

Abstract
Aims
To investigate the effects of semaglutide vs placebo on glucagon and other counterregulatory hormones during hypoglycaemia in type 2 diabetes (T2D).
Methods
In this double‐blind, placebo‐controlled, single‐centre trial, we randomized 38 men and women (treated only with metformin) 1:1 to 2 12‐week crossover periods of once‐weekly subcutaneous semaglutide or placebo, each followed by a hypoglycaemic clamp procedure. The primary endpoint was change in glucagon concentration from target plasma glucose (PG) level 5.5 mmol/L to nadir (target 2.5 mmol/L).
Results
The mean (range) participant age was 54.2 (41‐64) years, body mass index 29.4 (23.3‐36.1) kg/m2, glycated haemoglobin 60.8 (44.3‐83.6) mmol/mol (7.7 [6.2‐9.8]%), and diabetes duration 4.5 (0.3‐13.2) years. A total of 35 participants completed the trial and were included in the analyses. During the hypoglycaemic clamp from 5.5 mmol/L PG to nadir, the absolute change in mean glucagon concentration was similar for semaglutide vs placebo: 88.3 vs 83.1 pg/mL (estimated difference 5.2 pg/mL [95% confidence interval −7.7 to 18.1]). Concentrations of other counterregulatory hormones increased with both treatments, with a statistically significantly lower increase for noradrenaline and cortisol with semaglutide vs placebo. The glucose infusion rate to maintain constant clamp levels was similar for each treatment group, suggesting an overall similar counterregulatory response. The mean hypoglycaemic symptom score and proportion of participants recognizing hypoglycaemia during the study were lower for semaglutide vs placebo treatment at nadir, but cognitive function test results were similar. No new safety issues were observed for semaglutide.
Conclusions
Semaglutide treatment did not compromise the counterregulatory glucagon response during experimental hypoglycaemia in people with T2D.
Keywords: GLP‐1, GLP‐1 analogue, hypoglycaemia, type 2 diabetes
1. INTRODUCTION
Type 2 diabetes (T2D) is a complex metabolic disorder involving increased insulin resistance, coupled with progressive loss of β‐cell mass and function, and leading to dysregulation of insulin and glucagon secretion.1, 2 While many treatment options for T2D are available, one limitation is the risk of hypoglycaemia.3
Glucagon is a key hormone in the regulation of glucose homeostasis.4, 5 The counterregulation of hypoglycaemia depends largely on increases in glucagon and adrenaline secretion and decreased insulin secretion.6 In people with insulin‐deficient diabetes, the key counterregulatory responses of increased glucagon and suppressed insulin are impaired and the sympathoadrenal response, mainly adrenaline, becomes important.7, 8 Counterregulatory hormone deficiencies tend to be mild in the early stages of the T2D,9 developing only as the disease advances and endogenous insulin secretion declines.10
Glucagon‐like peptide‐1 (GLP‐1), an incretin hormone secreted by intestinal L cells in response to food intake,11, 12, 13 inhibits glucagon secretion and stimulates insulin secretion in a glucose‐dependent manner.12, 13 Semaglutide (Novo Nordisk, Bagsværd, Denmark), a GLP‐1 analogue, is approved in major markets for the treatment of T2D. With 94% homology to native GLP‐1, semaglutide has three structural modifications that prolong its half‐life to ~1 week, making it appropriate for once‐weekly administration.3, 12 In phase III trials, semaglutide led to improved glycaemic control and decreased body weight, with a safety profile similar to other GLP‐1 receptor agonists (GLP‐1RAs).14, 15, 16 Liraglutide, having a similar structure to semaglutide but a different pharmacokinetic profile, was found to have no effect on the counterregulatory responses to hypoglycaemia.17, 18, 19 These responses have not been evaluated in people treated with semaglutide. Because of the ability of semaglutide to suppress glucagon, it is important to establish that the glucagon counterregulatory response during hypoglycaemia is preserved.
The primary objective of the present study, therefore, was to investigate the effect of semaglutide vs placebo on the glucagon response during hypoglycaemia in people with T2D, and effects on other counterregulatory hormones (adrenaline, noradrenaline, cortisol and growth hormone), hypoglycaemic symptoms, cognitive function and vital signs.
2. PARTICIPANTS AND METHODS
This was a randomized single‐centre, double‐blind, crossover clinical pharmacology trial in 38 people with T2D treated only with metformin. The protocol was approved by the local ethics committee and participants were recruited at the Division of Endocrinology and Diabetology, Department of Internal Medicine at the Medical University of Graz, Austria. Written informed consent was obtained before trial‐related activities began. The trial was registered at http://clinicaltrials.gov (identifier NCT02147431) and conducted in accordance with the Declaration of Helsinki and Good Clinical Practice guidelines.20, 21
2.1. Trial population
Men and women (age 18‐64 years) with T2D were eligible for participation. Inclusion criteria were: on stable metformin treatment for 90 days before screening; glycated haemoglobin (HbA1c) 47 to 86 mmol/mol (6.5%‐10.0%); and body mass index (BMI) 20 to 35 kg/m2. Key exclusion criteria were: treatment with any glucose‐lowering agent(s) other than metformin within 90 days before screening, except for short‐term treatment (≤7 days in total) with insulin for intercurrent illness; history of chronic pancreatitis or idiopathic acute pancreatitis; personal/family history of medullary thyroid carcinoma or multiple endocrine neoplasia syndrome type 2; and recurrent severe hypoglycaemia (>1 event during the last 12 months), or hypoglycaemic unawareness as judged by the investigator.
2.2. Trial design
Eligible participants were randomized 1:1 to one of two treatment sequences: semaglutide 1.0 mg/placebo or placebo/semaglutide 1.0 mg (Figure 1A). Participants were assigned by the investigators at the trial site to the lowest available randomization number on a participant‐specific pre‐packed trial product (Novo Nordisk). The sponsor, trial participants and investigators remained blinded to treatment allocation during the trial. Once‐weekly treatment with the pre‐packed trial product was self‐administered subcutaneously using PDS290 pen‐injectors (Novo Nordisk). As per usual practice, the semaglutide dose was escalated, starting at a dose of 0.25 mg for 4 weeks, with subsequent dose escalation to 0.5 mg for 4 weeks, followed by the maintenance 1.0‐mg dose for 5 weeks. Throughout the trial, dosing was to occur on the same week day at any time of day. The participants' background metformin treatment remained unchanged. Each group underwent two treatment periods, each followed by a hypoglycaemic clamp procedure and 5 weeks of pharmacokinetic blood sampling, separated by a 1‐ to 3‐week washout period, with a follow‐up visit in the final week of the second sampling period (Figure 1A).
Figure 1.
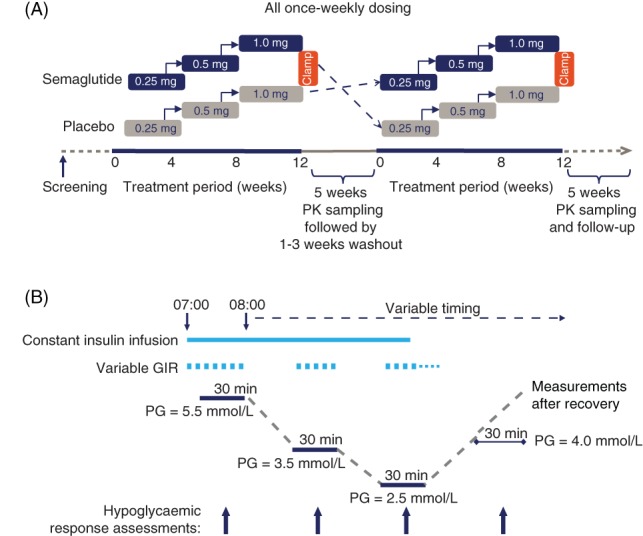
A, Trial design. Semaglutide was self‐injected once weekly with the starting dose of 0.25 mg for 4 weeks (starting at week 0), increasing to 0.5 mg for the next 4 weeks and subsequently to 1.0 mg for 5 weeks. Assessments were made after the 5th dose of 1.0 mg semaglutide. Placebo was given in doses volume‐matched to semaglutide. B, Hypoglycaemic clamp design. Response assessments included counterregulatory hormones, C‐peptide, hypoglycaemic symptoms, hypoglycaemic recognition, cognitive function tests, and vital signs. GIR, glucose infusion rate; PG, plasma glucose; PK, pharmacokinetics
2.3. Hypoglycaemic clamp
Approximately 48 hours after the last (13th) treatment dose, coinciding with the expected maximum concentration of semaglutide,13 a stepwise hypoglycaemic clamp was performed during a 4‐day in‐house visit (Figure 1B).17, 22 Participants were admitted to the clinical research centre where, on day 1, they received the last treatment dose. On the evening of day 2, participants received a variable intravenous infusion of glucose or human soluble insulin (Novo Nordisk) to maintain glucose levels at a 5.5 mmol/L target in a euglycaemic clamp.
The hypoglycaemic clamp was initiated in the morning of day 3 by increasing the insulin infusion to a constant rate of 2.5 mU/kg/min, based on the participant's body weight at randomization. The target plasma glucose (PG) level of 5.5 mmol/L was maintained continuously for 60 minutes before induction of hypoglycaemia, when PG was allowed to decline to 3.5 mmol/L and subsequently nadir (target 2.5 mmol/L). At each level, a variable glucose infusion rate (GIR) was used to maintain the target stable for 30 minutes and assessments of the hypoglycaemic response were made, including blood sampling. PG was not permitted to fall below 2.2 mmol/L. After 20 minutes at nadir, the insulin infusion was terminated and the variable GIR used to maintain stable PG levels for 10 minutes, after which the glucose infusion was tapered off to allow spontaneous recovery from the hypoglycaemia, if possible. If PG had not reached the level of 4.0 mmol/L within 40 minutes of termination of the insulin infusion, an intravenous glucose infusion was started at a constant rate of 5.5 mg/kg/min. The clamp was terminated when the PG concentration reached 5.5 mmol/L, or when deemed safe by the investigator. Throughout the clamp, participants remained fasted (water was allowed) in a supine or semi‐supine position.
2.4. Endpoints and assessments during the hypoglycaemic clamp
The primary endpoint was the change in mean glucagon concentration from the target PG level 5.5 mmol/L to nadir. Secondary endpoints included absolute concentrations of counterregulatory hormones, C‐peptide and insulin and changes from 5.5 to 3.5 mmol/L and nadir (Figure 1B). Changes from fasting PG levels (the morning of the day before the hypoglycaemic clamp) to each target level were evaluated for glucagon only, and changes from nadir to recovery (PG ≥4.0 mmol/L) were evaluated for each counterregulatory hormone. During each of the 4 30‐minute target levels of the clamp, the mean of each variable was calculated as an average of 3 measurements at 10, 20 and 30 minutes (the fasting value was based on one measurement). The area under the curve for GIR (AUCGIR) was assessed at each target PG level. The time to reach recovery was also recorded.
Other secondary endpoints assessed during the clamp at each target PG level included symptoms of hypoglycaemia, measured using the Edinburgh Hypoglycaemia Scale,23, 24 hypoglycaemic recognition, evaluated based on participants' responses (yes/no) to the question, “Do you feel hypo?” and cognitive function tests (the Trail Making‐B test,25 the Digit Symbol Substitution test26 and the Four‐Choice Reaction Time test27). Vital signs (heart rate and systolic and diastolic blood pressure [BP]) were measured at each target level, calculated as the mean of two measurements at 0 and 30 minutes.
2.5. Endpoints and assessments during the treatment period
Effects of semaglutide vs placebo on glucose metabolism in each of the 2 treatment periods were assessed by evaluating the concentration and change from baseline to end‐of‐treatment (1 week after the 13th dose) in HbA1c, fasting serum glucose and fasting C‐peptide. Because of the crossover design, baseline assessments were made before the first and second treatment periods.
Pharmacokinetic endpoints are described in the Supplemental Methods (File S1).
2.6. Safety assessments
The safety evaluation included assessments from baseline to follow‐up in the number of treatment‐emergent adverse events, defined as starting on or after the first treatment day and no later than the follow‐up visit. Self‐reported hypoglycaemic episodes, classified according to American Diabetes Association criteria,3 were also assessed. Nocturnal episodes had time of onset between 12:01 am and 5:59 am. Additional safety endpoints included haematology, biochemistry, amylase and lipase, calcitonin, urine analysis, vital signs, physical examination and ECG, body weight, and the development of anti‐semaglutide antibodies.
2.7. Laboratory assays
Plasma glucagon concentrations were analysed using an enzyme‐linked immunosorbent assay (Mercodia AB, Uppsala, Sweden).28 Measurements of other variables were made using standard methods, as described in the Supplemental Methods (File S1).
2.8. Statistical methods
A sample size of 30 completing participants was determined based on the criterion that the width of the 95% confidence interval (CI) for the treatment comparison of the primary endpoint was within ±23.3 pg/mL with 80% probability. This was based on a within‐participant SD of 40 pg/mL from a previous trial.17 Assuming a 20% dropout rate, at least 38 randomized participants were required to obtain 30 trial‐completing participants.
The pre‐specified analyses used data from the full analysis set of all randomized and exposed participants with at least one post‐baseline assessment. The safety analysis set included all exposed participants. The primary endpoint, absolute change in glucagon concentration from target level 5.5 mmol/L to nadir, was analysed using an analysis of covariance model, with treatment, period and participant as fixed effects. As an additional analysis of the primary endpoint, the relative change was calculated and analysed based on log‐transformed values, using a similar statistical model. Secondary clamp‐related endpoints were analysed as per the primary endpoint, except that hypoglycaemic recognition was summarized descriptively. The AUCGIR was calculated using the linear trapezoidal technique and analysed on the original scale. The time to reach recovery (PG ≥4.0 mmol/L) was log‐transformed and analysed as per the primary endpoint. The proportion of participants in each group who recovered spontaneously 40 minutes after termination of the insulin infusion was analysed by logistic regression, with treatment and period as fixed effects and participant as a random effect. Additional secondary endpoints were summarized descriptively. The statistical analyses were performed using SAS software, version 9.3 (SAS Institute, Cary, North Carolina).
3. RESULTS
3.1. Trial population
Of 81 screened participants, 38 were randomized and received treatment. Three withdrew from the trial: 2 as a result of adverse events while on placebo treatment and 1 after 1 day and a single dose of 0.25 mg semaglutide because of non‐adherence to the protocol (only the latter participant was excluded from the full analysis set and all were included in the safety analyses). Thirty‐five participants completed the trial, which was conducted between May 21, 2014 and May 20, 2015.
The baseline characteristics of the 37 participants who comprised the full analysis set are shown in Table 1.
Table 1.
Baseline demographics and participant characteristics (N = 37)
| Characteristic | |
|---|---|
| Women, n (%) | 12 (32.4) |
| Men, n (%) | 25 (67.6) |
| Age (years) | |
| Mean (SD) | 54.2 (6.4) |
| Minimum–maximum | 41–64 |
| Duration of diabetes (years) | |
| Mean (SD) | 4.5 (3.2) |
| Minimum–maximum | 0.3–13.2 |
| Daily metformin treatment (mg) | |
| Minimum–maximum | 850–3000 |
| Smoking habits, n (%) | |
| Current smoker | 7 (18.9) |
| BMI, kg/m2 | |
| Mean (SD) | 29.4 (3.4) |
| Minimum–maximum | 23.3–36.1 |
| Body weight (kg) | |
| Mean (SD) | 88.5 (11.1) |
| Minimum–maximum | 66.2–112.0 |
| HbA1c, mmol/mol | |
| Mean (SD) | 60.8 (11.4) |
| Minimum–maximum | 44.3–83.6 |
| HbA1c (%) | |
| Mean (SD) | 7.7 (1.0) |
| Minimum–maximum | 6.2–9.8 |
| Fasting serum glucose (mmol/L) | |
| Mean (SD) | 9.5 (2.6) |
| Minimum–maximum | 6.0–15.0 |
| Fasting C‐peptide (nmol/L) | |
| Mean (SD) | 1.2 (0.6) |
| Minimum–maximum | 0.4–3.2 |
Abbreviations: BMI, body mass index; HbA1c, glycated haemoglobin.
Data are presented as means (SD), unless otherwise stated, and were assessed at screening, except for body weight and diabetes‐related characteristics, which were assessed at baseline. All 37 participants were white and not Hispanic or Latino.
3.2. Results of the hypoglycaemic clamp
As expected, similar mean PG levels in the semaglutide and placebo groups were achieved at the 5.5‐ and 3.5‐mmol/L target PG levels. The mean time taken to achieve the target was similar with each treatment: 17.2 vs 18.0 minutes to reach 3.5 mmol/L for semaglutide vs placebo and 62.2 vs 61.3 minutes to reach nadir. The nadir target of 2.5 mmol/L was not reached by 13 participants (35%) with semaglutide and 15 (43%) with placebo; 9 participants (26%) did not achieve the target with both treatments. Hence the mean (range) PG level at nadir of 2.9 (2.4−3.9) mmol/L with semaglutide and 2.9 (2.4−4.4) mmol/L with placebo was slightly higher than the target level.
3.2.1. Counterregulatory hormones
Glucagon concentrations during the clamp are shown in Figure 2A. At the fasting PG level and at each target level, except nadir, the mean glucagon level was lower with semaglutide than with placebo. The estimated absolute change in mean glucagon concentration from 5.5 mmol/L to nadir (primary endpoint) was similar between treatments (Table 2); however, a higher relative increase in glucagon concentration was observed with semaglutide vs placebo, primarily driven by the lower mean glucagon concentration at 5.5 mmol/L with semaglutide. Mean absolute and relative changes in glucagon from 5.5 to 3.5 mmol/L glucose and from fasting glucose levels to each target level were not statistically significantly different for semaglutide vs placebo, except that the relative change from fasting to nadir was greater with semaglutide (estimated treatment ratio 1.23 [95% CI 1.01‐1.49]).
Figure 2.
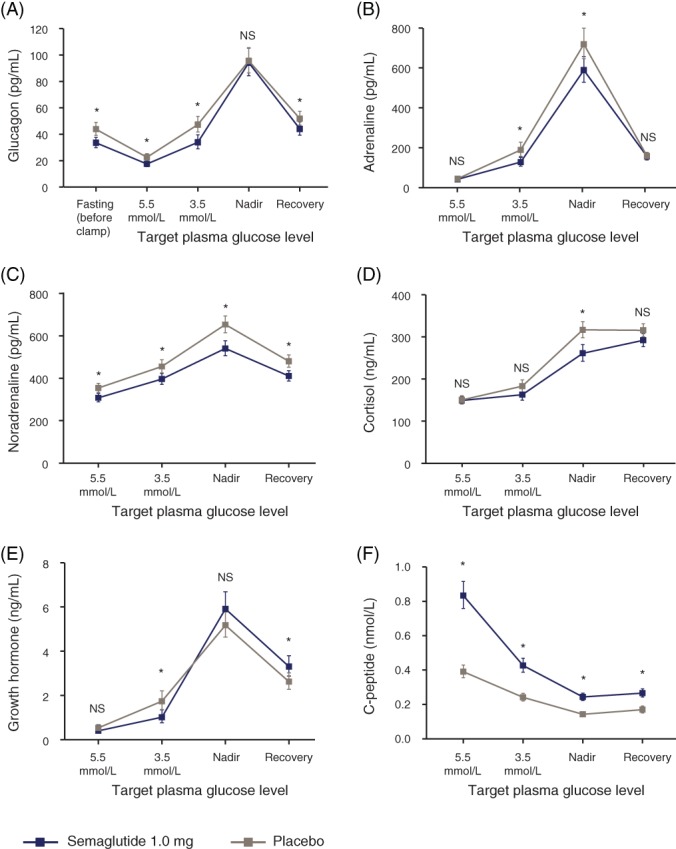
Counterregulatory responses to hypoglycaemic clamp: A, glucagon; B, adrenaline; C, noradrenaline; D, cortisol; E, growth hormone and F, C‐peptide. Graphs show geometric means and standard error bars. Of the 37 participants included in the full analysis set, 35 completed both treatment periods and are included in the analysis. Mean (SD) fasting plasma glucose concentrations (prior to the clamp) were 6.6 (1.3) mmol/L for semaglutide and 8.8 (2.6) mmol/L for placebo. For glucagon SI units (ng/L), please use the conversion factor 1.0. For adrenaline SI units (pmol/L), please use the conversion factor 5.459. For noradrenaline SI units (pmol/L), please use the conversion factor 5.911. For cortisol SI units (nmol/L), SI units (pmol/L), please use the conversion factor 27.588. For growth hormone SI units (μg/L), please use the conversion factor 1.0. *P < 0.05 for semaglutide vs placebo from analysis of covariance. NA, not analysed; NS, not significant
Table 2.
Counterregulatory responses to hypoglycaemia from target plasma glucose level 5.5 mmol/L to nadir
| Estimated mean change: semaglutide vs placebo | Estimated treatment difference (95% CI)a | P | |
|---|---|---|---|
| Glucagon (pg/mL) | 88.28 vs 83.07 | 5.21 (−7.72 to 18.14) | 0.418 |
| Adrenaline (pg/mL) | 667.7 vs 795.9 | −128.2 (−279.0 to 22.6) | 0.093 |
| Noradrenaline (pg/mL) | 246.0 vs 316.4 | −70.4 (−136.8 to −4.1) | 0.038 |
| Cortisol (ng/mL) | 125.5 vs 172.7 | −47.2 (−80.7 to −13.7) | 0.007 |
| Growth hormone (ng/mL) | 6.15 vs 4.87 | 1.28 (−0.14 to 2.70) | 0.075 |
| C‐peptide (nmol/L) | −0.68 vs −0.28 | −0.40 (−0.49 to −0.31) | <0.001 |
| Estimated mean ratiobSemaglutide vs placebo | Estimated treatment ratio (95% CI)a | P | |
|---|---|---|---|
| Glucagon | 5.39 vs 4.23 | 1.28 (1.04‐1.56) | 0.020 |
Abbrevation: CI, confidence interval.
Of the 37 participants included in the full analysis set, 35 completed both treatment periods and are included in the analysis. All P values are 2‐sided based on a null hypothesis of no treatment difference. For glucagon SI units (ng/L), please use the conversion factor 1.0. For adrenaline SI units (pmol/L), please use the conversion factor 5.459. For noradrenaline SI units (pmol/L), please use the conversion factor 5.911. For cortisol SI units (nmol/L), SI units (pmol/L), please use the conversion factor 27.588. For growth hormone SI units (μg/L), please use the conversion factor 1.0.
For semaglutide vs placebo.
Relative change for glucagon was an additional analysis of the primary endpoint. Relative changes in other variables than glucagon are included in Table S1 in File S1.
The mean concentrations of other counterregulatory hormones increased in both treatment groups in response to hypoglycaemia from 5.5 mmol/L glucose to nadir (Figure 2B–E), with lower increases in the absolute concentrations of noradrenaline and cortisol with semaglutide vs placebo (Table 2).
Mean C‐peptide concentrations decreased with both treatments in response to hypoglycaemia, although semaglutide vs placebo treatment resulted in significantly greater mean C‐peptide concentrations at each target PG level (Figure 2F). The estimated absolute decrease in mean C‐peptide from 5.5 mmol/L to nadir was greater for participants in the semaglutide group than the placebo group (Table 2).
3.2.2. Time to reach recovery
The proportion of participants who recovered spontaneously (reaching PG ≥4.0 mmol/L < 40 minutes after termination of the insulin infusion at nadir) was ~50% in each group. Recovery was achieved within a similar time for participants when treated with semaglutide (geometric mean 44 minutes) and placebo (mean 47 minutes).
3.2.3. Glucose infusion rate
The AUCGIR was similar in the semaglutide and placebo treatment groups throughout the clamp (Figure S1 in File S1).
3.2.4. Hypoglycaemia symptoms and recognition and cognitive function tests
No difference between treatments was observed in the estimated mean hypoglycaemic symptom score at the target PG level of 5.5 mmol/L, whereas at 3.5 mmol/L and nadir, the mean hypoglycaemic symptom score was 11% to 12% lower with semaglutide vs placebo (P = 0.002 and 0.019, respectively; Figure 3).
Figure 3.
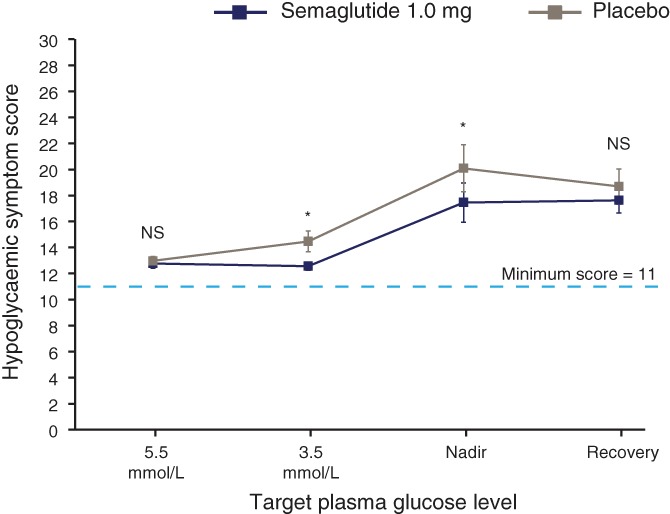
Hypoglycaemic symptom score. The score was based on responses to 11 questions, providing a total score of between 11 and 77, with a lower score indicating fewer symptoms. Graph shows geometric means and standard error bars. *P < 0.05 for semaglutide vs placebo from analysis of covariance. NS, not significant
Regarding hypoglycaemia recognition, all participants answered “no” to the question “Do you feel hypo?” at the target level of 5.5 mmol/L, except for one answering “yes” during treatment with placebo. A lower proportion of participants answered “yes’ during hypoglycaemia when treated with semaglutide than with placebo: 1 (2.9%) vs 5 (15.6%) participants at 3.5 mmol/L and 12 (32.4%) vs 18 participants (51.4%) at nadir.
The results of three cognitive function tests indicated no effect of semaglutide on cognitive function, with similar results across all PG levels in the semaglutide and placebo groups (Figure S2 in File S1).
3.2.5. Vital signs
During the clamp, there were no systematic differences between semaglutide and placebo in the response to hypoglycaemia for heart rate, systolic or diastolic BP, with no statistically significant treatment differences with respect to mean changes from target PG level 5.5 mmol/L to nadir and 5.5 to 3.5 mmol/L (Table S2 in File S1).
3.3. Pharmacodynamic changes during treatment
One week after the last (13th) treatment dose, the mean (SD) decreases from baseline for semaglutide in HbA1c, fasting serum glucose and body weight were greater than those observed with placebo (−1.1 [0.7]% vs 0.4 [0.8]%, −2.44 [1.85] mmol/L vs −0.11 [1.79] mmol/L and −3.7 [2.7] vs 0.3 [2.8] kg; Table S3 in File S1). The mean fasting C‐peptide levels increased by 27% from baseline to end‐of‐treatment with semaglutide vs placebo).
3.4. Semaglutide pharmacokinetics
Semaglutide pharmacokinetic variables were similar to those observed in previous trials,13, 29 supporting overall treatment compliance (see the Supplementary Results [File S1]). The terminal half‐life of semaglutide was 150 (113‐183) hours (geometric mean [range]).
3.5. Safety
The number of adverse events and the proportion of participants reporting events, which were all of mild or moderate severity, were greater for semaglutide vs placebo (Table S4 in File S1). The most common adverse events were gastrointestinal disorders and infections and infestations. Seven serious adverse events were reported by 5 participants (see the Supplemental Results for details in File S1). No deaths or pancreatitis cases were reported during the trial. Excluding episodes induced during the clamp, 4 participants on placebo treatment reported 6 hypoglycaemic episodes; one was severe or blood‐glucose confirmed symptomatic3 (Table S4 in File S1), and no nocturnal episodes occurred.
No safety findings were identified based on clinical laboratory assessments (including amylase, lipase and calcitonin), physical examination or ECG, and no participant developed anti‐semaglutide antibodies. A greater mean (SD) reduction in systolic BP from baseline to end‐of‐treatment was observed for participants treated with semaglutide vs placebo (−8 [16] vs −3 [12] mm Hg); diastolic BP was the same with both treatments (−2 [10] mm Hg). The mean heart rate increased with semaglutide and was unchanged with placebo (6 [8] vs 0 [8] beats/min).
4. DISCUSSION
Absolute concentrations of glucagon during induced hypoglycaemia from 5.5 mmol/L to nadir were similar for semaglutide and placebo, despite the lower glucagon concentration at fasting glucose levels, indicating that semaglutide did not alter the primary counterregulatory response in participants with T2D. Whilst the relative glucagon increase was higher with semaglutide vs placebo, this was primarily driven by the expected lower mean glucagon concentration at 5.5 mmol/L with semaglutide.
In general, the physiological response to hypoglycaemia depends on the primary corrective actions of decreased insulin and increased glucagon concentrations.6 Semaglutide, similarly to other GLP‐1 analogues, inhibits glucagon secretion at fasting glucose concentrations,12, 13, 29 as observed in the present trial, which helps to improve glycaemic control.30 It is, nevertheless, important that the glucagon inhibition does not impair the counterregulatory response under hypoglycaemic conditions. The effect of GLP‐1 and GLP‐1RAs on the counterregulatory response to hypoglycaemia has been investigated in several studies because impaired counterregulation could potentially limit the use of treatment. Native GLP‐1 had no effect on the response to hypoglycaemia in healthy volunteers,31 neither did other GLP‐1RAs in people with type 1 diabetes or T2D.17, 18, 19, 32, 33 The present trial included participants with T2D treated only with metformin, to ensure an adequate glucagon response during the clamp; inclusion of participants with more progressed disease would have lessened the likelihood of detecting an effect of semaglutide. The fact that participants exhibited a robust glucagon response might also explain why not all attained the nadir target of 2.5 mmol/L PG level, despite the constant insulin delivery. More than half of those participants who did not achieve the nadir target failed to do so with both treatments.
Absolute concentrations of other counterregulatory hormones exhibited a smaller increase with semaglutide than with placebo, statistically significantly so for noradrenaline and cortisol; however, the observed differences were unlikely to be of clinical relevance and may have been influenced by improvements in glycaemic control with semaglutide. Adrenaline, as a third defence against hypoglycaemia, normally becomes important only when glucagon is deficient; noradrenaline, cortisol and growth hormone play even smaller roles.8
The higher absolute concentrations of C‐peptide, a marker of endogenous insulin production, observed with semaglutide vs placebo at each target PG level during the clamp were consistent with the ability of GLP‐1RAs to stimulate insulin secretion in a glucose‐dependent manner.12, 13, 29 Importantly, C‐peptide levels decreased with progressing hypoglycaemia; the decrease being more pronounced for participants treated with semaglutide vs placebo. This probably illustrates the expected loss of insulinotropic activity of semaglutide as blood glucose fell into the hypoglycaemic range. The higher C‐peptide concentration with semaglutide at each hypoglycaemia target was influenced by the higher concentration at the start of the clamp procedure and the much longer half‐life of C‐peptide compared with insulin.
The GIR was similar for both treatments throughout the clamp, confirming no compromised counterregulatory response with semaglutide. Neither did semaglutide appear to interfere with recovery from hypoglycaemia; no treatment difference was observed with recovery‐related endpoints.
Participants on semaglutide had lower recognition of hypoglycaemic symptoms at 3.5 mmol/L and nadir vs placebo, as indicated by a lower hypoglycaemic symptom score and fewer positive responses to the question, “Do you feel hypo?” This might be related to lower concentrations of noradrenaline and, to a lesser extent, adrenaline with semaglutide (in the presence of a greater relative increase in glucagon). These are sympathoadrenal hormones which mediate the generation of autonomic symptoms such as palpitations, tremor and anxiety.8 Increases in circulating noradrenaline, however, reflect noradrenaline spillover at post‐ganglionic nerve endings and are not a reliable measure of sympathoadrenal activation.
Nevertheless, these observations raise the possibility that semaglutide might reduce hypoglycaemia awareness and so lead to an increased risk of severe hypoglycaemia in clinical practice. Reassuringly, the glucagon response during hypoglycaemia, the primary counterregulatory hormone, was unaffected in the presence of semaglutide. Indeed, the relative glucagon response was greater than that observed with placebo and, conceivably, a reduced sympathoadrenal response may reflect more efficient counterregulation during hypoglycaemia. This interpretation is supported by similar GIRs in the two arms and the similar rates of glucose recovery. Furthermore, in the studies of semaglutide conducted to date, no increases in rates of hypoglycaemia have been noted, although these (and indeed the present study) were not powered to evaluate differences in severe hypoglycaemia;14, 15, 16, 34 however, in the LEADER trial with the closely‐related molecule, liraglutide, a statistically significantly lower incidence of severe hypoglycaemia was seen for liraglutide compared with placebo.35 In summary, the clinical relevance of the observations in the present trial remains uncertain. Providing a definite answer would require a specifically designed, adequately powered trial.
As expected, semaglutide treatment led to reductions in HbA1c, fasting serum glucose and body weight, and increases in fasting C‐peptide, consistent with findings from larger phase III trials,14, 15, 16, 34 and concomitant with lower glucagon concentrations with semaglutide vs placebo at fasting glucose levels before the clamp. Collectively, data suggest that semaglutide provides effective glycaemic control with a low risk of hypoglycaemia,14, 15, 16, 34 consistent with its mode of action.12, 13
Semaglutide was generally well tolerated and reported side effects were consistent with other semaglutide trials.14, 15, 16, 34 No unexpected treatment‐related safety issues were identified. The increased heart rate with semaglutide is a known effect of treatment with GLP‐1RAs,36 although a large cardiovascular outcomes trial showed a significant reduced risk of cardiovascular events with semaglutide.34
A limitation of the present study is the artificial clamp setting in which maximum physiological concentrations of counterregulatory hormones might not have been obtained. Nevertheless, the clamp procedure is a well‐established method for investigating physiological responses to hypoglycaemia.22, 37 Also, many participants failed to reach the target nadir of 2.5 mmol/L (average 2.9 mmol/L), which may have affected the counterregulatory response measurements. A strength of the present trial is its crossover design; each participant underwent both treatment periods, acting as their own control.
In summary, semaglutide did not impair the overall counterregulatory response to experimental hypoglycaemia in people with T2D, consistent with its mode of action to provide effective glycaemic control with a low risk of hypoglycaemia.
Supporting information
File S1. Supporting information files.
ACKNOWLEDGMENTS
The authors thank the trial participants and trial site personnel who helped conduct the trial, as well as Mikkel Agersnap, MD, PhD and Anne Flint PhD of Novo Nordisk A/S for reviewing and providing input to the manuscript. Editorial and medical writing services were provided by Angela Stocks PhD (Larix A/S, Copenhagen, Denmark), funded by Novo Nordisk.
Conflict of interest
No potential conflicts of interest relevant to this article are reported by S.K., M.B. or S.S. S.R.H. has served on speaker panels for AstraZeneca, Eli Lilly, Novo Nordisk, Sanofi Aventis and Takeda, for which he has received personal remuneration. He has consulted for Boeringher Ingelheim, Eli Lilly, Novo Nordisk and Takeda, for which his institution has received remuneration. T.R.P. declares Board membership for Adocia, AstraZeneca, Eli Lilly, Novo Nordisk and Sanofi, and institutional grants from AstraZeneca and Novo Nordisk, and payment for lectures/speakers bureaus from Novo Nordisk. L.J., M.D.T. and A.G.H. are employed by and hold stock in Novo Nordisk.
Author contributions
S.K., L.J., S.R.H. and T.R.P. contributed to the design of the trial. S.K., M.B., S.S. and T.R.P. were involved in the acquisition of data for the trial and M.D.T. analysed the data. All authors contributed to the interpretation of the data and were involved in writing the manuscript, and all approved the final version and agreed to be accountable for the work.
Korsatko S, Jensen L, Brunner M, et al. Effect of once‐weekly semaglutide on the counterregulatory response to hypoglycaemia in people with type 2 diabetes: A randomized, placebo‐controlled, double‐blind, crossover trial. Diabetes Obes Metab. 2018;20:2565–2573. 10.1111/dom.13422
Funding information Novo Nordisk funded the trial (NN9535‐3684), provided the trial products and was involved in the design of the trial, including protocol development. Novo Nordisk also provided data management and statistical support, and reviewed the manuscript for scientific accuracy.
REFERENCES
- 1. Halban PA, Polonsky KS, Bowden DW, et al. ß‐Cell failure in type 2 diabetes: postulated mechanisms and prospects for prevention and treatment. Diabetes Care. 2014;37:1751‐1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ahren B. Glucagon‐early breakthroughs and recent discoveries. Peptides. 2015;67:74‐81. [DOI] [PubMed] [Google Scholar]
- 3. Seaquist ER, Anderson J, Childs B, et al. Hypoglycemia and diabetes: a report of a workgroup of the American Diabetes Association and the Endocrine Society. Diabetes Care. 2013;36:1384‐1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Christensen M, Bagger JI, Vilsbøll T, Knop FK. The alpha‐cell as target for type 2 diabetes therapy. Rev Diabet Stud. 2011;8:369‐381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lund A, Bagger JI, Christensen M, Knop FK, Vilsbøll T. Glucagon and type 2 diabetes: the return of the alpha cell. Curr Diab Rep. 2014;14:555. [DOI] [PubMed] [Google Scholar]
- 6. Sprague JE, Arbelaez AM. Glucose counterregulatory responses to hypoglycemia. Pediatr Endocrinol Rev. 2011;9:463‐473. [PMC free article] [PubMed] [Google Scholar]
- 7. Cryer PE. Hypoglycemia in type 1 diabetes mellitus. Endocrinol Metab Clin N Am. 2010;39:641‐654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cryer PE, Davis SN. Hypoglycemia Harrison's Principles of Internal Medicine. 18th ed. The McGraw‐Hill Companies, Inc., USA; 2012. [Google Scholar]
- 9. Zammit NN, Frier BM. Hypoglycemia in type 2 diabetes. Pathophysiology, frequency, and effects of different treatment modalities. Diabetes Care. 2005;28:2948‐2961. [DOI] [PubMed] [Google Scholar]
- 10. Cryer PE. The barrier of hypoglycemia in diabetes. Diabetes. 2008;57:3169‐3176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Flint A, Raben A, Astrup A, Holst JJ. Glucagon‐like peptide 1 promotes satiety and suppresses energy intake in humans. J Clin Invest. 1998;101:515‐520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lau J, Bloch P, Schaffer L, et al. Discovery of the once‐weekly glucagon‐like peptide‐1 (GLP‐1) analogue semaglutide. J Med Chem. 2015;58:7370‐7380. [DOI] [PubMed] [Google Scholar]
- 13. Kapitza C, Nosek L, Jensen L, Hartvig H, Jensen CB, Flint A. Semaglutide, a once‐weekly human GLP‐1 analog, does not reduce the bioavailability of the combined oral contraceptive, ethinylestradiol/levonorgestrel. J Clin Pharmacol. 2015;55:497‐504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sorli C, Harashima S, Tsoukas GM, et al. Efficacy and safety of once‐weekly semaglutide monotherapy versus placebo in patients with type 2 diabetes (SUSTAIN 1): a double‐blind, randomised, placebo‐controlled, parallel‐group, multinational, multicentre phase 3a trial. Lancet. 2017;5(4):251‐260. [DOI] [PubMed] [Google Scholar]
- 15. Bo A, Masmiquel L, Kumar H, et al. Efficacy and safety of once weekly semaglutide versus sitagliptin as add‐on to metformin and/or thiazolidinediones in subjects with type 2 diabetes (SUSTAIN 2): a 56‐week randomised, controlled clinical trial. Lancet Diabetes Endocrinol. 2017;5:341‐354. [DOI] [PubMed] [Google Scholar]
- 16. Aroda VR, Bain SC, Cariou B, et al. Efficacy and safety of once‐weekly semaglutide versus once‐daily insulin glargine in insulin‐naïve subjects with type 2 diabetes (SUSTAIN 4): a randomised open‐label clinical trial. Lancet Diabetes. 2017;5:355‐366. [DOI] [PubMed] [Google Scholar]
- 17. Pieber TR, Deller S, Korsatko S, et al. Counter‐regulatory hormone responses to hypoglycaemia in subjects with type 1 diabetes following 4 weeks of treatment with liraglutide adjunct to insulin: a randomized, placebo‐controlled, double‐blind, crossover trial. Diabetes Obes Metab. 2015;17:742‐750. [DOI] [PubMed] [Google Scholar]
- 18. Nauck MA, El Ouaghlidi A, Hompesch M, Jacobsen J, Elbroend B. No impairment of hypoglycemia counterregulation via glucagon with the long‐acting GLP‐1 derivative, NN2211, in subjects with type 2‐diabetes. Diabetologia. 2003;46(Supplement 2):A285. [Google Scholar]
- 19. Yabe D, Eto T, Shiramoto M, et al. Effects of DPP‐4 inhibitor linagliptin and GLP‐1 receptor agonist liraglutide on physiological response to hypoglycaemia in Japanese subjects with type 2 diabetes: a randomized, open‐label, 2‐arm parallel comparative, exploratory trial. Diabetes Obes Metab. 2017;19(3):442‐447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. World Medical Association Declaration of Helsinki . Ethical principles for medical research involving human subjects. JAMA. 2000;284:3043‐3045. [PubMed] [Google Scholar]
- 21. ICH Expert Working Group. International Conference on Harmonisation . ICH Harmonised Tripartite Guideline. Good Clinical Practice, 2003. https://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E6/E6_R1_Guideline.pdf. Accessed December 21, 2017. [Google Scholar]
- 22. Koehler G, Heller S, Korsatko S, et al. Insulin degludec is not associated with a delayed or diminished response to hypoglycaemia compared with insulin glargine in type 1 diabetes: a double‐blind randomised crossover study. Diabetologia. 2014;57:40‐49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. McAulay V, Deary IJ, Frier BM. Symptoms of hypoglycaemia in people with diabetes. Diabet Med. 2001;18:690‐705. [DOI] [PubMed] [Google Scholar]
- 24. Deary IJ, Hepburn DA, MacLeod KM, Frier BM. Partitioning the symptoms of hypoglycaemia using multi‐sample confirmatory factor analysis. Diabetologia. 1993;36:771‐777. [DOI] [PubMed] [Google Scholar]
- 25. Bowie CR, Harvey PD. Administration and interpretation of the Trail Making Test. Nat Protoc. 2006;1:2277‐2281. [DOI] [PubMed] [Google Scholar]
- 26. Wechsler D. Manual of the Wechsler Adult Intelligence Scale ‐ Revised. New York, NY: The Psychological Corporation Ltd; 1981. [Google Scholar]
- 27. Wilkinson RT, Houghton D. Portable four choice reaction time test with magnetic tape memory. Behav Res Methods Instrum. 1975;7:441‐446. [Google Scholar]
- 28. Wewer Albrechtsen NJ, Hartmann B, Veedfald S, et al. Hyperglucagonaemia analysed by glucagon sandwich ELISA: nonspecific interference or truly elevated levels? Diabetologia. 2014;57:1919‐1926. [DOI] [PubMed] [Google Scholar]
- 29. Kapitza C, Dahl K, Bonde Jacobsen J, Axelsen MB, Flint A. The effects of once‐weekly semaglutide on beta‐cell function in subjects with type 2 diabetes. Diabetes Res Clin Pract. 2016;120:S127‐S128. [Google Scholar]
- 30. Creutzfeldt WO, Kleine N, Willms B, Orskov C, Holst JJ, Nauck MA. Glucagonostatic actions and reduction of fasting hyperglycemia by exogenous glucagon‐like peptide I(7‐36) amide in type I diabetic patients. Diabetes Care. 1996;19:580‐586. [DOI] [PubMed] [Google Scholar]
- 31. Nauck MA, Heimesaat MM, Behle K, et al. Effects of glucagon‐like peptide 1 on counterregulatory hormone responses, cognitive functions, and insulin secretion during hyperinsulinemic, stepped hypoglycemic clamp experiments in healthy volunteers. J Clin Endocrinol Metab. 2002;87:1239‐1246. [DOI] [PubMed] [Google Scholar]
- 32. Hompesch M, Jones‐Leone A, Carr MC, et al. Albiglutide does not impair the counter‐regulatory hormone response to hypoglycaemia: a randomized, double‐blind, placebo‐controlled, stepped glucose clamp study in subjects with type 2 diabetes mellitus. Diabetes Obes Metab. 2015;17:82‐90. [DOI] [PubMed] [Google Scholar]
- 33. Farngren J, Persson M, Ahren B. Effect of the GLP‐1 receptor agonist lixisenatide on counterregulatory responses to hypoglyemia in subjects with insulin‐treated type 2 diabetes. Diabetes Care. 2016;39:242‐249. [DOI] [PubMed] [Google Scholar]
- 34. Marso SP, Bain SC, Consoli A, et al. Semaglutide and cardiovascular outcomes in patients with type 2 diabetes. N Engl J Med. 2016;375:1834‐1844. [DOI] [PubMed] [Google Scholar]
- 35. Marso SP, Daniels GH, Brown‐Frandsen K, et al. Liraglutide and cardiovascular outcomes in type 2 diabetes. N Engl J Med. 2016;375:311‐322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Robinson LE, Holt TA, Rees K, Randeva HS, O'Hare JP. Effects of exenatide and liraglutide on heart rate, blood pressure and body weight: systematic review and meta‐analysis. BMJ Open. 2013;3:e001986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Davis SN, Fowler S, Costa F. Hypoglycemic counterregulatory responses differ between men and women with type 1 diabetes. Diabetes. 2000;49:65‐72. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
File S1. Supporting information files.