, L. , Somers, R. , Vanderheydt, C. , Weeks, S. D. , Gunnarsson, J. , Nilsson, E. et al., MALT1 auto‐proteolysis is essential for NF‐κB‐dependent gene transcription in activated lymphocytes cebecauer M, ed. PLoS One. 2014. 9: e103774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ginster, S. , Bardet, M. , Unterreiner, A. , Malinverni, C. , Renner, F. , Lam, S. , Freuler, F. et al., Two antagonistic MALT1 auto‐cleavage mechanisms reveal a role for TRAF6 to unleash MALT1 activation. PLoS One. 2017. 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hori, S. ; Nomura, T. ; Sakaguchi, S. , Control of regulatory T Cell development by the transcription factor Foxp3. Science. 2003. 299: 1057–1061. [PubMed] [Google Scholar]
- 16. Amado, I. F. , Berges, J. , Luther, R. J. , Mailhé, M.‐P. , Garcia, S. , Bandeira, A. , Weaver, C. et al., IL‐2 coordinates IL‐2–producing and regulatory T cell interplay. J. Exp. Med. 2013. 210: 2707–2720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Liston, A. , Gray, D. H. D. , Homeostatic control of regulatory T cell diversity. Nat. Rev. Immunol. 2014. 14: 154–65. [DOI] [PubMed] [Google Scholar]
- 18. Schorle, H. , Holtschke, T. , Hünig, T. , Schimpl, A. , Horak, I. , Development and function of T cells in mice rendered interleukin‐2 deficient by gene targeting. Nature. 1991. 352: 621–4. [DOI] [PubMed] [Google Scholar]
- 19. Wolf, M. , Schimpl, A. , Hünig, T. , Control of T cell hyperactivation in IL‐2‐deficient mice by CD4+CD25‐ and CD4+CD25+ T cells: evidence for two distinct regulatory mechanisms. Eur. J. Immunol. 2001. 31: 1637–1645. [DOI] [PubMed] [Google Scholar]
- 20. Willerford, D. M. , Chen, J. , Ferry, J. A. , Davidson, L. , Ma, A. , Alt, F. W. , Interleukin‐2 receptor α chain regulates the size and content of the peripheral lymphoid compartment. Immunity. 1995. 3: 521–530. [DOI] [PubMed] [Google Scholar]
- 21. Suzuki, H. , Kündig, T. M. , Furlonger, C. , a, W. , Timms, E. , Matsuyama, T. , Schmits, R. et al., Deregulated T cell activation and autoimmunity in mice lacking interleukin‐2 receptor beta. Science 1995. 268: 1472–1476. [DOI] [PubMed] [Google Scholar]
- 22. Nishikawa, H. , Sakaguchi, S. , Regulatory T cells in cancer immunotherapy. Curr. Opin. Immunol. 2014. 27: 1–7. [DOI] [PubMed] [Google Scholar]
- 23. Brüstle, A. , Brenner, D. , Knobbe‐Thomsen, C. B. , Cox, M. , Lang, P. A. , Lang, K. S. , Mak, T. W. , MALT1 is an intrinsic regulator of regulatory T cells. Cell Death Differ. 2017. 24: 1214–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Siggs, O. M. , Miosge, L. A. , Yates, A. L. , Kucharska, E. M. , Sheahan, D. , Brdicka, T. , Weiss, A. et al., Opposing functions of the T Cell receptor kinase ZAP‐70 in immunity and tolerance differentially titrate in response to nucleotide substitutions. Immunity. 2007. 27:912–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pierson, W. , Cauwe, B. , Policheni, A. , Schlenner, S. M. , Franckaert, D. , Berges, J. , Humblet‐Baron, S. et al., Antiapoptotic Mcl‐1 is critical for the survival and niche‐filling capacity of Foxp3+ regulatory T cells. Nat. Immunol. 2013. 14:959–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rudra, D. , DeRoos, P. , Chaudhry, A. , Niec, R. E. , Arvey, A. , Samstein, R. M. , Leslie, C. et al., Transcription factor Foxp3 and its protein partners form a complex regulatory network. Nat. Immunol. 2012. 13: 1010–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Arvey, A. , van der Veeken, J. , Samstein, R. M. , Feng, Y. , Stamatoyannopoulos, J. A. , Rudensky, A. Y. , Inflammation‐induced repression of chromatin bound by the transcription factor Foxp3 in regulatory T cells. Nat. Immunol. 2014. 15: 580–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Levine, A. G. , Arvey, A. , Jin, W. , Rudensky, A. Y. , Continuous requirement for the TCR in regulatory T cell function. Nat. Immunol. 2014. 15: 1070–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chinen, T. , Kannan, A. K. , Levine, A. G. , Fan, X. , Klein, U. , Zheng, Y. , Gasteiger, G. et al., An essential role for the IL‐2 receptor in Treg cell function. Nat. Immunol. 2016. 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Almeida, A. R. M. , Zaragoza, B. , Freitas, A. A. , Indexation as a novel mechanism of lymphocyte homeostasis: The Number of CD4+CD25+ regulatory T Cells is indexed to the number of IL‐2‐producing Cells. J. Immunol. 2006. 177: 192–200. [DOI] [PubMed] [Google Scholar]
- 31. Boyman, O. , Kovar, M. , Rubinstein, M. P. , Surh, C. D. , Sprent, J. , Selective stimulation of T cell subsets with antibody‐cytokine immune complexes. Science. 2006. 311: 1924–1927. [DOI] [PubMed] [Google Scholar]
- 32. Malek, T. R. , Castro, I. , Interleukin‐2 receptor signaling: at the interface between tolerance and immunity. Immunity 2010. 33: 153–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Humblet‐Baron, S. , Franckaert, D. , Dooley, J. , Bornschein, S. , Cauwe, B. , Schönefeldt, S. , Bossuyt, X. et al., IL‐2 consumption by highly activated CD8 T cells induces regulatory T‐cell dysfunction in patients with hemophagocytic lymphohistiocytosis. J. Allergy Clin. Immunol. 2016. 138. [DOI] [PubMed] [Google Scholar]
- 34. Lal, G. , Bromberg, J. S. , Epigenetic mechanisms of regulation of Foxp3 expression. Blood. 2009. 114: 3727–3735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Luu, M. , Jenike, E. , Vachharajani, N. , Visekruna, A. , Transcription factor c‐Rel is indispensable for generation of thymic but not of peripheral Foxp3+ regulatory T cells. Oncotarget. 2017. 8: 52678–52689. Available at: http://www.oncotarget.com/fulltext/17079%0Ahttp://www.oncotarget.com/abstract/17079%5Cnhttp://www.ncbi.nlm.nih.gov/pubmed/28467968. https://doi.org/10.18632/oncotarget.17079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Punwani, D. , Wang, H. , Chan, A. Y. , Cowan, M. J. , Mallott, J. , Sunderam, U. , Mollenauer, M. et al., Combined immunodeficiency due to MALT1 mutations, treated by hematopoietic cell transplantation. J. Clin. Immunol. 2015. 35: 135–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Curiel, T. J. , Coukos, G. , Zou, L. , Alvarez, X. , Cheng, P. , Mottram, P. , Evdemon‐Hogan, M. et al., Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat. Med. 2004. 10: 942–949. [DOI] [PubMed] [Google Scholar]
- 38. Bates, G. J. , Fox, S. B. , Han, C. , Leek, R. D. , Garcia, J. F. , Harris, A. L. , Banham, A. H. , Quantification of regulatory T cells enables the identification of high‐risk breast cancer patients and those at risk of late relapse. J. Clin. Oncol. 2006. 24: 5373–5380. [DOI] [PubMed] [Google Scholar]
- 39. Shang, B. , Liu, Y. , Jiang, S. , Liu, Y. , Prognostic value of tumor‐infiltrating FoxP3+ regulatory T cells in cancers: a systematic review and meta‐analysis. Sci. Rep. 2015. 5: 15179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cossarizza, A. , Chang, H.‐D. , Radbruch, A. , Akdis, M. , Andrä, I. , Annunziato, F. , Bacher, P. et al., Guidelines for the use of flow cytometry and cell sorting in immunological studies. Eur. J. Immunol. 2017. 47: 1584–1797. Available at: http://www.ncbi.nlm.nih.gov/pubmed/29023707 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Peer review correspondence