

ABSTRACT
Our knowledge on sodium and water homeostasis and regulation continues to evolve. A considerable amount of new information in this area has emerged in recent years. This review summarizes existing and new literature and discusses complex multi‐organ effects of high‐salt and low‐water intake and role of arginine vasopressin in this process, as well as the potential clinical significance of non‐osmotic sodium storage pool and rhythmicity of urine sodium excretion. It has become clear that sodium and water dysregulation can exert profound effects on kidney and vascular health, far greater than previously recognized. Maladaptation to a combined high‐salt and low‐water intake can be linked to the growing epidemic of hypertension and chronic kidney disease.
Keywords: arginine vasopressin (AVP), chronic kidney disease (CKD), hypertension, non‐osmotic pool of sodium, salt and water regulation, skin
Summary at a Glance
• Na+ and water regulations are tightly connected, involving neurohumeral multi‐organ regulations of AVP, glucocorticoids, RAAS activation, muscle catabolism, urea generation and salt‐retention.
• High‐salt and low‐water intake, common in the general population, is potentially pathogenic and linked to the genesis of hypertension and CKD.
• The tissue (mainly skin and muscle) storage pool of Na+ likely participates in Na+ and water homeostasis; heavier tissue Na+ storage may be pathological and has been associated with resistant hypertension and left ventricular hypertrophy.
• When appropriate, a modest increase (∼0.5 ‐1.5 L/day) in water intake and reduction in dietary salt to a recommended range can blunt AVP increase and will likely be beneficial.
Although classically considered separately, sodium (Na+) is associated more with volume status in the body, and water more with body‐fluid tonicity, mounting evidence indicates a complex and tight link between Na+ and water regulation, involving multiple organ systems. An update on this topic with a focus on kidney‐related regulations is provided. The role of Na+‐overload and water insufficiency in the development of hypertension (HTN) and chronic kidney disease (CKD), as well as the potential benefits of dietary modifications, is discussed.
SALT AND WATER REGULATION, AN UPDATE
Regulation of salt and water is highly influenced by the circulating levels of vasopressin (arginine vasopressin [AVP], also known as antidiuretic hormone, ADH). AVP is produced by hypothalamus magnocellular neurons (MCN) of the paraventricular and supraoptic nuclei and parvocellular neurons. AVP is not only stored in the posterior pituitary and released upon stimulation (detailed below), but also travels through portal vessels to the anterior pituitary, where AVP stimulates adrenocorticotropic hormone (ACTH) release, thus participating in the regulation of glucocorticoids. Additionally, AVP stimulates cortisol release from the adrenal medulla.1, 2
Arginine vasopressin is secreted to the circulation in an equimolar ratio with copeptin, a 39‐animo acid glycopeptide, cleaved from the C‐terminal part of the AVP precursor. Compared to AVP, copeptin is more stable and easily measurable (median level in healthy adults, <5 pmol/L3, 4). Copeptin levels track nicely with the circulating levels of AVP, both at the baseline and in response to stimulations. It is considered a surrogate marker for AVP.
Arginine vasopressin exerts its functions through interaction with its receptors, which are a group of G‐protein‐coupled (V1 and V2) receptors. V1 receptors (V1R) signal through 1,2‐DAG signalling pathways and are further divided into V1aR and V1bR. V1aRs are predominantly expressed in the vascular smooth muscle cells, where they mediate vasoconstriction. V1aRs are also expressed in cardiac myocytes, kidney vasa recta, medullary interstitial cells and collecting duct principle cells. Precise functions of V1aRs in these systems are being actively investigated. Additionally, V1aR participates in habit‐forming circadian signals.5 V1bRs are expressed in the anterior pituitary cells and adrenal medulla, mediating AVP‐stimulated ACTH and cortisol release.6 V2Rs signal through adenylyl cyclase (primarily adenylyl cyclase type 6) signalling pathways. V2Rs are expressed mainly in the kidney collecting ducts, regulating water absorption through controlling aquaporin 2 (AQP2) gene expression and protein distribution (Figure 1A,C). V2R‐mediated water absorption is critical to body‐fluid tonicity and is functionally non‐redundant; loss‐of‐function and gain‐of‐function mutations in V2Rs induce clinically significant nephrogenic diabetes insipidus and syndrome of inappropriate ADH, respectively.7, 8, 9
Figure 1.
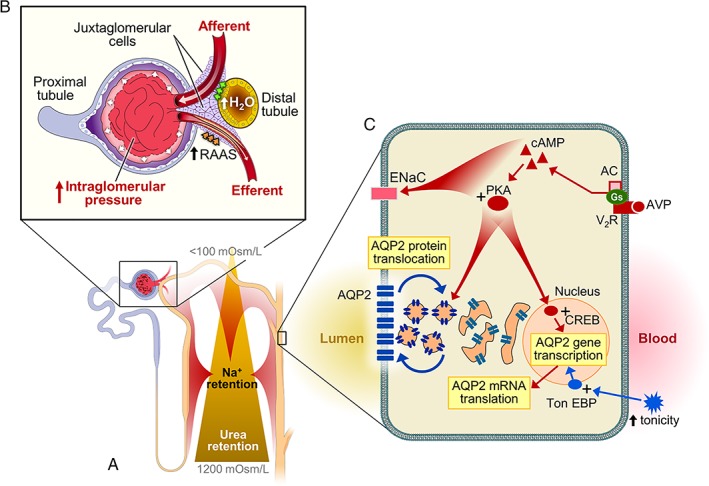
(A) and (B) Arginine vasopressin (AVP)‐mediated intraglomerular hypertension (HTN), a consequence of combined afferent vasodilation (modulation of tubuloglomerular feedback, TGF) and efferent vasoconstriction (activation of renin angiotensin‐aldosterone system, RAAS). AVP positively influences renal sodium absorption at the levels of thick‐ascending limb and distal tubules. It also increases urea cycling and intramedullary urea retention by upregulating urea transporters A1 (UT‐A1) and A3 (UT‐A3) expression. (C) AVP V2 receptor‐mediated regulation of aquaporin 2 (AQP2) and ENaC. Additionally, hypertonicity of extracellular fluids can efficiently activate tonicity response element binding protein (TonEBP) which upregulates AQP2 expression (independent of AVP). TonEBP also upregulates the expression of osmoprotective genes in the renal epithelial cells, critical in preserving medullary structural integrity.
Major physiological signals for the posterior pituitary AVP release are serum osmolality elevation, hypovolemia and circadian rhythmicity. Serum osmolality is an exquisitely sensitive but less potent AVP stimulant. A small rise in osmolality, even within physiological range (280–295 mOsm/kg), can trigger a brisk rise of circulating AVP. Hypovolemia, on the other hand, is a potent but much less sensitive stimulant. Greater than ~7% of volume depletion is needed to trigger AVP release. Once triggered, the rise can be sustained and amounts to a large magnitude. Circadian rhythmicity of AVP, peaking at around midnight, is habit‐forming, independent of serum osmolality and volume status. Insufficient AVP release can be associated with enuresis. In 2017, the Food and Drug Administration USA (FDA) approved the clinical use of desmopressin acetate (a selective V2 receptor agonist) for patients with nocturnal polyuria. An habitual V1aR‐mediated signal, triggering water‐intake prior to sleep in anticipation of a no‐water‐intake period (sleeping) has also been described.5
Arginine vasopressin‐mediated water absorption and Na+ retention are accomplished via V2‐mediated effects in the kidneys (Figure 1A,C). In the distal nephrons, through engaging V2 receptors, AVP activates adenylyl cyclase‐mediated cyclic AMP/PKA pathways, positively influencing AQP2 gene transcription, mRNA translation and protein translocation to the apical membrane. AVP also participates in the building of intramedullary hyperosmolality to facilitate osmotic‐gradient‐driven water absorption via AQP2. AVP enhances Na+ absorption at multiple tubular segments including the thick‐ascending limb of Henle (NKCC2), distal convoluted tubules (NCC) and collecting ducts (ENaC). It also increases urea cycling and medullary urea retention through upregulating the expression of urea transporters A1 (UT‐A1) and A3 (UT‐A3).10 AVP thus not only stimulates water conservation, but also retains Na+ and urea. The Na+ retentive effect, demonstrated in both animals and humans,11 can result in a positive Na+ balance. Although V1aR‐mediated counter V2R force can mitigate the Na+ retention, the risk for AVP‐provoked deleterious effects remains.12
In glomeruli, AVP exerts multiple effects (Figure 1A,B). As a result of heightened tubular Na+ and urea retention induced by AVP, tubular fluids passing through the distal nephrons contain more water/less solutes (Na+ and urea). Such a fluid composition modulates tubuloglomerular feedback (TGF) at the macula densa, causing afferent arteriole vasodilation. Simultaneously, AVP stimulates renin production in AT‐1 receptor‐dependent and ‐independent (via PKA/CREB13) manners in the macula densa and in the collecting ducts, respectively. The activation of the renin and its downstream angiotensin‐aldosterone system (RAAS) constricts efferent arterioles, resulting in glomerular hyperfiltration.14 Moreover, AVP has also been shown to stimulate mesangial contraction and proliferation.15 These collective AVP‐mediated effects are consistent with epidemiological data showing concentrated urine (high AVP state) and serum copeptin levels proportionally correlated with the occurrence and severity of proteinuria,16 a marker of long‐term kidney damage.
From an evolutionary view, AVP, mediating water and Na+ conservation, fits well with limited dietary sources of water and salt. With modernization and industrialization; however, salt has become a readily‐available and inexpensive commodity. Our daily salt intake has risen from of 1–3 g by our ancestors to an average of 10–15 g. Coupled with insufficient water intake,17 such a dietary habit creates a challenge that demands the kidneys to conserve water but dispose of excess salt. The kidneys, in order to conserve water, have to retain extra Na+ (and urea) to strengthen the intramedullary hypertonicity. During this process, extra Na+ is being ‘saved’. The surplus of body‐fluid Na+ raises osmolality,18 stimulating AVP release. It is conceivable that the combined effects of high‐salt and low‐water intake exert a major role in the epidemics of non‐communicable chronic diseases including HTN and CKD (Figure 2).
Figure 2.
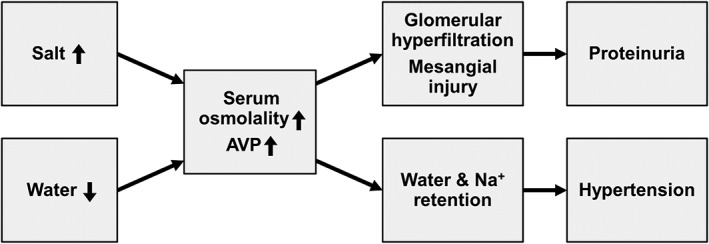
Combined effects of high‐salt and low‐water intake and their link to hypertension (HTN) and kidney dysfunction.
Na+ homeostasis has been viewed classically as almost exclusively regulated by the kidneys. This notion has been challenged; however, in light of new evidence showing multiple organ system involvement in Na+ regulation and the discovery of a non‐osmotic storage pool of Na+, potentially contributing to a unique circaseptan rhythmicity of urine Na+ excretion. Valuable information was obtained through examining a simulated space‐flight to Mars in which intake and output of healthy adults (n = 10) were precisely recorded. Two studies of 105 and 520 days yielded a tight correlation between salt intake and blood pressure elevation, as well as changes in serum and urine aldosterone (reduction) and free cortisol products (elevation).19, 20 High‐salt diet (12 g/day) raised urine Na+ without triggering extra water intake (lacking thirst stimulation). The kidneys achieve the salt‐unloading and water‐conservation through activating an array of metabolic processes, including an elevated hepatic urea genesis. Moreover, through activating glucocorticoid receptors in the skeletal muscle, high‐salt intake accelerates muscle catabolism, presumably providing building blocks and energy equivalents to facilitate hepatic urea genesis.
The discovery of a circaseptan rhythmicity of urine Na+ excretion is clinically important. It negates the value of a single 24 h urine collection as the gold‐standard for judging an individual's Na+ intake. In fact, a single 24 h urine study has a mere 50–50 predictive value. It is clear that, depending on the timing in the circaseptan, a single 24 h urine Na+ excretion could substantially deviate from one's Na+ intake, despite being in a stable condition.21 Urine Na+ excretion derived from an average of seven consecutive days yields a more accurate estimation of daily Na+ consumption.22, 23
With the advent of Na+ imaging technique, an osmolality‐ and volume‐independent pool of Na+ in the skin (epidermal and dermal regions) and muscle has been revealed,24, 25 potentially contributing to Na+ homeostasis/circaseptan rhythm. Heavy Na+ signal in the skin seems deleterious and has been shown to be an independent risk factor for refractory HTN and left ventricular hypertrophy.21, 26, 27 Along the same line, recent studies have linked high‐salt intake to vascular inflammation, impairment of endothelial surface glycocalyx layer28 and endothelial nitric oxide response, contributing to artery stiffness,29 reduced brain oxygen delivery and cognitive dysfunction.30
COMBINED SALT OVERCONSUMPTION AND UNDER‐HYDRATION (SUBOPTIMAL WATER INTAKE), CULPRITS FOR HTN AND CKD?
Except for several methodologically‐challenged studies,31 both animal and human studies have demonstrated a positive correlation between salt intake and blood pressure.32, 33 Knowledge derived in the last few years from studies of salt‐triggered metabolic reactions and maladaptive signalling mechanisms has afforded a better understanding of the underlying pathogenesis. With short‐term (<7 days) high‐salt intake, the rise in serum osmolality and, hence, AVP, is short‐lived. This is because serum osmolality/AVP‐mediated vasoconstriction is promptly sensed by baroreceptors, activating inhibitory GABAergic neurons in the hypothalamus and turning off AVP production. Chronic high‐salt intake, on the contrary, activates an aberrant pathway involving BDNF/Trk activation, which leads to KCC2 channel inhibition; the latter collapses the transmembrane Cl− gradient, causing the AVP‐producing MCN to erroneously respond to the inhibitory GABAergic signal as an AVP stimulatory signal.34, 35 In essence, sustained high‐salt intake turns the negative feedback loop into a positive one, resulting in an exaggerated AVP production and vasoconstriction.36, 37 A superimposed under‐hydration (signified by a high‐normal serum Na+ concentration) would further augment AVP secretion. Moreover, high‐salt intake has been shown to elevate endothelin‐1 (ET‐1) which participates in the regulation of the skin storage pool of Na+, influencing blood pressure and diurnal pressure rhythm.30, 38 These results are consistent with the observations of an elevated HTN risk in adults with high plasma copeptin37 and a remarkable blood pressure reductive response to long‐term dietary salt reduction.39
Turning to the contribution of salt excess and water inadequacy to the risk of kidney dysfunction, the detrimental effects have also been tied to AVP. Studies in 5/6 nephrectomized Brattleboro rats (genetically lacking AVP genes) show that, compared to their wild‐type counterparts, these rats exhibit a milder degree of kidney dysfunction.40 After given back dDAVP (a V2 receptor agonist), they developed more severe kidney damage with worsening serum creatinine, proteinuria and anaemia. Similarly, water‐drinking lowers AVP and reduces proteinuria and interstitial inflammation.41, 42 The AVP effects on albuminuria and renal function have also been demonstrated in humans.43, 44 Due to structural defects in the kidney medulla, ADPKD patients typically exhibit a degree of obligatory polyuria. The water loss, consequently, is associated with higher levels of serum osmolality and AVP (copeptin), especially after fasting. Even with preserved kidney function, an abnormal urine protein excretion is evident.44 The pathogenic role of AVP has also been demonstrated in several multicentre prospective interventional trials, showing renal protection with V2R blockade in ADPKD.45, 46
Arginine vasopressin‐mediated kidney dysfunction is not unique to ADPKD. Rather, it exerts negative effects in the non‐ADPKD population.47 Elevated copeptin in kidney transplant recipients is associated with an accelerated decline of kidney function.48 In a prospective study from 2002 to 2008 (n = 2148), concentrated urine (a high AVP state) was significantly associated with a more rapid and progressive loss of kidney function.49 Similarly, Ponte et al. examined data from December 2009 to March 2013 (n = 1128), showing that circulating copeptin is positively correlated with the occurrence of proteinuria, declining kidney function and kidney shrinkage.50 Recently, in a 5 year prospective study of non‐CKD and non‐diabetes adults (n = 12 041), Kuwabara et al. show that serum Na+ concentration (in the range of 137–147 mmol/L) and calculated serum osmolality, despite falling within reference range, are positively associated with a cumulative incidence of CKD (eGFR of <60 mL/min per BSA).51 A recent multicenter interventional study of CKD stage 3 patients (n = 631) showed a smaller decline in serum creatinine clearance (no significant change in the eGFR) resulting from a one‐year increase in self‐reported fluid intake by 0.6 to 0.8 L/day; this was associated with a reduction in serum copeptin (‐2.2 pmoL/L).52
SUMMARY
Na+ and water regulation is complex, involving neuro‐humeral multi‐organ regulations of AVP, glucocorticoids, RAAS activation, muscle catabolism, urea generation and salt‐retention. Several lines of evidence lend strong support to a pathogenic role of high‐salt and low‐water intake, common in the general population,17 in the genesis of HTN and CKD. Moreover, although detailed regulatory mechanisms are yet to be elucidated, the non‐osmotic storage pool of Na+ in the skin/muscle likely participates in the Na+ and water homeostasis; heavier tissue Na+ storage may be pathological and has recently been suggested to be a risk for resistant HTN and left ventricular hypertrophy.26, 27 Given even a modest increase (~0.5–1.5 L/day) in water intake and reduction in dietary salt can blunt increments of AVP and blood pressure,39, 53, 54 public education to raise awareness and promote these preventive measures would be cost effective and beneficial.
DISCLOSURE
The author has no conflict of interest to report.
REFERENCES
- 1. Christ‐Crain M, Fenske W. Copeptin in the diagnosis of vasopressin‐dependent disorders of fluid homeostasis. Nat. Rev. Endocrinol. 2016; 12: 168–76. [DOI] [PubMed] [Google Scholar]
- 2. Torres VE. Vasopressin receptor antagonists, heart failure, and polycystic kidney disease. Annu. Rev. Med. 2015; 66: 195–210. [DOI] [PubMed] [Google Scholar]
- 3. Morgenthaler NG, Struck J, Alonso C, Bergmann A. Assay for the measurement of copeptin, a stable peptide derived from the precursor of vasopressin. Clin. Chem. 2006; 52: 112–9. [DOI] [PubMed] [Google Scholar]
- 4. Bhandari SS, Loke I, Davies JE, Squire IB, Struck J, Ng LL. Gender and renal function influence plasma levels of copeptin in healthy individuals. Clin. Sci. (Lond.) 2009; 116: 257–63. [DOI] [PubMed] [Google Scholar]
- 5. Gizowski C, Zaelzer C, Bourque CW. Clock‐driven vasopressin neurotransmission mediates anticipatory thirst prior to sleep. Nature 2016; 537: 685–8. [DOI] [PubMed] [Google Scholar]
- 6. Thibonnier M, Preston JA, Dulin N, Wilkins PL, Berti‐Mattera LN, Mattera R. The human V3 pituitary vasopressin receptor: Ligand binding profile and density‐dependent signaling pathways. Endocrinology 1997; 138: 4109–22. [DOI] [PubMed] [Google Scholar]
- 7. Fujiwara TM, Bichet DG. Molecular biology of hereditary diabetes insipidus. J. Am. Soc. Nephrol. 2005; 16: 2836–46. [DOI] [PubMed] [Google Scholar]
- 8. Feldman BJ, Rosenthal SM, Vargas GA et al Nephrogenic syndrome of inappropriate antidiuresis. N. Engl. J. Med. 2005; 352: 1884–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Decaux G, Vandergheynst F, Bouko Y, Parma J, Vassart G, Vilain C. Nephrogenic syndrome of inappropriate antidiuresis in adults: High phenotypic variability in men and women from a large pedigree. J. Am. Soc. Nephrol. 2007; 18: 606–12. [DOI] [PubMed] [Google Scholar]
- 10. Fenton RA, Flynn A, Shodeinde A, Smith CP, Schnermann J, Knepper MA. Renal phenotype of UT‐A urea transporter knockout mice. J. Am. Soc. Nephrol. 2005; 16: 1583–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bankir L, Bichet DG, Bouby N. Vasopressin V2 receptors, ENaC, and sodium reabsorption: A risk factor for hypertension? Am. J. Physiol. Renal Physiol. 2010; 299: F917–28. [DOI] [PubMed] [Google Scholar]
- 12. Bankir L, Perucca J, Weinberger MH. Ethnic differences in urine concentration: Possible relationship to blood pressure. Clin. J. Am. Soc. Nephrol. 2007; 2: 304–12. [DOI] [PubMed] [Google Scholar]
- 13. Gonzalez AA, Cifuentes‐Araneda F, Ibaceta‐Gonzalez C et al Vasopressin/V2 receptor stimulates renin synthesis in the collecting duct. Am. J. Physiol. Renal Physiol. 2016; 310: F284–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bouby N, Ahloulay M, Nsegbe E, Déchaux M, Schmitt F, Bankir L. Vasopressin increases glomerular filtration rate in conscious rats through its antidiuretic action. J. Am. Soc. Nephrol. 1996; 7: 842–51.8793792 [Google Scholar]
- 15. Ganz MB, Pekar SK, Perfetto MC, Sterzel RB. Arginine vasopressin promotes growth of rat glomerular mesangial cells in culture. Am. J. Physiol. 1988; 255: F898–906. [DOI] [PubMed] [Google Scholar]
- 16. Meijer E, Bakker SJL, Halbesma N, de Jong PE, Struck J, Gansevoort RT. Copeptin, a surrogate marker of vasopressin, is associated with microalbuminuria in a large population cohort. Kidney Int. 2010; 77: 29–36. [DOI] [PubMed] [Google Scholar]
- 17. Ferreira‐Pego C, Guelinckx I, Moreno LA et al Total fluid intake and its determinants: Cross‐sectional surveys among adults in 13 countries worldwide. Eur. J. Nutr. 2015; 54: 35–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Suckling RJ, He FJ, Markandu ND, MacGregor GA. Dietary salt influences postprandial plasma sodium concentration and systolic blood pressure. Kidney Int. 2012; 81: 407–11. [DOI] [PubMed] [Google Scholar]
- 19. Kitada K, Daub S, Zhang Y et al High salt intake reprioritizes osmolyte and energy metabolism for body fluid conservation. J. Clin. Invest. 2017; 127: 1944–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rakova N, Kitada K, Lerchl K et al Increased salt consumption induces body water conservation and decreases fluid intake. J. Clin. Invest. 2017; 127: 1932–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Titze J, Dahlmann A, Lerchl K et al Spooky sodium balance. Kidney Int. 2014; 85: 759–67. [DOI] [PubMed] [Google Scholar]
- 22. Rakova N, Jüttner K, Dahlmann A et al Long‐term space flight simulation reveals infradian rhythmicity in human Na(+) balance. Cell Metab. 2013; 17: 125–31. [DOI] [PubMed] [Google Scholar]
- 23. Titze J, Rakova N, Kopp C, Dahlmann A, Jantsch J, Luft FC. Balancing wobbles in the body sodium. Nephrol. Dial. Transplant. 2016; 31: 1078–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Machnik A, Neuhofer W, Jantsch J et al Macrophages regulate salt‐dependent volume and blood pressure by a vascular endothelial growth factor‐C‐dependent buffering mechanism. Nat. Med. 2009; 15: 545–52. [DOI] [PubMed] [Google Scholar]
- 25. Wiig H, Schröder A, Neuhofer W et al Immune cells control skin lymphatic electrolyte homeostasis and blood pressure. J. Clin. Invest. 2013; 123: 2803–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kopp C, Linz P, Dahlmann A et al 23Na magnetic resonance imaging‐determined tissue sodium in healthy subjects and hypertensive patients. Hypertension 2013; 61: 635–40. [DOI] [PubMed] [Google Scholar]
- 27. Schneider MP, Raff U, Kopp C et al Skin sodium concentration correlates with left ventricular hypertrophy in CKD. J. Am. Soc. Nephrol. 2017; 28: 1867–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Schierke F, Wyrwoll MJ, Wisdorf M et al Nanomechanics of the endothelial glycocalyx contribute to Na(+)‐induced vascular inflammation. Sci. Rep. 2017; 7: 46476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. London GM, Marchais SJ, Guerin AP, Metivier F, Adda H. Arterial structure and function in end‐stage renal disease. Nephrol. Dial. Transplant. 2002; 17: 1713–24. [DOI] [PubMed] [Google Scholar]
- 30. Faraco G, Brea D, Garcia‐Bonilla L et al Dietary salt promotes neurovascular and cognitive dysfunction through a gut‐initiated TH17 response. Nat. Neurosci. 2018; 21: 240–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Campbell N, Correa‐Rotter R, Neal B, Cappuccio FP. New evidence relating to the health impact of reducing salt intake. Nutr. Metab. Cardiovasc. Dis. 2011; 21: 617–9. [DOI] [PubMed] [Google Scholar]
- 32. Fernandes S, Bruneval P, Hagege A, Heudes D, Ghostine S, Bouby N. Chronic V2 vasopressin receptor stimulation increases basal blood pressure and exacerbates deoxycorticosterone acetate‐salt hypertension. Endocrinology 2002; 143: 2759–66. [DOI] [PubMed] [Google Scholar]
- 33. Denton D, Weisinger R, Mundy NI et al The effect of increased salt intake on blood pressure of chimpanzees. Nat. Med. 1995; 1: 1009–16. [DOI] [PubMed] [Google Scholar]
- 34. Choe KY, Han SY, Gaub P et al High salt intake increases blood pressure via BDNF‐mediated downregulation of KCC2 and impaired baroreflex inhibition of vasopressin neurons. Neuron 2015; 85: 549–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kim YB, Kim YS, Kim WB et al GABAergic excitation of vasopressin neurons: Possible mechanism underlying sodium‐dependent hypertension. Circ. Res. 2013; 113: 1296–307. [DOI] [PubMed] [Google Scholar]
- 36. Afsar B. Pathophysiology of copeptin in kidney disease and hypertension. Clin. Hypertens. 2017; 23: 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Schoen T, Hohmann EM, van der Lely S et al Plasma copeptin levels and ambulatory blood pressure characteristics in healthy adults. J. Hypertens. 2015; 33: 1571–9. [DOI] [PubMed] [Google Scholar]
- 38. Speed JS, Heimlich JB, Hyndman KA et al Endothelin‐1 as a master regulator of whole‐body Na+ homeostasis. FASEB J. 2015; 29: 4937–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sacks FM, Svetkey LP, Vollmer WM et al Effects on blood pressure of reduced dietary sodium and the Dietary Approaches to Stop Hypertension (DASH) diet. DASH‐Sodium Collaborative Research Group. N. Engl. J. Med. 2001; 344: 3–10. [DOI] [PubMed] [Google Scholar]
- 40. Bouby N, Hassler C, Bankir L. Contribution of vasopressin to progression of chronic renal failure: Study in Brattleboro rats. Life Sci. 1999; 65: 991–1004. [DOI] [PubMed] [Google Scholar]
- 41. Bouby N, Bachmann S, Bichet D, Bankir L. Effect of water intake on the progression of chronic renal failure in the 5/6 nephrectomized rat. Am. J. Physiol. 1990; 258: F973–9. [DOI] [PubMed] [Google Scholar]
- 42. Sugiura T, Yamauchi A, Kitamura H et al High water intake ameliorates tubulointerstitial injury in rats with subtotal nephrectomy: Possible role of TGF‐beta. Kidney Int. 1999; 55: 1800–10. [DOI] [PubMed] [Google Scholar]
- 43. Bardoux P, Bichet DG, Martin H et al Vasopressin increases urinary albumin excretion in rats and humans: Involvement of V2 receptors and the renin‐angiotensin system. Nephrol. Dial. Transplant. 2003; 18: 497–506. [DOI] [PubMed] [Google Scholar]
- 44. Zittema D, Boertien WE, van Beek AP et al Vasopressin, copeptin, and renal concentrating capacity in patients with autosomal dominant polycystic kidney disease without renal impairment. Clin. J. Am. Soc. Nephrol. 2012; 7: 906–13. [DOI] [PubMed] [Google Scholar]
- 45. Torres VE, Higashihara E, Devuyst O et al Effect of tolvaptan in autosomal dominant polycystic kidney disease by CKD stage: Results from the TEMPO 3:4 trial. Clin. J. Am. Soc. Nephrol. 2016; 11: 803–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Torres VE, Chapman AB, Devuyst O et al Multicenter, open‐label, extension trial to evaluate the long‐term efficacy and safety of early versus delayed treatment with tolvaptan in autosomal dominant polycystic kidney disease: The TEMPO 4:4 trial. Nephrol. Dial. Transplant. 2017; 32: 1262. [DOI] [PubMed] [Google Scholar]
- 47. Corradi V, Martino F, Gastaldon F et al Copeptin levels and kidney function in ADPKD: Case‐control study. Clin. Nephrol. 2016; 86: 147–53. [DOI] [PubMed] [Google Scholar]
- 48. Meijer E, Bakker SJL, de Jong PE et al Copeptin, a surrogate marker of vasopressin, is associated with accelerated renal function decline in renal transplant recipients. Transplantation 2009; 88: 561–7. [DOI] [PubMed] [Google Scholar]
- 49. Clark WF, Sontrop JM, Macnab JJ et al Urine volume and change in estimated GFR in a community‐based cohort study. Clin. J. Am. Soc. Nephrol. 2011; 6: 2634–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ponte B, Pruijm M, Ackermann D et al Copeptin is associated with kidney length, renal function, and prevalence of simple cysts in a population‐based study. J. Am. Soc. Nephrol. 2015; 26: 1415–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kuwabara M, Hisatome I, Roncal‐Jimenez CA et al Increased serum sodium and serum osmolarity are independent risk factors for developing chronic kidney disease; 5 year cohort study. PLoS One 2017; 12: e0169137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Clark WF, Sontrop JM, Huang SH, Gallo K, Moist L, House AA, Cuerden, MS, Weir MA, Bagga A, Brimble S et al Effect of Coaching to Increase Water Intake on Kidney Function Decline in Adults With Chronic Kidney Disease: The CKD WIT Randomized Clinical Trial. JAMA 2018, 319, 1870–1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lemetais G, Melander O, Vecchio M, Bottin JH, Enhörning S, Perrier ET. Effect of increased water intake on plasma copeptin in healthy adults. Eur. J. Nutr. 2017, 57(5): 1883–1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sontrop JM, Huang SH, Garg AX et al Effect of increased water intake on plasma copeptin in patients with chronic kidney disease: Results from a pilot randomised controlled trial. BMJ Open 2015; 5: e008634. [DOI] [PMC free article] [PubMed] [Google Scholar]