

Abstract
Phenylphenalenones, polycyclic aromatic natural products from some monocotyledonous plants, are known as phytoalexins in banana (Musa spp.). In this study, 1H nuclear magnetic resonance (NMR)‐based metabolomics along with liquid chromatography and mass spectrometry were used to explore the chemical responses of the susceptible ‘Williams’ and the resistant ‘Khai Thong Ruang’ Musa varieties to the ascomycete fungus Mycosphaerella fijiensis, the agent of the black leaf Sigatoka disease. Principal component analysis discriminated strongly between infected and non‐infected plant tissue, mainly because of specialized metabolism induced in response to the fungus. Phenylphenalenones are among the major induced compounds, and the resistance level of the plants was correlated with the progress of the disease. However, a virulent strain of M. fijiensis was able to overcome plant resistance by converting phenylphenalenones to sulfate conjugates. Here, we report the first metabolic detoxification of fungitoxic phenylphenalenones to evade the chemical defence of Musa plants.
Keywords: Musa, detoxification, metabolomics, sulfate conjugates
Short abstract
Both the susceptible ‘Williams’ and the resistant ‘Khai Thong Ruang’ varieties of Musa acuminata produced enhanced levels of phenylphenalenones in response to the infection with the ascomycete fungus Mycosphaerella fijiensis, the agent of the Black Leaf Sigatoka Disease.
The resistant Musa variety ‘Khai Thong Ruang’ produced a larger number and higher concentrations of phenylphenalenones than the susceptible variety ‘Williams’.
The virulent M. fijiensis strain Ca10_13 was able to overcome the resistance of the Musa variety ‘Khai Thong Ruang’ by metabolic deactivation of phenylphenalenones to sulfate conjugates.
Introduction
Bananas and plantains constitute the fourth most important staple fruit worldwide with a global production of more than 100 million metric tons in 2012; they rank first among all varieties of fruits produced (FAO 2015). The revenues from trading these fruits internationally represent an important economical income for several tropical and subtropical developing countries (FAO 2014). However, their production is often affected not only by herbivores such as Radopholus similis, which cause the burrowing nematode disease (Chabrier & Queneherve 2003), but also by several pathogens: Ralstonia solanacearum (Race 2) causes the Moko's disease (Sequeira 1998); Fusarium oxysporum f. sp. cubense causes Panama's disease (Ploetz 2006); and the Sigatoka disease complex of banana, involving three related ascomycetous fungi Mycosphaerella musicola, M. eumusae and M. fijiensis. Among these fungi, M. fijiensis causes the most detrimental disease in banana, black leaf streak disease (BLSD) (Arzanlou et al. 2007; Etebu & Young‐Harry 2011).
Cavendish bananas, belonging to the subgroup AAA, are among the most frequently consumed commercial banana crops because of their sweetness and testability and the nutritional value of the fruit. However, they are highly susceptible to BLSD. Although several efforts have been made to breed Musa hybrids resistant to pathogens, the banana fruit for each cultivar type still does not meet consumers' expectations (Heslop‐Harrison 2011; Kovacs et al. 2013; Perrier et al. 2011; Vishnevetsky et al. 2011). Therefore, chemical treatments through the application of fungicides (such as benomyl, propiconazole and azoxystrobin) remain the most effective method to control this disease. As a consequence of poor agricultural management practices, namely, the blanket application of fungicides to avoid reduction in crop yields by fungal infections, banana plants have developed widespread resistance to fungicides. As a result, such reinforced microbial resistance has brought enormous losses in the commercial banana trade (Etebu & Young‐Harry 2011; Martinez‐Bolanos et al. 2012; Ploetz 2000).
Understanding the physiological responses triggered in resistant Musa plants during their interaction with pathogens, especially with M. fijiensis, will allow scientists to search for new strategies that can lead towards the control of pests without damaging the environment. Thus, several biochemical traits in Musa have been recognized to play a role during the pathogenesis. Among these, strengthened physical barriers through lignin biosynthesis, along with induction of pathogenesis‐related proteins, hypersensitivity response and production of phytoalexins, constitute the basis for the broad resistance of Musa plants to pathogens and herbivores (Dhakshinamoorthy et al. 2014; Hölscher et al. 2014; Otálvaro et al. 2007; Torres et al. 2012; Vaganan et al. 2014; Wang et al. 2015). Phenylphenalenones, a group of polycyclic aromatic compounds found in Haemodoraceae, Pontederiaceae, Strelitziaceae and Musaceae plant families (Munde et al. 2013), act as phytoalexins in the Musa genus because they are induced either in the leaves or in the roots of Musa plants that are affected by biotic or abiotic factors. In addition, their strong antimicrobial and nematicidal properties have made them valuable to researchers attempting to explain the chemical basis for the resistance of some cultivars of Musa (Hölscher et al. 2014; Jitsaeng & Schneider 2010; Quiñones et al. 2000).
As some phenylphenalenones have recently been identified at the cellular level in the stomata of Musa leaves (Hölscher et al. 2015), this experimental system has become attractive for exploring the plant–pathogen interaction in more detail. Furthermore, metabolomic analyses that use nuclear magnetic resonance (NMR) and mass spectrometry (MS) have been demonstrated to be powerful tools for analysing host metabolic pathways that are triggered by pathogens (Ali et al. 2009; López‐Gresa et al. 2010; Parker et al. 2009). The research reported in this study aimed to elucidate and explore the metabolic changes behind the Musa–M. fijiensis interaction, which is correlated with host resistance and pathogen virulence. Thus, chemical responses from the susceptible ‘Williams’ (Musa spp. AAA group) (Carlier et al. 2000) and the resistant ‘Khai Thong Ruang’ (‘KTR’) (Musa spp. AAA Ibota group) varieties in the interaction with M. fijiensis, strain E22 ‘sensitive’ and strain Ca10_13 ‘tolerant’ to the fungicide propiconazole, were probed by using 1H NMR‐based metabolomics along with high‐performance liquid chromatography (HPLC)–diode array detector (DAD) analysis.
Materials and Methods
Plant material
In vitro plants of Musa acuminata var. ‘Williams’ (AAA, Cavendish subgroup) were provided by Universidad Católica de Oriente – Rio Negro (Antioquía, Colombia), and in vitro plants of M. acuminata var. ‘KTR’ (AAA, Ibota subgroup) were originally provided by the International Transit Centre (ITC), Bioversity International, Katholieke Universiteit Leuven, Belgium (Leuven, Belgium). Plants were propagated and cultured individually in medium B5Z according to the standard procedures (Banerjee & Delanghe 1985) and maintained at 26 °C under 12 h photoperiod conditions until they reached a height of 10–12 cm (4–5 weeks). Plants were further transferred to 10 cm diameter plant pots containing sterile soil of a mixture of Klasmann‐Ton‐Substrat (KTS), vermiculite 2–3 mm, sand 0.7–1.2 mm and perlite (in a ratio 2:1:1:2) with fertilizer Oscomote Exact 19‐9‐12 (333 mg L−1 soil), and each plant received a dose of Ferty 3 (0.1%; Planta Düngemittel GmbH, Regenstauf, Germany) every week. The plants were maintained in an incubation chamber, Percival Model AR‐95HILX (CLF Plant Climatics, Emersacker, Germany), at a temperature of 26/23 °C (day/night), 70% humidity and 12 h photoperiod for 1 month. Then the plants were transferred to 4 L plant pots under the same soil conditions and kept for 3 months before the infection experiments began.
Fungal organisms
Mycosphaerella fijiensis strains voucher E22 and voucher Ca10_13 were provided by Dr Gert Kema from the Plant Research International (Wageningen, the Netherlands) and maintained in solid potato dextrose agar medium (PDA, emended with 100 μg mL−1 ampicillin), at 25 °C under dark conditions for 15 d. They were further cryo‐preserved according with the International Network for the Improvement of Banana and Plantain (INIBAP) technical guidelines (Carlier et al. 2002) until the inoculum for the infection protocol and bioassays was prepared.
Analytical equipment
One‐dimensional (1D) and two‐dimensional (2D) NMR experiments were carried out using a Bruker Avance 500 NMR spectrometer (Bruker BioSpin, Karlsruhe, Germany), operating frequency 500.13 MHz for 1H and 125.75 MHz for 13C. A triple resonance inverse (TCI) cryoprobe (5 mm) was used to measure spectra at 300 K. Tetramethyl silane (TMS) (0.05%) was used as an internal standard for referencing 1H and 13C NMR spectra unless otherwise indicated. One‐dimensional Nuclear Overhauser Effect Spectroscopy (NOESY) experiments were performed using a Bruker Avance III HD 700 NMR spectrometer (Bruker BioSpin, Karlsruhe, Germany), operating frequency 700 MHz for 1H. A TCI H‐C/N‐D 1.7 mm microcryoprobe was used to measure spectra at 298 K. NMR spectrometers were controlled with bruker topspin 2.1 and 3.2 software (Bruker BioSpin, Karlsruhe, Germany) for the 500 and 700 MHz instruments, respectively. Electrospray ionization mass spectra (ESIMS) and LC–ESIMS were recorded on a Bruker Esquire 3000 ion trap mass spectrometer (Bruker Daltonics, Bremen, Germany). High‐resolution ESIMS was recorded on ultra high performance liquid chromatography (UHPLC) system of the Ultimate 3000 series RSLC (Dionex, Sunnyvale, CA, USA) connected to an LTQ‐Orbitrap XL mass spectrometer (Thermo Fisher Scientific, Bremen, Germany). Analytical and semipreparative HPLC was performed on an Agilent series HP1100 (binary pump G1312A, autosampler G1367A, diode array detector G1315A; Agilent Technologies, Waldbronn, Germany). The ultraviolet (UV) spectra were recorded by the DAD during analytical HPLC. The semipreparative HPLC system was coupled with the fraction collector CHF 122SB ADVANTEC (Dublin, CA, USA).
Metabolomics analysis
Fungal inoculum preparation
The standard protocol reported by Abadie et al. (2008) was used with slight modifications. Briefly, M. fijiensis strain E22 kept in glycerol (15%) at −80 °C was placed in PDA medium and incubated at 25 °C under dark conditions for 20 d. Afterwards, the fungal mycelium was cut off from the agar medium and transferred to Petri dishes with sterile V8 juice agar (containing 300 mL of vegetable juice V8, 3 g CaCO3, 20 g of agar and 700 mL water) and incubated for 15 d under 12 h photoperiod. Four plugs of fungal mycelium were cut off from the Petri dish and placed in a Falcon tube (50 mL) containing 25 mL of sterile water for further shaking in a vortex mixer for 2 min followed by ultrasound for 1 min. An aliquot of 2 mL was transferred to Petri dishes with sporulation V8 juice agar (containing 100 mL of vegetable juice V8, 0.2 g CaCO3, 20 g of agar and 900 mL water, pH adjusted at 6.0) and incubated at 25 °C under continuous cool white light conditions for 15 d. Sterile water (~2 mL × 3) was added to the sporulation culture, and conidia were removed by brushing the colonies' surface with a sterile camel's hair brush. The concentration of conidia was calculated by using a Neubauer chamber and adjusted to a final concentration of 1 × 104 conidia mL−1 with sterile water (containing 0.1% of Tween 20) and used to further inoculate the Musa plants.
Artificial mock‐inoculation of Musa plants
Three‐month‐old M. acuminata plants of the susceptible variety ‘Williams’ and the resistant variety ‘KTR’ were used for the infection experiment. A volume of 2 mL of the fungal inoculum was spread on the left‐half side of the lower surface (abaxial side) of the second youngest unfolded leaf of each Musa plant using a camel's hair brush. The mock‐inoculated plants were kept at room temperature (~20 °C) for 2 h, while the microbial suspension was dried and then returned to the climate chamber under the conditions described in the preceding text. The relative humidity was initially set at 90% and after the first week of incubation was maintained at 80%. A sterile solution of 0.1% Tween 20 (without fungal inoculum) was spread to Musa plants of each variety and used as a control. Four biological replicates of both treated and control Musa plants of each variety were used for the study.
Harvesting plant tissue
The infected Musa plants of each variety were harvested at 35 d post inoculation (dpi) once the plants developed visual BLSD symptoms on the upper side of the leaf. Four sections of each infected plant were collected as follows (Fig. 2): (1) necrotic plant tissue ‘sample A’; (2) asymptomatic tissue from the treated half of the leaf ‘sample B’; (3) healthy tissue from the non‐treated half of the infected leaf ‘sample D’; and (4) non‐treated leaves from the treated plant ‘sample E’. Plants without fungal treatment were used as a control, ‘sample C’. Each specimen was cut off under visual inspection (using a binocular loupe), immediately frozen in liquid nitrogen and stored in a pre‐cooled sterile Eppendorf tube (2 mL lock‐safe Eppendorf tubes) until further lyophilized in a freeze drier for 72 h. Ceramic beads (0.7 mm in diameter) were added to each tube, and the plant material was ground by shaking it for 3 min in a paint shaker (Skandex SO‐10M; Fluid Management Europe, Sassenheim, The Netherlands). The samples were then stored in a desiccator until they were used for analysis (not longer than 48 h).
Figure 2.
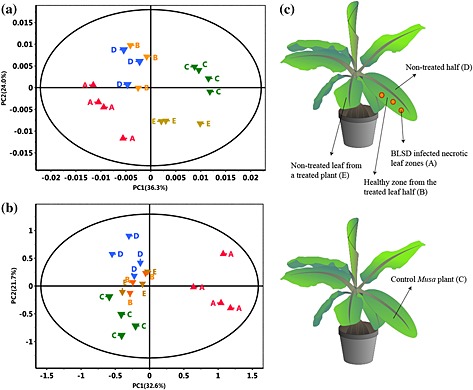
Score plots of PC1 versus PC2 of eight‐component principal component analysis (PCA) model of the region 0.5–8.7 ppm of the 1H NMR spectra (500 MHz) of samples A to E (0.005 ppm buckets). Regions 3.25–3.32 ppm and 4.8–5.0 ppm were excluded; data were normalized to total intensity of the spectrum. (a) ‘Williams’ variety (Pareto scaling was applied with r 2 = 0.903 of PCA model). (b) ‘Khai Thong Ruang’ (‘KTR’) variety (unit variance scaling was applied with r 2 = 0.854 of PCA model). (c) Sketch of the sampling procedure: A, black leaf streak disease (BLSD)‐infected necrotic leaf zones; B, healthy zones from the treated half of a leaf; C, control Musa plant; D, non‐treated half of a treated leaf; and E, non‐treated leaf from a treated plant.
Sample preparation for NMR analysis
A protocol established by Kim et al. (2010) was adapted with some modifications. Briefly, 25 mg of dried and ground plant specimens were transferred to an Eppendorf tube to which was added 0.8 mL of MeOD‐d 4 with 0.2 mL of KH2PO4 buffer in D2O (pH 6.0) containing 0.05% trimethylsilyl propanoic acid (TSP) as internal standard. The solution was vortexed (1 min) and sonicated (15 min) at room temperature followed by a centrifugation step at 20,000 × g for 15 min. The supernatant (600 μL) was directly transferred to a 5 mm NMR tube for further analysis.
NMR data analysis
1H NMR spectra were recorded at 500 MHz using a standard pulse sequence with water suppression (PURGE), with 256 scans with 64k data points and a spectral width of 15 ppm using a 30° flip angle, a 4.43 s acquisition time and a recycle delay of 2.0 s. Fourier transformation was applied to cumulative free induction decay (FID) with exponential line broadening of 0.3 Hz. All spectra were manually phased, baseline‐corrected and calibrated to TSP (δ 0.0). The 1H NMR spectra were reduced to 1586 sequentially integrated regions (‘buckets’) of 0.005 ppm width, from δ 0.5–8.7, and the data were scaled to total intensity using amix version 3.9.11 software (Bruker BioSpin, Karlsruhe, Germany). Regions from δ 3.32 to 3.25 and δ 5.0 to 4.8 were excluded before the bucketing analysis, and the whole dataset was exported as a text file (.txt). Principal component analysis (PCA) of the data was performed with simca‐p version 13.0 software (UMETRICS Company, Umeå, Sweden), and eight PCs were processed to meet 96% of the explained variance. Pareto or unit variance scaling was applied to the data before PCA analysis.
Analysis of inducible phenylphenalenones in Musa during pathogen attack
Phytochemical analysis of infected Musa plants
The crude extract from sample ‘A’ of the variety ‘KTR’ (42.1 mg) and variety ‘Williams’ (34.3 mg) 35 dpi with the fungus was used to isolate the major metabolites induced during the pathogenesis. Briefly, the crude extract was cleaned up by using solid‐phase extraction (SPE, Chromabond C18ec‐octadecyl modified silica, 500 mg absorbent weight, endcapped; Macherey‐Nagel, Düren, Germany) before analysis by HPLC. The methanolic extract after SPE was dried under a stream of nitrogen and redissolved in methanol to a final concentration of 10 mg mL−1. An aliquot of 5 μL of methanolic solution was injected into semipreparative HPLC. Separation was achieved on a Hibar column RP‐18e (Purospher STAR, 250–4.6 mm, 5 μm particle size) (Merck, Darmstadt, Germany). Water with 0.05% trifluoroacetic acid (TFA) (solvent A) and a mixture of methanol:acetonitrile (1:1) with 0.05% trifluoroacetic acid (TFA) (solvent B) were used as a mobile phase. The elution profile was from 0 min (0% B), 60 min (45% B), 90 min (65% B), 110 min (65% B), 115 min (80% B), 125 min (80% B), 130 min (100% B) to 140 min (100% B). The mobile phase flow rate was 1 mL min−1. Column temperature was maintained at 30 °C. A wavelength at 254 nm was used for monitoring the chromatographic traces (HPLC method A). Further purification was carried out on a LiChrospher 100 RP‐18 column (250 × 4 mm, 5 μm particle size) (Merck, Darmstadt, Germany) with water (0.1% TFA) and acetonitrile (solvent B) as a mobile phase. Flow rate of 1 mL min−1 and a linear gradient from 0 min (35% B) to 40 min (100% B) was used.
The crude extract of sample ‘D’ (asymptomatic area of infected plant) of the variety ‘Williams’ (18.2 mg) was processed following the same procedure as described before. Separation of the major compound was achieved by semipreparative HPLC using a LiChrospher 100 RP‐18 column (250 × 4 mm, 5 μm particle size). Water with 0.1% TFA (solvent A) and methanol (solvent B) were used as the mobile phase. The elution gradient was from 0 min (0% B), 12 min (45% B), 17 min (100% B) to 25 min (100% B). Column temperature was kept at 30 °C. The monitored wavelength was 254 nm. One‐dimensional and 2D NMR experiments were carried out for the metabolites isolated.
Infection, sample preparation and disease rating
Musa plants of both varieties (‘Williams’ and ‘KTR’) were raised and treated with the fungus according to the conditions of the infection protocol with the fungus as described in the preceding text with few modifications. In this case, 5 mL of the fungal inoculum was spread on the whole (abaxial) lower surface of the second youngest unfolded leaf of each Musa plant using a camel's hair brush. Three areas of 4 × 4 cm2 were randomly traced with a permanent marker (Multimark 1523 Permanent, Faber‐Castell, Stein, Germany) on the upper surface of the infected leaf for determining the severity of the fungal pathogen over time. The average number and area of the symptoms were evaluated on the delimited area at 25 and 50 dpi with the fungus in order to determine the progress of the disease. Conidia of M. fijiensis strains E22 and Ca10_13, differing in their tolerance to the fungicide propiconazole, were chosen as infective propagules for both Musa varieties in separate experiments. The infected plant tissue (sample A, Fig. 2) was collected and processed as described in the preceding text. The experimental design consisted of six biological replicates of each plant variety per time assessed. Three plants without fungal inoculation were used as control (5 mL of water with 0.1% Tween 20).
Quantification by HPLC‐DAD analysis
9‐Phenylphenalenone was synthesized according to the protocol reported by Otálvaro et al. (Otálvaro et al. 2004), extensively purified by chromatographic techniques (column chromatography, preparative thin‐layer chromatography, semipreparative HPLC) up to a purity >98% (UV 254 and 280 nm) and used as internal standard for quantification of phenylphenalenones. A total of 25 mg of the infected dried and ground tissue was transferred into 0.8 mL methanol with 0.2 mL phosphate buffer and 0.1 mL of a 9‐phenylphenalenone solution (final concentration: 0.05 mg mL−1) for extraction and cleaning procedures as described in the preceding text, except that an SPE cartridge of 200 mg was used instead of a 500 mg SPE cartridge. The final extract was resuspended in 1 mL of a mixture of methanol:acetonitrile (1:1) and passed through a filter (0.45 μm); the final solution (5 μL) was injected into the HPLC using method A. The average of the peak integral value of three injections from each sample was used for quantification. Calibration curves of the phenylphenalenones and structural analogues (compounds 1–5, 7, 9–13 and 15 isolated and in‐house synthetic standards, purity > 98% checked by LC–MS, UV 254–280 nm) were generated using six standard solutions of each compound in the range of 0.1–1.5 mm (each solution containing the internal standard at a concentration of 0.05 mg mL−1). An aliquot of 5 μL of each standard solution was injected in triplicate into the HPLC using method A. The average area of the peak integral value of each concentration was calculated from the triplicate data, and a linear regression equation was obtained by plotting the peak areas (y) versus the injected amounts (x) of each standard compounds. The linearity for each compound was evaluated by the correlation coefficient (r 2 ≥ 0.9967). The lower limit of quantification (LOQ) was 0.013 nmol per injection (based on the signal/noise ratio of approximately 10:1 extracted from the HPLC trace at 254 nm). Compounds 6, 8 and 14 (Table 1) were quantified based on the relative response factor of hydroxyanigorufone (5), because the UV absorption of this compound at 254 nm was very similar to compounds 6, 8 and 14. The relative response factor of hydroxyanigorufone (5) was 0.5, as calculated relative to the internal standard, 9‐phenylphenalenone.
Table 1.
Phenylphenalenones isolated by HPLC and identified by 1D and 2D NMR spectroscopy and HRMS from ‘KTR’ leaves after infection with M. fijiensis strain E22
| No. | R t (min) | Compound | Chemical structure | Occurrence in Musa | |
|---|---|---|---|---|---|
| ‘Williams’ | ‘KTR’ | ||||
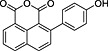
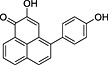
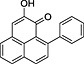
| 1 | 74.77 | 2‐(4′‐Hydroxyphenyl)‐1,8‐naphthalic anhydride |
|
+ | + |
| 2 | 82.12 | trans‐2,3‐Dihydro‐2,3‐dihydroxy‐9‐phenylphenalenone |
|
+ | + |
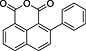
| 3 | 85.30 | 2‐Phenyl‐1,8‐naphthalic anhydride |
|
+ | + |
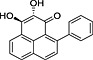
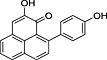
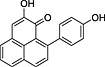
| 4 | 87.81 | 2‐Hydroxy‐4‐(4′‐hydroxyphenyl)‐1H‐phenalen‐1‐one (irenolone) |
|
+ | + |
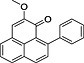
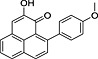
| 5 | 89.35 | 2‐Hydroxy‐9‐(4′‐hydroxyphenyl)‐1H‐phenalen‐1‐one (hydroxyanigorufone) |
|
+ | + |
| 6 | 89.99 | 2‐Methoxy‐9‐(4′‐hydroxyphenyl)‐1H‐phenalen‐1‐one |
|
− | + |
| 7 | 101.62 | Methoxyanigorufone |
|
− | + |
| 8 | 102.92 | Dihydroxyanigorootin |
|
+ | + |
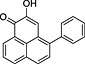
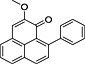
| 9 | 104.20 | Anigorufone |
|
+ | + |
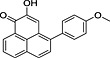
| 10 | 105.89 | 2‐Hydroxy‐9‐(4′‐methoxyphenyl)‐1H‐phenalen‐1‐one (4′‐O‐methylanigorufone) |
|
− | + |
| 11 | 107.55 | Isoanigorufone |
|
− | + |
| 12 | 108.84 | 2‐Hydroxy‐4‐(4′‐methoxyphenyl)‐1H‐phenalen‐1‐one (4′‐O‐methylirenolone) |
|
+ | + |
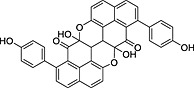
| 13 | 118.98 | 3,3′‐bis‐Hydroxyanigorufone |
|
+ | + |
| 14 | 120.64 | Hydroxyanigorootin |
|
− | + |
| 15 | 121.94 | Anigorootin |
|
+ | + |

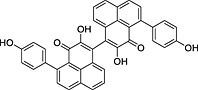
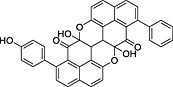
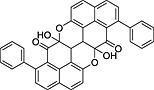
Occurrence of the metabolites is represented by (+) or absence/not detectable (−).
HPLC, high‐performance liquid chromatography; KTR, Khai Thong Ruang; NMR, nuclear magnetic resonance; 1D, one‐dimensional; 2D, two‐dimensional.
Antifungal bioassay
A microtiter well method developed by Peláez et al. (2006) was used with some modifications (Hidalgo et al. 2009). Briefly, M. fijiensis strains (vouchers E22 and Ca10_13) grown for 15–17 d at 26 °C ± 1 in PDA (Fluka Analytical, Steinheim, Germany) were used for preparing the fungal inoculum. Mycelium of the culture was scraped with a smear loop and sterile water, and the dense suspension obtained was then fragmented in a vortex mixer with glass beads of 4 ± 0.3 mm diameter (Carl Roth GmbH, Karlsruhe, Germany) followed by filtration through four layers of sterile cheesecloth to obtain uniform myceliar fragments. The concentration of the inoculum was counted under a light microscope using a Neubauer haemocytometer (Marienfeld, Lauda‐Königshofen, Germany) and adjusted to 2 × 105 mycelial fragments mL−1 with sterile water. Sterile microtiter plates 96 well‐F (Sarstedt AG & Co, Nümbrecht, Germany) were used for the biological incubation. Each well was filled with 50 μL of Sabouraud broth (Fluka Analytical), 50 μL of the fungal inoculum (described in the preceding text) and 50 μL of the corresponding compound to be assessed. Concentrations ranging from 0.001 to 0.1 mg mL−1 of each compound were prepared in aqueous dimethyl sulfoxide (12% DMSO, BioReagent for molecular biology, ≥99.9%; Sigma‐Aldrich, Steinheim, Germany) and passed through a filter (0.1 μm, hydrophilic polyethersulfone membrane sterile; PALL Life Sciences, Ann Arbor, MI, USA) before being administered to the incubation system. The culture medium (50 μL; Sabouraud), 50 μL of inoculum and 50 μL of aqueous DMSO (12%, sterile) were used as a reference for growth control. Propiconazole (Fluka Analytical, 98.4%) was used as a positive control. The same procedure was applied for the negative control except that the inoculum solution was replaced by sterile water. The microplates were incubated under darkness or a photoperiod for 8 d (logarithmic growth phase of the fungi) at 25 °C in an incubation chamber. For the light‐controlled experiments, the plates were placed at a distance of 1 m and irradiated by two cool white light tubes (Sylvania Luxline Plus T8 36W/840, Havells Sylvania Großschirma, Germany) during 12 h d−1. Both experiments (darkness and photoperiod) were carried out simultaneously. The absorbance for the optical density (OD595 nm) was recorded by a spectrophotometer (TECAN Infinite M200 built‐in with multiple reads per well; TECAN, Crailsheim, Germany) at time 0 and 8 d after incubation in order to measure the mycelia growth. The experimental design consisted of three biological replicates (including three technical replicates). The half maximal inhibitory concentration (IC50) value was calculated by plotting the mycelial growth (data previously normalized to the reference control) against the logarithm of each concentration of the compound assessed.
Phenylphenalenone metabolism
The antimicrobial activity of phenylphenalenones and derivatives depends on the chemical structure of the compound (Hidalgo et al. 2009; Otálvaro et al. 2007). In order to test whether the survival of M. fijiensis to the incubation with certain phenylphenalenones is in part due to the ability to metabolize or degrade them, an experimental approach was set up including several parameters to be analysed during in vitro interaction of the fungal pathogen and the phenylphenalenone. Thus, seven phenylphenalenone‐type compounds covering a range from weak to strong antifungal activity against M. fijiensis were selected for this analysis. The protocol described in the preceding text for the antifungal bioassay was used with some modifications. The fungal inoculum was prepared and adjusted to a final concentration of 3 × 105 mycelial fragments mL−1 in identical conditions as has been described in the preceding text. Sabouraud broth (600 μL) with 600 μL of the fungal inoculum and 60 μL of each concentration of the compound to be assessed was incubated in a sterile Safe‐Lock Eppendorf tube (2.0 mL; Eppendorf, Hamburg, Germany) containing five sterile ceramic beads (0.7 mm in diameter). Three solutions of each compound (0.21, 0.42 and 0.84 mg mL−1) were prepared in dimethyl sulfoxide (DMSO, BioReagent for molecular biology, ≥99.9%; Sigma‐Aldrich) to achieve a final concentration of 0.01, 0.02 and 0.04 mg mL−1 in the incubation system (maximum concentration of DMSO in the system: 4.7%). Sabouraud broth (600 μL) with 600 μL of the fungal inoculum and 60 μL of DMSO (0.1 μm filtered) was used as control growth. Similarly, 600 μL of Sabouraud broth with 600 μL of sterile water and 60 μL of each compound solution (at each concentration) were prepared in order to ensure a comparable analysis with the treated samples. Ten replicates of control growth, reference compounds and treated samples were used for reproducibility purposes and additional analysis. The samples were maintained at 25 °C, shaken at 150 rpm under dark conditions and incubated for 8 d (logarithmic growth phase). Fungal biomass, soluble protein, ergosterol production and quantification of each phenylphenalenone either in the culture medium as well as in fungal mycelium were analysed as follows.
Biomass quantification
After the microbial incubation, the fungal mycelium was completely removed from each tube and transferred to a new sterile Eppendorf tube. The fungal biomass was washed with 500 μL (×3) phosphate‐buffered saline (sterile PBS, pH 7.4, 10% DMSO) and centrifuged at 20,000 × g for 10 min, and the supernatant was poured off and collected for further analysis. A final rinse was followed with 500 μL of sterile water, and the mycelium was immediately frozen in liquid nitrogen and lyophilized in a freeze drier for 48 h. The biomass was left for 24 h in a desiccator before being weighed on a microbalance Mettler Toledo XP26 (0.001 mg readability; Giessen, Germany).
Soluble protein quantification
The protein quantification was carried out by the Bradford method. Briefly, after 8 d of incubation with the fungus, 120 μL of each culture medium (control growth, negative controls and treated samples) was placed in a sterile microtiter 96‐well plate and mixed with 120 μL of Coomassie reagent (Coomassie Protein Assay Reagent; Thermo Scientific, Rockford, IL, USA) and then allowed to react for 15 min followed by reading the absorbance at 595 nm as indicated by the manufacturer. Three technical replicates of each sample were measured, and the average was used for quantification. The protein concentration was calculated from a calibration curve with bovine gamma globulin as standard.
Ergosterol quantification
The fungal biomass obtained either from control as well as material treated with phenylphenalenones (n = 4) was ground in a paint shaker using stainless steel beads (1.4 mm in diameter; Carl Roth, Karlsruhe, Germany), and 0.7 mg was weighed, transferred to a Safe‐Lock Eppendorf tube (2.0 mL) and subjected to saponification with 1.5 mL of methanolic solution of KOH (0.1 m) followed by heating at 70 °C and shaking at 600 rpm for 10 min (Eppendorf Thermomixer Comfort 1.5 mL; Eppendorf, Hamburg, Germany). The reaction was left at room temperature, and then the tubes were centrifuged at 15,000 × g for 10 min and the supernatant transferred to a 4 mL glass vial. Distilled water (1 mL) was added to the methanolic solution along with 1 mL of diethyl ether for liquid–liquid extraction. The ether phase was poured off, and the extraction with diethyl ether was repeated twice. The organic phases were pooled and evaporated under a stream of nitrogen. The raw extract was resuspended in 60 μL of CDCl3. 1H NMR spectra were recorded in a Bruker Avance 700 MHz spectrometer (Bruker Biospin, Karlsruhe, Germany) using a standard 1D NOESY pulse sequence with water suppression, with 512 scans with 32k data points. Other parameters were used as described in the preceding text for metabolomics analysis, and the chemical shifts were referenced to internal TMS (δ 0.0). Ergosterol was quantified using eretic2 software (Bruker Biospin, Karlsruhe, Germany) built‐in in topspin 3.2 (calibration method). Ergosterol (13.35 μ m; purity >98%) in CDCl3 was used as standard for the calibration procedure. The singlet signal at δ 0.63, which integrates for the protons of the methyl group attached to C‐13, was used for quantification. A minimal signal–noise ratio of 80:1 was calculated. A T1 relaxation time of 349.3 ms was determined for the signal at δ 0.63, and 5 × T1 was set up as a repetition time. Five standard solutions of ergosterol in the range 0.01–2.0 mm were prepared and evaluated for the accuracy of the molar quantification method. An area of at least five times the full width at half height (FWHH) of the 1H NMR signal was integrated and considered for the quantification. Absolute error below 2.34% was determined. This value is in agreement with validation methods using NMR spectroscopy (Pauli et al. 2005).
Quantification of metabolized phenylphenalenones
To quantify phenylphenalenones, the culture medium was extracted with ethyl acetate (500 μL × 5) by shaking for 10 min and centrifuged at 10,000 × g for 5 min. The pooled organic phase was evaporated under a stream of nitrogen gas. The extract was resuspended in 50 μL of methanol, and an aliquot of 5 μL of this solution was injected into HPLC. Separation was achieved in a Hibar column RP‐18e (Purospher STAR, 250–4.6 mm, 5 μm particle size) using water (0.1% TFA, solvent A) and methanol (solvent B) as a mobile phase. A linear gradient was used from 0 min (40% B), 20 min (100% B) and 27 min (100% B). The monitor wavelength was set at 254 nm (HPLC method B). The analysis was carried out by triplicate injections of each sample and reference controls. A coefficient correlation r 2 ≥ 0.9997 was determined by plotting the peak area (average data) versus the concentration of each reference compound. The data were normalized by comparing the peak area of the compounds (average data) of the treated sample against the peak area (average data) of the reference compound. The LOQ calculated for the phenylphenalenones was used as described in the preceding text.
To quantify phenylphenalenones in the mycelium, fungal biomass was collected and lyophilized as described in the preceding text. Methanol (1 mL) was added to the fungal mycelium (0.7 mg), shaken at 650 rpm for 15 min at room temperature and centrifuged at 15,000 × g during 10 min at 4 °C. The extraction was repeated twice, and the organic extracts were pooled and evaporated under stream of nitrogen gas. The extract was resuspended in 50 μL of methanol, and an aliquot of 5 μL was injected into the HPLC system using method B. Triplicate analysis was conducted, and the data were normalized to the reference controls as was described in the preceding text.
Analysis of phenylphenalenone metabolites
To investigate the products derived from the metabolism of phenylphenalenones by the fungus M. fijiensis, an up‐scaled incubation system with anigorufone (9) as reference compound was implemented. Thus, the fungal inoculum (3 × 105 mycelial fragments mL−1) of the M. fijiensis strain Ca10_13 was prepared as described in the preceding text. A volume of 5 mL of inoculum was added to 50 mL of Sabouraud medium (sterile, incubation in Erlenmeyer flask volume 100 mL), and 2.38 mL of a solution of 1.5 m of anigorufone (prepared in 20% DMSO and passed through 0.1 μm sterile filter) was added. A parallel experiment without any treatment was used to control fungal growth (2.38 mL of a solution 20% of sterile DMSO was added). The incubation was carried out at 25 °C, shaken at 150 rpm in a rotatory shaker under darkness for 12 d. Afterwards, the fungal biomass together with the culture medium was transferred to sterile Falcon tubes (50 mL) for centrifugation at 18,000 × g during 15 min. The supernatant was poured off to a new sterile Falcon tube (50 mL), and the mycelium was washed twice with 30 mL of PBS (pH 7.4) and rinsed with 30 mL sterile water (10% DMSO). A centrifugation step was used in each wash, and the supernatants were transferred to new sterile Falcon tube (volume 50 mL). Both fungal mycelium and the supernatants were frozen in liquid nitrogen, lyophilized for 72 h and then kept in a desiccator until further analysis. The lyophilized supernatants were resuspended in 5 mL of methanol and injected into LC–MS system using a LiChrospher 100 column RP‐18 (250–4 mm, 5 μm particle size) with water (0.1% formic acid, solvent A) and methanol (0.1% formic acid, solvent B) as a mobile phase. A linear gradient was used from 0 min (40% B), 20 min (100% B) and 30 min (100% B). The fungal biomass [1.3 g dry weight (DW)] contained in a Falcon tube was ground using stainless steel beads (1.4 mm in diameter) and shaken in a paint shaker for 5 min. Methanol (10 mL × 3) was used for extraction by shaking for 10 min followed by centrifugation. The methanolic fraction was passed through a sterile membrane filter (0.45 μm) and evaporated under a stream of nitrogen gas. The crude extract was resuspended in methanol (5 mg mL−1), and 5 μL was injected into LC–MS system using the parameters described in the preceding text.
The methanolic extracts either from mycelium or from culture medium incubated with phenylphenalenones 5, 7 and 9–12 (Table 1) were analysed by UHPLC–ESIMS. Phenylphenalenones were separated using an Acclaim C18 column (150 × 2.1 mm, 2.2 μm; Dionex, Sunnyvale, CA, USA) with a flow rate of 300 μL min−1 in a binary solvent system of water (solvent A) and acetonitrile (solvent B) (hypergrade for LC–MS; Merck, Darmstadt, Germany), both containing 0.1% (v/v) formic acid (eluent additive for LC–MS; Sigma‐Aldrich, Steinheim, Germany). Separation was accomplished using a linear gradient from 0 min (0% B), 15 min (100% B) and 20 min (100% B) and an equilibration time at 0% B for 5 min. ESI source parameters were set to 4 kV for spray voltage and 35 V for transfer capillary voltage at a capillary temperature of 275 °C. The samples were measured in positive and negative ion mode in the mass range of m/z 100 to 2000 using 30 000 m Δm−1 resolving power in the Orbitrap mass analyser. Data were interpreted using xcalibur software (Thermo Fisher Scientific, Waltham, MA, USA).
Statistical analysis
Unless otherwise stated, statistical tests were carried out with sigma plot 12.0 (SYSTAT Software Inc., San Jose, CA, USA) using analysis of variance (anova). Leven's and Shapiro–Wilk tests were applied to determine error variance and normality of the data, respectively. Holm–Sidak post hoc test was used for pairwise or multiple comparison. Datasets that did not fulfil the assumptions for anova were natural log, decimal log, root square or rank transformed before analysis. A non‐parametric Kruskal–Wallis one‐way anova on ranks was applied for variables that did not meet the normality assumption.
Results
Differential responses of the interaction of Musa–M. fijiensis detected by principal component analysis
In order to explore whether the susceptible Musa variety ‘Williams’ and the resistant variety ‘KTR’ respond differently to infection with the phytopathogenic ascomycete fungus M. fijiensis, in vitro plants of the two varieties were treated with conidia of the strain E22 as described in Section on Materials and Methods. The progress of the BLSD development was monitored by inspecting the leaf surface of the two Musa varieties using a binocular loupe. Initially, both the susceptible variety ‘Williams’ and the resistant variety ‘KTR’ developed similar symptoms at the site of treatment (Table S1). When BLSD symptoms of the treated plants, according to the classification by Fouré (1986), reached a dimension similar to stages 2 and 3, the necrotic zones (sample A) and healthy zones (B) of treated half of the leaves were probed (Fig. 2c). Simultaneously, further samples were taken from the non‐treated half of each treated leaf (D), non‐treated leaves of the treated plants (E) and untreated plants (control, C). When 1H NMR spectra of crude extracts of necrotic tissue (A) were compared with spectra of non‐necrotic tissue (D) from the same treated leaf and the control plant (C) (Fig. 1), signals of aromatic metabolites were more pronounced in the extracts of leaf areas where the fungal infection had taken place than in extracts of samples taken from ‘healthy zones’. This was observed both for ‘Williams’ and ‘KTR’. 1H NMR spectra of samples B and E (not shown in Fig. 1) closely resembled those of (C).
Figure 1.
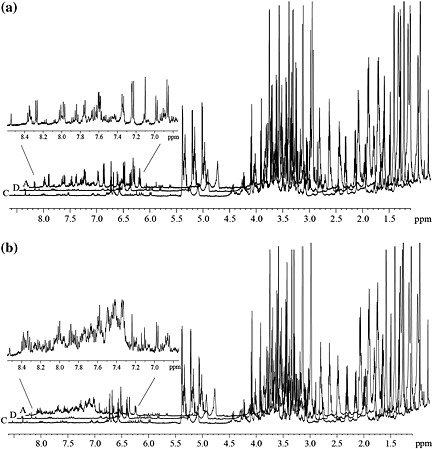
1H NMR spectra (500 MHz, MeOH‐d 4) of crude extracts of Musa leaf tissues from varieties ‘Williams’ (a) and ‘KTR’ (b). Extracts were prepared from necrotic areas of the infected half (Fig. 2, A) and the non‐infected half (D) of a leaf treated with M. fijiensis strain E22, and a leaf of an untreated control plant (C) was taken 35 d after inoculation with fungal conidia. The extensions show the 1H NMR signals of induced phenolics in the necrotic leaf areas (A). Spectral intensities were scaled with the signal of trimethylsilyl propanoic acid (TSP), which was added as an internal standard.
The 1H NMR spectra of all extracts are not much different in the regions of primary metabolites such as carbohydrates and amino acids (0.5–5.0 ppm, Fig. 1). However, the aromatic regions (6.0–9.0 ppm, Fig. 1) of the spectra exhibited not only largely enhanced signals obtained from the necrotic zones of infected leaves (A) but, compared with the spectra from extracts of non‐treated tissues, also a significantly increased number of signals. Thus, qualitative and quantitative gains in metabolic activity were observed in the infected zones of the plant and could be interpreted as a response to the pathogen attack. Therefore, it was thought that identification and quantification of the major metabolites produced while the BLSD ran its course would provide insight into the role and turnover of inducible natural products in this plant.
An NMR‐based metabolomic analysis was carried out to gain information about metabolic changes occurring in Musa plants under attack by the pathogen. The full 1H NMR spectra were used for understanding whether differences other than in the aromatic compounds exist between infected (A), non‐infected (B, D and E) and control (C) Musa plant samples (Fig. 2c). According to the score plots of PCA (Fig. 2a,b), the samples from BLSD‐infected necrotic leaf zones (A, red) were clearly discriminated from those of the control group (C, green) by the first principal component (Fig. 2a,b), mainly owing to aromatic compounds. Interestingly, the other non‐infected plant sections (asymptomatic areas B, non‐treated leaf areas D and E) obtained from plants treated with conidia of M. fijiensis E22 (Fig. 2c) appeared also separated in the PCA (blue, orange and gold) from the group of control Musa plants (green).
With the intention of detecting the signals in the spectra that contribute to the differences between samples, 1D and 2D NMR analysis was carried out along with comparisons with reference compounds and previously reported data (Franzyk et al. 2004; Huang et al. 2004; Kashiwada et al. 1988; Kim et al. 2010; Liang et al. 2006; Lu & Foo 2003; Najbjerg et al. 2011; Ramsay et al. 2014; Shimomura et al. 1987; Strack et al. 1989). This allowed the identification of the metabolites summarized in Table S2 (for chemical structures, see Fig. S1). Taking the information from the PCA, including consideration of the metabolites identified, the corresponding loading plots were constructed (Fig. 3). For both Musa varieties, the red‐orange color in the scale of the loading plots demonstrated that the phenolic metabolites, which are up‐regulated in infected tissue, permit the infected tissue to be discriminated from the non‐infected plant sections (Fig. 3). Interestingly, the level of the 1‐O‐((E)‐p‐coumaroyl)‐β‐d‐glucose was lower in infected tissue than in healthy tissue, perhaps because of consumption in specialized metabolism, which was triggered to produce phenylphenalenones (Table S2) such as irenolone and hydroxyanigorufone, already reported as phytoalexins of Musa plants (Luis et al. 1993, 1994, 1995). Further, among the identified metabolites, dopamine, a well‐known catecholamine in banana fruits (Kanazawa & Sakakibara 2000), and 2‐(3,4‐dihydroxyphenyl)ethyl β‐d‐glucopyranoside (common name dopaol β‐d‐glucoside), which is reported here for a first time as a metabolite from Musa plants, allowed the treated tissue to be discriminated from the untreated Musa tissue (for relevant resonances in the 1H NMR spectrum, see the region between 6.5 and 6.9 ppm in Fig. S2a,b).
Figure 3.
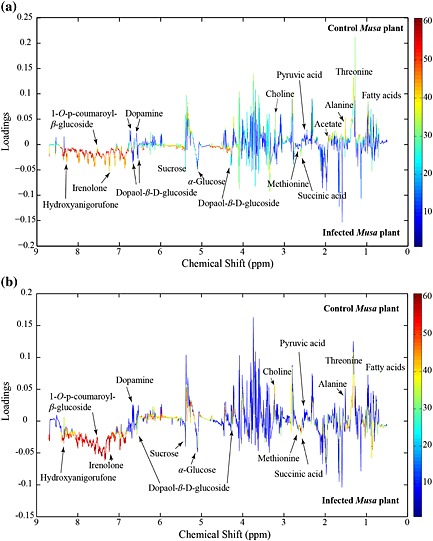
Loading plots for principal component analysis (PCA) model shown in Fig. 2a,b displaying the regions of the metabolites with strong influence (in red and orange) and the metabolites with less influence (light blue and dark blue) during the group discrimination observed in the PCA analysis. (a) ‘Williams’ and (b) ‘Khai Thong Ruang’ (‘KTR’) Musa varieties.
A detailed analysis of 1H NMR spectra of all extracts of ‘Williams’ and ‘KTR’ showed that dopamine was present in infected leaves and in control plants. While control samples contained only traces of dopaol β‐d‐glucoside (Fig. S2c), its level almost equaled that of dopamine in the necrotic tissue of treated plants (Fig. S2a). In non‐infected zones from (B), (D) and (E) samples of both varieties, the level of the glucoside clearly exceeded the level of dopamine, which was detectable only in very low levels (Fig. S2b). This finding confirms the significance of dopaol β‐d‐glucoside in the differences observed by the PCA analysis.
According to the accumulation of dopaol β‐d‐glucoside in the different leaf areas of Musa plants treated with conidia of M. fijiensis but not in the control plant, this compound could be considered a phytoalexin; phytoalexins are produced in those areas of a plant where the pathogen has not been yet spread but which might already have received electrophysiological or chemical signals. Therefore, the antifungal property of dopaol β‐d‐glucoside was assayed against M. fijiensis in an in vitro experiment. However, antimicrobial inhibition was not observed, and the weak activity was comparable with that obtained for dopamine in the same assay (Fig. S3), ruling out the function as a phytoalexin. Thus, the biological function of this compound in the Musa–M. fijiensis pathosystem remains to be investigated, including its potential role in signalling.
In addition, the roots of infected and control plants were evaluated in both Musa varieties in order to check if specialized metabolites were observed in the roots as they were in the leaves. However, no differences between control and treated plants were found (data not shown), supporting the hypothesis that after Musa leaves are treated with the conidia of M. fijiensis, chemical defences are activated mainly in the above‐ground plant parts, especially at the infection site.
Identification and quantification of induced defence metabolites in Musa after infection with M. fijiensis
Phytochemical analysis of infected Musa plants
Among the up‐regulated metabolites found by means of the previously described metabolomic approach, only a small number of phenolic compounds were identified (Table S2, entry 12–19). However, the many overlapping signals in the overcrowded aromatic region in the 1H NMR spectra (Fig. 1) suggested that more up‐regulated phenolics occur in these crude samples, especially in the ‘KTR’ variety, and made the identification of the signals difficult even using 2D NMR experiments. Therefore, a conventional phytochemical analysis of the infected leaves of both Musa cultivars showing BLSD symptoms (35 dpi) was carried out in order to identify the metabolites produced in response to the pathogen attack. HPLC‐guided isolation together with 1D and 2D NMR spectroscopy and high‐resolution mass spectrometry (HRMS) analysis allowed the identification of 15 phenylphenalenone‐type compounds or structural analogues in the resistant variety ‘KTR’ while 10 were found in the susceptible variety ‘Williams’ (Table 1). Among these compounds are irenolone (4) and hydroxyanigorufone (5), which have been identified in the metabolomic approach. Further compounds such as 2‐(4′‐hydroxyphenyl)‐naphthalic anhydride (1), methoxyanigorufone (7) and anigorufone (9) have been reported to be strongly toxic to fungi (Hirai et al. 1994; Kamo et al. 1998b; Lazzaro et al. 2004; Luis et al. 1995; Otálvaro et al. 2002a, 2002b, 2007). The 1H NMR spectra of the compounds from the infected Musa leaves matched those of authentic in‐house standards, and HRMS spectra were fully consistent with the suggested structures. All compounds, except compound 10, are known natural products and have been identified from Musa plants (Kamo et al. 1998a, 1998b; Liu et al. 2014; Luis et al. 1993, 1994, 1995, 1999; Otálvaro et al. 2002a, 2002b). Unfortunately, some minor compounds remained unidentified because of the limited amount isolated and/or of insufficient chemical stability during the isolation procedure (data not shown). Their role in the plant defence therefore remains unclear.
Quantification of phenylphenalenone‐type compounds based on the progress of the disease
In order to study the hypothetical correlation between the virulence of M. fijiensis to Musa plants and the formation of phenylphenalenones, in vitro plants of ‘Williams’ and ‘KTR’ were treated in independent experiments with the fungal strains, E22 and Ca10_13, after which the symptoms were evaluated and the leaf tissues quantitatively analysed for the presence of phenylphenalenones. The two M. fijiensis strains were chosen according to their different tolerance (Ca10_13) or susceptibility (E22) to the fungicide propiconazole, and their conidia were used as infective propagules for exploring differential responses of susceptible and resistant Musa varieties. The virulence of the two strains was evaluated by visually inspecting the number and area of the lesions on the surface of the Musa leaves at different time points after infection (Fig. 4). At early stages of infection, the resistant variety ‘KTR’ exhibited some symptoms characterized by small red specks (~1 mm in diameter) on the lower surface of the leaf in response to each M. fijiensis strain, E22 and Ca10_13; these symptoms were stronger compared with symptoms observed in the susceptible ‘Williams’ variety (Table S1). This was evident by symptom‐free leaves in ‘Williams’ at 8 dpi. The further development of the BLSD symptoms is clearly different for the infection of the two Musa varieties treated with the strain E22 or Ca10_13. While the number and area of the lesions constantly increased in ‘Williams’, these symptoms remained almost unaltered on the leaves of ‘KTR’ after infection with E22 but increased after infection with Ca10_13, reaching severe damage after 50 dpi (Fig. 4, Table S1).
Figure 4.
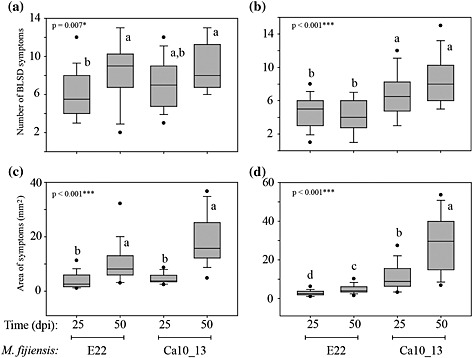
Number and area of black leaf streak disease (BLSD) symptoms determined for Musa ‘Williams’ (a,c) and ‘Khai Thong Ruang’ (‘KTR’) (b,d) varieties during the interaction with M. fijiensis strains E22 and Ca10_13. Letters a–d in the box plots indicate significant differences among the treatments. One‐way analysis of variance (anova), P < 0.001 = ***, P < 0.05 = *.
After evaluating the BLSD symptoms, an HPLC–DAD approach was applied to quantify the inducible phenylphenalenone‐type compounds produced by the two Musa varieties ‘Williams’ and ‘KTR’ during their response to the pathogen. The concentration of phenylphenalenones was determined either in an early stage (25 dpi) or in a late stage (50 dpi) of the disease. The results for the ‘KTR’ variety (Fig. 5a) showed irenolone (4) as the major metabolite, with a concentration exceeding 25 nmol mg−1 DW. Metabolites 2, 4, 7, 9, 11 and 12 were produced in a concentration of >10 nmol mg−1 DW. All other compounds were produced in a concentration range between 1.5 and 9.5 nmol mg−1 DW.
Figure 5.
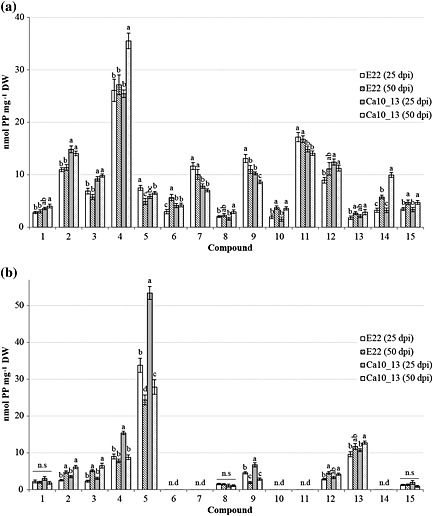
Quantification of the phenylphenalenones [nmol phenylphenalenone (PP) mg−1 dry weight (DW)] identified in ‘Khai Thong Ruang’ (‘KTR’) (a) and ‘Williams’ (b) varieties after infection with M. fijiensis strain E22 at 25 and 50 d post inoculation (dpi) and with M. fijiensis strain Ca10_13 at 25 and 50 dpi. Letters a–d indicate significant differences among treatments within the specified time (dpi) and compound [three‐way analysis of variance (anova), Holm–Sidak post hoc test: P < 0.05; n.s: not significant], n.d: not detectable. For chemical structures of compounds 1–15, see Table 1.
A significant increase in the concentration of compounds 6, 10 and 13–15 (P < 0.001) during the observed period of time from 25 to 50 dpi was found when the ‘KTR’ plants were treated with M. fijiensis strain E22. In contrast, compounds 5 (hydroxyanigorufone) and 9 (anigorufone) were significantly reduced in concentration between 25 and 50 dpi (P < 0.001) (Fig. 5a). When the plants were treated with M. fijiensis strain Ca10_13, mainly compounds 4 and 14 but also 8, 10 and 15 were significantly induced at 50 dpi, while metabolite 9 again was found to be significantly reduced (P < 0.001).
The Musa ‘KTR’ variety reacted differently to the infection by each fungal strain. Overall, a significant increase in the production of compounds 2, 3, 4, 12 and 14 were observed during the response against the strain Ca10_13, while compounds 6, 7, 9 and 11 were more induced by the infection with the strain E22 when compared with Ca10_13 (P < 0.001).
Like ‘KTR’, the variety ‘Williams’ also exhibited one dominant metabolite in addition to a number of medium‐concentrated and minor compounds. However, unlike ‘KTR’, which mainly produced irenolone (4), its positional isomer, hydroxyanigorufone (5) was the major compound in ‘Williams’ (25–52 nmol mg–1 DW) (Fig. 5b). Only two further compounds, 4 and 13, also exceeded the concentration of 10 nmol mg−1 DW, while compounds 1–3, 8, 9, 12 and 15 remained below that level and were produced in the range of 1.3–8.5 nmol mg−1 DW.
According to the statistical analysis, a significant increase in the time‐dependent induction of metabolites 2 and 3 were observed at 50 dpi, independently of the fungal strain used for the infection. Compounds 12 and 13 followed an induction pattern similar to that of 2 and 3 when the plant was treated with M. fijiensis strains E22 and Ca10_13, respectively. In contrast, the concentration of compounds 4, 5 and 9 was dramatically reduced during the time BLSD developed, when the strain Ca10_13 was used (P < 0.001). A remarkable drop of the concentration of hydroxyanigorufone (5) from ~52 to ~27 nmol mg−1 DW was observed between 25 and 50 dpi. Similar results were found for compounds 5 and 9 after infection with strain E22 (Fig. 5b). The de novo biosynthesis of metabolites 1, 8 and 15 remained almost unaffected both by the time point evaluated and by the fungal strain.
An overall overview of the total content of phenylphenalenones (and structural analogues) by the two Musa varieties was obtained by adding up the amount of each metabolite per milligram of plant dry material. Thus, both after inoculation with the strain E22 and the strain Ca10_13, the susceptible variety ‘Williams’ produced lower levels of phenylphenalenones compared with the resistant variety ‘KTR’ (Fig. S4). For treatment with E22, ~68 nmol mg−1 DW was found in ‘Williams’, and ~120 nmol mg−1 DW was found in ‘KTR’. No significant statistical differences in the total phenylphenalenone content were detected between plants at 25 dpi showing early BLSD symptoms and plants at 50 dpi showing stronger symptoms. However, large differences in total phenylphenalenone concentration between the early (25 dpi) and the late stage of the disease (50 dpi) were observed for both Musa varieties when infected with the M. fijiensis strain Ca10_13. ‘Williams’ and ‘KTR’ responded differently as the disease progressed. While the phenylphenalenone content in ‘Williams’ dropped significantly from ~100 nmol mg−1 DW at 25 dpi to ~70 nmol mg−1 DW at 50 dpi, the phenylphenalenone content in ‘KTR’ increased from ~120 to ~140 nmol mg−1 DW (Fig. S4).
Antifungal bioassay of the major phenylphenalenones
The major phenylphenalenone‐type compounds identified in both Musa plants were assayed for their antifungal properties against the two M. fijiensis strains E22 and Ca10_13 used in this study. The IC50 values were generated using the method reported by Peláez et al. (2006) with modifications as described in the Section on Materials and Methods and using propiconazole as a positive control. The results showed that both fungal strains were sensitive not only to the commercial fungicide propiconazole but, though to a different extent, also to most of the phenylphenalenone‐type compounds assessed. Among compounds 3–5, 7, 9–11, 14 and 15 tested, only hydroxyanigorootin (14) and 2‐phenyl‐1,8‐naphthalic anhydride (3) did not display significant inhibitory activity against both M. fijiensis strains. Most of the IC50 values were found to be in the range between ~0.05 and ~0.15 mm (Fig. 6). Isoanigorufone (11), a 4‐phenylphenalenone found in the resistant variety ‘KTR’ but not in susceptible ‘Williams’, displayed antimycotic activity in IC50‐E22 = 0.032 ± 0.002 mm under light conditions. This value represented the best antifungal bioactivity among all the compounds tested. The antimicrobial activity of irenolone (4), hydroxyanigorufone (5) and 4′‐methoxyirenolone (12) against both M. fijiensis strains also increased significantly when the experiments were carried out under light‐controlled conditions (P < 0.05). The antifungal activity of anigorufone (9) and methoxyanigorufone (7) was also significantly light‐dependent but only in response to M. fijiensis Ca10_13 (P < 0.05) (Fig. 6). The results also showed that M. fijiensis Ca10_13 was not only tolerant to propiconazole but also to the phenylphenalenones assessed in this study. This became especially evident by comparing the growth of the strain E22 treated with anigorufone (9), irenolone (4) (under darkness only, P < 0.05) and methoxyanigorufone (7) (under darkness and photoperiod treatments, P < 0.001) with the growth of the strain Ca10_13 under the same conditions (Fig. 6).
Figure 6.

Half maximal inhibitory concentration (IC50) of the major inducible metabolites found in both Musa varieties during infection with M. fijiensis. Letters a–c indicate significant differences among treatments within the specified compound [two‐way analysis of variance (anova), Holm–Sidak post hoc test: P < 0.001; n.s: not significant]. For chemical structures of compounds 3–5, 7, 9–12, 14 and 15, see Table 1.
Metabolism of phenylphenalenones by the fungus
Motivation
The finding that, despite the induced production of antifungal phenylphenalenones, the BLSD symptoms caused by M. fijiensis strains E22 and Ca10_13 increased not only on the leaves of the susceptible Musa variety ‘Williams’ but also on the leaves of the resistant variety ‘KTR’ (for the case when the infection experiments were carried out with the strain Ca10_13) raised the question about the mechanism that enables the fungus to counteract the plant defence. The metabolism of defence chemicals is one of the most important ways the resistance of a plant is overcome by the pathogen (Morrissey & Osbourn 1999; Pedras & Ahiahonu 2005). The decreasing concentration of some of the most active antifungal compounds, especially anigorufone (9), during the progressing disease supported the suggestion that metabolic deactivation might be involved in the virulence mechanism of M. fijiensis. Therefore, selected phenylphenalenones were incubated under in vitro conditions with the fungus, and the mycelium and the fungal medium were screened for potential metabolites. Strain Ca10_13 was used in these experiments because its growth rate was inhibited to a lesser extent than the growth rate of strain E22 when incubated with phenylphenalenones (except 2‐phenyl‐1,8‐naphthalic anhydride (3) and hydroxyanigorufone (5), Fig. S5). Additionally, strain Ca10_13 was more virulent to banana plants compared with strain E22 (Fig. 4). Phenylphenalenones were applied at a concentration of 50% of the IC50. In addition to chemical analysis of phenylphenalenone‐derived metabolites, clues for the plant–fungus interaction such as the fungal biomass, the extracellular protein level and ergosterol concentration were determined.
Fungal biomass production during incubation with phenylphenalenones
Seven phenylphenalenone‐type compounds were incubated at three different concentrations (0.01, 0.02 and 0.04 mg mL−1) with the M. fijiensis strain Ca10_13. The results showed that anigorufone (9), 4′‐O‐methylirenolone (12) and 2‐phenyl‐1,8‐naphthalic anhydride (3) did not affect the production of biomass in comparison with the control (Fig. S6). However, incubation with isoanigorufone (11), hydroxyanigorufone (5) and 4′‐O‐methylanigorufone (10) resulted in a biomass production of up to ~80% in comparison with the control. The inhibition of the growth of fungal mycelium by compounds 5 and 10 was significantly dose‐dependent (Fig. S6). Interestingly, already the lowest applied concentration (0.01 mg mL−1) of methoxyanigorufone (7) significantly inhibited the biomass production to approximately 80% of the control. At concentrations of 0.02 and 0.04 mg mL−1, biomass production was dramatically reduced to approximately 30% of the control, demonstrating that the microorganism was enormously affected by this particular compound but not the other compounds.
Extracellular protein production
Extracellular microbial proteins can be involved in the degradation and deactivation of chemical defences of competing organisms (Morrissey & Osbourn 1999; Pedras & Ahiahonu 2005). Therefore, the production of soluble extracellular proteins was analysed in the culture medium in which the fungus was incubated with phenylphenalenone‐type compounds found in both Musa varieties. The aim of the analysis was to find out whether individual phenylphenalenones have a promoting or inhibiting effect on the protein production of the microorganism. The results showed clearly that isoanigorufone (11) and methoxyanigorufone (7) significantly induced the production of extracellular proteins in a dose‐dependent manner and caused pronounced increase when concentrations of 0.02 and 0.04 mg mL−1 were applied (Fig. S7). However, no significant differences in comparison with the control were found when the fungal culture was incubated with anigorufone (9), 4′‐O‐methylirenolone (12), 4′‐O‐methylanigorufone (10) and 2‐phenyl‐1,8‐naphthalic anhydride (3). Interestingly, hydroxyanigorufone (5) reduced the production of extracellular proteins to approximately 50% that of the control.
Quantification of ergosterol by 1H NMR analysis
Ergosterol is the major sterol of the fungal cell membranes and an important target molecule for many fungicides, which exert their mode of action through the inhibition of its biosynthesis (Debieu et al. 1998, 2013; Spotts & Cervantes 1986). In order to test whether phenylphenalenones affect ergosterol biosynthesis in M. fijiensis, a 1H NMR‐based quantification approach was used. Again, isoanigorufone (11) and methoxyanigorufone (7) were the metabolites that differ in their activity from the other compounds assessed. For 11, a significant increase was observed when the compound was assessed at 0.01 and 0.02 mg mL−1, but this was not the case for the concentration of 0.04 mg mL−1, where the production of ergosterol was only slightly above the control level (Fig. S8). During the incubation with hydroxyanigorufone (5), an increase of the ergosterol concentration of 40% in comparison with that of the control was observed only when 0.02 mg mL−1 of the phenylphenalenone was applied. A similar increase of the ergosterol concentration was detected with 4′‐O‐methylirenolone (12), but no dose dependence was observed in this case. In contrast to treatment with compounds 5 and 12, and especially with isoanigorufone (11), which all resulted in an enhanced ergosterol concentration, incubation of the fungus with methoxyanigorufone (7) drastically inhibited the production of ergosterol because it was not detectable at concentrations of 0.02 and 0.04 mg mL−1 Anigorufone (9) and 2‐phenyl‐1,8‐naphthalic anhydride (3) did not show significant differences in comparison with the control, and no dose dependence was detected.
Quantification of phenylphenalenones in M. fijiensis cultures
The question of whether phenylphenalenone‐type compounds are taken up by the mycelial tissue or persist in the medium during incubation with the fungus was assessed by HPLC. Fungal cultures of strain Ca10_13 were incubated for 8 d with three different concentrations (0.01, 0.02 and 0.04 mg mL−1) of the corresponding phenylphenalenones, as described in the preceding text. Each phenylphenalenone was then quantitatively determined in the medium as well as in the mycelium. The results showed a very different distribution of the phenylphenalenones between the medium, the mycelium and a non‐recovered portion of the compounds (Fig. 7). A considerable portion of most compounds persisted in the medium. For compounds 9–12, this portion was between 27 and 45% at the lowest concentration tested and increased up to approximately 70 to 80% at the highest concentration. Simultaneously, a certain percentage of compounds 9–12 accumulated in the mycelium biomass and showed a tendency to decline at higher doses of the applied compounds. According to the data shown in Fig. 7, the portions of compounds 9–12 detected in the medium and in the fungal mycelium do not sum up to 100%: an essential portion was missing, exceeding 40% in some cases. Again, the non‐recovered portion tends to decline with increasing doses of the applied compounds. However, generalizing with regard to the data is problematic, and an individualized consideration of the distribution pattern of most compounds seems to be reasonable, at least in the case of hydroxyanigorufone (5) and compounds 3 and 7. Remarkably, compound 5 was no longer detectable after incubating a dose of 0.01 mg mL−1 with M. fijiensis strain Ca10_13, and even more than 70% of the highest applied dose of 0.04 mg mL−1 remained unrecovered. Unlike all other phenylphenalenones applied, methoxyanigorufone (7) and 2‐phenyl‐1,8‐naphthalic anhydride (3) were taken up by the mycelium only to a relatively small (~10% for 7) or even negligible extent (3); that is, they remained mostly unchanged in the medium. For compound 3, this finding coincided with its inactivity on the biomass production of M. fijiensis strain Ca10_13 as shown in Fig. S6.
Figure 7.
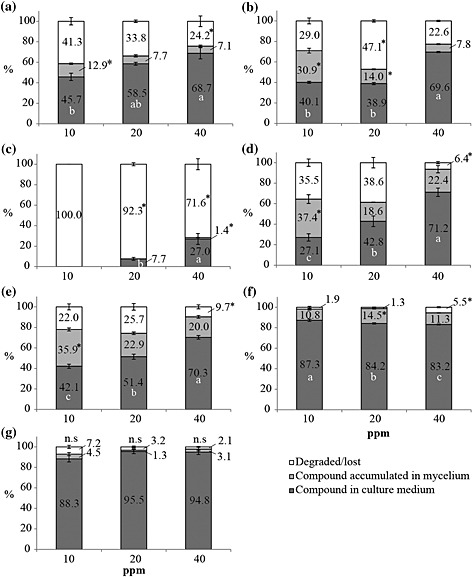
Quantification of phenylphenalenones after incubation of M. fijiensis strain Ca10_13 with different doses (0.01, 0.02 and 0.04 mg mL−1) of the specified compound. Data were normalized as a percentage based on the peak area generated by high‐performance liquid chromatography (HPLC)–diode array detector (DAD) analysis of the negative controls of each compound. Phenylphenalenone recovery from culture medium (dark‐grey bars), mycelium (light‐grey bars) and the unrecovered portions of phenylphenalenones were determined as the difference between the applied dose and the concentration determined in the medium and the mycelium after 8 d of incubation (white bars). (a) Isoanigorufone (11); (b) anigorufone (9); (c) hydroxyanigorufone (5); (d) 4′‐O‐methylirenolone (12); (e) 4′‐O‐methylanigorufone (10); (f) methoxyanigorufone (7); and (g) 2‐phenyl‐1,8‐naphthalic anhydride (3). Different letters or asterisks indicate significant differences among treatments (concentrations) of the compound analysed [one‐way analysis of variance (anova), Holm–Sidak post hoc test: P < 0.05; n.s: not significant].
Metabolism of phenylphenalenones by the fungus
The extensive (compounds 9–12) or even complete (compound 5 at 0.01 mg mL−1) disappearance of phenylphenalenones (Fig. 7) during incubation with the fungus could be explained by metabolic processes. Therefore, in the next step of this study, the mycelium and the culture medium were analysed for potential metabolites of phenylphenalenones. In an up‐scaled incubation experiment with M. fijiensis strain Ca10_13, synthetic anigorufone (9) was used as a model compound to study the metabolism. Except for the parent compound 9, no phenylphenalenone‐related compound was detected in the liquid culture medium, and the HPLC trace of the medium was identical with that of the control sample (data not shown). However, an additional peak, which was not present in the control sample, emerged in the UV and selected ion chromatograms (LC–MS, positive ion mode) of the methanolic mycelium extract (Fig. S9). The mass spectrum of this peak (R t = 7.95 min) displayed a molecular ion of m/z 353.2 [M + H]+ and a fragment ion of m/z 273.2, suggesting a metabolite of compound 9. HRMS analysis confirmed the fragment ion as anigorufone (9) ([M + H]+ found: m/z 273.0908, calc.: m/z 273.0916), and the molecular ion peak m/z 353.0475 ([M + H]+) was consistent with a sulfated metabolite of 9 ([M + H]+ calc.: m/z 353.0484). Because the hydroxyl group in position 2 of anigorufone (9) is the only place where a sulfate group could be attached to the molecule, the new metabolite must be anigorufone‐2‐sulfate (Fig. S9).
After anigorufone‐2‐sulfate was identified as a metabolite of anigorufone (9), the mycelia and extracts obtained after incubation of further phenylphenalenones were also analysed by UHPLC–HRMS for sulfate metabolites. While no such compounds were detected in the culture media, the mass spectra of the methanolic extracts of mycelia incubated with compounds 10 to 12 displayed not only peaks of the parent compounds but also characteristic peaks of corresponding sulfate conjugates (Table 2). Like anigorufone (9), compounds 10 to 12 have a single hydroxyl group at C‐2, and therefore, the sulfate unit of their conjugates must be attached to this particular position. A sulfate conjugate was also found for hydroxyanigorufone (5) (Table 2). As in the conjugates of compounds 10 to 12, the position of the sulfate in the hydroxyanigorufone conjugate was arbitrarily assigned to the ortho‐hydroxyketone functionality, although two hydroxyl groups, 2‐OH and 4′‐OH, are available in this special case. No sulfate conjugate was produced during incubation of the methoxyanigorufone (7) with the fungus. This was not unexpected because no free hydroxyl group was available in this structure. Furthermore, as shown in Fig. 7, the uptake of compound 7 into the mycelia was very limited.
Table 2.
Phenylphenalenone sulfates identified in methanolic extracts of mycelia after incubation of the parent phenylphenalenones with M. fijiensis Ca10_13
| No. | Parent compound | Product | R t (min)a | HRMS (m/z) | |
|---|---|---|---|---|---|
| Calc. | Found | ||||
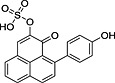
| 5 |
|
|
9.52 | [M + H]+ = 369.0433 | [M + H]+ = 369.0428 |
| 7 |
|
No metabolite detected | — | — | — |
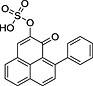
| 9 |
|
|
13.27 | [M + H]+ = 353.0484 | [M + H]+ = 353.0475 |
| 10 |
|
|
14.21 | [M − H]− = 381.0438 | [M − H]−3 = 81.0435 |
| 11 |
|
|
11.78 | [M + H]+ = 353.0484 | [M + H]+ = 353.0475 |
| 12 |
|
|
14.53 | [M − H]− = 381.0438 | [M − H]− = 381.0436 |

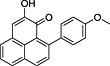
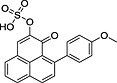
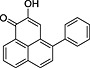
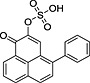
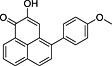
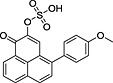
Retention time of the phenylphenalenone sulfate [data from ultra‐high‐performance liquid chromatography–electrospray ionization mass spectra (UHPLC–ESIMS) analysis].
The isomeric hydroxyanigorufone‐4′‐sulfate may be an alternative structure.
Discussion
Monitoring Musa's metabolic responses during M. fijiensis attack
1H NMR‐based metabolomics revealed similar biochemical profiles for both Musa varieties, ‘Williams’ (susceptible) and ‘KTR’ (resistant), in response to their interaction with the fungus M. fijiensis. Indeed, infection by this pathogen not only triggered the primary metabolism but also strongly affected the biosynthesis of specialized metabolites, especially aromatic compounds. These effects of infection were inferred from evaluating the group discrimination of each Musa plant part after the PCA (Fig. 2) and the corresponding loading plots (Fig. 3). While the levels of carbohydrates, represented mainly by glucose and sucrose, rose in the infected plant tissue, the levels of most amino acids (such as l‐alanine and l‐threonine) declined. l‐Methionine was an exception and tended to be more up‐regulated in the infected Musa plants than in the non‐treated plants. These findings confirm previous reports of plant–pathogen interactions that suggest that the translocation of sugars from the roots or other plant organs to the infected tissue may help counteract the metabolic imbalances caused by microbial invaders (Heil & Bostock 2002).
Carbohydrates are also involved in the biosynthesis of a broad spectrum of specialized metabolites and can mediate the gene expression of pathogenesis‐related proteins (Aliferis et al. 2014; Herbers et al. 1996; Herbers et al. 2000; Scharte et al. 2005). Gene expression demands a substantial pool of amino acids for the synthesis of proteins commonly associated with the defence against pathogens. For example, chitinases and glucanases have been reported to play a role in the interaction between Musa and M. fijiensis and in other plant pathosystems (Harish et al. 2009; Torres et al. 2012). The production of defence‐related proteins could explain the reduced content of amino acids found in the infected plant tissue of both Musa varieties. Surprisingly, l‐methionine, which was the only amino acid positively regulated during the infection, may be associated with enzymatic reactions that make up the methionine S‐methyltransferases involved in the O‐methylation of some phenylpropanoids and phenylphenalenone‐type compounds, as has been reported by Otálvaro et al. (2010).
An enhanced number of signals in the 1H NMR region from δ 6.0 to 9.0 were the most relevant features observed in the loading plots (Fig. 3) of both Musa varieties. Phenolic metabolites were more strongly up‐regulated in the infected local tissue of the resistant variety ‘KTR’ than in the infected local tissue of the susceptible variety ‘Williams’, thus providing the first hints that the two varieties respond differently to the infection with the pathogen.
Dopamine, an animal neurotransmitter also occurring in plants (Kulma & Szopa 2007; Li et al. 2015), is a catecholamine identified previously in banana fruits (Kanazawa & Sakakibara 2000). In our experiments, it was found in Musa leaves. We observed that it declined in the non‐treated areas of infected plants (Fig. 2, samples B and D). Simultaneously, a positive correlation was observed for dopaol‐β‐d‐glucoside, identified for the first time in healthy Musa plant tissue. Dopamine was thought to serve as a substrate in the biosynthesis of its glucoside, which then played a role as a phytoalexin. However, this assumption was ruled out because dopaol‐β‐d‐glucoside (and dopamine as well) did not show any antifungal properties against M. fijiensis (Fig. S3). Thus, the role of those metabolites in Musa plant defence remains to be studied.
A group of phenylpropanoid conjugates was identified for the first time as constituents of Musa plants (Table S2, entries 14–17). 1‐O‐((E)‐p‐Coumaroyl)‐β‐d‐glucose may function as a depot compound, which, after being hydrolyzed to p‐coumaric acid by a putative cell wall‐associated β‐glucosidase (Chong et al. 1999; Chong et al. 2002), could be used as a precursor for the biosynthesis of phenylphenalenone‐type compounds (Hölscher & Schneider 1995; Schmitt et al. 2000). This phenomenon could explain the reduced levels of such phenylpropanoids in the infected tissue. Moreover, phenylpropanoids are not only involved in the biosynthesis of phytoalexins of the phenylphenalenone‐type but also play a role in lignification processes in order to counteract the invading pathogen (Liang et al. 2006).
Irenolone (4) and hydroxyanigorufone (5), the two major phenylphenalenones identified from the metabolomic analysis (Table S2), were induced only in the infected plant tissue; along with other phenolic compounds, these major phenylphenalenones constituted the chemical basis for the Musa response to M. fijiensis. Unfortunately, because of the overlapping 1H NMR signals, metabolic changes of the other phenolics could be not observed in the loading plots (Fig. 3) but, rather, required a conventional phytochemical approach.
Incompatible Musa–M. fijiensis interaction depends on the pathogen virulence
Phytochemical analysis of the infected Musa tissue led to the identification of 15 phenylphenalenone‐type compounds and structural analogues from the resistant variety ‘KTR’. A smaller number (10) of such metabolites appeared in the defence response of the susceptible variety ‘Williams’. With the exception of 2‐hydroxy‐9‐(4′‐methoxyphenyl)‐1H‐phenalen‐1‐one (10), all other compounds had been previously reported in different varieties of Musa, and their occurrence has been correlated with the resistance phenotype exhibited by some Musa varieties against pathogens and herbivores (Hölscher et al. 2014; Otálvaro et al. 2007). Nonetheless, there has been little quantitative analysis of phenylphenalenones as BLSD progresses, and resistant traits have not been studied with isolates of M. fijiensis that differ in their virulence. Therefore, two strains of M. fijiensis, E22 and Ca10_13, were used as inoculum. According to the method proposed by Cañas‐Gutierrez et al. (2009), the strain E22 was classified as ‘sensitive’ and Ca10_13 as ‘tolerant’ to the fungicide propiconazole. It is important to note that sensitivity to the fungicide propiconazole does not necessarily correlate with a high degree of virulence of the pathogen. However, fungicide tolerance could help the microorganism overcome the plant's chemical defence and/or synthetic fungicides.
For the variety ‘KTR’, an early response was observed by the appearance of small specks on the lower surface of the infected leaf 7–9 dpi with the fungus, regardless of which M. fijiensis strain was used; no symptoms were detectable in the variety ‘Williams’ up to 15–17 dpi (Table S1). This difference between varieties is not surprising because an early recognition of virulence effectors from the pathogen triggers the defence mechanism in Musa (as they do in other plant–pathogen systems), conferring resistance on plants that are attacked (Hoss et al. 2000; Torres et al. 2012).
The resistance conferred on Musa ‘KTR’ in its interaction with M. fijiensis strain E22 was confirmed by the slow progress of BLSD determined at the two points of time. In contrast, M. fijiensis Ca10_13 not only exhibited a higher degree of virulence than E22 but also was able to overcome the resistance of ‘KTR’ and cause severe foliar damages in the infected leaf (Fig. 4, Table S1). According to previous reports, the breakdown of the resistance in Musa caused by M. fijiensis has also been observed in other Musa varieties such as the variety ‘Yangambi Km5’, which is classified as highly resistant to this particular pathogen (Fullerton & Olsen 1995; Mouliom‐Pefoura 1999).
For the variety ‘Williams’, the necrotic symptoms of BLSD were larger than those observed for ‘KTR’ during the progress of the fungal infection; this difference was expected for a susceptible variety. Nonetheless, leaf damage was increased when M. fijiensis Ca10_13 was used as a pathogen, confirming the strong virulence of this pathogen.
Priming defence mechanisms in plants at early stages of infection, after the recognition of the effectors signals produced by a pathogen, are crucial in the incompatible plant–pathogen interactions (Conrath et al. 2006; Frost et al. 2008). As demonstrated for some resistant Musa varieties, the production of pathogenesis‐related proteins, lignification processes, hypersensitivity response and accumulation of phytoalexins must work in a concerted action to enable plants to block pathogenesis and herbivory (Harelimana et al. 1997; Hölscher et al. 2014; Otálvaro et al. 2002a, 2002b; Quiñones et al. 2000; Torres et al. 2012).
Here, the BLSD symptoms appearing on the leaves of the ‘KTR’ variety after infection with the strain Ca10_13 clearly demonstrated that resistance in this Musa variety could be overcome by a highly virulent pathogen. Analysis of the chemical composition of both Musa varieties during their responses to the two fungal strains showed that phenylphenalenone‐type compounds constituted the major induced metabolites both in ‘Williams’ and in ‘KTR’. The susceptible variety ‘Williams’ produced not only a smaller number but also lower levels of these metabolites (less than 10 nmol mg−1 DW) compared with ‘KTR’. Hydroxyanigorufone (5) appeared to be the exception: it was the most prominent compound synthesized in ‘Williams’ in response to the strain Ca10_13. Nevertheless, as the disease developed, the concentration of hydroxyanigorufone (5) and anigorufone (9) decreased, while that of other compounds (e.g. 1–4) stayed constant or even slightly increased. The reduced levels of compounds 5 and 9 at 50 dpi could be due to an oxidative metabolic reaction and may result in the naphthalic anhydrides 1 and 3, respectively. Reactive oxygen species like hydrogen peroxide, which have been reported to be produced in the infected tissue of Musa plants (Torres et al. 2012), could function as the oxidizing agent. In ‘Williams’, the susceptibility seemed to be associated with a late recognition of the pathogen, which then successfully colonizes the plant tissue. This is consistent with a relatively low total level of phenylphenalenones (Fig. S4), although hydroxyanigorufone (5), which is one of the active phenylphenalenones (Fig. 6), was overproduced in comparison with the other metabolites (Fig. 5b).
In contrast, the ‘KTR’ variety not only rapidly recognized the pathogen but also responded with a burst of phenylphenalenones, providing an enhanced chemical response to both fungal strains. This variety produced a number of phenylphenalenones in concentrations over 10 nmol mg−1 DW; irenolone (4) was the most abundant compound (>25 nmol mg−1 DW) (Fig. 5a). Compound 4 and its congeners seemed to have a strong inhibiting effect on the fungal colonization of plant tissue infected by the strain E22 but not on tissue infected by Ca10_13 (Fig. 5a). The response of the two Musa varieties to the two fungal strains E22 and Ca10_13 produced slight quantitative differences but no qualitative differences in the metabolic patterns (data not shown).
In ‘KTR’, the concentrations of compounds 2–4, 12 and 14 increased significantly in response to Ca10_13 at 25 or 50 dpi but not in the response to the strain E22. Irenolone (4) was the metabolite with the highest response against Ca10_13 at 50 dpi in comparison with all other induced metabolites (Fig. 5a). This finding suggests that, at least from the chemical point of view, ‘KTR’ was able to distinguish between the virulence capacities of the fungal strains.
Pathogen virulence associated with metabolism of phenylphenalenones
The breakdown in the resistance of ‘KTR’ caused by the virulent strain Ca10_13 encouraged us to explore whether some phenylphenalenones (e.g. compounds 3, 9, 11, 12), although up‐regulated in response to M. fijiensis, did not significantly inhibit the growth of this particular fungus in planta. The antifungal bioassay showed that phenylphenalenones were active against both fungal strains (Fig. 6). The fungal pathogen E22 was more sensitive in comparison with Ca10_13 when irenolone (4), anigorufone (9), isoanigorufone (11) and methoxyanigorufone (7) were assessed under dark conditions. Furthermore, these compounds displayed an enhanced antifungal activity when light conditions were applied to the experimental system (Fig. 6). Phenylphenalenones (Flors & Nonell 2006; Lazzaro et al. 2004) and perinaphthenones (Hidalgo et al. 2009) have been reported to be light‐dependent singlet oxygen producers, and these could significantly affect the fungal growth rate. Overall, the hypothesis that M. fijiensis strain Ca10_13 could tolerate higher concentrations of phenylphenalenones than the strain E22 was found to hold only for some compounds and under darkness.
Differences in the fungal growth rate between both M. fijiensis strains became more apparent when the incubation experiments were scaled up to almost 10 times the volume of liquid culture used for the microtitre bioassay. The growth rate of M. fijiensis strain E22 was much slower than that of strain Ca10_13 when phenylphenalenones in the range of 0.01 to 0.04 mg mL−1 were supplied in the incubation medium; hydroxyanigorufone (5) and 2‐phenyl‐1,8‐naphthalic anhydride (3) were the exceptions (Fig. S5). Therefore, the strain Ca10_13 was selected for further experiments. Because some plant pathogens have developed strategies to evade plant defences through a complex detoxification process (Barz & Welle 1992; Jeandet et al. 2010; Pedras & Taylor 1993), we wondered whether the resistance of M. fijiensis to some phenylphenalenones could be attributed to metabolic detoxification.
In fact, the strain Ca10_13 was able not only to metabolize most of the compounds into sulfate‐derivatives (compounds 5 and 9–12; Table 2) but also to further degrade them into undetectable metabolites or even decompose them completely as in the case of hydroxyanigorufone (5) at the lowest concentration assessed. In general, the formation of sulfate conjugates through sulfotransferases constitutes a biochemical reaction, which is involved in such physiological processes as intracellular signalling, extracellular interactions and detoxification of xenobiotics (Chapman et al. 2004; Zhang et al. 1996). The present work is the first report of an inactivation or detoxification of phenylphenalenones by M. fijiensis through sulfate conjugation. In addition, to the best of our knowledge, ours is the first report of a detoxification reaction of any phenylphenalenone by a fungal organism. New strategies to control BLSD through inhibitors that target sulfotransferases in the microorganism may result from this new understanding of the role of sulfate conjugates.
Interestingly, only phenylphenalenones containing a hydroxyl group attached to C‐2 in the molecule were found to be sulfated (for hydroxyanigorufone, a sulfation in the free hydroxyl group at C‐4′ cannot be ruled out). Because conversion to sulfates seems to minimize the toxic effects of most phenylphenalenones, the fungal biomass production remained almost unaffected (Fig. S6). However, methoxyanigorufone (7), a metabolite which is missing a hydroxyl group (instead of the OH, an O‐methyl group is attached to C‐2), enormously affected growth and biomass production (Fig. S6). This supports the hypothesis that an α‐hydroxyketone feature helps the molecule to be recognized by the aryl sulfotransferase. In addition, the chemical pressure generated by compound (7) on the metabolism of the strain Ca10_13 was confirmed by the overproduction of soluble extracellular proteins (Fig. S7) and the inhibited production of ergosterol (Fig. S8). Because no metabolites derived from phenylphenalenones were found in the liquid culture medium during the incubation with the fungus, the overexpression of extracellular proteins observed in response to compounds 7 and 11 could not be associated with the external enzymatic detoxification of these metabolites. Instead, detoxification may suggest that these metabolites increase the physiological stress in the fungus, which then results in the overexpression of fungal proteins involved in the suppression of the plant defences (Dodds & Rathjen 2010; Escobar et al. 2015). Identification of the proteins, which were overexpressed by the interaction with specific phenylphenalenones, would help explain the pathophysiological processes taking place during the interaction between Musa and M. fijiensis. Because of their role in the defence of Musa plants against M. fijiensis, methoxyanigorufone (7) and isoanigorufone (11) are considered the most relevant metabolites for further exploration of the molecular mode of phenylphenalenones.
Supporting information
Figure S1. Chemical structures of the metabolites identified by 1D‐ and 2D NMR analysis in ‘Williams’ and ‘KTR’ Musa varieties.
Figure S2. Partial 1H NMR spectra showing the aromatic signals of dopamine and dopaol‐β‐D‐glucoside in a: infected leaf areas (A, see Fig. 2); b: non‐infected areas from (D); c: control Musa ‘Williams’ variety.
Figure S3. Fungal growth (data are normalized to the average growth of untreated control cultures) of M. fijiensis strain Ca10_13 after treatment with different concentrations of dopamine and dopaol‐β‐D‐glucoside.
Figure S4. Total content of phenylphenalenone‐type compounds (PP) produced by the Musa varieties ‘Williams’ and ‘KTR’ during 25 and 50 dpi with two different M. fijiensis strains. Letters a ‐ d indicate significant differences among treatments (One‐Way ANOVA, Holm‐Sidak post hoc test: P<0.001).
Figure S5. Data obtained from incubation of M. fijiensis strain E22 with different doses (0.01, 0.02 and 0.04 mg mL−1) of phenylphenalenones for 8 d.
Figure S6. Biomass produced by M. fijiensis strain Ca10_13 under in vitro treatment with different phenylphenalenone‐type compounds assessed at 0.01, 0.02 and 0.04 mg mL−1 for 8 d.
Figure S7. Soluble extracellular protein in in vitro culture medium of M. fijiensis strain Ca10_13 after incubation with phenylphenalenone‐type compounds at 0.01, 0.02 and 0.04 mg mL−1.
Figure S8. Ergosterol production by M. fijiensis strain Ca10_13 after in vitro incubationwith different phenylphenalenone‐type compounds at 0.01, 0.02 and 0.04 mg mL−1.
Figure S9. LC‐ESIMS analysis of anigorufone (9) and its metabolite in themethanolic extract of M. fijiensis Ca10_13mycelium. Panel a:UVchromatogramat 254 nm displaying peaks of anigorufone (R t = 15.3 min) and an anigorufone‐derived metabolite (R t = 7.8 min) identified as anigorufone‐2‐sulfate. Panel b: Extracted ion chromatogram (EIC) at m/z 353.2 [M +1]+, the mass corresponding to anigorufone‐2‐sulfate; Panel c: Mass spectrum of anigorufone‐2‐sulfate obtained at R t = 7.8 min showing the molecular ion peak m/z 353.2 [M+H]+ and the fragment ion peak of m/z 273.2, corresponding to anigorufone (9).
Table S1. Black leaf streak disease symptoms developed by both M. fijiensis strains (E22 and Ca10_13) in ‘Williams’ and ‘KTR’ Musa varieties.
Table S2. Metabolites identified by 1D‐ and 2D‐NMR analysis in Musa varieties ‘Williams’ and ‘KTR’ and their up‐ and downregulation after infection with M. fijiensis E22 as inferred from the corresponding loading plots.
Supporting Information
Acknowledgments
The authors thank Tamara Krügel and the greenhouse team of the Max Planck Institute for Chemical Ecology (Jena, Germany) for plant cultivation, Gert Kema (Plant Research International, Wageningen, the Netherlands) for kindly providing M. fijiensis strains and Emily Wheeler for the editorial assistance. This study was supported by the Max Planck Institute for Chemical Ecology, COLCIENCIAS (111565842551), and CODI (IN651CE). WH gratefully acknowledges COLCIENCIAS for a scholarship.
Hidalgo, W. , Chandran, J. N. , Menezes, R. C. , Otálvaro, F. , and Schneider, B. (2016) Phenylphenalenones protect banana plants from infection by Mycosphaerella fijiensis and are deactivated by metabolic conversion. Plant, Cell & Environment, 39: 492–513. 10.1111/pce.12630.
References
- Abadie C., Zapater M.F., Pignolet L., Carlier J. & Mourichon X. (2008) Artificial inoculation on plants and banana leaf pieces with Mycosphaerella spp. responsible for Sigatoka leaf spot diseases. Fruits 63, 319–323. [Google Scholar]
- Ali K., Maltese F., Zyprian E., Rex M., Choi Y.H. & Verpoorte R. (2009) NMR metabolic fingerprinting based identification of grapevine metabolites associated with Downy mildew resistance. Journal of Agricultural and Food Chemistry 57, 9599–9606. [DOI] [PubMed] [Google Scholar]
- Aliferis K.A., Faubert D. & Jabaji S. (2014) A metabolic profiling strategy for the dissection of plant defense against fungal pathogens. Plos One 9, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arzanlou M., Abeln E.C.A., Kema G.H.J., Waalwijk C., Carlier J., de Vries I., … Crous P.W. (2007) Molecular diagnostics for the Sigatoka disease complex of banana. Phytopathology 97, 1112–1118. [DOI] [PubMed] [Google Scholar]
- Banerjee N. & Delanghe E. (1985) A tissue‐culture technique for rapid clonal propagation and storage under minimal growth‐conditions of Musa (banana and plantain). Plant Cell Reports 4, 351–354. [DOI] [PubMed] [Google Scholar]
- Barz W. & Welle R. (1992) Biosynthesis and metabolism of isoflavones and pterocarpan phytoalexins in chickpea, soybean and phytopathogenic fungi. Phenolic Metabolism in Plants 26, 139–164. [Google Scholar]
- Cañas‐Gutierrez G.P., Angarita‐Velasquez M.J., Restrepo‐Florez J.M., Rodriguez P., Moreno C.X. & Arango R. (2009) Analysis of the CYP51 gene and encoded protein in propiconazole‐resistant isolates of Mycosphaerella fijiensis . Pest Management Science 65, 892–899. [DOI] [PubMed] [Google Scholar]
- Carlier J., De Waele D. & Escalant J.V. (2002) Global Evaluation of Musa Germplasm for Resistance to Fusarium Wilt, Mycosphaerella Leaf Spot Diseases and Nematodes: In‐depth Evaluation. INIBAP, Montpellier (FRA) [Google Scholar]
- Carlier J., Fouré E., Gauhl F., Jones D.R., Lepoivre P., Mourichon X., … Romero R.A. (2000) Fungal disease of the foliage In Diseases of Banana, Abacá and Enset. (ed. Jones D.R.), pp. 37–141. CABI Publishing, Wallingford, Oxon (UK). [Google Scholar]
- Chabrier C. & Queneherve P. (2003) Control of the burrowing nematode (Radopholus similis Cobb) on banana: impact of the banana field destruction method on the efficiency of the following fallow. Crop Protection 22, 121–127. [Google Scholar]
- Chapman E., Best M.D., Hanson S.R. & Wong C.H. (2004) Sulfotransferases: structure, mechanism, biological activity, inhibition, and synthetic utility. Angewandte Chemie‐International Edition 43, 3526–3548. [DOI] [PubMed] [Google Scholar]
- Chong J., Baltz R., Fritig B. & Saindrenan P. (1999) An early salicylic acid‐, pathogen‐ and elicitor‐inducible tobacco glucosyltransferase: role in compartmentalization of phenolics and H2O2 metabolism. FEBS Letters 458, 204–208. [DOI] [PubMed] [Google Scholar]
- Chong J., Baltz R., Schmitt C., Beffa R., Fritig B. & Saindrenan P. (2002) Downregulation of a pathogen‐responsive tobacco UDP‐Glc: phenylpropanoid glucosyltransferase reduces scopoletin glucoside accumulation, enhances oxidative stress, and weakens virus resistance. Plant Cell 14, 1093–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrath U., Beckers G.J.M., Flors V., Garcia‐Agustin P., Jakab G., Mauch F., … Mauch‐Mani B. (2006) Priming: getting ready for battle. Molecular Plant‐Microbe Interactions 19, 1062–1071. [DOI] [PubMed] [Google Scholar]
- Debieu D., Bach J., Lasseron A., Malosse C. & Leroux P. (1998) Effects of sterol biosynthesis inhibitor fungicides in the phytopathogenic fungus, Nectria haematococca: ergosterol depletion versus precursor or abnormal sterol accumulation as the mechanism of fungitoxicity. Pesticide Science 54, 157–167. [Google Scholar]
- Debieu D., Bach, J. , Montesinos, E. , Fillinger, S. & Leroux, P. (2013) Role of sterol 3‐ketoreductase sensitivity in susceptibility to the fungicide fenhexamid in Botrytis cinerea and other phytopathogenic fungi. Pest Management Science 69, 642–651. [DOI] [PubMed] [Google Scholar]
- Dhakshinamoorthy S., Mariama K., Elsen A. & De Waele D. (2014) Phenols and lignin are involved in the defence response of banana (Musa) plants to Radopholus similis infection. Nematology 16, 565–576. [Google Scholar]
- Dodds P.N. & Rathjen J.P. (2010) Plant immunity: towards an integrated view of plant‐pathogen interactions. Nature Reviews Genetics 11, 539–548. [DOI] [PubMed] [Google Scholar]
- Escobar L., Guzmán M., Sandoval J. & Gómez M. (2015) Comparative analysis of the in vitro and in planta secretomes from Mycosphaerella fijiensis isolates. British Mycological Society 119, 447–470. [DOI] [PubMed] [Google Scholar]
- Etebu E. & Young‐Harry W. (2011) Control of black Sigatoka disease: challenges and prospects. African Journal of Agricultural Research 6, 508–514. [Google Scholar]
- FAO (2014) Banana market review and banana statistic 2012‐2013. Intergovernmental group on bananas and tropical fruits, pp. 39.
- FAO (2015) http://www.statista.com/statistics/264001/worldwide-production-of-fruit-by-variety/ accessed June 2015.
- Flors C. & Nonell S. (2006) Light and singlet oxygen in plant defense against pathogens: phototoxic phenalenone phytoalexins. Accounts of Chemical Research 39, 293–300. [DOI] [PubMed] [Google Scholar]
- Fouré E. (1986) Varietal reactions of bananas and plantains to black leaf streak disease In Banana and Plantain Breeding Strategies. Proceedings of an International Workshop Held at Cairns, Australia 13‐17 October 1986 (eds Persley G.J. & De Langhe E.A.) pp. 110–113. ACIAR Proceedings, Cairns, Australia. [Google Scholar]
- Franzyk H., Olsen C.E. & Jensen S.R. (2004) Dopaol 2‐keto‐ and 2,3‐diketoglycosides from Chelone obliqua . Journal of Natural Products 67, 1052–1054. [DOI] [PubMed] [Google Scholar]
- Frost C.J., Mescher M.C., Carlson J.E. & De Moraes C.M. (2008) Plant defense priming against herbivores: getting ready for a different battle. Plant Physiology 146, 818–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fullerton R.A. & Olsen T.L. (1995) Pathogenic variability in Mycosphaerella fijiensis Morelet, cause of black Sigatoka in banana and plantain. New Zealand Journal of Crop and Horticultural Science 23, 39–48. [Google Scholar]
- Harelimana G., Lepoivre P., Jijakli H. & Mourichon X. (1997) Use of Mycosphaerella fijiensis toxins for the selection of banana cultivars resistant to black leaf streak. Euphytica 96, 125‐128. [Google Scholar]
- Harish S., Kavino M., Kumar N. & Samiyappan R. (2009) Differential expression of pathogenesis‐related proteins and defense enzymes in banana: interaction between endophytic bacteria, Banana bunchy top virus and Pentalonia nigronervosa . Biocontrol Science and Technology 19, 843–857. [Google Scholar]
- Heil M. & Bostock R.M. (2002) Induced systemic resistance (ISR) against pathogens in the context of induced plant defences. Annals of Botany 89, 503–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbers K., Meuwly P., Frommer W.B., Metraux J.P. & Sonnewald U. (1996) Systemic acquired resistance mediated by the ectopic expression of invertase: possible hexose sensing in the secretory pathway. Plant Cell 8, 793–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbers K., Takahata Y., Melzer M., Mock H.P., Hajirezaei M. & Sonnewald U. (2000) Regulation of carbohydrate partitioning during the interaction of potato virus Y with tobacco. Molecular Plant Pathology 1, 51–59. [DOI] [PubMed] [Google Scholar]
- Heslop‐Harrison J.S. (2011) Genomics, banana breeding and superdomestication. International Ishs‐Promusa Symposium on Global Perspectives on Asian Challenges 897, 55–62. [Google Scholar]
- Hidalgo W., Duque L., Saez J., Arango R., Gil J., Rojano B., … Otálvaro F. (2009) Structure activity relationship in the interaction of substituted perinaphthenones with Mycosphaerella fijiensis . Journal of Agricultural and Food Chemistry 57, 7417–7421. [DOI] [PubMed] [Google Scholar]
- Hirai N., Ishida H. & Koshimizu K. (1994) A phenalenone‐type phytoalexin from Musa acuminata . Phytochemistry 37, 383–385. [Google Scholar]
- Hölscher D., Dhakshinamoorthy S., Alexandrov T., Becker M., Bretschneider T., Buerkert, A. , …, Swennen R.L. (2014) Phenalenone‐type phytoalexins mediate resistance of banana plants (Musa spp.) to the burrowing nematode Radopholus similis . Proceedings of the National Academy of Sciences of the United States of America 111, 105–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hölscher D., Fuchser J., Knop K., Menezes R., Buerkert A., Svatoš A., Schubert U. & Schneider B. (2015) High resolution mass spectrometry imaging reveals the occurrence of phenylphenalenone‐type compounds in red paracytic stomata and red epidermis tissue of Musa acuminata ssp. zebrina cv. ‘Rowe Red’. Phytochemistry 116, 239–245. [DOI] [PubMed] [Google Scholar]
- Hölscher D. & Schneider B. (1995) The biosynthetic origin of the central one‐carbon unit of phenylphenalenones in Anigozanthos preissii . Natural Product Letters 7, 177–182. [Google Scholar]
- Hoss R., Helbig J. & Bochow H. (2000) Function of host and fungal metabolites in resistance response of banana and plantain in the black Sigatoka disease pathosystem (Musa spp. Mycosphaerella fijiensis). Journal of Phytopathology 148, 387–394. [Google Scholar]
- Huang S.X., Liao X., Nie Q.J., Ding L.S. & Peng S.L. (2004) Phenyl and phenylethyl glycosides from Picrorhiza scrophulariiflora . Helvetica Chimica Acta 87, 598–604. [Google Scholar]
- Jeandet P., Delaunois B., Conreux A., Donnez D., Nuzzo V., Cordelier S., … Courot E. (2010) Biosynthesis, metabolism, molecular engineering and biological functions of stilbene phytoalexins in plants. Biofactors 36, 331–341. [DOI] [PubMed] [Google Scholar]
- Jitsaeng K. & Schneider B. (2010) Metabolic profiling of Musa acuminata challenged with Sporobolomyces salmonicolor . Phytochemistry Letters 3, 84–87. [Google Scholar]
- Kamo T., Kato N., Hirai N., Tsuda M., Fujioka D. & Ohigashi H. (1998a) A biosynthetic intermediate of phytoalexins in banana fruits. Phytochemistry 49, 1617–1621. [DOI] [PubMed] [Google Scholar]
- Kamo T., Kato N., Hirai N., Tsuda M., Fujioka D. & Ohigashi H. (1998b) Phenylphenalenone‐type phytoalexins from unripe Bungulan banana fruit. Bioscience Biotechnology and Biochemistry 62, 95–101. [DOI] [PubMed] [Google Scholar]
- Kanazawa K. & Sakakibara H. (2000) High content of dopamine, a strong antioxidant, in Cavendish banana. Journal of Agricultural and Food Chemistry 48, 844–848. [DOI] [PubMed] [Google Scholar]
- Kashiwada Y., Nonaka G.I., Nishioka I. & Yamagishi T. (1988) Galloyl and hydroxycinnamoylglucoses from Rhubarb. Phytochemistry 27, 1473–1477. [Google Scholar]
- Kim H.K., Choi Y.H. & Verpoorte R. (2010) NMR‐based metabolomic analysis of plants. Nature Protocols 5, 536–549. [DOI] [PubMed] [Google Scholar]
- Kovacs G., Sagi L., Jacon G., Arinaitwe G., Busogoro J.P., Thiry E., … Remy S. (2013) Expression of a rice chitinase gene in transgenic banana (‘Gros Michel’, AAA genome group) confers resistance to black leaf streak disease. Transgenic Research 22, 117–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulma A. & Szopa J. (2007) Catecholamines are active compounds in plants. Plant Science 172, 433–440. [Google Scholar]
- Lazzaro A., Corominas M., Marti C., Flors C., Izquierdo L.R., Grillo T.A., … Nonell S. (2004) Light‐ and singlet oxygen‐mediated antifungal activity of phenylphenalenone phytoalexins. Photochemical & Photobiological Sciences 3, 706–710. [DOI] [PubMed] [Google Scholar]
- Li C., Sun X.K., Chang C., Jia D.F., Wei Z.W., Li C.Y. & Ma F.W. (2015) Dopamine alleviates salt‐induced stress in Malus hupehensis . Physiologia Plantarum 153, 584–602. [DOI] [PubMed] [Google Scholar]
- Liang Y.S., Kim H.K., Lefeber A.W.M., Erkelens C., Choi Y.H. & Verpoorte R. (2006) Identification of phenylpropanoids in methyl jasmonate treated Brassica rapa leaves using two‐dimensional nuclear magnetic resonance spectroscopy. Journal of Chromatography A 1112, 148–155. [DOI] [PubMed] [Google Scholar]
- Liu F., Zhang Y., Sun Q.Y., Yang F.M., Gu W., Yang J., …, Long C.L. (2014) Diarylheptanoids and phenylphenalenones from Musa itinerans fruits. Phytochemistry 103, 171–177. [DOI] [PubMed] [Google Scholar]
- López‐Gresa M.P., Maltese F., Belles J.M., Conejero V., Kim H.K., Choi Y.H. & Verpoorte R. (2010) Metabolic response of tomato leaves upon different plant–pathogen interactions. Phytochemical Analysis 21, 89–94. [DOI] [PubMed] [Google Scholar]
- Lu Y.R. & Foo L.Y. (2003) Polyphenolic constituents of blackcurrant seed residue. Food Chemistry 80, 71–76. [Google Scholar]
- Luis J.G., Echeverri F., Quiñones W., Brito I., Lopez M., Torres F. … Rojas M. (1993) Irenolone and emenolone – two new types of phytoalexin from Musa paradisiaca . Journal of Organic Chemistry 58, 4306–4308. [Google Scholar]
- Luis J.G., Fletcher W., Echeverri F., Abad T., Kishi M. & Perales A. (1995) New phenalenone‐type phytoalexins from Musa acuminata (Colla AAA) Grand Nain. Natural Product Letters 6, 23–30. [Google Scholar]
- Luis J.G., Fletcher W.Q., Echeverri F. & Grillo T.A. (1994) Phenalenone‐type phytoalexins from Musa acuminata. Synthesis of 4‐phenylphenalenones. Tetrahedron 50, 10963–10970. [Google Scholar]
- Luis J.G., Lahlou E.H. & Andres L.S. (1999) 4′‐Dehydroxy‐irenolone. A new phytoanticipin from the resistant Musa selected hybrid SH‐3481. Natural Product Letters 13, 299–304. [Google Scholar]
- Martinez‐Bolanos L., Teliz‐Ortiz D., Rodriguez‐Maciel J.C., Mora‐Aguilera J.A., Nieto‐Angel D., Cortes‐Flores J.I., …, Silva‐Aguayo G. (2012) Fungicides resistance on Mycosphaerella fijiensis populations of southeastern Mexico. Agrociencia 46, 707–717. [Google Scholar]
- Morrissey J.P. & Osbourn A.E. (1999) Fungal resistance to plant antibiotics as a mechanism of pathogenesis. Microbiology and Molecular Biology Reviews 63, 708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouliom‐Pefoura A. (1999) First observation of the breakdown of high resistance in Yangambi km 5 (Musa sp.) to the black leaf Sigatoka disease in Cameroon. Plant Disease 83, 78. [DOI] [PubMed] [Google Scholar]
- Munde T., Brand S., Hidalgo W., Maddula R.K., Svatos A. & Schneider B. (2013) Biosynthesis of tetraoxygenated phenylphenalenones in Wachendorfia thyrsiflora . Phytochemistry 91, 165–176. [DOI] [PubMed] [Google Scholar]
- Najbjerg H., Young J.F. & Bertram H.C. (2011) NMR‐based metabolomics reveals that conjugated double bond content and lipid storage efficiency in HepG2 cells are affected by fatty acid cis/trans configuration and chain length. Journal of Agricultural and Food Chemistry 59, 8994–9000. [DOI] [PubMed] [Google Scholar]
- Otálvaro F., Echeverri F., Quiñones W., Torres F. & Schneider B. (2002a) Correlation between phenylphenalenone phytoalexins and phytopathological properties in Musa and the role of a dihydrophenylphenalene triol. Molecules 7, 331–340. [Google Scholar]
- Otálvaro F., Görls H., Hölscher D., Schmitt B., Echeverri F., Quiñones W. & Schneider B. (2002b) Dimeric phenylphenalenones from Musa acuminata and various Haemodoraceae species. Crystal structure of anigorootin. Phytochemistry 60, 61–66. [DOI] [PubMed] [Google Scholar]
- Otálvaro F., Jitsaeng K., Munde T., Echeverri F., Quiñones W. & Schneider B. (2010) O‐Methylation of phenylphenalenone phytoalexins in Musa acuminata and Wachendorfia thyrsiflora . Phytochemistry 71, 206–213. [DOI] [PubMed] [Google Scholar]
- Otálvaro F., Nanclares J., Vasquez L.E., Quiñones W., Echeverri F., Arango R. & Schneider B. (2007) Phenalenone‐type compounds from Musa acuminata var. “Yangambi km 5” (AAA) and their activity against Mycosphaerella fijiensis . Journal of Natural Products 70, 887–890. [DOI] [PubMed] [Google Scholar]
- Otálvaro F., Quiñones W., Echeverri F. & Schneider B. (2004) Synthesis of [phenyl‐13C6]lachnanthocarpone and other 13C‐labelled phenylphenalenones. Journal of Labelled Compounds & Radiopharmaceuticals 47, 147–159. [Google Scholar]
- Parker D., Beckmann M., Zubair H., Enot D.P., Caracuel‐Rios Z., Overy D.P., … Draper J. (2009) Metabolomic analysis reveals a common pattern of metabolic re‐programming during invasion of three host plant species by Magnaporthe grisea . Plant Journal 59, 723–737. [DOI] [PubMed] [Google Scholar]
- Pauli G.F., Jaki B.U. & Lankin D.C. (2005) Quantitative H‐1 NMR: development and potential of a method for natural products analysis. Journal of Natural Products 68, 133–149. [DOI] [PubMed] [Google Scholar]
- Pedras M.S.C. & Ahiahonu P.W.K. (2005) Metabolism and detoxification of phytoalexins and analogs by phytopathogenic fungi. Phytochemistry 66, 391–411. [DOI] [PubMed] [Google Scholar]
- Pedras M.S.C. & Taylor J.L. (1993) Metabolism of the phytoalexin brassinin by the blackleg fungus. Journal of Natural Products 56, 731–738. [Google Scholar]
- Peláez J., Vásquez, L. , Díaz, T. , Castañeda, D. , Rodríguez, E. & Arango, R. (2006) Use of micro titer plate dilution assay to measure activity of antifungal compounds against Mycosphaerella fijiensis, Morelet. Revista Facultad Nacional de Agronomía Medellín 59, 3425–3433. [Google Scholar]
- Perrier X., De Langhe E., Donohue M., Lentfer C., Vrydaghs L., Bakry F., …, Denham T. (2011) Multidisciplinary perspectives on banana (Musa spp.) domestication. Proceedings of the National Academy of Sciences of the United States of America 108, 11311–11318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ploetz R.C. (2000) Black Sigatoka. Pesticide Outlook 11, 19–23. [Google Scholar]
- Ploetz R.C. (2006) Fusarium wilt of banana is caused by several pathogens referred to as Fusarium oxysporum f. sp. cubense . Phytopathology 96, 653–656. [DOI] [PubMed] [Google Scholar]
- Quiñones W., Escobar G., Echeverri F., Torres F., Rosero Y., Arango V., … Gallego A. (2000) Synthesis and antifungal activity of Musa phytoalexins and structural analogs. Molecules 5, 974–980. [Google Scholar]
- Ramsay A., Fliniaux O., Fang J.J., Molinie R., Roscher A., Grand E., …, Mesnard F. (2014) Development of an NMR metabolomics‐based tool for selection of flaxseed varieties. Metabolomics 10, 1258–1267. [Google Scholar]
- Scharte J., Schoen H. & Weis E. (2005) Photosynthesis and carbohydrate metabolism in tobacco leaves during an incompatible interaction with Phytophthora nicotianae . Plant, Cell and Environment 28, 1421–1435. [Google Scholar]
- Schmitt B., Hölscher D. & Schneider B. (2000) Variability of phenylpropanoid precursors in the biosynthesis of phenylphenalenones in Anigozanthos preissii . Phytochemistry 53, 331–337. [DOI] [PubMed] [Google Scholar]
- Sequeira L. (1998) Bacterial wilt: the missing element in international banana improvement programs In Bacterial Wilt Disease (eds Prior P., Allen C. & Elphinstone J.), pp. 6–14. Springer, Berlin. [Google Scholar]
- Shimomura H., Sashida Y. & Adachi T. (1987) Phenolic glucosides from Prunus grayana . Phytochemistry 26, 249–251. [Google Scholar]
- Spotts R.A. & Cervantes L.A. (1986) Effects of fungicides that inhibit ergosterol biosynthesis on apple Powdery mildew control, yield, and fruit‐growth factors. Plant Disease 70, 305–306. [Google Scholar]
- Strack D., Heilemann J., Wray V. & Dirks H. (1989) Structures and accumulation patterns of soluble and insoluble phenolics from Norway spruce needles. Phytochemistry 28, 2071–2078. [Google Scholar]
- Torres J.M., Calderon H., Rodriguez‐Arango E., Morales J.G. & Arango R. (2012) Differential induction of pathogenesis‐related proteins in banana in response to Mycosphaerella fijiensis infection. European Journal of Plant Pathology 133, 887–898. [Google Scholar]
- Vaganan M.M., Ravi I., Nandakumar A., Sarumathi S., Sundararaju P. & Mustaffa M.M. (2014) Phenylpropanoid enzymes, phenolic polymers and metabolites as chemical defenses to infection of Pratylenchus coffeae in roots of resistant and susceptible bananas (Musa spp.). Indian Journal of Experimental Biology 52, 252–260. [PubMed] [Google Scholar]
- Vishnevetsky J., White T.L., Palmateer A.J., Flaishman M., Cohen Y., Elad Y., …, Perl A. (2011) Improved tolerance toward fungal diseases in transgenic Cavendish banana (Musa spp. AAA group) cv. Grand Nain. Transgenic Research 20, 61–72. [DOI] [PubMed] [Google Scholar]
- Wang Z., Jia C.H., Li J.Y., Huang S.Z., Xu B.Y. & Jin Z.Q. (2015) Activation of salicylic acid metabolism and signal transduction can enhance resistance to Fusarium wilt in banana (Musa acuminata L. AAA group, cv. Cavendish). Functional & Integrative Genomics 15, 47–62. [DOI] [PubMed] [Google Scholar]
- Zhang D.L., Yang Y.F., Leakey J.E.A. & Cerniglia C.E. (1996) Phase I and phase II enzymes produced by Cunninghamella elegans for the metabolism of xenobiotics. FEMS Microbiology Letters 138, 221–226. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Chemical structures of the metabolites identified by 1D‐ and 2D NMR analysis in ‘Williams’ and ‘KTR’ Musa varieties.
Figure S2. Partial 1H NMR spectra showing the aromatic signals of dopamine and dopaol‐β‐D‐glucoside in a: infected leaf areas (A, see Fig. 2); b: non‐infected areas from (D); c: control Musa ‘Williams’ variety.
Figure S3. Fungal growth (data are normalized to the average growth of untreated control cultures) of M. fijiensis strain Ca10_13 after treatment with different concentrations of dopamine and dopaol‐β‐D‐glucoside.
Figure S4. Total content of phenylphenalenone‐type compounds (PP) produced by the Musa varieties ‘Williams’ and ‘KTR’ during 25 and 50 dpi with two different M. fijiensis strains. Letters a ‐ d indicate significant differences among treatments (One‐Way ANOVA, Holm‐Sidak post hoc test: P<0.001).
Figure S5. Data obtained from incubation of M. fijiensis strain E22 with different doses (0.01, 0.02 and 0.04 mg mL−1) of phenylphenalenones for 8 d.
Figure S6. Biomass produced by M. fijiensis strain Ca10_13 under in vitro treatment with different phenylphenalenone‐type compounds assessed at 0.01, 0.02 and 0.04 mg mL−1 for 8 d.
Figure S7. Soluble extracellular protein in in vitro culture medium of M. fijiensis strain Ca10_13 after incubation with phenylphenalenone‐type compounds at 0.01, 0.02 and 0.04 mg mL−1.
Figure S8. Ergosterol production by M. fijiensis strain Ca10_13 after in vitro incubationwith different phenylphenalenone‐type compounds at 0.01, 0.02 and 0.04 mg mL−1.
Figure S9. LC‐ESIMS analysis of anigorufone (9) and its metabolite in themethanolic extract of M. fijiensis Ca10_13mycelium. Panel a:UVchromatogramat 254 nm displaying peaks of anigorufone (R t = 15.3 min) and an anigorufone‐derived metabolite (R t = 7.8 min) identified as anigorufone‐2‐sulfate. Panel b: Extracted ion chromatogram (EIC) at m/z 353.2 [M +1]+, the mass corresponding to anigorufone‐2‐sulfate; Panel c: Mass spectrum of anigorufone‐2‐sulfate obtained at R t = 7.8 min showing the molecular ion peak m/z 353.2 [M+H]+ and the fragment ion peak of m/z 273.2, corresponding to anigorufone (9).
Table S1. Black leaf streak disease symptoms developed by both M. fijiensis strains (E22 and Ca10_13) in ‘Williams’ and ‘KTR’ Musa varieties.
Table S2. Metabolites identified by 1D‐ and 2D‐NMR analysis in Musa varieties ‘Williams’ and ‘KTR’ and their up‐ and downregulation after infection with M. fijiensis E22 as inferred from the corresponding loading plots.
Supporting Information