

Abstract
Amenamevir (formerly ASP2151) induces cytochrome P450 (CYP)2B6 and CYP3A4 and inhibits CYP2C8. We conducted 2 studies, 1 using montelukast as a probe to assess CYP2C8 and the other bupropion to assess CYP2B6. The montelukast study examined the effect of amenamevir on the pharmacokinetics of montelukast in 24 healthy men: each subject received montelukast 10 mg alone, followed by montelukast 10 mg with amenamevir 400 mg, or vice versa after a washout period. In the bupropion study, 24 subjects received a single dose of 150 mg bupropion on days 1, 15, 22, and 29, and repeated once‐daily doses of 400 mg amenamevir on days 6‐15. Amenamevir increased peak concentration and area under the concentration‐time curve of montelukast by about 22% (ratio 121.7%, 90%CI [114.8, 129.1]; 121% [116.2, 128.4], respectively) with a similar increase in hydroxymontelukast (ratio 121.4%, 90%CI [106.4, 138.5]; 125.6 % [111.3, 141.7]). Amenamevir reduced peak concentration and area under the concentration‐time curve of bupropion by 16% (84.29%, 90%CI [78.00, 91.10]; 84.07%, 90%CI [78.85, 89.63]), with recovery after 1 week; the pharmacokinetics of the primary metabolite hydroxybupropion was unaffected. Thus, amenamevir increased plasma concentrations of montelukast and decreased those of bupropion, but it did not do so enough to require dose adjustment of coadministered substrates of either CYP2C8 or CYP2B6.
Keywords: amenamevir, montelukast, bupropion, drug‐drug interaction
Amenamevir is an orally available non–nucleoside analogue inhibitor of the helicase‐primase enzyme complex essential for herpesvirus DNA replication. It has activity against herpes simplex types 1 and 2 and varicella zoster virus and has shown promise in early clinical trials.1 Herpes simplex infection is extremely common and can lead to serious complications such as encephalitis and hepatitis, especially in immunocompromised patients. All other currently licensed medicines for herpesvirus infections act by inhibiting DNA polymerase and are first activated by viral enzymes so that they are active only in virus‐infected cells.2 The treatment of choice since the late 1970s has been acyclovir, which is activated by viral thymidine kinase, which some strains lack, leading to resistance in about 1% in the general population and 3.5% to 14% in immunocompromised patients.3 Alternative treatment options are therefore required.
The pharmacokinetics and metabolism of amenamevir have previously been investigated in preclinical and clinical studies undertaken by Maruho (unpublished data held by the company). In healthy volunteers amenamevir was rapidly absorbed following single oral doses with maximum plasma concentration (Cmax) 1.33‐2.5 hours independent of dose. During repeated dosing, Cmax and the area under the concentration‐time curve (AUC) decreased between the first dose (day 1), an intermediate dose (day 9), and the last dose (day 16), suggesting autoinduction of metabolism, probably via cytochrome P450 (CYP)3A4/5. AUC and Cmax increased less than dose proportionally in dose‐ranging studies. Plasma protein binding of 14C‐amenamevir in humans was about 75%, and t½ about 7‐8 hours. After a single oral dose of 200 mg 14C‐amenamevir, 74.6% of 14C‐radioactivity was recovered in feces. These data are consistent with later pharmacokinetic analyses of studies in healthy volunteers4 and patients.5
The metabolic profile of amenamevir was evaluated in vitro using pooled liver microsomes and cryopreserved hepatocytes of mouse, rat, rabbit, dog, and human origin. The major human metabolite was a monohydroxy derivative (AS1955888‐00, Mo4, R5), which was also detected in all other species tested.
CYP isoforms involved in amenamevir metabolism were studied in vitro using human liver microsomes. Amenamevir's metabolism correlated significantly with CYP2B6, CYP2C19, and CYP3A4/5 activity. The correlation was strongest with CYP3A4/5 (0.9236 r2 coefficient of determination, P < .0001), suggesting a major role for CYP3A4/5 in the metabolism of amenamevir, whereas correlation with CYP2B6 (0.3578 r2, P = .0144) and CYP2C19 was less marked (0.3489 r2, P = .0160). Correlation with CYP2C8 was weaker (0.1967 r2, P = .0853).
The potential for amenamevir to inhibit cytochrome metabolism was evaluated in vitro using human liver microsomes and CYP‐selective substrates. Amenamevir had weak direct inhibitory activity against CYP2C8 (IC50 69 μmol/L) but no activity against CYP2B6 in the range of concentrations studied (0.1 to 100 μmol/L) (IC50 >100 μmol/L).
To investigate the potential of amenamevir to induce CYP2B6, CYP2B6 activity and gene expression were measured in human hepatocytes with or without pretreatment for 72 hours with amenamevir. After pretreatment with 2, 20, and 200 μmol/L amenamevir, CYP2B6 activity increased 1.4‐, 1.9‐, and 2.9‐fold, respectively, and CYP2B6 gene expression increased by 1.9‐, 4.5‐, and 4.8‐fold, respectively, compared with the negative control. Those results suggest that amenamevir has the potential to induce CYP2B6.
We report here 2 studies in healthy volunteers that were part of a series of investigations to elucidate potential interactions. Two probe substrates were selected: montelukast to investigate effects of amenamevir on CYP2C8 and bupropion to test effects on CYP2B6.
Montelukast is an orally available leukotriene receptor antagonist used for the preventive treatment of asthma and seasonal allergic rhinitis. It is metabolized to its primary metabolite methyl‐hydroxymontelukast (M6) via 36‐hydroxylation by CYP2C8, which is also responsible for subsequent conversion to the secondary metabolite montelukast dicarboxylic acid (M4). CYP2C8 is thought to account for 70% to 80% of montelukast's metabolism in vivo, with most of the remainder accounted for by CYP3A4‐mediated conversion to M5a and M5b metabolites: less than 0.2% is eliminated in urine.6, 7, 8, 9, 10, 11 This, together with its benign safety profile, makes it an appropriate choice as a CYP2C8 probe in healthy subjects.
Bupropion is an orally available antidepressant and nonnicotine smoking cessation aid that is thought to exert its activity by reuptake inhibition of norepinephrine and dopamine and by noncompetitive antagonism of nicotinic acetylcholine receptors.12, 13 It is metabolized in vivo to 3 primary active metabolites: hydroxybupropion, threohydrobupropion, and erythrohydrobupropion. Hydroxybupropion has around 50% of the activity of bupropion, but, as its Cmax is 4‐7 times greater and AUC about 10‐fold greater, it is responsible for most of bupropion's activity.14, 15 Threohydrobupropion concentrations are about 5‐fold greater than that of bupropion, and erythrohydrobupropion concentrations are similar to those of bupropion, but they are only 20% as potent as the parent.16 Hydroxybupropion formation is closely correlated with CYP2B6 activity in human microsomes and with CYP2B6‐specific N‐demethylation of S‐mephenytoin and can be 95% inhibited by a CYP2B6‐specific monoclonal antibody. Bupropion's other metabolites are formed independently of cytochrome activity.17 Thus, measurement of hydroxybupropion formation can be used to investigate CYP 2B6 metabolism of bupropion.
Subjects and Methods
Both studies were done concurrently at Hammersmith Medicines Research (HMR), London, after approval by both the Medicines and Healthcare products Regulatory Agency (MHRA) and the London‐Brent Ethics Committee. The studies were conducted in accordance with the International Conference on Harmonisation Good Clinical Practice Guidelines and the ethical principles outlined in the Declaration of Helsinki. The montelukast study (EudraCT no 2014‐003955‐73) lasted from December 2014 to April 2015, and the bupropion study (EudraCT no 2014‐004656‐59) from February to April 2015.
Subjects
Each study recruited the planned number of 24 healthy male volunteers aged 18‐45 years, deemed healthy on the basis of medical history, medical examination, vital signs, electrocardiogram, laboratory safety tests on blood and urine, and urine tests for drugs of abuse (Table 1). During the study, smoking, alcohol, caffeine, all enzyme‐inducing foodstuffs, and concomitant medications were prohibited. Subjects fasted overnight until a standardized light breakfast, which they finished 30 minutes before dosing. No food or drink was then allowed until 4 hours after dosing. Subjects took standard meals and drinks at 4, 10, and 24 hours after dosing and then at standard meal times on nondosing days. Safety tests on blood and urine, vital signs, and medical examination were done at appropriate intervals throughout each study.
Table 1.
Subject Demographics
| Bupropion Subjects (N = 24) | Montelukast Subjects (N = 24) | ||
|---|---|---|---|
| Age (y) | Mean (SD) range | 32.6 (7.14) 20‐45 | 30.8 (7.1) 20‐43 |
| Race n (%) | White | 18 (75.0) | 19 (79.2) |
| Black | 3 (12.5) | 4 (16.7) | |
| Asian | 1 (4.2) | 0 | |
| Mixed race | 2 (8.4) | 1 (4.2) | |
| Ethnicity n (%) | Not Hispanic or Latino | 23 (95.8) | 24 (100.0) |
| Hispanic or Latino | 1 (4.2) | 0 | |
| Mean (SD) | 178.7 (5.32) | 180.2 (8.1) | |
| Height (cm) | Range | 169‐188 | 164‐199 |
| Mean (SD) | 78.07 (9.46) | 81.22 (12.24) | |
| Weight (kg) | Range | 64.3‐99.8 | 61.9‐104.9 |
| Mean (SD) | 24.46 (2.80) | 25.00 (3.38) | |
| BMI (kg/m2) | Range | 20.0‐30.1 | 19.4‐30.0 |
| History of smoking | n (%) | 4 (16.7) | 4 (16.7) |
| Consumes any alcohol (%) | 15 (62.5) | 17 (70.8) | |
| Units/week mean (SD) | 5.8 (3.8) | 5.7 (4.2) | |
| Alcohol consumption | Rangea | 1‐12 | 1‐17 |
BMI indicates body mass index.
Includes only those subjects who drink alcohol.
Montelukast Study
This was a randomized, single‐center, open‐label, 2‐way crossover drug‐drug interaction study to investigate the effect of a single oral dose of amenamevir on the pharmacokinetics of a single oral dose of montelukast in 24 healthy men.
Each subject received montelukast 10 mg alone, followed 2 weeks later by montelukast 10 mg with amenamevir 400 mg, or vice versa; 400 mg amenamevir was selected because it was the projected therapeutic dose daily dose in Japan. Likewise, 10 mg montelukast was selected on the basis that it is the adult daily dose for treatment of asthma and seasonal allergies in adults and is well tolerated in healthy volunteers.18
Subjects were resident on the ward from the afternoon of the day before dosing (day –1) until day 4 and subsequently returned to give blood samples for pharmacokinetic analysis at 7 and 14 days after their dose of amenamevir. Plasma samples for analysis of montelukast and methyl‐hydroxymontelukast were obtained predose and at 0.5, 1, 1.5, 2, 2.5, 3, 4, 5, 6, 8, 10, 12, 16, 20, 24, 30, 36, 48, and 72 hours after dosing. Subjects returned for a final follow‐up visit about 30 days after their last dose.
Bupropion Study
This was a single‐center, open‐label drug‐drug interaction study to investigate the effect of amenamevir‐mediated CYP2B6 induction on the probe substrate bupropion. Subjects received a single dose of 150 mg bupropion on days 1, 15, 22, and 29 and once‐daily doses of 400 mg amenamevir on days 6‐15 (Figure 1). Subjects were resident on the ward on 3 occasions: from the day before the first dose (day –1) until day 19; from day 21 until day 26; and from day 28 until day 33. Subjects returned for an outpatient visit on day 36 and a final follow‐up visit on day 45. Blood samples for assay of bupropion and hydroxybupropion were taken before each dose of bupropion and at 0.5, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 12, 24, 36, 48, 60, 72, and 96 hours afterward (Supplementary Table S1).
Figure 1.
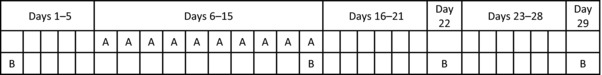
Bupropion dosing intervals. A, dosing with amenamevir 400 mg once daily; B, dosing with bupropion 150 mg.
Blood samples for assay of amenamevir were taken at predose on days 6‐14, and before and frequently up to 24 hours after dosing with amenamevir on day 15.
Safety and Tolerability Assessments
Safety and tolerability assessments included adverse events, vital signs, 12‐lead electrocardiogram, physical examination, and clinical laboratory tests.
Assays
Plasma concentrations of all compounds and metabolites were determined using validated liquid chromatography tandem mass spectrometry by Shin Nippon Biomedical Laboratories, Ltd (Tokyo, Japan) and Analytical Services International (London, UK). The lower limit of quantification (LLQ) was 5 ng/mL for amenamevir, 10 ng/mL for montelukast, 1 ng/mL for methyl‐hydroxymontelukast, 2.5 ng/mL for bupropion, and 10 ng/mL for hydroxybupropion.
Blood samples for bupropion, hydroxybupropion, montelukast, and methyl‐hydroxymontelukast were collected in lithium heparin tubes. Plasma was separated by centrifugation at ∼1500g for 10 minutes at 4°C, then stored at –20°C or below until analysis by Analytical Services International as described below.
Blood samples for amenamevir and AS1955888‐00 were collected in sodium heparin tubes and prepared as above before assay by Shin Nippon Biomedical Laboratories, as described by Adeloye et al.19
Bupropion
Preparation of Internal Standard and Calibrators
Bupropion, hydroxybupropion (calibrators), internal standard 1 (bupropion‐d9), and internal standard 2 (hydroxybupropion‐d6) were extracted from 100 μL of human plasma by protein precipitation. The internal standards were prepared by diluting 50 μL of bupropion‐d9 stock + 200 μL hydroxybupropion‐d6 stock with 20 mL acetonitrile + 20 mL deionized water to yield a final concentration of 125 ng/mL bupropion and 500 ng/mL hydroxybupropion. For calibration, a substock solution containing bupropion and hydroxybupropion was prepared by diluting 50 μL of bupropion stock (1 mg/mL) and 200 μL hydroxybupropion stock (1 mg/mL) in 20 mL of human plasma.
Analytical Method
We then added to a 2‐mL polypropylene tube 100 μL of calibrator or plasma sample, 50 μL of internal standard (125 ng/mL bupropion‐d9 and 500 ng/mL hydroxybupropion‐d6), 200 μL precipitating agent (acetonitrile), and 200 μL of deionized water.
The sample was vortexed for 2 minutes before centrifugation and 150 μL supernatant was transferred to an autosampler tube and submitted to analysis by liquid chromatography tandem mass spectrometry.
HPLC Conditions
The analytic column was an Onyx Monolithic C18 column (100 mm × 3 mm) (Phenomenex, Torrance, California). The mobile phase comprised methanol 1000 mL, deionized water 1000 mL, plus 10 g ammonium formate. Elution was isocratic at a flow rate of 300 μL/min.
MS Settings
An Applied Biosystems (Foster City, California) API4000 mass spectrometer was used with turbo ion spray and positive ionization. Mass ratios were 240.0/184.0 amu for bupropion, 249.0/185.0 amu for the bupropion standard, 256.0/167.0 amu for hydroxybupropion, and 282.0/130.0 amu for the hydroxybupropion standard.
Variability
Variabilities of bupropion and hydroxybupropion determinations are shown in Table 2.
Table 2.
Bupropion and Hydroxybupropion Variability
| Concentration | Mean | Precision | Accuracy | |
|---|---|---|---|---|
| (ng/mL) | (ng/mL) | (%CV) | (%) | |
| Bupropion | ||||
| QC1 | 5 | 4.640 | 5.1 | 92.9 |
| QC2 | 75 | 69.86 | 4.2 | 93.2 |
| QC3 | 200 | 192.5 | 3.6 | 96.3 |
| Hydroxybupropion | ||||
| QC1 | 20 | 19.24 | 7.5 | 96.2 |
| QC2 | 300 | 286.1 | 5.0 | 95.4 |
| QC3 | 800 | 781.1 | 4.9 | 97.3 |
QC indicates quality control.
Montelukast
Preparation of Internal Standard and Calibrators
Montelukast, montelukast 1,2‐diol (calibrators), and montelukast‐d6 (internal standard) were extracted from 100 μL of human plasma by protein precipitation. Working solutions at a concentration of 1000 ng/mL were prepared by diluting an aliquot (25 μL) of stock solution (1000 μg/mL) with analyte‐free plasma up to a final volume of 25 mL.
Analytical Method
We added to a 2‐mL polypropylene tube 100 μL of calibrator or plasma sample, 25 μL of internal standard (montelukast‐d6), and 500 μL of precipitating solution (acetonitrile:methanol, 1:1). The sample was vortexed for 2 minutes before centrifugation. The supernatant was transferred into a polypropylene autosampler tube, and 10 μL was injected onto the high‐precision liquid chromatography tandem mass spectrometer. Because the drug, metabolite and internal standard are all light sensitive, steps were taken to minimize exposure to light throughout.
HPLC Conditions
The column used was a Monolith PR‐18e column (100 mm × 3.0 mm). The mobile phase comprised 1000 mL acetonitrile, 1000 mL deionized water, and ammonium formate 5 g. Elution was isocratic at a flow rate of 300 μL/min.
MS Settings
An Applied Biosystems API4000 mass spectrometer was used with turbo ion spray and positive ionization. Mass ratios were 586.4/422.1 amu for montelukast, 592.3/427.1 amu for the motelukast standard, 602.4/438.0 amu for montelukast 1,2‐diol, and for hydroxybupropion, and 592.3/427.1 amu for the montelukast 1,2‐diol standard.
Variability
Variabilities of montelukast and montelukast 1,2‐diol are shown in Table 3.
Table 3.
Montelukast and Montelukast 1,2‐diol variability
| Concentration | Mean | Precision | Accuracy | |
|---|---|---|---|---|
| (ng/mL) | (ng/mL) | (%CV) | (%) | |
| Montelukast | ||||
| QC1 | 20.1 | 20.14 | 8.7 | 100.2 |
| QC2 | 150.4 | 149.6 | 7.0 | 99.5 |
| QC3 | 752.2 | 727.7 | 7.889 | 96.747 |
| Montelukast 1,2‐diol | ||||
| QC1 | 2.0 | 1.951 | 13.3 | 97.6 |
| QC2 | 14.7 | 13.70 | 13.4 | 93.2 |
| QC3 | 73.5 | 70.80 | 15.0 | 96.3 |
QC indicates quality control.
Sample Size
A sample size of 24 subjects for the montelukast study was selected based on a statistical power calculation. From published studies, estimates of between‐subject percentage coefficient of variation (%CV) for Cmax and AUC0‐∞ were 20% to 30%.7 Statistical power was estimated by 10,000 simulations using the model below. The model included treatment (montelukast in combination with amenamevir and montelukast alone), sequence, and session as fixed effects and subject as a random effect.
Given a %CV of 25, a true mean ratio of 1.0, and an acceptance limit of 80% to 125% for 90%CI, a sample size of 24 subjects would provide at least 86% power to detect an interaction, as defined by the criteria above. In practice, within‐subject variability was likely to be less than between‐subject variability.
A sample size of 24 subjects was selected for the bupropion study based on a statistical power calculation. Two previous studies of single doses of 150 mg bupropion gave within‐subject estimates of %CV for Cmax and AUC0–∞ between 15% and 30%.20, 21 Statistical power was estimated by simulation (the number of simulations was 10,000) using the model below. The model included treatment (bupropion in combination with amenamevir and bupropion alone) as a fixed effect and subject as a random effect.
Given a %CV of 22.5 (the average of 15% and 30%), a true mean ratio of 1.0, and an acceptance limit of 80% to 125% for the 90%CI, a sample size of 24 subjects would provide 93% power.
Pharmacokinetic and Statistical Analysis
Pharmacokinetic parameters were calculated using WinNonlin (Certara, Princeton, New Jersey) version 6.3. Interactions were tested using an equivalence analysis.
To assess the effect of amenamevir on montelukast, montelukast in combination with amenamevir was compared with montelukast alone. The AUC0‐∞ and Cmax were logarithmically transformed and subjected to ANOVA with treatment (montelukast alone or montelukast in combination with amenamevir), sequence, and session as fixed effects and subject as a random effect. Absence of a clinically significant effect of amenamevir on the pharmacokinetics of montelukast was concluded if the 90%CI for both AUC0‐∞ and Cmax ratios fell within the prespecified interval of 80% to 125%. To assess the effect of amenamevir on methyl‐hydroxymontelukast, montelukast in combination with amenamevir was compared with montelukast alone using the method described above.
To assess the effect of amenamevir on CYP2B6 activity, bupropion in combination with amenamevir (Test) was compared with bupropion alone on day 1 (Reference). Briefly, AUC0–∞ and Cmax were logarithmically transformed and subjected to ANOVA with treatment (bupropion alone or bupropion in combination with amenamevir) as a fixed effect and subject as a random effect. The least‐squares mean of the treatment difference and its 90%CI, were transformed to the original scale in order to obtain the mean AUC0‐∞ and Cmax ratios (bupropion in combination with amenamevir relative to bupropion alone). Absence of a clinically significant drug‐drug effect of amenamevir on bupropion was concluded if the 90%CI for both AUC0–∞ and Cmax ratios fell within the prespecified interval of 80% to 125%. To assess the effect of amenamevir on hydroxybupropion, bupropion in combination with amenamevir (Test) was compared with bupropion alone (Reference) using the method described above.
To assess the recovery of CYP2B6 activity, the effect of amenamevir on bupropion was assessed on days 22 and 29 using the method described above. The 95%CIs for the difference in means (day 22 vs day 1 and day 29 vs day 1) were used to determine how long it took for CYP2B6 activity to return to normal.
Results
Montelukast Study
Mean age, weight, and body mass index were similar across treatment sequences. There were no notable differences in race, ethnicity, or the subjects’ usual cigarette smoking or alcohol intake habits.
From about 1 hour after dosing until 30 hours postdose, mean plasma concentrations of montelukast were about 1.2‐fold higher when montelukast was coadministered with amenamevir than when given alone (Figure 2, Supplementary Table S1). After both treatments, mean montelukast plasma concentrations approached LLQ at 24 hours after dosing, and were below the limit of quantification by 30–36 hours after dosing.
Figure 2.
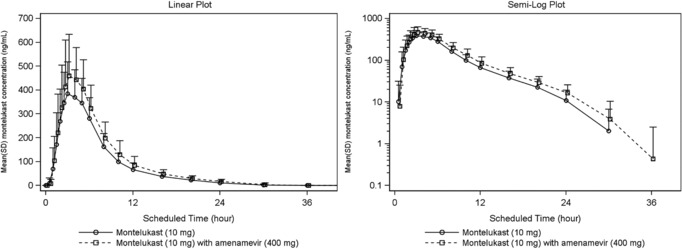
Mean montelukast plasma concentration‐time plots (linear and semilogarithmic, N = 24). Values below the limit of quantification were entered as 0 and included in the calculation of means.
At all time points from 1 to 30 hours after dosing, mean plasma concentrations of methyl‐hydroxymontelukast were 1.2‐ to 2.2‐fold higher when montelukast was taken with amenamevir than when given alone (Figure 3, Supplementary Table S2).
Figure 3.
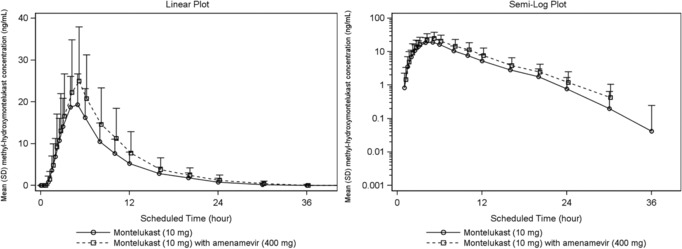
Mean methyl‐hydroxymontelukast plasma concentration‐time plots (linear and semilogarithmic, N = 24). Values below the limit of quantification were entered as 0 and included in the calculation of means.
Both after montelukast alone and combined with amenamevir, mean methyl‐hydroxymontelukast plasma concentration approached LLQ at 20 hours after dosing, and was below the limit of quantification by 36–48 hours after dosing.
Both Cmax and AUC0‐∞ of montelukast increased significantly, by about 22% (Tables 4 and 5), when combined with amenamevir (121.7%, 90%CI [114.8, 129.1]; 122.1% [116.2, 128.4] respectively), as did Cmax and AUC0‐∞ of its primary metabolite hydroxymontelukast (121.4%, 90%CI [106.4, 138.5]; 125.6 % [111.3, 141.7]). After coadministration of amenamevir, the time to peak montelukast concentration remained unchanged. The elimination half‐life of montelukast was slightly increased, and apparent total body clearance was slightly reduced after coadministration of amenamevir (neither was statistically significant).
Table 4.
Summary of Effects of Amenamevir on Cmax and AUC0‐∞ of Montelukast and Methyl‐Hydroxymontelukast Using Log‐Transformed Values (N = 24)
| Least‐Squares Means | Montelukast With Amenamevir vs Montelukast Alone | ||||
|---|---|---|---|---|---|
| Analyte | Parameter | Montelukast With Amenamevir | Montelukast | Ratio (%) | 90%CI |
| Montelukast | Cmax (ng/mL) | 505.9 | 415.6 | 121.7 | 114.8, 129.1 |
| AUC0‐∞ (h·ng/mL) | 3418.5 | 2799.2 | 122.1 | 116.2, 128.4 | |
| Methyl‐hydroxymontelukast | Cmax (ng/mL) | 23.3 | 19.2 | 121.4 | 106.4, 138.5 |
| AUC0‐∞ (h·ng/mL) | 188.6 | 150.2 | 125.6 | 111. 3, 141.7 | |
AUC0‐∞ indicates area under concentration‐time curve extrapolated to infinite time; Cmax, peak concentration.
Table 5.
Summary of Montelukast Pharmacokinetic Parameters
| Parameter | Montelukast Alone (N = 24) | Montelukast With Amenamevir (N = 24) | |
|---|---|---|---|
| Cmax (ng/mL) | Mean | 427.5 | 521.5 |
| SD | 103.2 | 131.5 | |
| AUC0‐tn (h·ng/mL) | Mean | 2820.6 | 3460.2 |
| SD | 869.6 | 1114.0 | |
| AUC0‐∞ (h·ng/mL) | Mean | 2916.3 | 3567.5 |
| SD | 878.9 | 1115.7 | |
| Tmax (h) | Median | 3.00 | 3.00 |
| Range | 2.00‐6.00 | 1.50‐5.00 | |
| t½ (h) | Mean | 5.05 | 5.45 |
| SD | 1.40 | 0.92 | |
| CL/F (L/h) | Mean | 3.72 | 3.04 |
| SD | 1.08 | 0.86 | |
AUC0‐∞ indicates area under concentration‐time curve extrapolated to infinite time; AUC0‐tn, area under concentration‐time curve up to last nonzero value; CL/F, apparent total body clearance from plasma; Cmax, peak concentration; t½, half‐life; Tmax, time of peak concentration.
Overall, 7 subjects (29.2%) reported 9 adverse events (AEs): each of those subjects reported an AE after 10 mg montelukast alone, and 2 subjects (8.3%) also reported an AE after 10 mg montelukast with 400 mg amenamevir. Most AEs occurred at least 72 hours after dosing, and only 1 subject required concomitant medication within 72 hours after dosing (paracetamol for pharyngitis).
All AEs were of mild or moderate intensity. Moderate AEs were more frequent after 10 mg montelukast alone (16.7% of subjects) than after 10 mg montelukast with 400 mg amenamevir (4.2% of subjects). Mild AEs were reported by more subjects (12.5%) after 10 mg montelukast alone than after 10 mg montelukast with 400 mg amenamevir (4.2% of subjects).
Bupropion Study
From about 2 to 6 hours after dosing, mean plasma concentrations of bupropion were lower when bupropion was taken with amenamevir than when it was taken alone, whether on day 1 (before amenamevir had been given), or on days 22 and 29 (a week or more after repeated amenamevir dosing had ended (Figure 4, Supplementary Table S3).
Figure 4.
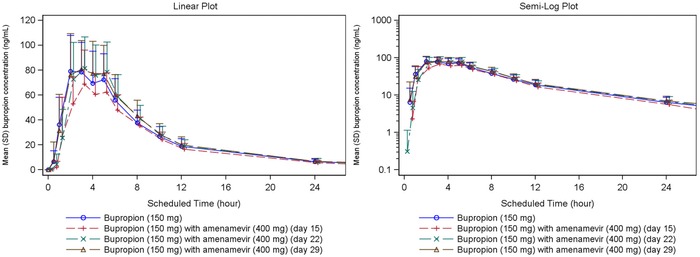
Mean bupropion plasma concentration‐time plots (linear and semilogarithmic, N = 24). Values below the limit of quantification were entered as 0 and included in the calculation of means.
After all treatments, mean bupropion plasma concentrations were approaching the LLQ at 36 hours after dosing and were below LLQ by 96 hours after dosing.
Plasma concentrations of bupropion after a single 150‐mg dose were about 16% lower after 10 days’ dosing with amenamevir than when it was given alone, as evidenced by the reduction in both Cmax and AUC0–∞ to about 84% (84.29%, 90%CI [78.00, 91.10]; 84.07%, 90%CI [78.85, 89.63], respectively) (Table 6). Plasma concentrations then recovered to pretreatment levels on days 22 and 29.
Table 6.
Summary of the Effect of 10 Days’ Pretreatment With Amenamevir on Cmax and AUC0‐∞ on a Single Dose of Bupropion (Day 15, N = 24) with 90%CIs
| Least‐Squares Means | Bupropion With Amenamevir vs Bupropion Alone | |||
|---|---|---|---|---|
| Bupropion Parameter | Bupropion With Amenamevir | Bupropion | Ratio (%) | 90%CI |
| Cmax (ng/mL) | 76.94 | 91.28 | 84.29 | 78.00, 91.10 |
| AUC0‐∞ (h·ng/mL) | 653.70 | 777.61 | 84.07 | 78.85, 89.63 |
AUC0–∞ indicates area under concentration‐time curve extrapolated to infinite time; Cmax, peak concentration.
Parameters have been log‐transformed.
Bupropion concentrations on day 22 were similar to those on day 1 (ratio 104.07, 95%CI [94.74, 114.32]), indicating that the induction of CYP2B6 by repeated doses of amenamevir had remitted by 1 week after the last dose of amenamevir (Table 7).
Table 7.
Summary of Bupropion Concentrations Before, During, and After Induction of CYP2B6 by Amenamevir (N = 24) With 95%CIs
| Parameter | Day 1 (Control) | Day 15 | Day 22 | Day 29 | |
|---|---|---|---|---|---|
| Cmax | LS mean | 91.28 | 76.94 | 92.21 | 95.00 |
| (ng/mL) | ratio vs control (%) (95%CI) | N/A | 84.29 (76.74, 92.60) | 101.02 (91.96, 110.97) | 104.07 (94.74, 114.32) |
| AUC0‐∞ | LS mean | 777.61 | 653.70 | 785.52 | 841.36 |
| (h·ng/mL) | ratio vs control (%) (95%CI) | N/A | 84.07 (78.12, 90.46) | 101.02(93.88, 108.70) | 108.20 (100.55, 116.43) |
AUC0–∞ indicates area under concentration‐time curve extrapolated to infinite time; Cmax, peak concentration; LS mean, least‐squares mean; N/A, not applicable. Parameters have been log‐transformed.
Figure 5 shows the mean plasma concentrations of hydroxybupropion plotted against time. On day 1, the AUC of the primary metabolite hydroxybupropion was about 15‐fold that of the parent molecule (Table 8). Coadministration of amenamevir with bupropion had no significant effect on plasma concentrations of hydroxybupropion.
Figure 5.
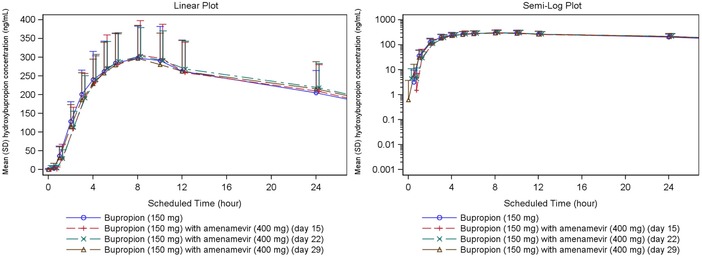
Hydroxybupropion plasma concentration‐time plots (linear and semilogarithmic, N = 24). Values below the limit of quantification were entered as 0 and included in the results.
Table 8.
Summary of Hydroxybupropion Pharmacokinetic Parameters
| Bupropion Parameter | Day 1 Bupropion Alone (N = 24) | Day 15 Bupropion + Amenamevir (N = 24) | Day 22 Bupropion Alone (N = 24) | Day 29 Bupropion Alone (N = 24) | |
|---|---|---|---|---|---|
| Cmax (ng/mL) | Mean | 308.0 | 315.1 | 310.8 | 304.2 |
| SD | 87.8 | 96.8 | 79.5 | 86.5 | |
| AUC0‐tn (h·ng/mL) | Mean | 11,045.1 | 10,807.2 | 11,725.4 | 11,623.6 |
| SD | 3471.9 | 3828.3 | 3814.3 | 3858.9 | |
| AUC0‐∞ (h·ng/mL) | Mean | 11,784.3 | 11,556.5 | 12,754.5 | 12,559.6 |
| SD | 3882.3 | 4263.4 | 4423.4 | 4238.0 | |
| Tmax (h) | Median | 8.00 | 8.00 | 8.00 | 8.00 |
| Range | 4.00‐10.08 | 5.00‐24.00 | 5.00‐12.00 | 3.00‐12.02 | |
| t½ (h) | Mean | 21.83 | 21.42 | 23.70 | 23.19 |
| SD | 4.70 | 4.94 | 5.76 | 3.74 | |
AUC0‐∞ indicates area under concentration‐time curve extrapolated to infinite time; AUC0‐tn, area under concentration‐time curve up to last nonzero value; Cmax, peak concentration; t½, half‐life; tmax, time of peak concentration.
Median time to peak concentration of bupropion was 3 hours on days 1, 15, 22, and 29 (Table 9). Mean t½ was shortened by amenamevir on day 15 by around 2 hours compared with that on day 1; consistent with that finding and with the reduction in AUC of bupropion, apparent total body clearance of bupropion was slightly higher when given with amenamevir than when given alone.
Table 9.
Summary of Bupropion Pharmacokinetic Parameters
| Bupropion Parameter | Day 1 Bupropion Alone (N = 24) | Day 15 Bupropion + Amenamevir (N = 24) | Day 22 Bupropion Alone (N = 24) | Day 29 Bupropion Alone (N = 24) | |
|---|---|---|---|---|---|
| Cmax (ng/mL) | Mean | 94.5 | 80.3 | 95.6 | 97.8 |
| SD | 25.8 | 24.4 | 26.9 | 24.5 | |
| AUC0‐tn (h·ng/mL) | Mean | 764.5 | 629.8 | 777.5 | 825.7 |
| SD | 246.9 | 175.5 | 242.5 | 235.7 | |
| AUC0‐∞ (h·ng/mL) | Mean | 812.6 | 675.7 | 820.9 | 873.7 |
| SD | 269.0 | 188.7 | 253.1 | 245.5 | |
| Tmax (h) | Median | 3.00 | 3.00 | 3.00 | 3.00 |
| Range | 2.00‐6.00 | 1.00‐5.98 | 2.00‐5.00 | 1.00‐5.00 | |
| t½ (h) | Mean | 10.92 | 8.97 | 9.76 | 11.87 |
| SD | 6.09 | 5.08 | 4.01 | 5.24 | |
| CL/F (L/h) | Mean | 200.64 | 236.46 | 199.59 | 185.59 |
| SD | 56.33 | 57.20 | 62.04 | 57.71 | |
AUC0–∞ indicates area under the concentration‐time curve extrapolated to infinite time; AUC0‐tn, area under concentration‐time curve up to last nonzero value; CL/F, apparent total body clearance from plasma; Cmax, peak concentration; t½, half‐life; tmax, time of peak concentration.
Overall, 12 subjects (50.0%) reported AE. Headache was the most frequently reported AE. Cannula site pain, rhinitis, back pain, and anxiety were reported by 2 subjects each. No other AE was reported by more than 1 subject.
Discussion
Montelukast
Coadministration of amenamevir with montelukast caused a 22% increase in both Cmax and AUC of montelukast. The data failed to exclude a clinically significant drug‐drug effect because the 90%CI of the log ratio montelukast versus amenamevir plus montelukast did not fall within the range 80% to 125% for both Cmax and AUC. The increase in Cmax is consistent with a reduction in first‐pass intestinal and hepatic extraction. Apparent clearance and t½ showed a trend toward reduction in rate of elimination of montelukast. Our results are consistent with the in vitro finding that amenamevir is a weak inhibitor of CYP2C8. An increase of 22% in plasma concentration of a drug that is a substrate of CYP2C8 would be of importance only for medicines with a very narrow therapeutic window, so reduction of the dose of concurrent medication is unlikely to be necessary in clinical practice.
Coadministration of amenamevir with montelukast was associated with a mean 22% increase in plasma Cmax and AUC of montelukast's major metabolite, methyl‐hydroxymontelukast. That increase mirrored the 22% increase in Cmax and AUC of parent montelukast and is likely due to the metabolite also being dependent on CYP2C8 for subsequent conversion. Thus, rather than concentration decreasing as might be expected, it increased in parallel due to inhibition in a manner consistent with the effect on the parent compound.
Montelukast is metabolized not only by CYP2C8 but also by CYP3A4, of which amenamevir is both a substrate and inducer. However, CYP2C8 is believed to account for about 80% of montelukast's metabolism: inhibition of CYP2C8 by gemfibrozil increases the AUC of montelukast 4‐fold.9 The same study also showed that inhibition of CYP3A4 by itraconazole did not affect the metabolism of montelukast, in contrast with later findings7, 9: in 1 study, inhibition of CYP3A4 increased AUC of montelukast by 144%.7 Thus, current evidence is contradictory, but it is certainly conceivable that induction of CYP3A4 activity by amenamevir might reduce plasma concentrations of montelukast. However, amenamevir was given only as a single dose together with a single dose of montelukast in this study, and it is unlikely that CYP3A4 induction by amenamevir could have developed quickly enough to have influenced the results.
In respect to safety and tolerability, both single and combined dosing were equally well tolerated in the healthy men in this study.
Bupropion
Amenamevir 400 mg once daily for 10 days decreased both Cmax and AUC0‐∞ of bupropion by about 16%. The results did not exclude a significant effect of amenamevir on bupropion exposure, as the 90%CI of the least‐squares geometric mean ratios (bupropion with amenamevir to bupropion) of Cmax and AUC0‐∞ did not fall within the prespecified interval of 80% to 125%. At 1 and 2 weeks (days 22 and 29) after the final dose of amenamevir, Cmax, and AUC0‐∞ of bupropion were similar to those on day 1, indicating that CYP2B6 activity had returned to pretreatment levels within 1 week after the last amenamevir dose. That is consistent with the observation that even extensive induction of CYP2B6 by rifampicin is fully reversed by 2 weeks after cessation of rifampicin treatment.22
Prior treatment with amenamevir 400 mg once daily for 10 days did not affect Cmax or AUC0‐∞ of hydroxybupropion, which is bupropion's main metabolite. Because amenamevir treatment did reduce AUC0‐∞ of bupropion parent drug, there was a small change in the ratio of AUC0‐∞ for hydroxybupropion:bupropion; before amenamevir the ratio was 15.2; after treatment with amenamevir, the corresponding ratio was 17.7. Those results are consistent with the findings of Laizure et al, who showed that the more powerful induction of CYP2B6 by rifampicin reduced AUC0‐∞ of bupropion by 3‐fold, but doubled AUC0‐∞ of the hydroxybupropion metabolite.15
The minor reduction (by 16%) in plasma concentrations of bupropion, coupled with no change in concentrations of the active metabolite hydroxybupropion, would be unlikely to warrant dose adjustment when amenamevir is coadministered with CYP2B6 substrates.
Our AE data showed that repeated doses of 400 mg amenamevir, given alone or with a single dose of 150 mg bupropion, were well tolerated in healthy men.
Conclusions
The minor increase (22%) of the concentration of montelukast and the similarly marginal reduction (16%) in plasma concentration of bupropion suggest that dose adjustment is unlikely to be necessary when amenamevir is coadministered with CYP2C8 or CYP2B6 substrates.
Supporting information
Supporting Information
Declaration of Conflict of Interest and Financial Disclosure
The studies were sponsored by Maruho. The authors are employees of either Maruho or the clinical unit. The authors confirm that they have no conflict of interest.
None of the authors is a Fellow of the American College of Clinical Pharmacology.
References
- 1. Tyring S, Wald A, Zadeikis N, Dhadda S, Takenouchi K, Rorig R. ASP2151 for the treatment of genital herpes: a randomized, double‐blind, placebo‐ and valacyclovir‐controlled, dose‐finding study. J Infect Dis. 2012;205(7):1100–1110. [DOI] [PubMed] [Google Scholar]
- 2. Morfin F, Thouvenot D. Herpes simplex virus resistance to antiviral drugs. J Clin Virol. 2003;26(1):29–37. [DOI] [PubMed] [Google Scholar]
- 3. Strasfeld L, Chou S. Antiviral drug resistance: mechanisms and clinical implications. Infect Dis Clin North Am. 2010;24(2):413–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kusawake T, Keirns JJ, Kowalski D, et al. Pharmacokinetics and safety of amenamevir in healthy subjects: analysis of four randomized phase 1 studies. Ad Ther. 2017;34(12):2625–2637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Takada A, Katashima M, Kaibara A, Sawamoto T, Zhang W, Keirns J. Statistical analysis of amenamevir (ASP2151) between pharmacokinetics and clinical efficacies with non‐linear effect model for the treatment of genital herpes. Clin Pharmacol Drug Dev. 2014;3(5):365–370. [DOI] [PubMed] [Google Scholar]
- 6. Cardoso JdO, Oliveira RV, Lu JBL, Desta Z. In vitro metabolism of montelukast by cytochrome P450s and UDP‐glucuronosyltransferases. Drug Metab Dispos. 2015;43(12):1905–1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hegazy SK, Mabrouk MM, Elsisi AE, Mansour NO. Effect of clarithromycin and fluconazole on the pharmacokinetics of montelukast in human volunteers. Eur J Clin Pharmacol. 2012;68(9):1275–1280. [DOI] [PubMed] [Google Scholar]
- 8. VandenBrink BM, Foti RS, Rock DA, Wienkers LC, Wahlstrom JL. Evaluation of CYP2C8 inhibition in vitro: utility of montelukast as a selective CYP2C8 probe substrate. Drug Metab Dispos. 2011;39(9):1546. [DOI] [PubMed] [Google Scholar]
- 9. Karonen T, Neuvonen PJ, Backman JT. CYP2C8 but not CYP3A4 is important in the pharmacokinetics of montelukast. Br J Clin Pharmacol. 2012;73(2):257–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Filppula AM, Laitila J, Neuvonen PJ, Backman JT. Reevaluation of the microsomal metabolism of montelukast: major contribution by CYP2C8 at clinically relevant concentrations. Drug Metab Dispos. 2011;39(5):904–911. [DOI] [PubMed] [Google Scholar]
- 11. Balani SK, Xu X, Pratha V, et al. Metabolic profiles of montelukast sodium (Singulair), a potent cysteinyl leukotriene receptor antagonist, in human plasma and bile. Drug Metab Dispos. 1997;25(11):1282–1287. [PubMed] [Google Scholar]
- 12. Stahl SM, Pradko JF, Haight BR, Modell JG, Rockett CB, Learned‐Coughlin S. A review of the neuropharmacology of bupropion, a dual norepinephrine and dopamine reuptake inhibitor. Prim Care Companion J Clin Psychiatry. 2004;6(4):159–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Slemmer JE, Martin BR, Damaj MI. Bupropion is a nicotinic antagonist. J Pharmacol Exp Ther. 2000;295(1):321–327. [PubMed] [Google Scholar]
- 14. Jefferson JW, Pradko JF, Muir KT. Bupropion for major depressive disorder: pharmacokinetic and formulation considerations. Clin Ther. 2005;27(11):1685–1695. [DOI] [PubMed] [Google Scholar]
- 15. Laizure SC, DeVane CL, Stewart JT, Dommisse CS, Lai AA. Pharmacokinetics of bupropion and its major basic metabolites in normal subjects after a single dose. Clin Pharmacol Ther. 1985;38(5):586–589. [DOI] [PubMed] [Google Scholar]
- 16. Loboz KK, Gross AS, Williams KM, et al. Cytochrome P450 2B6 activity as measured by bupropion hydroxylation: effect of induction by rifampin and ethnicity. Clin Pharmacol Ther. 2006;80(1):75–84. [DOI] [PubMed] [Google Scholar]
- 17. Hesse LM, Venkatakrishnan K, Court MH, et al. CYP2B6 mediates the in vitro hydroxylation of bupropion: potential drug interactions with other antidepressants. Drug Metab Dispos. 2000;28(10):1176–1183. [PubMed] [Google Scholar]
- 18. Schoors DF, De Smet M, Reiss T, et al. Single dose pharmacokinetics, safety and tolerability of MK‐0476, a new leukotriene D4‐receptor antagonist, in healthy volunteers. Br J Clin Pharmacol. 1995;40(3):277–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Adeloye T, Sahgal O, Puri A, Warrington S, Endo T, Dennison J, et al. Amenamevir: studies of potential CYP3A‐mediated pharmacokinetic interactions with midazolam, cyclosporine and ritonavir in healthy volunteers. Clin Pharmacol Drug Dev 2018;7(8):844–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hsyu P‐H, Singh A, Giargiari TD, Dunn JA, Ascher JA, Johnston JA. Pharmacokinetics of bupropion and its metabolites in cigarette smokers versus nonsmokers. J Clin Pharmacol. 1997;37(8):737–743. [DOI] [PubMed] [Google Scholar]
- 21. Qin W‐J, Zhang W, Liu Z‐Q, et al. Rapid clinical induction of bupropion hydroxylation by metamizole in healthy Chinese men. Br J Clin Pharmacol. 2012;74(6):999–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Reitman ML, Chu X, Cai X, et al. Rifampin's acute inhibitory and chronic inductive drug interactions: experimental and model‐based approaches to drug‐drug interaction trial design. Clin Pharmacol Ther. 2011;89(2):234–242. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information