

Abstract
Objective
Fcγ receptor IIb (FcγRIIb) is an essential negative regulator of B cells that blocks B cell receptor (BCR) signaling and triggers c‐Abl–dependent apoptosis of B cells. FcγRIIb‐deficient mice display splenomegaly with expansion of B cells, leading to lupus. FcγRIIb‐I232T is a hypofunctional polymorphism associated with lupus susceptibility in humans, an autoimmune disease linked to diminished deletion of autoreactive B cells. In the context of the FcγRIIb‐I232T polymorphism, we investigated the role of FcγRIIb in the deletion of low‐affinity germinal center (GC) B cells, an important mechanism for preventing autoimmunity.
Methods
We generated FcγRIIb232T/T mice to mimic human FcγRIIb‐I232T carriers and immunized mice with chicken gamma globulin (CGG)–conjugated NP, a T cell–dependent antigen, to examine the response of GC B cells.
Results
Compared to wild‐type (WT) mice, FcγRIIb232T/T mice showed increased numbers of low‐affinity NP‐specific IgG and NP‐specific B cells and plasma cells; additionally, the expression of a somatic mutation (W33L) in their VH186.2 genes encoding high‐affinity BCR was reduced. Notably, FcγRIIb232T/T mice had a higher number of GC light zone B cells and showed less apoptosis than WT mice, despite having equivalent follicular helper T cell numbers and function. Moreover, phosphorylation of c‐Abl was reduced in FcγRIIb232T/T mice, and treatment of WT mice with the c‐Abl inhibitor nilotinib during the peak of GC response resulted in reduced affinity maturation reminiscent of FcγRIIb232T/T mice.
Conclusion
Our findings provide evidence of a critical role of FcγRIIb/c‐Abl in the negative selection of GC B cells in FcγRIIb232T/T mice. Importantly, our findings indicate potential benefits of up‐regulating FcγRIIb expression in B cells for treatment of systemic lupus erythematosus.
Fcγ receptor IIb (FcγRIIb) is a low‐affinity Fcγ receptor for IgG. The Fc portion of IgG binds to the second Ig domain located near the transmembrane region of FcγRIIb proteins 1, 2. In B cells, FcγRIIb is an indispensable inhibitory regulator. FcγRIIb‐deficient mice exhibit splenomegaly due to expansion of B cells and eventually develop lupus‐like disease 3, 4. Depending on the affinity of antigens to the B cell receptor (BCR), FcγRIIb can transduce 2 distinct inhibitory signals upon stimulation of IgG immune complexes (ICs) to block B cell function 5. When FcγRIIb is co‐ligated to the BCR, FcγRIIb blocks BCR signaling for proliferation and differentiation, and when independently engaged, FcγRIIb triggers B cell apoptosis by a c‐Abl–dependent mechanism 1, 2, 5. When the BCR and FcγRIIb are co‐engaged, the cytoplasmic immunoreceptor tyrosine‐based inhibition motif of FcγRIIb is phosphorylated by the Lyn kinase, followed by recruitment of the lipid phosphatase SH2 domain–containing inositol‐5′‐phosphatase (SHIP), which hydrolyzes PI3, 4, 5P3 to antagonize phosphatidylinositol 3‐kinase signals for activation and proliferation of B cells 6, 7, 8. On the other hand, when the antigen in IgG ICs has low or no affinity for BCRs, FcγRIIb can directly trigger apoptosis of B cells via c‐Abl kinase 5. The FcγRIIb‐dependent apoptosis of B cells has been proposed to play a role in the elimination of autoreactive B cells, which emerge as low‐affinity B cells in the germinal center (GC) 9, but evidence from in vivo studies is largely lacking.
The human FcγRIIb‐I232T polymorphic variant, in which the isoleucine at position 232 of FcγRIIb is replaced by threonine, is a risk allele for systemic lupus erythematosus (SLE). The prevalence of FcγRIIb‐I232T carriers has been reported to be up to 40% of SLE patients in Africans and Southeast Asians 10, 11, 12. Biochemical and imaging analyses have revealed a decreased association of FcγRIIb‐232T proteins with lipid microdomains on the plasma membranes, resulting in blocking the association with BCR that results in inhibitory signaling 13, 14, 15. Nevertheless, people carrying the FcγRIIb‐232T allele are protected against malaria infection owing to enhanced antibody response 12, 16, 17. Conversely, these subjects are susceptible to autoimmune diseases, e.g., SLE 12. Consistent with these findings, the surface expression of wild‐type (WT) FcγRIIb in memory B cells and plasma cells (PCs) is down‐regulated in patients with SLE 18, 19, 20. Furthermore, a failure to up‐regulate FcγRIIb expression on GC B cells has been found in lupus‐prone mice regardless of their genetic background 21. These findings strongly suggest a role of FcγRIIb in the GC response and raise the question of whether the hypofunctional FcγRIIb‐232T allele might result in abnormality in the clonal selection of B cells in GCs, particularly in the deletion of low‐affinity autoreactive B cells.
The GC is a critical site for antigen‐driven selection of GC B cells for differentiation into PCs to generate high‐affinity antibodies for protective immunity. In response to antigen, GC B cells first undergo V(D)J gene hypermutation of their BCRs in the dark zone, followed by migration of GC B cells to the adjacent light zone for selection of cells with high affinity to antigen, a critical process known as affinity maturation 22, 23, 24. Importantly, while high‐affinity GC B cells are positively selected for further development into memory B cells and PCs, GC B cells carrying mutated BCRs of low or no antigenic affinity are negatively selected for apoptosis 25, 26. To investigate the pathogenesis of human lupus associated with the FcγRIIb‐I232T polymorphism, we generated FcγRIIb232T/T mice to mimic human FcγRIIb‐I232T carriers. Given that IgG ICs are readily formed after secondary immunization 27, 28, the surface expression level of FcγRIIb in GC B cells is up‐regulated 21, and FcγRIIb activation can trigger apoptosis of B cells via c‐Abl 5, we reasoned that the FcγRIIb‐232T allele with reduced inhibitory function might result in abnormal negative selection of GC B cells. Whether the dysfunction of the FcγRIIb‐I232T polymorphism is linked to a GC defect is virtually unexplored. In addition, the consequences of abnormal GC reaction in the pathogenesis of autoimmune diseases are incompletely understood. Importantly, new insights into the causal relationship between the FcγRIIb‐I232T polymorphism and the pathogenesis of SLE may provide valuable implications for therapeutic exploitation of FcγRIIb for patients with SLE and perhaps other autoimmune diseases.
Materials and Methods
Reagents. Chicken gamma globulin–conjugated NP20 (NP20‐CGG), bovine serum albumin (BSA)–conjugated NP7, BSA‐conjugated NP30, and phycoerythrin (PE)–conjugated NP were purchased from LGC Biosearch Technologies. Imject Alum adjuvant was acquired from Thermo Scientific. F(ab′)2 goat anti‐mouse IgG and IgM antibodies were purchased from Jackson ImmunoResearch. Mouse IgG isotypes and monoclonal antibodies (mAb) specific for CD16/32 (clone 2.4G2), PE–Cy7–conjugated CD19 (clone 1D3), BV421‐conjugated CD138 (clone 281‐2), PerCP–Cy5.5–conjugated CD11b (clone M1/70), Alexa Fluor 700–conjugated CD11c (clone HL3), and fluorescein isothiocyanate (FITC)–conjugated inducible costimulator (ICOS) (clone 7E.17G9) were purchased from BioLegend. Allophycocyanin (APC)–Cy7–conjugated B220 (clone RA3‐6B2), BV605‐conjugated CD86 (clone GL1), Alexa Fluor 647–conjugated GL‐7 (clone GL‐7), BV421‐conjugated CXCR4 (clone 2B11), PE–Cy7–conjugated CD95 (clone Jo2), Alexa Fluor 647–conjugated CD4 (clone RM4‐5), BV421‐conjugated programmed death 1 (PD‐1; clone J43), PE‐conjugated CXCR5 (clone 2G8), and 7‐aminoactinomycin D (7‐AAD) were acquired from BD Biosciences. Ninety‐six–well MultiScreen‐HTS filter plates were acquired from Merck Millipore. Blood lancets were obtained from MEDIpoint. Mouse reference serum was acquired from Bethyl Laboratories. Vectastain ABC kits containing biotinylated goat anti‐rabbit IgG and rabbit anti‐goat IgG mAb were purchased from Vector. Horseradish peroxidase (HRP)–conjugated isotype IgG and polyclonal antibodies specific to phospho–c‐Abl (Y245) were obtained from Santa Cruz Biotechnology. The active caspase 3 mAb was purchased from Cell Signaling Technology. Nilotinib and DMSO were obtained from Selleckchem.
FcγRIIb 232T/T mice and immunization protocols. FcγRIIb232T/T mice on a C57BL/6J background were generated at the gene knockout mouse core facility at the Center of Genomic Medicine of National Taiwan University (NTU). The ATT codon of isoleucine 231 in exon 5 of the Fcgr2b gene was mutated to ACT to encode threonine using a recombineering approach. A neo gene cassette flanked with loxP sequences was inserted into the intron 5 region. The targeting vector was then linearized for electroporation into JM8A3 embryonic stem cells (ESCs). Correctly targeted ESC clones were subsequently injected into C57BL/6 blastocysts to produce chimeras. Chimeric males were bred with C57BL/6 females to produce FcγRIIb232I/T mice. To remove the neo cassette, FcγRIIb232I/T mice were crossed with Sox2‐Cre mice (Tg(Sox2‐cre)1Amc/J), which were kindly provided by Dr. Ming‐Ji Fann (National Yang‐Ming University, Taipei, Taiwan). Male and female FcγRIIb232I/T mice were bred to generate offspring carrying FcγRIIb232I/I (WT), FcγRIIb232I/T (heterozygote), or FcγRIIb232T/T (homozygote) genotypes for experiments. All mice were maintained in specific pathogen–free conditions at the Center for Laboratory Animals in the College of Medicine of NTU. The protocols of animal use were reviewed, and the experiments were performed according to the guidelines approved by the Institutional Animal Care and Use Committee of the College of Medicine of NTU. Female mice (7–8 weeks old) were immunized with 50 μg NP20‐CGG per mouse by intraperitoneal injection. Nilotinib (2 mg/kg/day) was administered intraperitoneally once a day on days 7–9 after secondary immunization. Mice were killed the day after the last injection.
Flow cytometric analysis. Mouse splenocytes were stained with FITC‐conjugated CD16/32 mAb for 10 minutes at 4°C, followed by addition of an antibody cocktail containing PE–Cy7–conjugated CD19, BV421‐conjugated CD138, PerCP–Cy5.5–conjugated CD11b, Alexa Fluor 700–conjugated CD11c, Alexa Fluor 647–conjugated GL‐7, and PE‐conjugated NP for 20 minutes on ice. To distinguish GC light zone from dark zone B cells, mouse splenocytes were stained with FITC‐conjugated CD16/32, APC–Cy7–conjugated B220, BV605‐conjugated CD86, Alexa Fluor 647–conjugated GL‐7, BV421‐conjugated CXCR4, PE–Cy7–conjugated CD95, and PE‐conjugated NP. Splenic follicular helper T (Tfh) cells were stained with the following mAb: APC–Cy7–conjugated B220, Alexa Fluor 647–conjugated CD4, BV421‐conjugated PD‐1, PE‐conjugated CXCR5, FITC‐conjugated ICOS, and BV605‐conjugated CD69. Bone marrow cells were stained with FITC‐conjugated CD16/CD32, BV421‐conjugated CD138, PE–Cy7–conjugated CD19, PerCP–Cy5.5–conjugated CD11b, Alexa Fluor 700–conjugated CD11c, and PE‐conjugated NP. Dead splenocytes and bone marrow cells were stained with 7‐AAD for 5 minutes before being washed. Cells were processed for analysis using a multicolor LSRFortessa cytometer (BD Biosciences). Data were analyzed using FlowJo version 10.
Confocal microscopy. Splenic B cells from 8‐week‐old WT and FcγRIIb232T/T mice were isolated (>98% purity) using a mouse B lymphocyte enrichment kit (catalog no. 557792; BD Biosciences) according to the manufacturer's instructions. Purified cells (2 × 106/ml) were incubated with rabbit anti‐mouse IgM (25 μg/ml) for 10 minutes on ice followed by Cy3‐labeled goat anti‐rabbit IgG (50 μg/ml) and FITC‐labeled cholera toxin B (10 μg/ml), which binds the lipid raft resident protein ganglioside GM1, for an additional 10 minutes. Cells were then placed on a shaker (200 revolutions per minute) and incubated at 25°C for the indicated times. After a brief wash, cells were immediately fixed with 4% paraformaldehyde for 10 minutes at room temperature before mounting on slides. Images were acquired, analyzed, and quantified using a Zeiss LSM 880 confocal microscope.
Enzyme‐linked immunosorbent assay (ELISA). BSA‐conjugated NP7 or BSA‐conjugated NP30 (5 μg/ml) was added to 96‐well high bind plates (100 μl/well; Corning) and incubated at 4°C overnight. Mouse serum samples were diluted to detect IgG (1:200,000) and IgM (1:15,000). After blocking and incubation at 4°C overnight, plates were washed, followed by addition of HRP‐conjugated rabbit anti‐mouse IgG (Fcγ‐specific) (catalog no. 115‐035‐071; Jackson ImmunoResearch) or goat anti‐mouse IgM (μ‐specific) (catalog no. 115‐035‐075; Jackson ImmunoResearch) for a 1‐hour incubation at room temperature. After washes, plates were developed with tetramethylbenzidine substrate and the reaction was quenched with 2N H2SO4. Plates were read at an optical density of 450 nm (OD450 nm) and OD570 nm using an ELISA plate reader (BioTek). The reading values of HRP activities were calculated using OD450 nm minus OD570 nm. Standard curves of IgM and IgG concentrations were generated using serially diluted samples of mouse reference serum.
Enzyme‐linked immunospot (ELISpot) assay. ELISpot assay was performed as previously described 29, 30, except that BSA‐conjugated NP7 and BSA‐conjugated NP30 were used as the immobilized antigens to capture NP‐specific PCs. Briefly, HRP‐conjugated goat anti‐mouse IgG and IgM were used for detection of PCs. Approximately 2 × 104 cells/well and 1.6 × 105 cells/well with 2‐fold serial dilutions for the detection of IgG and IgM PCs, respectively, were incubated overnight at 37°C. Spots in wells were developed by addition of 50 μl per well of 3‐amino‐9‐ethylcarbazole substrates and incubation for 30 minutes. After washes and complete air dry, plates were scanned to enumerate spots using a CTL S6 universal analyzer (Cellular Technology).
Immunohistochemical examination. Immunohistochemical analysis of mouse spleen sections was performed using standard procedures with Vectastain ABC kits. Deparaffinized sections (4 μm thick) were stained overnight at 4°C with antibodies specific to active caspase 3 or phospho–c‐Abl according to the manufacturer's instructions. After washes with phosphate buffered saline (PBS)–Tween (PBS buffer with 0.1% [volume/volume] Tween 20), sections were incubated with a species‐specific antibody conjugated with HRP for 1 hour at ambient temperature. Slides were washed thoroughly, followed by addition of 3,3′‐diaminobenzidine substrates for development. Sections were counterstained with hematoxylin before mounting. Slides were photographed using an Axioplan 2 light microscope (Zeiss).
Sequence analysis of the V H 186.2 region of BCRs from NP+ GC B cells. Genomic DNA of sorted GC B cells (NP+B220+GL‐7+IgG+;˜1,000 cells per mouse) was extracted using a QIAamp DNA micro kit (Qiagen). For the amplification of VH186.2–JH2 segments, 1 ng of genomic DNA was used as template, and polymerase chain reaction (PCR) products were generated from high‐fidelity PrimeStar DNA polymerase (Takara Bio). The PCR primers have been described previously 31: forward primer, 5′‐AGCTGTATCATGCTCTTCTTGGCA‐3′ and reverse primer, 5′‐AGATGGAGGCCAGTGAGGGAC‐3′ 31. Illumina libraries were generated from PCR products using a TruSeq library preparation kit and were sequenced using Illumina MiSeq to generate paired‐end reads of 300 nucleotides. Raw sequencing data were aligned to mouse germline VH186.2 sequences using Burrows‐Wheeler aligner and SAMtools 32, 33. The W33L mutation percentages in first complementarity‐determining region sequences were compared with those in WT mice.
Statistical analysis. Bar graphs were plotted and analyzed using GraphPad Prism software version 6.0. Student's unpaired 2‐tailed t‐test was used for statistical analysis. The t‐test was modified by Welch's correction in case of unequal variance. Tukey's test with one‐way analysis of variance was used to compare multiple groups. Results are presented as the mean ± SEM. P values less than 0.05 were considered significant.
Results
FcγRIIb 232T/T mice exhibit enhanced antibody production with reduced affinity maturation. Because the isoleucine 232 residue of human FcγRIIb is conserved in mice (NCBI accession nos. NP_001070657.1 and NP_003992.3), we generated a mouse line, termed the FcγRIIb232T/T mouse line, which carries a point mutation of the isoleucine residue at position 231 (232 in humans) replaced by threonine (Figure 1A). Consistent with previous findings of live cell imaging 15, the surface FcγRIIb‐232T proteins and lipid rafts were neither stably associated nor co‐clustered to form cap structures in splenic B cells (see Supplementary Figure 1, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.40555/abstract).
Figure 1.
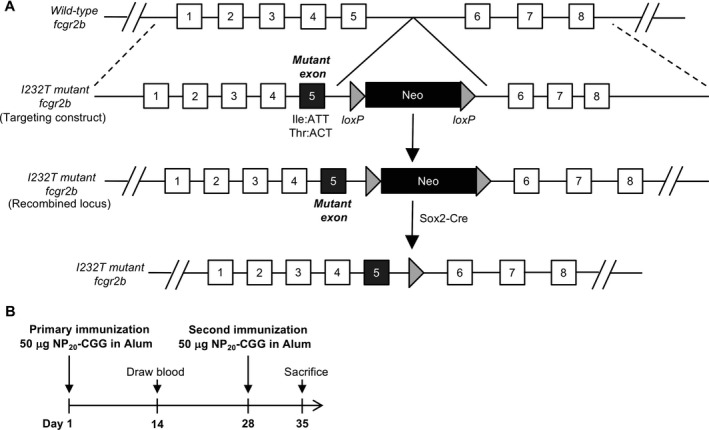
Generation of Fcγ receptor IIb (FcγRIIb)232T/T mice and immunization schedule. A, Schematic diagram of the generation of FcγRIIb‐I232T mutant mice. B, Schedule of immunization of mice with chicken gamma globulin–conjugated NP (NP20‐CGG). Wild‐type and FcγRIIb232T/T mice 7–8 weeks of age were each injected intraperitoneally with 50 μg of NP20‐CGG proteins mixed with 50 μl of alum. A second immunization was performed 4 weeks later.
To investigate GC response, we immunized each WT and FcγRIIb232T/T mouse by intraperitoneal injection with 50 μg of NP20‐CGG mixed with an equal volume of alum. All mice received a booster injection with the same amount of NP20‐CGG on day 28 after primary immunization and were killed on day 35, when clonal selection was actively proceeding 24, 25 (Figure 1B). Serum samples were collected on day 14 and day 35 and tested in ELISA plates coated with either IgG‐specific antibodies to detect total serum IgG, NP30 to detect NP‐specific antibodies of all affinities, or NP7 to detect high‐affinity NP‐specific antibodies 30, 31. The serum levels of total IgG were significantly higher in FcγRIIb232T/T mice than in WT mice following both primary immunization (day 14) (P = 0.0035) and secondary immunization (day 35) (P = 0.032) (Figure 2A).
Figure 2.
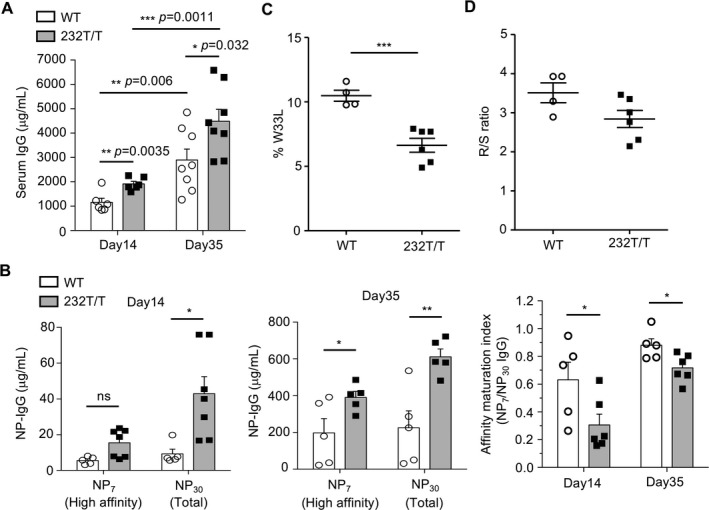
Enhanced NP‐specific IgG production in immunized Fcγ receptor IIb (FcγRIIb)232T/T mice. A and B, Serum levels of total IgG (A) and high‐affinity NP7 IgG and total NP30 IgG (B) in wild‐type (WT) mice and FcγRIIb232T/T mice 2 weeks after primary immunization (day 14) (P = 0.0035 for IgG, P = 0.5033 for NP7, and P = 0.0469 for NP30) and 1 week after secondary immunization (day 35) (P = 0.032 for IgG, P = 0.0411 for NP7, and P = 0.0068 for NP30). In B, the ratios of NP7 to NP30 IgG levels in serum on day 14 (P = 0.0427) and day 35 (P = 0.0277) after immunization are also shown. Symbols represent individual mice; bars show the mean ± SEM (n = 5–8 mice per group). C, Frequency of W33L mutation in the VH186.2 region of individual B cell receptor (BCR) genes of NP+ germinal center (GC) B cells in WT mice and FcγRIIb232T/T mice 7–8 days after primary immunization (P = 0.0008). D, Ratio of replacement mutations to silent mutations (R:S) in the VH186.2 region in NP+ GC B cells in WT mice and FcγRIIb232T/T mice (P = 0.0845). In C and D, symbols represent individual mice; horizontal and vertical lines show the mean ± SEM (n = 4 WT mice and 6 FcγRIIb232T/T mice). * = P < 0.05; ** = P < 0.01; *** = P < 0.001. NS = not significant.
Fourteen days after the primary immunization, FcγRIIb232T/T mice produced similar levels of high‐affinity NP‐specific antibodies (P = 0.5033) but more than double the amount of low‐affinity NP‐specific antibodies compared to WT mice (P < 0.05) (Figure 2B). By day 35, seven days after secondary immunization, FcγRIIb232T/T mice produced significantly more high‐affinity and low‐affinity NP‐specific antibodies than WT mice (P < 0.05 for NP7; P < 0.01 for NP30) (Figure 2B). The ratio of high‐affinity to total (low affinity plus high affinity) NP‐specific IgG (NP7‐bound IgG:NP30‐bound IgG) was lower in the serum of FcγRIIb232T/T mice than in that of WT mice, indicating reduced affinity maturation of antibodies (P < 0.05) (Figure 2B). Moreover, sequencing of the NP‐specific VH186.2 region of B cells expressing NP‐specific BCRs showed a decreased percentage of W33L replacement, which gives rise to high‐affinity BCR variants, in FcγRIIb232T/T mice (P < 0.001) (Figure 2C). In addition, we found a trend toward a lower ratio of replacement to silent hypermutation in FcγRIIb232T/T mice than in WT mice 8 days after primary immunization (Figure 2D). These results indicate a dysfunction in the affinity maturation of GC B cells in FcγRIIb232T/T mice.
Retention of low‐affinity B cells in FcγRIIb 232T/T mice after secondary immunization. Because affinity maturation was reduced in FcγRIIb232T/T mice compared to WT mice, we investigated whether the elimination of low‐affinity antigen‐specific B cells was abnormal in GCs. As shown in Figure 3A, the percentages of splenic CD19+ B cells in lymphocytes were comparable in WT and FcγRIIb232T/T mice. However, the percentage of NP+CD19+ B cells was substantially increased in FcγRIIb232T/T mice compared to WT mice after secondary immunization. Similarly, the percentage of splenic CD19+CD138+ PCs in FcγRIIb232T/T mice was not different from that in WT mice, but the percentage of NP+CD19+CD138+ PCs was significantly increased in FcγRIIb232T/T mice (P < 0.001) (Figure 3B).
Figure 3.
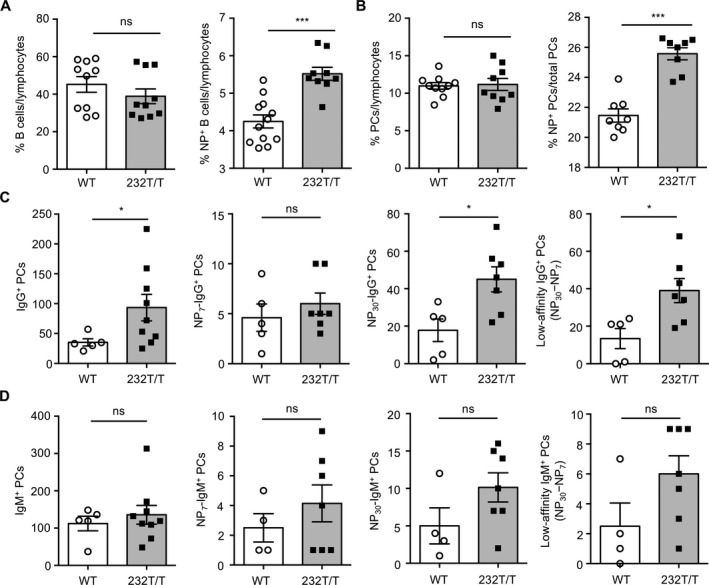
Increased frequency of low‐affinity NP+IgG+ plasma cells (PCs) in the spleen in FcγRIIb232T/T mice after secondary immunization. A and B, Percentages of CD19+ B cells (P = 0.2846) and NP+CD19+ B cells (P = 0.0007) (A) and percentages of CD19+CD138+ PCs (P = 0.8432) and NP+CD19+CD138+ PCs (P = 0.0009) (B) in splenic lymphocytes from wild‐type (WT) mice and FcγRIIb232T/T mice on day 35, analyzed by flow cytometry. C, Splenic total IgG+ PCs (P = 0.0315), high‐affinity NP7‐specific IgG+ PCs (P = 0.4318), total NP30‐specific IgG+ PCs (P = 0.0168), and low‐affinity NP+IgG+ (NP30 minus NP7) PCs (P = 0.0159) in WT mice and FcγRIIb232T/T mice on day 35, determined using enzyme‐linked immunospot assays (using 6 × 103 cells to detect total PCs and 2.4 × 104 cells to detect NP+ PCs). D, Numbers of total IgM+ PCs (P = 0.5412), NP7‐specific IgM+ PCs (P = 0.355), NP30‐specific IgM+ PCs (P = 0.116), and low‐affinity NP+IgM+ (NP30 minus NP7) PCs (P = 0.0864) in WT mice and FcγRIIb232T/T mice. Symbols represent individual mice; bars show the mean ± SEM (n = 8–12 WT mice and 8–10 FcγRIIb232T/T mice in A and B; n = 5 WT mice and 7–9 FcγRIIb232T/T mice in C; n = 4–5 WT mice and 7–9 FcγRIIb232T/T mice in D). * = P < 0.05; *** = P < 0.001. NS = not significant.
We next used ELISpot assays to quantify the numbers of splenic NP+ PCs. Consistent with an increased level of circulating NP30+ IgG, a greater number of NP30+IgG+ PCs was detected in FcγRIIb232T/T mice than in WT mice (P < 0.05) (Figure 3C). Further analysis of splenic NP+IgG+ PCs revealed no significant differences in the numbers of high‐affinity NP7+ PCs between WT and FcγRIIb232T/T mice. In contrast, compared to WT mice, the numbers of low‐affinity IgG+ PCs (NP30+ PCs minus NP7+ PCs) increased ˜3‐fold in FcγRIIb232T/T mice (P < 0.05) (Figure 3C). These differences were not observed when the numbers of NP+IgM+ PCs were compared between WT and FcγRIIb232T/T mice (Figure 3D), suggesting a specific IgG‐associated effect through FcγRIIb. Of interest, we found that the numbers of low‐affinity NP+IgG+ PCs were also significantly increased in heterozygous FcγRIIb‐232T mice compared to WT mice (see Supplementary Figure 2, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.40555/abstract). These findings suggest that FcγRIIb‐232T might impede recruitment of sufficient amounts of WT receptors to reach the threshold required for efficient induction for apoptosis. Consistent with this notion, we previously demonstrated that FcγRIIb‐mediated apoptosis is dependent on the signal strength transduced from the receptor oligomers 5.
Tfh cell number and function in switching IgG isotypes are not altered in FcγRIIb 232T/T mice. Because the generation of PCs is influenced by Tfh (CD4+B220+PD‐1+ICOS+) cells, which provide help, e.g., interleukin‐21, to GC B cells to further their differentiation into PCs and to class‐switch Ig isotypes 32, 33, we examined the numbers of splenic Tfh cells to determine their contributions to the increase in NP+IgG+ PCs in FcγRIIb232T/T mice. As shown in Figure 4A, the numbers of Tfh cells were comparable between immunized WT and FcγRIIb232T/T mice (P = 0.8622) despite an increased number of NP+ PCs in FcγRIIb232T/T mice (Figure 3). Consistent with increased serum levels of total IgG in FcγRIIb232T/T mice (Figure 2A), the serum concentrations of IgG1, IgG2a, and IgG3 isotypes were all significantly higher in immunized FcγRIIb232T/T mice than WT mice (P < 0.05) (Figure 4B). Similarly, the serum levels of high‐affinity (NP7)–specific and total NP+ (NP30)–specific IgG isotypes remained higher in FcγRIIb232T/T mice than in WT mice (Figures 4C and D).
Figure 4.
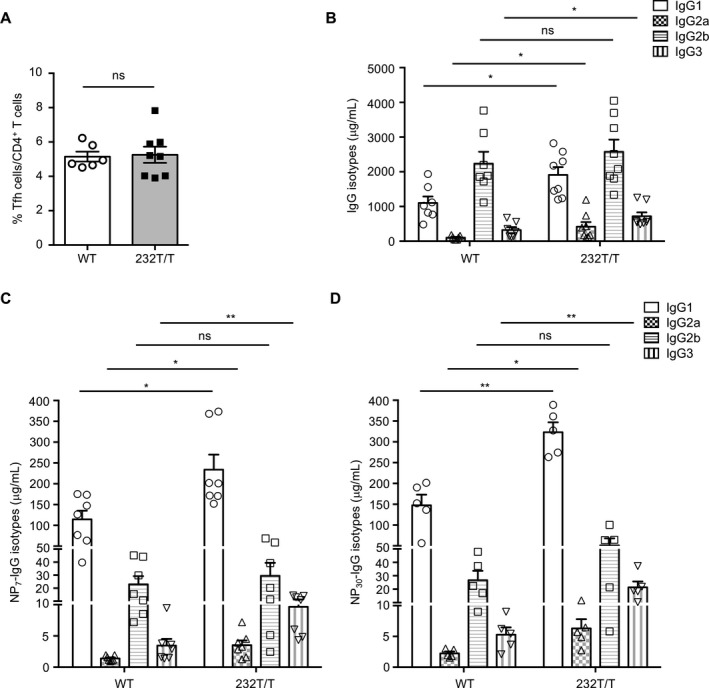
Splenic follicular helper T (Tfh) cell numbers and serum titers of class‐switched NP+ IgG isotypes in wild‐type (WT) and Fcγ receptor IIb (FcγRIIb)232T/T mice. A, Percentages of Tfh cells (CD4+B220− programmed death 1–positive inducible costimulator–positive) in splenic T cells in WT mice and FcγRIIb232T/T mice on day 35 (P = 0.8622). B, Serum concentrations of IgG1 (P = 0.0098), IgG2a (P = 0.0076), IgG2b (P = 0.9813), and IgG3 (P = 0.045) in immunized WT mice and FcγRIIb232T/T mice. C and D, Serum levels of high‐affinity NP7‐specific IgG isotypes (IgG1 [P = 0.0137], IgG2a [P = 0.0178], IgG2b [P = 0.5956], and IgG3 [P = 0.0091]) (C) and total NP30‐specific IgG isotypes (IgG1 [P = 0.0011], IgG2a [P = 0.0283], IgG2b [P = 0.217], and IgG3 [P = 0.0079]) (D) in WT and FcγRIIb232T/T mice. Symbols represent individual mice; bars show the mean ± SEM (n = 6 WT mice and 8 FcγRIIb232T/T mice in A; n = 7 WT mice and 8 FcγRIIb232T/T mice in B; n = 5–7 mice per group in C and D). * = P < 0.05; ** = P < 0.01. NS = not significant.
Reduced apoptosis of B cells in GC light zones of immunized FcγRIIb 232T/T mice. To further delineate the abnormality of GC response in FcγRIIb232T/T mice, we quantified NP+CD19+GL‐7+ GC B cells and found that FcγRIIb232T/T mice exhibited more splenic NP+ GC B cells than WT mice (Figure 5A). When GC dark zone (CD86lowCXCR4high) and light zone (CD86highCXCR4low) B cells were analyzed and compared, we found a decreased percentage of GC dark zone B cells (P < 0.001) (Figure 5A) but an increased percentage of GC light zone B cells (P < 0.001) (Figure 5A) in FcγRIIb232T/T mice. Moreover, we detected an increase in the size of GCs in splenic sections from immunized FcγRIIb232T/T mice (Figure 5B). It has been shown that the surface expression of FcγRIIb in GC B cells is up‐regulated in normal mice 16. We examined the FcγRIIb expression levels in GC B cells and found no significant differences in FcγRIIb expression between WT and FcγRIIb232T/T mice either before or after immunization (Figure 5C). Because clonal selection of GC B cells occurs primarily in the light zone of GCs 17, 18, 19, 20, 21, 22, we next investigated the apoptosis of GC B cells to determine the extent of negative selection of low‐affinity B cells after secondary immunization. Consistent with an increase in GC B cell numbers in FcγRIIb232T/T mice, the percentage of dead GC B cells was significantly decreased in these mice (P < 0.05) (Figure 5D). Significantly fewer apoptotic GC B cells, which were stained by active caspase 3 mAb, were detected in FcγRIIb232T/T mice after secondary immunization (Figure 5D).
Figure 5.
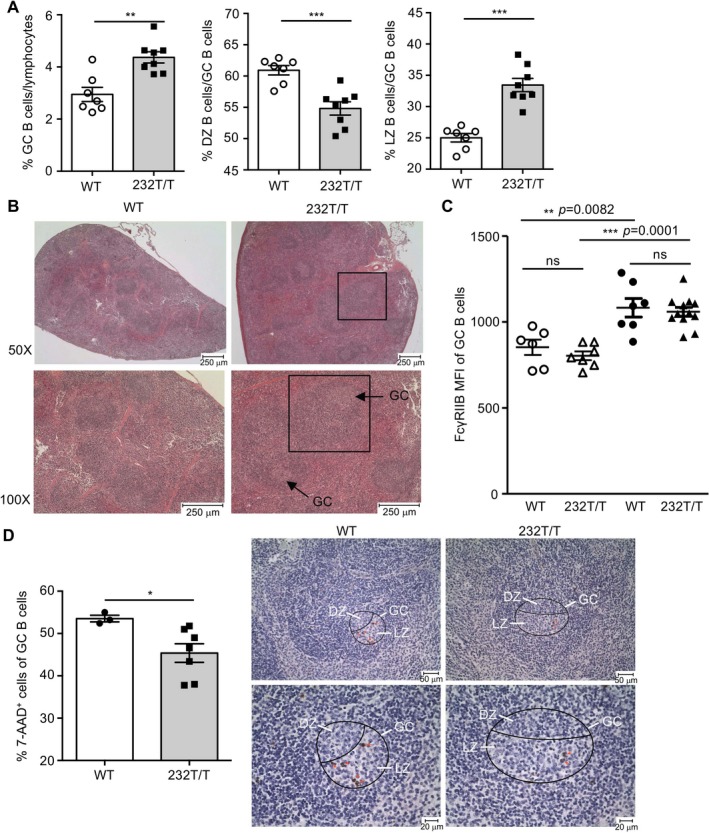
Increased numbers of light zone (LZ) germinal center (GC) B cells and reduced numbers of apoptotic GC B cells in Fcγ receptor IIb (FcγRIIb)232T/T mice after secondary immunization. A, Percentages of splenic CD19+GL‐7+ GC B cells (P = 0.0011), dark zone (DZ) NP+ GC B cells (P = 0.0005), and light zone NP+ GC B cells (P = 0.0002) in WT mice and FcγRIIb232T/T mice. Symbols represent individual mice; bars show the mean ± SEM (n = 7 WT mice and 8 FcγRIIb232T/T mice). B, Representative splenic sections from WT and FcγRIIb232T/T mice showing the size of GCs. Bottom panels are higher‐magnification views of the top panels. The boxed areas show follicles. C, Surface expression levels of FcγRIIb on GC B cells in age‐matched nonimmunized WT and FcγRIIb232T/T mice (open symbols) (P = 0.3613) and immunized WT and FcγRIIb232T/T mice on day 35 (solid symbols) (P = 0.7026). There was a significant difference in expression of FcγRIIb in nonimmunized WT mice versus immunized WT mice (P = 0.0082) and in nonimmunized FcγRIIb232T/T mice versus immunized FcγRIIb232T/T mice (P = 0.0001). Symbols represent individual mice; horizontal and vertical lines show the mean ± SEM (n = 6 nonimmunized WT mice, 7 nonimmunized FcγRIIb232T/T mice, 7 immunized WT mice, and 11 immunized FcγRIIb232T/T mice). MFI = mean fluorescence intensity. D, Left, Percentages of 7‐aminoactinomycin D (7‐AAD)+GL‐7+ GC B cells in WT mice and FcγRIIb232T/T mice (P = 0.0484). Symbols represent individual mice; bars show the mean ± SEM (n = 3 WT mice and 7 FcγRIIb232T/T mice). Right, Staining for active caspase 3 to detect apoptotic GC B cells in the light zone of GCs (encircled areas) in splenic sections from WT and FcγRIIb232T/T mice. * = P < 0.05; ** = P < 0.01; *** = P < 0.001. NS = not significant.
Blocking c‐Abl activity in WT mice during clonal selection recapitulates the GC phenotype of FcγRIIb 232T/T mice. Because FcγRIIb is known to mediate apoptosis via c‐Abl kinase in response to IgG ICs in B cells 5, we investigated the expression of active c‐Abl (p‐Y245) proteins in GC B cells. In WT mice, phospho–c‐Abl proteins were readily detectable and mainly localized in the light zone of GCs, where affinity maturation and clonal selection occur. In contrast, the levels of phospho–c‐Abl proteins were substantially decreased in the GCs of FcγRIIb232T/T mice (Figure 6A). It has been reported that the apoptosis of GC B cells peaks during days 7–9 after secondary immunization 34. Thus, to determine whether c‐Abl activity is crucial for FcγRIIb to negatively regulate GC B cells, we treated WT mice with nilotinib (2 mg/kg/day) to block c‐Abl kinase activity during the peak period when GC B cells undergo apoptosis for selection after secondary immunization. Indeed, the serum titers of NP+ IgG displayed reduced affinity maturation in nilotinib‐treated mice (P < 0.05) (Figure 6B). Moreover, the number of low‐affinity NP+IgG+ PCs (NP30 PCs minus NP7 PCs) was significantly increased after c‐Abl kinase activity was blocked in WT mice (P < 0.01) (Figure 6C). Thus, the findings in antibodies produced from nilotinib‐treated WT mice are reminiscent of the GC defect observed in FcγRIIb232T/T mice after secondary immunization (Figures 2 and 3).
Figure 6.
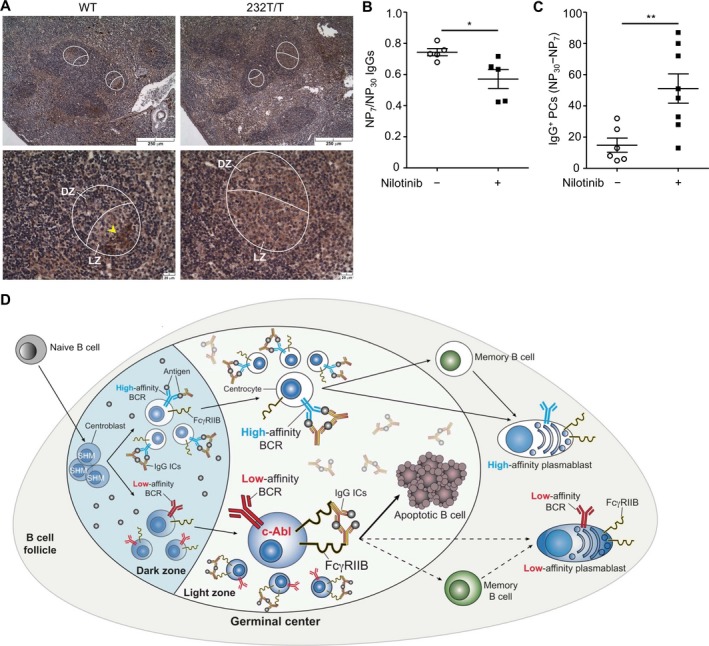
Association of reduced c‐Abl activation with retention of low‐affinity germinal center (GC) B cells in mice after secondary immunization. A, Reduced staining of phospho–c‐Abl (Y245)+ GC B cells (arrowhead) in Fcγ receptor IIb (FcγRIIb)232T/T mice compared to wild‐type (WT) mice. Female WT mice (7–8 weeks old) were immunized as described in Figure 1B. Mice were treated with either vehicle or nilotinib (2 mg/kg/day) on days 7–9 after the second immunization. Encircled areas show GCs. Bottom panels show higher‐magnification views of the top panels. B and C, Ratio of NP7 to NP30 serum IgG (P = 0.0283) (B) and number of low‐affinity NP+IgG+ (NP30 minus NP7) plasma cells in splenocytes (2.4 × 104) (P = 0.0087) in vehicle‐treated WT mice and nilotinib‐treated WT mice. Symbols represent individual mice; horizontal and vertical lines show the mean ± SEM (n = 5–6 vehicle‐treated mice and 5–8 nilotinib‐treated mice). * = P < 0.05; ** = P < 0.01. D, Illustration of the crucial role of FcγRIIb in the negative selection of low‐affinity GC B cells via c‐Abl in the light zone of GCs in response to IgG immune complexes (ICs) in secondary immunization in WT mice (solid lines). Broken lines represent FcγRIIb232T/T mice. BCR = B cell receptor; SHM = somatic hypermutation.
Discussion
In this study, we showed for the first time that compromised inhibitory activity of FcγRIIb‐232T proteins results in insufficient deletion of low‐affinity antigen‐specific GC B cells and reduces affinity maturation in clonal selection. Consequently, the resultant increase in low‐affinity antigen‐specific GC B cells leads to a corresponding increase in low‐affinity antigen‐specific IgG in circulation in FcγRIIb232T/T mice (Figures 2 and 3). Compared to WT mice, the number of GC light zone B cells was increased in FcγRIIb232T/T mice due to a decrease in the apoptosis of GC B cells and in the phosphorylation of c‐Abl proteins (Figures 5 and 6). Furthermore, administration of nilotinib to WT mice to block c‐Abl kinase activity at the peak of apoptosis of GC B cells in clonal selection resulted in reduced affinity maturation reminiscent of the phenotype of immunized FcγRIIb232T/T mice. The involvement of c‐Abl in the clonal selection of GC B cells is a novel and important finding. The newly identified, crucial role of FcγRIIb in regulating the stringency of affinity maturation by triggering apoptosis to delete low‐affinity GC B cells via c‐Abl is illustrated in Figure 6D.
Because FcγRIIb232T/T mice display a higher serum level of high‐affinity NP‐specific IgG, the reduced inhibition of FcγRIIb‐232T on BCRs might have the potential to promote the proliferation of GC B cells. Indeed, numbers of GC B cells increased in FcγRIIb232T/T mice after secondary immunization (Figure 5A). Nevertheless, the increased number of B cells was largely due to an increase in low‐affinity B cells, since no increase in high‐affinity IgG+ PCs was observed in FcγRIIb232T/T mice (Figure 3). This leads us to conclude that the retention of low‐affinity B cells is more a consequence of insufficient apoptosis rather than insufficient inhibition of proliferation of GC B cells in FcγRIIb232T/T mice. In addition, because low‐affinity B cells are intrinsically less competitive than high‐affinity cells for antigen stimulation, the increased survival of low‐affinity FcγRIIb232T/T GC B cells is positively associated with reduced apoptosis. One important caveat is that low‐affinity antigen‐specific GC B cells may have a chance to undergo further affinity maturation when the competition with high‐affinity B cells becomes reduced in late GCs 35. However, in the persistence of FcγRIIb‐232T dysfunction, repeated immunization generates a new pool of low‐affinity B cells, which normally should have been deleted. Consistent with this finding, the serum level of low‐affinity antigen‐specific IgG and the number of IgG+ PCs remained significantly increased after secondary immunization in FcγRIIb232T/T mice (Figures 2 and 3).
These findings raise the possibility of undesired consequences of enhanced immune response due to the presence of the FcγRIIb‐232T allele. For example, low‐affinity memory B cells might persist in peripheral lymphoid organs, and low‐affinity PCs emigrated from GCs into circulation might be able to become long‐term residents in the bone marrow to secrete low‐affinity antibodies 36. An additional caveat is that because serum NP+ IgG display increasing affinity maturation in WT mice over time but remain reduced in FcγRIIb232T/T mice after primary immunization, the contribution of extrafollicular response to influence the outcome of low‐affinity B cells is likely limited (Supplementary Figure 3, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.40555/abstract).
Consistent with previous findings in living cells 14, 15, FcγRIIb‐I232T proteins appear to form small raft‐associated clusters rather than coalesced caps for signal amplification as compared to receptors in WT mice at 30–60 minutes (see Supplementary Figure 1, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.40555/abstract). The resultant diminished recruitment of SHIP to FcγRIIb‐232T therefore can account for reduced inhibition on B cells in response to IgG ICs. However, mice deficient in the Ship gene show no differences from WT mice in antibody production after immunization with a T cell–dependent antigen 37. Nevertheless, when the Ship gene is specifically deleted in B cells, they are indeed more sensitive to antigen activation than WT cells in vitro 38.
Surprisingly, the number of NP+ GC B cells and the serum level of NP+ IgG decreased markedly after NP‐CGG immunization. It appears that hyperactive BCR signaling in SHIP‐deficient GC B cells directly induces apoptosis of both low‐ and high‐affinity B cells 38. Thus, a tightly regulated balance between FcγRIIb/SHIP and FcγRIIb/c‐Abl pathways in response to IgG ICs is crucial for normal outcome of GC reaction. Consistent with this notion, an increased sensitivity to FcγRIIb‐dependent apoptosis might contribute in part to the GC phenotype in mice with SHIP deficiency in B cells. Indeed, loss of SHIP in DT40 B cells enhances FcγRIIb‐induced apoptosis 9. We previously showed that apoptosis of B cells induced by FcγRIIb is dependent on c‐Abl, but independent of SHIP, suggesting a decisive role of c‐Abl when activated 5.
In the GC, low‐affinity B cells are outcompeted by high‐affinity B cells for antigen stimulation and Tfh cell help, thereby lacking survival advantages 39, 40. In the present study, we provided evidence of a new role of the FcγRIIb/c‐Abl signaling pathway to participate in the negative selection of low‐affinity B cells. Consistent with our findings, enhancing survival of GC B cells by overexpression of the Bcl‐xL gene in mice results in reduced affinity maturation 41. Similarly, mice overexpressing Bcl‐2 show decreased negative selection of GC B cells 42, 43. These findings indicate that enhanced BCR signaling can increase the survival of low‐affinity B cells to avoid negative selection. Whether these low‐affinity B cells might have overcome the apoptotic induction from FcγRIIb to escape deletion is of interest for future studies. Meanwhile, it has been demonstrated that low‐affinity autoreactive B cells are more sensitive to CpG double‐stranded DNA–induced differentiation into PCs than high‐affinity B cells 44, 45. Thus, low‐affinity autoreactive B cells are able to efficiently expand independent of antigen stimulation if they can escape from elimination in GCs. Because autoreactive B cells are routinely generated in response to a foreign antigen in normal mice, e.g., resulting from host immune response against pathogens 46, low‐affinity autoreactive B cells, which are likely generated from time to time, need to be deleted when they emerge in GCs to prevent autoimmunity.
Our findings indicate that in FcγRIIb‐232T allele carriers, the persistent presence of low‐affinity B cells and especially PCs may gradually become a key contributor that puts them at risk of developing autoimmune diseases over time. Reduced surface expression levels of WT FcγRIIb in the B cells of SLE patients may result in susceptibility to the presence of low‐affinity B cells 18, 19, 20. Accordingly, because transgenic mice overexpressing Fcgr2b in B cells exhibit reduced SLE disease severity 47, it will be of interest to investigate whether regimens that up‐regulate the surface expression level of FcγRIIb‐232T proteins to enhance their inhibition can restore competency to negatively regulate low‐affinity GC B cells for therapeutic exploitation.
Author Contributions
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Dr. Tzeng had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design
Jhou, Tzeng.
Acquisition of data
Jhou, Yu, Hwai, C.‐S. Chen, Tzeng.
Analysis and interpretation of data
Jhou, Hwai, P.‐L. Chen, Tzeng.
Supporting information
Acknowledgments
We thank Miss Yu‐Syuan You and Mr. Tsung‐Chih Tseng for excellent technical assistance. We acknowledge Dr. Shu‐Wha Lin for the technical assistance and service provided by the Gene Knockout Mouse Core of the NTU Center for Genomic Medicine, College of Medicine, National Taiwan University. We also appreciate the services provided by the staff of the First Core Laboratory at the College of Medicine, National Taiwan University and the RCF7 Laboratory of the Department of Medical Research at National Taiwan University Hospital.
Supported by the Ministry of Science and Technology of the Executive Yuan of Taiwan (grant NSC99‐2320‐B‐002‐011).
References
- 1. Daeron M. Fc receptors as adaptive immunoreceptors. Curr Top Microbiol Immunol 2014;382:131–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Espéli M, Smith KG, Clatworthy MR. FcγRIIB and autoimmunity. Immunol Rev 2016;269:194–211. [DOI] [PubMed] [Google Scholar]
- 3. Bolland S, Ravetch JV. Spontaneous autoimmune disease in FcγRIIB–deficient mice results from strain–specific epistasis. Immunity 2000;13:277–85. [DOI] [PubMed] [Google Scholar]
- 4. Tiller T, Kofer J, Kreschel C, Busse CE, Riebel S, Wickert S, et al. Development of self–reactive germinal center B cells and plasma cells in autoimmune FcγRIIB–deficient mice. J Exp Med 2010;207:2767–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tzeng SJ, Bolland S, Inabe K, Kurosaki T, Pierce SK. The B cell inhibitory Fc receptor triggers apoptosis by a novel c–Abl family kinase–dependent pathway. J Biol Chem 2005;280:35247–54. [DOI] [PubMed] [Google Scholar]
- 6. Chacko GW, Tridandapani S, Damen JE, Liu L, Krystal G, Coggeshall KM. Negative signaling in B lymphocytes induces tyrosine phosphorylation of the 145–kDa inositol polyphosphate 5–phosphatase, SHIP. J Immunol 1996;157:2234–8. [PubMed] [Google Scholar]
- 7. D'Ambrosio D, Fong DC, Cambier JC. The SHIP phosphatase becomes associated with FcγRIIB1 and is tyrosine phosphorylated during ‘negative’ signaling. Immunol Lett 1996;54:77–82. [DOI] [PubMed] [Google Scholar]
- 8. Ono M, Bolland S, Tempst P, Ravetch JV. Role of the inositol phosphatase SHIP in negative regulation of the immune system by the receptor FcγRIIB. Nature 1996;383:263–6. [DOI] [PubMed] [Google Scholar]
- 9. Pearse RN, Kawabe T, Bolland S, Guinamard R, Kurosaki T, Ravetch JV. SHIP recruitment attenuates FcγRIIB–induced B cell apoptosis. Immunity 1999;10:753–60. [DOI] [PubMed] [Google Scholar]
- 10. Siriboonrit U, Tsuchiya N, Sirikong M, Kyogoku C, Bejrachandra S, Suthipinittharm P, et al. Association of Fcγ receptor IIb and IIIb polymorphisms with susceptibility to systemic lupus erythematosus in Thais. Tissue Antigens 2003;61:374–83. [DOI] [PubMed] [Google Scholar]
- 11. Chen JY, Wang CM, Ma CC, Luo SF, Edberg JC, Kimberly RP, et al. Association of a transmembrane polymorphism of Fcγ receptor IIb (FCGR2B) with systemic lupus erythematosus in Taiwanese patients. Arthritis Rheum 2006;54:3908–17. [DOI] [PubMed] [Google Scholar]
- 12. Willcocks LC, Carr EJ, Niederer HA, Rayner TF, Williams TN, Yang W, et al. A defunctioning polymorphism in FCGR2B is associated with protection against malaria but susceptibility to systemic lupus erythematosus. Proc Natl Acad Sci U S A 2010;107:7881–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kono H, Kyogoku C, Suzuki T, Tsuchiya N, Honda H, Yamamoto K, et al. FcγRIIB Ile232Thr transmembrane polymorphism associated with human systemic lupus erythematosus decreases affinity to lipid rafts and attenuates inhibitory effects on B cell receptor signaling. Hum Mol Genet 2005;14:2881–92. [DOI] [PubMed] [Google Scholar]
- 14. Sohn HW, Pierce SK, Tzeng SJ. Live cell imaging reveals that the inhibitory FcγRIIB destabilizes B cell receptor membrane–lipid interactions and blocks immune synapse formation. J Immunol 2008;180:793–9. [DOI] [PubMed] [Google Scholar]
- 15. Xu L, Xia M, Guo J, Sun X, Li H, Xu C, et al. Impairment on the lateral mobility induced by structural changes underlies the functional deficiency of the lupus–associated polymorphism FcγRIIB–T232. J Exp Med 2016;213:2707–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Clatworthy MR, Willcocks L, Urban B, Langhorne J, Williams TN, Peshu N, et al. Systemic lupus erythematosus–associated defects in the inhibitory receptor FcγRIIb reduce susceptibility to malaria. Proc Natl Acad Sci U S A 2007;104:7169–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Waisberg M, Tarasenko T, Vickers BK, Scott BL, Willcocks LC, Molina‐Cruz A, et al. Genetic susceptibility to systemic lupus erythematosus protects against cerebral malaria in mice. Proc Natl Acad Sci U S A 2011;108:1122–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mackay M, Stanevsky A, Wang T, Aranow C, Li M, Koenig S, et al. Selective dysregulation of the FcγIIB receptor on memory B cells in SLE. J Exp Med 2006;203:2157–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Su K, Yang H, Li X, Li X, Gibson AW, Cafardi JM, et al. Expression profile of FcγRIIb on leukocytes and its dysregulation in systemic lupus erythematosus. J Immunol 2007;178:3272–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Isaák A, Gergely P Jr, Szekeres Z, Prechl J, Poór G, Erdei A, et al. Physiological up‐regulation of inhibitory receptors FcγRII and CR1 on memory B cells is lacking in SLE patients. Int Immunol 2008;20:185–92. [DOI] [PubMed] [Google Scholar]
- 21. Rahman ZS, Manser T. Failed up‐regulation of the inhibitory IgG Fc receptor FcγRIIB on germinal center B cells in autoimmune–prone mice is not associated with deletion polymorphisms in the promoter region of the FcγRIIB gene. J Immunol 2005;175:1440–9. [DOI] [PubMed] [Google Scholar]
- 22. MacLennan IC, Gray D. Antigen‐driven selection of virgin and memory B cells. Immunol Rev 1986;91:61–85. [DOI] [PubMed] [Google Scholar]
- 23. French DL, Laskov R, Scharff MD. The role of somatic hypermutation in the generation of antibody diversity. Science 1989;244:1152–7. [DOI] [PubMed] [Google Scholar]
- 24. Gitlin AD, Shulman Z, Nussenzweig MC. Clonal selection in the germinal centre by regulated proliferation and hypermutation. Nature 2014;509:637–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. MacLennan IC. Germinal centers. Annu Rev Immunol 1994;12:117–39. [DOI] [PubMed] [Google Scholar]
- 26. De Silva NS, Klein U. Dynamics of B cells in germinal centres. Nat Rev Immunol 2015;15:137–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Goins CL, Chappell CP, Shashidharamurthy R, Selvaraj P, Jacob J. Immune complex–mediated enhancement of secondary antibody responses. J Immunol 2010;184:6293–8. [DOI] [PubMed] [Google Scholar]
- 28. Qin D, Wu J, Vora KA, Ravetch JV, Szakal AK, Manser T, et al. Fcγ receptor IIB on follicular dendritic cells regulates the B cell recall response. J Immunol 2000;164:6268–75. [DOI] [PubMed] [Google Scholar]
- 29. Tzeng SJ. The isolation, differentiation, and quantification of human antibody–secreting B cells from blood: ELISpot as a functional readout of humoral immunity. J Vis Exp 2016;118:e54582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tzeng SJ, Li WY, Wang HY. FcγRIIB mediates antigen‐independent inhibition on human B lymphocytes through Btk and p38 MAPK. J Biomed Sci 2015;22:87–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dominguez‐Sola D, Victora GD, Ying CY, Phan RT, Saito M, Nussenzweig MC, et al. The proto–oncogene MYC is required for selection in the germinal center and cyclic reentry. Nat Immunol 2012;13:1083–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Li H, Durbin R. Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics 2009;25:1754–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/map format and SAMtools. Bioinformatics 2009;25:2078–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Saitoh HA, Maeda K, Yamakawa M. In situ observation of germinal center cell apoptosis during a secondary immune response. J Clin Exp Hematop 2006;46:73–82. [DOI] [PubMed] [Google Scholar]
- 35. Dal Porto JM, Haberman AM, Kelsoe G, Shlomchik MJ. Very low affinity B cells form germinal centers, become memory B cells, and participate in secondary immune responses when higher affinity competition is reduced. J Exp Med 2002;195:1215–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jacob J, Kelsoe G. In situ studies of the primary immune response to (4‐hydroxy‐3‐nitrophenyl)acetyl. Part II. A common clonal origin for periarteriolar lymphoid sheath–associated foci and germinal centers. J Exp Med 1992;176:679–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Helgason CD, Kalberer CP, Damen JE, Chappel SM, Pineault N, Krystal G, et al. A dual role for Src homology 2 domain–containing inositol‐5‐phosphatase (SHIP) in immunity: aberrant development and enhanced function of B lymphocytes in ship –/– mice. J Exp Med 2000;191:781–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Leung WH, Tarasenko T, Biesova Z, Kole H, Walsh ER, Bolland S. Aberrant antibody affinity selection in SHIP–deficient B cells. Eur J Immunol 2013;43:371–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Shulman Z, Gitlin AD, Targ S, Jankovic M, Pasqual G, Nussenzweig MC, et al. T follicular helper cell dynamics in germinal centers. Science 2013;341:673–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Shulman Z, Gitlin AD, Weinstein JS, Lainez B, Esplugues E, Flavell RA, et al. Dynamic signaling by T follicular helper cells during germinal center B cell selection. Science 2014;345:1058–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Takahashi Y, Cerasoli DM, Dal Porto JM, Shimoda M, Freund R, Fang W, et al. Relaxed negative selection in germinal centers and impaired affinity maturation in bcl–xL transgenic mice. J Exp Med 1999;190:399–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hande S, Notidis E, Manser T. Bcl‐2 obstructs negative selection of autoreactive, hypermutated antibody V regions during memory B cell development. Immunity 1998;8:189–98. [DOI] [PubMed] [Google Scholar]
- 43. Smith KG, Light A, O'Reilly LA, Ang SM, Strasser A, Tarlinton D. Bcl‐2 transgene expression inhibits apoptosis in the germinal center and reveals differences in the selection of memory B cells and bone marrow antibody–forming cells. J Exp Med 2000;191:475–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rui L, Vinuesa CG, Blasioli J, Goodnow CC. Resistance to CpG DNA–induced autoimmunity through tolerogenic B cell antigen receptor ERK signaling. Nat Immunol 2003;4:594–600. [DOI] [PubMed] [Google Scholar]
- 45. Viglianti GA, Lau CM, Hanley TM, Miko BA, Shlomchik MJ, Marshak‐Rothstein A. Activation of autoreactive B cells by CpG dsDNA. Immunity 2003;19:837. [DOI] [PubMed] [Google Scholar]
- 46. Ray SK, Putterman C, Diamond B. Pathogenic autoantibodies are routinely generated during the response to foreign antigen: a paradigm for autoimmune disease. Proc Natl Acad Sci U S A 1996;93:2019–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Brownlie RJ, Lawlor KE, Niederer HA, Cutler AJ, Xiang Z, Clatworthy MR, et al. Distinct cell–specific control of autoimmunity and infection by FcγRIIb. J Exp Med 2008;205:883–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials