

Abstract
The transcription factor Nrf2 (nuclear factor‐erythroid 2 p45‐related factor 2) functions at the interface of cellular redox and intermediary metabolism. Nrf2 target genes encode antioxidant enzymes, and proteins involved in xenobiotic detoxification, repair and removal of damaged proteins and organelles, inflammation, and mitochondrial bioenergetics. The function of Nrf2 is altered in many neurodegenerative disorders, such as Huntington's disease, Alzheimer's disease, amyotrophic lateral sclerosis, and Friedreich's ataxia. Nrf2 activation mitigates multiple pathogenic processes involved in these neurodegenerative disorders through upregulation of antioxidant defenses, inhibition of inflammation, improvement of mitochondrial function, and maintenance of protein homeostasis. Small molecule pharmacological activators of Nrf2 have shown protective effects in numerous animal models of neurodegenerative diseases, and in cultures of human cells expressing mutant proteins. Targeting Nrf2 signaling may provide a therapeutic option to delay onset, slow progression, and ameliorate symptoms of neurodegenerative disorders.
Keywords: neurodegeneration, neuroinflammation, neuroprotection, Nrf2 activator, oxidative stress
Abbreviations
- AD
Alzheimer's disease
- ALS
amyotrophic lateral sclerosis
- AMPK
AMP‐activated protein kinase
- APP
amyloid precursor protein
- ARE/EpRE
antioxidant/electrophile response element
- Aβ
amyloid‐β
- CAT
catalase
- ERK
extracellular signal‐regulated kinases
- FRDA
Friedreich's ataxia
- FTLD
frontotemporal lobar degeneration
- GPX
glutathione peroxidase
- GSH
reduced glutathione
- GSK3
glycogen synthase kinase 3
- GST
glutathione S‐transferase
- HD
Huntington's disease
- HFD
high‐fat diet
- HO‐1
heme oxygenase 1
- HTT
huntingtin
- IL
interleukin
- Keap1
Kelch‐like ECH‐associated protein 1
- MPP+
positively charged 1‐methyl‐4‐phenylpyridinium
- MPTP
1‐methyl‐4‐phenyl‐1, 2, 3, 6‐tetrahydropyridine
- NQO1
NAD(P)H: quinone oxidoreductase 1
- Nrf2
nuclear factor‐erythroid 2 p45‐related factor 2
- PD
Parkinson's disease
- PGC‐1α
proliferator‐activated receptor gamma coactivator 1α
- ROS
reactive oxygen species
- SCF
Skp, Cullin, F‐box containing complex
- SOD1
superoxide dismutase 1
- tBHQ
tert‐butyl‐hydroquinone
- TNF‐α
tumor necrosis factor alpha
Introduction
The transcription factor Nrf2 (nuclear factor erythroid 2 p45‐related factor 2) regulates the expression of genes involved in cellular protection against damage by oxidants, electrophiles, and inflammatory agents, and in the maintenance of mitochondrial function, cellular redox, and protein homeostasis 1. Nrf2 protein comprises seven functional domains termed Nrf2‐ECH homology (Neh) 1–7 domains (Fig. 1). Nrf2 binds one of its major negative regulators, Kelch‐like ECH‐associated protein 1 (Keap1) through its N‐terminal Neh2 domain. Neh4 and Neh5 are transactivation domains that recruit cAMP response element‐binding protein (CREB)‐binding protein (CBP) and/or receptor‐associated coactivator 3 (RAC3). The Neh7 domain mediates binding to retinoid X receptor alpha (RXRα), another negative regulator of Nrf2. The Neh6 domain mediates interaction with a third negative regulator, β‐transducin repeat‐containing protein (β‐TrCP). Neh1 is responsible for the formation of a heterodimer with small musculoaponeurotic fibrosarcoma (sMaf) proteins, and mediates binding to antioxidant/electrophile response element (ARE/EpRE) sequences in the promoter regions of Nrf2 target genes. Finally, the C terminus Neh3 is another transactivation domain that recruits chromo‐ATPase/helicase DNA‐binding protein 6 (CHD6).
Figure 1.
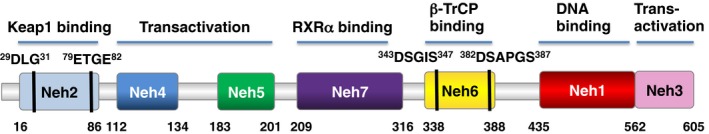
Domain structure of Nrf2. The low affinity binding ‘DLG’ motif and the high affinity binding ‘ETGE’ motif in the N‐terminal Neh2 domain, through which Nrf2 binds to Keap1, are indicated. Also shown is the phosphodegron in the Neh6 domain through which, following phosphorylation by glycogen synthase kinase 3 (GSK3), Nrf2 binds to β‐transducin repeat‐containing protein (β‐TrCP).
Nrf2 levels are regulated primarily by ubiquitination and proteasomal degradation (Fig. 2). After binding to the Neh2 domain, Keap1 mediates the Cullin3 (Cul3)/Rbx1‐dependent ubiquitination of Nrf2 2, 3, 4. Additionally, the Neh6 domain contains a phosphodegron for β‐TrCP/Cullin1‐mediated ubiquitination 5, 6. Synoviolin (Hrd1) and WDR23‐DDB1‐Cul4 are two other ubiquitin ligases that have been shown to participate in the proteasomal degradation of Nrf2 7, 8.
Figure 2.

Regulation of Nrf2 by ubiquitination and proteasomal degradation. Four known ubiquitin ligase systems mediate the degradation of Nrf2: Keap1, a substrate adaptor protein for Cul3/Rbx1‐based ubiquitin ligase and a cysteine‐based sensor for Nrf2 inducers; β‐TrCP, a substrate adaptor for Skp1‐Cul1/Rbx1‐based ubiquitin ligase, WDR23‐DDB1‐Cul4//Roc1‐based ubiquitin ligase, and Hrd1. Keap1‐mediated degradation requires that its cysteine sensors are in reduced state. β‐TrCP‐mediated degradation requires formation of a phosphodegron catalyzed by glycogen synthase kinase 3 (GSK3), which in turn requires phosphorylation by a priming kinase. Hrd1‐mediated degradation of Nrf2 occurs during ER stress.
At homeostatic conditions, Nrf2 is a short‐lived protein. Under stress conditions, Nrf2 is stabilized and translocates to the nucleus, where it binds (as a heterodimer with a member of the small Maf family of transcription factors) to the ARE/EpRE sequences in the promoter of its target genes, and activates their transcription. Nrf2 targets include genes that encode detoxification, antioxidant, and anti‐inflammatory proteins as well as proteins involved in the regulation of autophagy and clearance of damaged proteins, such as proteasomal subunits 9, 10, 11. Activation of Nrf2 leads to the upregulation of proteins involved in the synthesis of glutathione, the main intracellular small molecule antioxidant 12, 13, and NADPH, which provides reducing equivalents for the regeneration of reduced glutathione (GSH) from its oxidized form, GSSG 14, 15, 16, 17. Nrf2 also participates in the maintenance of mitochondrial function 18, 19 and quality control, through activation of mitophagy 20, 21. Nrf2 inhibits the transcription of genes encoding proinflammatory cytokines and suppresses proinflammatory responses following exposure to ultraviolet radiation 22 or lipopolysaccharide (LPS) 23. Such comprehensive cytoprotective functions suggest potential benefits of therapeutic targeting of Nrf2 to counteract neurodegeneration.
Nrf2 in neurodegenerative diseases
Aging is the primary risk factor for developing neurodegenerative diseases 24. The prevalence of age‐dependent neurodegenerative disorders, such as Alzheimer's disease (AD) and Parkinson's disease (PD), is projected to increase due to population aging 25, with a corresponding increase in the associated socioeconomic impact. Genetic and environmental risk factors also contribute to the development of neurodegenerative diseases. Currently, there is no disease‐modifying therapy for any neurodegenerative disease; and even in conditions where symptomatic treatments are available, the therapeutic benefit is limited. Although the clinical manifestations and genetic causes of neurodegenerative diseases are distinct, several of these disorders share strikingly similar pathological mechanisms, including mitochondrial impairment, excessive levels of reactive oxygen species (ROS), neuroinflammation, and disturbances in protein homeostasis (proteostasis) 24. Additionally, there is an age‐associated increase in oxidative damage to the brain, leading to mitochondrial dysfunction, dopamine auto‐oxidation, α‐synuclein aggregation, glial cell activation, alterations in calcium signaling, and excess free iron 26. Dopaminergic neurons show linear decline of 5–10% per decade with aging, and this process is accelerated in PD patients. A meta‐analysis of PD and AD microarray datasets identified 31 common downregulated genes containing the ARE/EpRE consensus sequence in their promoters, in addition to increased levels of Nrf2 27. Increased oxidative stress was observed in human peripheral blood mononuclear cells isolated from individuals with mild cognitive impairment and from 3‐month‐old 3xTg‐AD male mice, which was due to increased levels of nuclear Nrf2 and reduced superoxide dismutase 1 (SOD1) mRNA in the brain cortex 28. The AT‐Nrf2‐knockout mouse model, that combines amyloidopathy and tauopathy with Nrf2 deficiency, presents increased markers of oxidative stress and neuroinflammation in the brain tissue compared to AT‐Nrf2‐wild‐type mice 29. Furthermore, young adult AT‐Nrf2‐knockout mice have deficits in spatial learning and memory and reduced long‐term potentiation in the perforant pathway. Transcriptomic analysis has shown that Nrf2‐knockout mouse brains share 7 and 10 of the most dysregulated pathways with aging human and AD brains, respectively 29.
Oxidative stress in neurodegenerative disease and the counteracting role of Nrf2
The brain is especially sensitive to changes in cellular redox status; therefore, maintaining redox homeostasis in the brain is critical for preventing oxidative stress‐induced cellular damage 24, 25. Nrf2 regulates the expression of a variety of antioxidant enzymes and proteins that exert cytoprotective effects, and there is mounting evidence demonstrating improvement of neurological phenotypes in disease models following the induction of Nrf2‐dependent antioxidant activities.
Synaptic degeneration and neuronal death that characterize AD are caused by several deregulated processes including: (a) increased oxidative stress, (b) chronic inflammation (c) mitochondrial dysfunction, (d) accumulation of amyloid‐β (Aβ) 1–42 peptides, generated from the amyloid precursor protein (APP), (e) proteasome inhibition, (f) mutations in APP, presenilin‐1 and presenilin‐2 genes, and (g) aggregation of hyper‐phosphorylated tau protein 30. Oxidative stress is, at least partially, induced by oligomeric Aβ peptides, and precedes other biochemical changes, while oxidative damage in turn induces chronic inflammation.
Carnosic acid (Fig. 3) is a natural pro‐electrophilic compound that is converted to its active form by oxidative stress, which in turn activates Nrf2‐dependent transcription 31. In vitro, carnosic acid reduced dendritic spine loss in rat neurons exposed to neurotoxic oligomeric Aβ. In vivo, carnosic acid treatment of human amyloid precursor protein (hAPP)‐J20 mice improved learning and memory. Histological analysis showed that carnosic acid increased dendritic and synaptic markers, and decreased astrogliosis, Aβ plaque number, and phospho‐tau staining in the hippocampus. Sulfuretin (Fig. 3), a flavonoid glycoside, significantly attenuated the decrease in cell viability and accumulation of ROS associated with Aβ25‐35‐induced neurotoxicity in neuronal cells 32. Sulfuretin stimulated the activation of Nrf2, resulting in increased expression of the antioxidant Nrf2‐target gene heme oxygenase‐1 (HO‐1). The neuroprotective effects of sulfuretin were diminished following Nrf2 small interfering RNA (siRNA) injection and the addition of HO‐1 inhibitor zinc protoporphyrin IX 32.
Figure 3.

Chemical structures of naturally occurring (A) and synthetic (B) Nrf2 activators with demonstrated efficacy in models of neurological conditions.
Parkinson's disease is characterized by progressive loss of dopaminergic neurons and profound reduction of dopamine in the striatum. Although multiple genetic and environmental factors have been implicated in the etiology of PD, the majority of the clinical cases are sporadic. The 1‐methyl‐4‐phenyl‐1, 2, 3, 6‐tetrahydropyridine (MPTP) mouse model of PD recapitulates the effects of the neurotoxin MPTP‐induced Parkinsonism in humans. Nrf2 activation showed neuroprotective effects in MPTP mice, and these were associated with a reduction of oxidative damage and neuroinflammation 33, 34, 35, 36. Notably, astrocytes from adult and old rats are more susceptible to MPP+ toxicity than astrocytes from newborn rats, but following pretreatment with the Nrf2 activator tert‐butyl‐hydroquinone (Fig. 3), Nrf2‐dependent expression of antioxidant enzymes is increased, resulting in cytoprotection 37.
Huntington's disease (HD) is an autosomal dominant and highly penetrant neurodegenerative disorder, which results from the pathological expansion of trinucleotide CAG repeats – encoding polyglutamine – in the huntingtin (HTT) protein 38. At the time of diagnosis, brains from HD patients show marked striatal and cortical atrophy. Although the pathogenesis of HD is complex, oxidative stress is an important driver of pathology. Compared to nondisease controls, the levels of Nrf2 targets, including glutathione peroxidase, catalase, and SOD1, are increased in human HD brains 39. This suggests a partial activation of the Nrf2‐dependent antioxidant defense. Based on studies using various animal models, pharmacological Nrf2 activation holds promise for combating oxidative stress in HD.
Role of Nrf2 in mitochondrial function
Nrf2 regulates cellular defense through the modulation of mitochondrial function. Nrf2 induction counterbalances mitochondrial production of ROS and defends against mitochondria‐produced toxins 19, 40. Nrf2 function is decreased in mitochondria‐related disorders, such as AD, PD, and Friedreich's ataxia (FRDA). Studies using isolated mitochondria and cultured cells have demonstrated that Nrf2 deficiency leads to impaired mitochondrial fatty acid oxidation, respiration, and ATP production 18, 41. Chemical activators of Nrf2 promote mitochondrial integrity either by inducing mitophagy 20, 21 or by conferring resistance to the oxidative stress‐mediated opening of the mitochondrial permeability transition pore 42, 43. Emerging evidence suggests that Nrf2 also influences mitochondrial biogenesis, particularly under stress conditions. Nrf2 activates proliferator‐activated receptor gamma coactivator 1α (PGC‐1α) and nuclear respiratory factor 1, thereby stimulating mitochondrial biogenesis 44. A recent report has shown that increased proteasomal activity, one of the consequences of Nrf2 activation, leads to enhanced degradation of the mitochondrial fission protein dynamin‐related protein 1 (Drp1), promoting mitochondrial hyperfusion 45. The lower levels of Drp1 under conditions of Nrf2 activation could be beneficial in AD as partial reduction of Drp1 decreases the levels of phosphorylated Tau, a major microtubule‐associated protein (MAPT) of mature neurons, and improves mitochondrial dynamics and synaptic activity in Tau transgenic (P301L) mice 46.
Mitochondrial mass and oxygen consumption increase in primary glial cultures following treatment with the α7 acetylcholine nicotinic receptor (nAChR) agonist PNU282987 in the absence of oxidative stress 47. Importantly, these changes were abolished when either Nrf2 was absent, HO‐1 was inhibited, or PGC‐1α was silenced. Moreover, microglia of PNU282987‐treated animals showed a significant increase in mitochondrial content, while in HO‐1 mutant (LysMcre‐Hmox1Δ/Δ) and PGC‐1α‐deficient strains, the animals showed lower mitochondrial levels in microglia and treatment with PNU282987 was ineffective. These results suggest that α7 nAChR signaling increases glial mitochondrial mass via Nrf2‐dependent stimulation of HO‐1 and PCG‐1α.
Transcriptomic and metabolomic analyses of mutant Drosophila deficient for the mitochondrial serine/threonine‐protein kinase PTEN‐induced putative kinase 1 (PINK1), a model of PD, showed that PINK1 deficiency leads to alterations in nucleotide metabolism, suggesting that enhancing nucleotide biosynthetic pathways could be a strategy to reverse mitochondrial dysfunction in PD 48. Activation of Nrf2 increases the glucose flux through the pentose phosphate pathway and affects the metabolism of folate and glutamine, which in addition to upregulating NADPH and GSH biosynthesis (mentioned above), results in enhanced purine biosynthesis 17. Thus, enhancing nucleotide biosynthesis via Nrf2 activation might reverse mitochondrial dysfunction in PINK1 deficiency. This idea is supported by experiments showing that pharmacological activation of Nrf2 in PINK1‐knockout cells restores the mitochondrial membrane potential (∆ψm) and protects against dopamine toxicity 49.
Friedreich's ataxia is an autosomal recessive neurodegenerative disorder, caused by the expansion of intronic GAA repeats resulting in reduced levels of the mitochondrial protein frataxin – an essential protein for the biogenesis of iron‐sulfur clusters and proper functioning of mitochondrial respiratory chain complexes 50. Several studies using mouse models and cultured cells from patients with FRDA, have reported impaired Nrf2 signaling in FRDA 50, 51, 52. The sensitivity to oxidative stress of YG8R and KIKO fibroblasts from FRDA mouse models can be prevented by Nrf2 inducers, such as the isothiocyanate sulforaphane (Fig. 3) and the tricyclic cyanoenone TBE‐31 (Fig. 3) 53. The pentacyclic cyanoenone RTA‐408 (Omaveloxolone) (Fig. 3) is currently in a Phase 2 clinical trial in FRDA patients (ClinicalTrials.gov). Interestingly, Nrf2 binds an upstream response element in the frataxin locus, and the anesthetic dyclonine has been shown to activate Nrf2, increase the mRNA and protein levels of frataxin and rescue frataxin‐dependent enzyme deficiencies in the iron‐sulfur enzymes aconitase and succinate dehydrogenase 54.
Role of Nrf2 in neuroinflammation
A bidirectional communication takes place between the brain and the peripheral immune system in both physiological and pathological conditions 55. Brain inflammation results from acute injury or following the accumulation of mutant proteins or endogenous neurotoxic metabolites such as those associated with neurodegenerative diseases, including AD and PD. Microglia, in particular, play a key role in brain inflammation via the release of proinflammatory cytokines. Increased neuroinflammation and oxidative stress following microglial activation are associated with age‐related cognitive impairment 55, 56.
The anti‐inflammatory properties of Nrf2 signaling are well established, and recent evidence suggests a mechanism of transcriptional repression of proinflammatory cytokines (TNF‐α, IL‐1, IL‐6, IL‐8, MCP‐1) in microglia, macrophages, monocytes, and astrocytes following Nrf2 activation 23, 57. The Nrf2 activator sulforaphane increased Nrf2 DNA‐binding activity and upregulated Nrf2 target genes in RAW264.7 cells, BV2 microglia cells and primary mouse microglia, while reducing LPS‐induced interleukin IL‐1β, IL‐6, and inducible nitric oxide synthase (iNOS) 58, 59, 60. Furthermore, representatives of seven distinct chemical classes of Nrf2 activators show highly correlated upregulation of NQO1 – a prototypic Nrf2 target – and suppression of iNOS and COX‐2 expression in cell lines and primary mouse peritoneal macrophages 61, 62.
Oral administration of the kavalactone methysticin (Fig. 3) activated the Nrf2 pathway in the hippocampus and cortex of AD (APP/Psen1) mice 63. This treatment reduced microgliosis, astrogliosis, and the secretion of the proinflammatory cytokines TNF‐α and IL‐17A, as well as oxidative damage. Most importantly, the long‐term memory decline of AD mice was significantly attenuated.
Several human studies have demonstrated the protective effects of n‐3 polyunsaturated fatty acids (n‐3 PUFA), particularly in the early stages of mild cognitive impairment preceding AD 64. The Nrf2‐dependent anti‐inflammatory and antioxidant effects of n‐3 PUFA have been linked to a lower activation of microglia. It has been suggested that n‐3 PUFA shift microglia from the macrophage M1 to an M2 phenotype, resulting in lower oxidative stress and enhanced phagocytic activity toward Aβ peptide 64.
Nrf2 signaling‐mediated oxidative stress and neuroinflammation have also been implicated as the key therapeutic targets for amyotrophic lateral sclerosis (ALS). Genetic studies in ALS mouse models have shown a significant therapeutic effect of elevated Nrf2 levels in astrocytes, the major GSH suppliers for neighboring neurons 65. Moreover, Nrf2 signaling is critical for attenuating neuroinflammation in ALS through repression of the deleterious effects of activated microglia on neuronal survival. Consistent with the therapeutic potential of Nrf2 signaling, treatment with small molecule activators, including the potent cyanoenone triterpenoids, has shown efficacy in mouse models of ALS 66.
Role of Nrf2 in autophagy
Autophagy recycles macromolecular aggregates, resulting from oxidative stress, and may also reduce the mitochondrial production of ROS through recycling of old and damaged mitochondria 67, 68. The deregulation of autophagy initiation or progression results in the accumulation of aggregation‐prone proteins in neuronal and extraneural tissues.
Nrf2 promotes autophagy via induction of the autophagosome cargo protein p62/sequestosome‐1 (p62/SQSTM1) 69, 70 and other autophagy‐related genes 10. Knockdown of Nrf2 reduces autophagic flux, whereas pharmacological Nrf2 activation promotes autophagy, and this closely correlates with changes in the levels of p62/SQSTM1 69, 70. This area of research is of particular importance given the strong association between neurological phenotypes and the presence of accumulated misfolded proteins in affected brains, and deficiency of the protein degradation machinery in neurodegenerative diseases. The role of Nrf2 in the clearance of misfolded proteins has received much attention in recent studies, and the relationship with other Nrf2‐controlled stress responses has been evaluated in neurodegenerative and healthy conditions.
Redox mechanisms of autophagy regulation have been documented at the level of crosstalk between the Nrf2 pathway and p62‐associated autophagy. In a Drosophila model of FRDA, genetic or pharmacologic inhibition of the mechanistic target of rapamycin complex 1 (TORC1) signaling led to an increase in the cellular levels of ATP and organismal lifespan, which was in part due to enhanced transcription of antioxidant genes mediated by the Drosophila ortholog of Nrf2 71. Notably, under condition of oxidative stress, the protective effect of TORC1 inhibition was abolished upon inhibition of autophagy.
A recent study has demonstrated that nine autophagy genes containing ARE/EpRE sequence are responsive to Nrf2 activation by sulforaphane in human and mouse cells 10. Mouse embryonic fibroblasts from Nrf2‐knockout mice have reduced expression of autophagy genes, and this deficiency can be rescued by an Nrf2‐expressing lentivirus 10. Compared to their wild‐type counterparts, the neuronal levels of p62/SQSTM1, CALCOCO2/NDP52, ULK1, ATG5, and GABARAPL1 are low in Nrf2‐deficient mice co‐expressing mutant APP (HsAPPV717I) and MAPT (HsMAPTP301L), and these animals have more intracellular aggregates of the mutant proteins. Additionally, in the absence of Nrf2, the colocalization of the mutant APP and MAPT proteins with p62/SQSTM1 was impaired. The same study showed that in AD patient samples, neurons expressing high levels of APP or MAPT also have high levels of nuclear Nrf2 and its transcriptional target p62/SQSTM1 10.
A hallmark of the AD brain is the presence of senile plaques containing insoluble amyloid‐β peptide, a cleavage product of the APP, which is physiologically degraded by autophagy. The neurotoxic amyloid‐β peptide is the result of the enzymatic activity of β and/or γ secretase. The effect of genetic removal of Nrf2 has been investigated in double transgenic mice expressing a chimeric mouse/hAPP (Mo/HuAPP695swe) and a mutant human γ secretases (presenilin 1 (PS1‐dE9) 72. Compared to Nrf2‐proficient animals, APP/PS1 mice that are also deficient for Nrf2 had higher intracellular levels of APP, Aβ (1–42), and Aβ (1–40), which was accompanied by enhanced inflammation. In these Nrf2 mutant mice, the levels of poly‐ubiquitin‐conjugated proteins were increased, APP and Aβ were primarily found in the insoluble fraction, and autophagy was impaired – as evidenced by accumulation of multivesicular bodies, endosomes, and lysosomes.
In certain cases of PD, rare mutations in leucine‐rich repeat kinase 2 (LRRK2) and α‐synuclein (α‐Syn) lead to neurodegeneration, associated with accumulation of misfolded proteins 73, 74. Using a longitudinal imaging platform, the fate and location of mutant LRRK2 and α‐Syn were recently investigated in single live neurons 75. Activation of Nrf2 protected against the toxicity of the mutant proteins in a cell‐autonomous and time‐dependent manner. Interestingly, the protective effect of Nrf2 was different depending on the mutant proteins: Nrf2 activation led to a decrease in the steady‐state levels of α‐Syn by increasing its degradation, whereas misfolded diffuse LRRK2 was sequestered into inclusion bodies 75.
Genetic studies have identified rare mutations in SQSTM1 gene, which causes susceptibility to frontotemporal lobar degeneration (FTLD) and ALS, which further emphasizes a relationship between Nrf2 and autophagy. The term FTLD describes a spectrum of neurodegenerative disorders, characterized, by the deposition of misfolded proteins and focal atrophy of frontal and/or temporal lobes 76. Mutations in genes encoding three autophagy adaptor proteins, p62/SQSTM1, ubiquilin 2, and optineurin, represent a known causative factor for FTLD, implicating autophagy in FTLD pathogenesis. In the majority of FTLD cases, the accumulated protein aggregates contain either microtubule‐associated protein tau or TAR DNA‐binding protein (TDP)‐43 77. Intracellular deposition of TDP‐43 is characterized by prominent neuronal granular cytoplasmic immunoreactivity and abundant oligodendroglial inclusions 78. Aggregates are also positive for ubiquitin and p62/SQSTM1, indicating that these aggregates are targeted for degradation 79. During autophagy, the recognition of the lipidated form of microtubule‐associated protein light chain 3 isoform B (LC3B‐II) within the phagophore membrane by p62/SQSTM1 represents a critical protein–protein interaction that is mediated through its LC3‐interacting region, and notably, some FTLD and ALS mutations of p62/SQSTM1 map to this region, leading to reduction in LC3B binding affinity and limiting the recruitment of p62/SQSTM1 to the phagophore 80.
Mutations in SQSTM1 have been found in patients with ALS, FTLD or with FTLD/ALS when both syndromes are present in the same person 81. Analysis of 348 ALS and FTLD missense mutations in 14 genes focusing on protein stability based on available 3D structures predicted that most of the missense mutations with destabilizing energies occur in the regions that control protein–protein interactions, and that the predicted destabilization is greater for ALS compared to FTLD mutations and correlates with disease progression. However, other ALS causative mutations, which span throughout the SQSTM1 gene, have been identified, namely E81K, N239K, G297S, E372D, P388S, and P392L, suggesting more complex functions of the protein in disease pathogenesis 82. Intriguingly, some SQSTM1 mutations are associated with reduced Nrf2 activation in FTLD/ALS 83, with pharmacological Nrf2 activators showing beneficial effects 84.
Nrf2 signaling in aging
Although some of the downstream biochemical pathways regulated by Nrf2 are directly implicated in neurodegeneration, it is important to evaluate the role of Nrf2 and its impact on the aging processes in the brain as a conceptual paradigm. Aging is tightly associated with redox events, and the free radical theory of aging indicates that redox imbalance may be a promoting factor. A study using Caenorhabditis elegans and human fibroblasts compared differential responses to oxidative stress challenge in young and old individuals 85. In young individuals, higher levels of ROS were generated, and signaling pathways, including p‐ERK, p‐AKT, and p‐AMPKα/β, were activated in response to oxidative stress. Nrf2 translocated to the nucleus and induced the expression of antioxidant and detoxification enzymes, including SOD1, CAT, GPX, HO‐1, GSTP‐1, to maintain redox homeostasis. Moreover, young individuals had a better capacity to degrade damaged proteins by upregulating the expression of molecular chaperones and enhancing the activity of the proteasome 85.
Compelling evidence for Nrf2 suppression in aging came from a study of Hutchinson‐Gilford progeria syndrome (HGPS), a rare fatal premature aging disorder 86. The cause for the disease is progerin, a mutant form of the nuclear architectural protein lamin A. The constitutive production of progerin leads to morphological, genomic, and epigenetic changes, and to mesenchymal stem cell attrition. The study found that progerin sequesters Nrf2, resulting in subnuclear mislocalization and impaired activity of Nrf2 leading to chronic oxidative stress. The aging defects that occur in HGPS fibroblasts can be recapitulated by Nrf2 knockdown or increased oxidative stress caused by treatment with hydrogen peroxide of wild‐type fibroblasts. Conversely, activation of Nrf2 by genetic or pharmacological (by oltipraz treatment) means reverses the nuclear aging defects in HGPS patient cells and restores the viability of HGPS mesenchymal stem cells implanted into the tibialis anterior muscle of immunodeficient mice. These findings strongly suggest that repression of Nrf2 signaling is an essential contributor to the premature aging phenotype.
The regenerative function of neural stem/progenitor cells (NSPCs) continuously declines with advancing age. This specific temporal pattern of NSPC decline is functionally relevant at a behavioral level and correlates with decreased levels of Nrf2 87. Moreover, newborn Nrf2‐knockout mice exhibit a lower number of NSPCs in comparison with their wild‐type counterparts, and the proliferative and neurogenic potential of these NSPCs is compromised in these Nrf2‐deficient animals.
The age‐dependent decline in stem cell proliferation is most likely due to dysregulated signaling in the neurogenic niche and increased levels of inflammatory cytokines 88. Treatment with NT‐020, a proprietary blend of polyphenols, led to an increase in nuclear localization of Nrf2 and expression of its downstream transcriptional target HO‐1 in all subsets of cell types in the subgranular zone of the dentate gyrus and the subventricular zone in brains of both young and aged rats. Analyses of hippocampal tissue showed that the levels of a number of proinflammatory factors, including TNF‐α and IL‐1β, were reduced, whereas the levels of the anti‐inflammatory cytokines IL‐24, IL‐4, and IL‐10 increased. Furthermore, NT‐020 treatment improved performance on a spatial learning task in aged rats 89.
An important question is which aging brain cells lose their Nrf2 activation responses? Markers of Nrf2 activity can be observed in neurons in postmortem brain tissue and animal models of disease; however, recent evidence demonstrates that Nrf2 activity is low in iPS‐derived human neurons 90, and that during mouse cortical neuronal development in vitro and in vivo, the expression of Nrf2 is repressed by epigenetic inactivation of its promoter 91. In contrast, non‐neuronal cells such as astrocytes demonstrate consistent in vitro and in vivo Nrf2 activation responses 92. Astrocytes are highly dynamic cells that have roles in the maintenance of brain homeostasis and energy metabolism, regulation of neurotransmission, and processing of synaptic information and inflammatory triggers 93. One essential function of astrocytes is to provide antioxidant support to neighboring neurons 94. Consistently with their critical role, an astrocyte‐specific, but not neuronal‐specific, Nrf2‐activation rescued the neurological phenotype of mutant SOD1 mouse model of ALS 95. Astrocytes show an age‐dependent increase in oxidative/nitrosative stress, mitochondrial dysfunction, RNA oxidation, NADPH oxidase activity, superoxide levels, and iNOS expression. Astrocytes also display age‐dependent inflammatory response with higher levels of proinflammatory cytokines, such as TNF‐α, IL‐1β, IL‐6, IL‐18, as well as increased mRNA levels of cyclooxygenase 2 (COX‐2) 93. Nevertheless, aged astrocytes are amenable for Nrf2 activation and represent a target cell type for therapeutic intervention 37.
Long‐term consumption of high fat diet (HFD) contributes to cognitive impairment in aged mice 96. A 16‐week HFD increased age‐related oxidative damage (protein carbonyls) in the brain and impaired cognitive performance in behavioral tests of 20‐month‐old male mice. This selective increase in oxidative damage and cognitive decline was associated with decreased levels and activity of Nrf2, suggesting the involvement of lower antioxidant responses. In contrast to HFD consumption, moderate caloric restriction (CR) confers antioxidative and anti‐inflammatory effects, preserving a youthful phenotype in rodents 97. CR prevents age‐related increases in oxidative stress and inflammation and age‐related dysfunction of Nrf2. Intriguingly, exposure of aged cells to resveratrol, a CR mimicking compound and an activator of the cellular energy sensor AMP‐activated protein kinase (AMPK) 98, results in rapid activation of Nrf2 signaling 99, consistent with early reports on the ability of this compound to induce drug metabolizing enzymes and inhibit inflammation 100. Oral administration of another natural product, thymol (Fig. 3), a monoterpene phenol, has been shown to exhibit neuroprotective effects in C57BL/6J mice fed a HFD for 12 weeks 101. Thymol treatment significantly reversed the gain of body weight, improved cognitive impairments and decreased HFD‐induced Aβ deposition and tau hyperphosphorylation in the hippocampus, and these protective effects of thymol were associated with activation of Nrf2 signaling.
Chronic administration of testosterone propionate to aged rats has been shown to ameliorate the decline of balancing reactions and muscular strength associated with aging 102. Interestingly, these beneficial effects are also correlated with decreased oxidative stress, increased protein levels of Nrf2 and its transcriptional targets HO‐1 and NQO1, and greater number of tyrosine hydroxylase immunoreactive cells in substantia nigra, implicating Nrf2 as an important mediator of some of the physiological and behavioral effects of testosterone.
Nrf2‐dependent amelioration of age‐dependent neurological phenotype
A number of pharmacological Nrf2 activators are currently in clinical trials, and the fumaric acid ester dimethylfumarate (Fig. 3) is an FDA‐approved drug (Tecfidera, Biogen‐Idec) for the treatment of relapsing multiple sclerosis (MS). Dimethylfumarate blocks the degradation of Nrf2 by covalent modification of cysteine 151 in Keap1 103, which in addition to serving as a substrate adaptor for Cul3‐mediated degradation for Nrf2, acts as the protein sensor for electrophilies and oxidants 104. Following stereotaxic delivery of recombinant adeno‐associated viral vector expressing human α‐synuclein to the ventral midbrain of mice, daily oral administration of dimethylfumarate protected nigral dopaminergic neurons against the toxicity of α‐synuclein, and decreased astrocytosis and microgliosis; notably, this protective effect was not observed in Nrf2‐knockout mice 105. Dimethylfumarate is, however, of a relatively low potency and specificity. Other molecules with similar mechanism of action or with the ability to directly disrupt the Keap1/Nrf2 interaction are emerging as potential protective agents for the prevention and treatment of a broad range of currently incurable neurodegenerative diseases 57, 59, 106. Inhibition of Keap1 has also been shown to prevent neuronal toxicity in response to the AD‐initiating Aβ42 peptide, in correlation with Nrf2 activation 106. Consistent with this notion, pharmacological activation of Nrf2 by the cyanoenone triterpenoids CDDO‐ethyl amide and CDDO‐trifluoroethyl amide (Fig. 3) induces broad antioxidant effects in HD mouse brain and ameliorates the neurological phenotype 107. The benefits of Nrf2 activation can be extended to other polyglutamine diseases such as spinal and bulbar muscular atrophy (SBMA, also known as Kennedy's disease) 108.
Concluding remarks
In summary, it is becoming increasingly clear that Nrf2 activation counteracts multiple pathogenic processes involved in neurodegenerative disorders through upregulation of antioxidant defenses, inhibition of inflammation, improvement of mitochondrial function, and maintenance of protein homeostasis (Fig. 4). Finally, it is worthwhile pointing out that there appears to be a notable inverse relationship between the expression of mutant proteins that are causally associated with a number of neurodegenerative diseases and Nrf2‐dependent cytoprotective responses. Increased nuclear translocation of Nrf2 is found in a mutant transactive response DNA‐binding protein 43 (TDP‐43) transgenic model of ALS, but the expression of downstream antioxidant enzymes is decreased 109. A gene expression profiling has revealed an upregulation of Nrf2‐target gene expression in the spinal cord of TDP‐43Q331K mice compared to their control counterparts; however, the corresponding protein levels were not increased 110. In TDP‐43M337V patient fibroblasts and astrocyte cell cultures from TDP‐43Q331K mice, the levels of glutathione are low even though the mRNA levels of antioxidant enzymes are high 110. An increase of nuclear Nrf2 in brain cortex, but a decreased expression of Nrf2‐responsive targets was reported in an AD mouse model 28. The activation of Nrf2 by proteasomal inhibition is inhibited in cells expressing PINK1 G309D 111, a mutant PINK1 causally associated with hereditary early‐onset PD 112. The repressive effects of mutant HTT on Nrf2 responses have been observed in immortalized murine striatal cells expressing mutant HTT 113 and in human HD neural stem cells 57. Importantly, pharmacological Nrf2 activation, which was essentially absent in HD neural stem cells with an extreme (72) CAG repeat length, was restored upon isogenic genetic correction of the CAG expansion to a nonpathological (21) CAG repeat length 57. Collectively, these findings suggest a negative effect of a number of mutant proteins across a large spectrum of neurodegenerative diseases on functional Nrf2‐dependent responses and thus a possible exacerbation of the neurological phenotype through reduced Nrf2 signaling. Nonetheless, there is a growing body of convincing experimental evidence demonstrating that pharmacological Nrf2 activation is beneficial in counteracting many of the pathological processes that occur in neurodegenerative diseases, which although of distinct clinical manifestations, share similar molecular mechanisms. Thus, targeting Nrf2 signaling by pharmacological entities, some of which are currently in clinical trials, may provide a therapeutic option to delay onset, slow progression, and ameliorate symptoms of neurodegenerative disorders, including clinical cases of unknown etiology.
Figure 4.
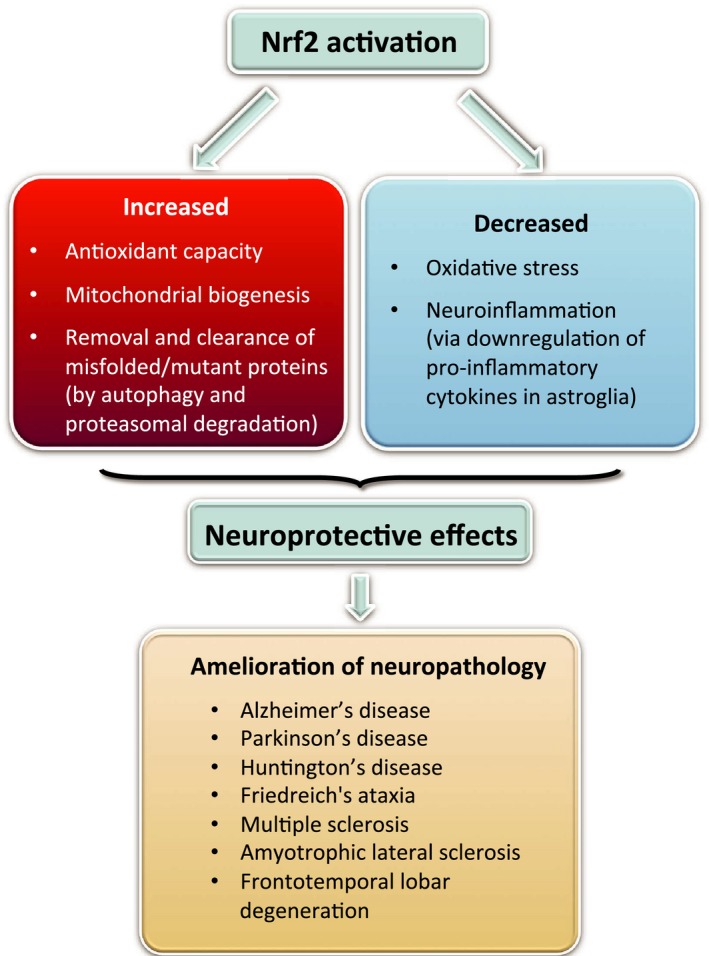
The multiple neuroprotective effects of Nrf2 and human pathologies, in which Nrf2 activation is envisioned to be therapeutically beneficial.
Author contributions
ATD‐K and AGK discussed and developed the concept of the review. AGK wrote the first draft. RVK prepared the figures. All authors participated in writing and editing of the manuscript.
Acknowledgements
We thank Cancer Research UK (C20953/A18644) and the BBSRC (BB/L01923X/1) for financial support.
References
- 1. Hayes JD & Dinkova‐Kostova AT (2014) The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem Sci 39, 199–218. [DOI] [PubMed] [Google Scholar]
- 2. Zhang DD, Lo SC, Cross JV, Templeton DJ & Hannink M (2004) Keap1 is a redox‐regulated substrate adaptor protein for a Cul3‐dependent ubiquitin ligase complex. Mol Cell Biol 24, 10941–10953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cullinan SB, Gordan JD, Jin J, Harper JW & Diehl JA (2004) The Keap1‐BTB protein is an adaptor that bridges Nrf2 to a Cul3‐based E3 ligase: oxidative stress sensing by a Cul3‐Keap1 ligase. Mol Cell Biol 24, 8477–8486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kobayashi A, Kang MI, Okawa H, Ohtsuji M, Zenke Y, Chiba T, Igarashi K & Yamamoto M (2004) Oxidative stress sensor Keap1 functions as an adaptor for Cul3‐based E3 ligase to regulate proteasomal degradation of Nrf2. Mol Cell Biol 24, 7130–7139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rada P, Rojo AI, Chowdhry S, McMahon M, Hayes JD & Cuadrado A (2011) SCF/{beta}‐TrCP promotes glycogen synthase kinase 3‐dependent degradation of the Nrf2 transcription factor in a Keap1‐independent manner. Mol Cell Biol 31, 1121–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chowdhry S, Zhang Y, McMahon M, Sutherland C, Cuadrado A & Hayes JD (2013) Nrf2 is controlled by two distinct beta‐TrCP recognition motifs in its Neh6 domain, one of which can be modulated by GSK‐3 activity. Oncogene 32, 3765–3781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wu T, Zhao F, Gao B, Tan C, Yagishita N, Nakajima T, Wong PK, Chapman E, Fang D & Zhang DD (2014) Hrd1 suppresses Nrf2‐mediated cellular protection during liver cirrhosis. Genes Dev 28, 708–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lo JY, Spatola BN & Curran SP (2017) WDR23 regulates NRF2 independently of KEAP1. PLoS Genet 13, e1006762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kwak MK, Wakabayashi N, Itoh K, Motohashi H, Yamamoto M & Kensler TW (2003) Modulation of gene expression by cancer chemopreventive dithiolethiones through the Keap1‐Nrf2 pathway. Identification of novel gene clusters for cell survival. J Biol Chem 278, 8135–8145. [DOI] [PubMed] [Google Scholar]
- 10. Pajares M, Jimenez‐Moreno N, Garcia‐Yague AJ, Escoll M, de Ceballos ML, Van Leuven F, Rabano A, Yamamoto M, Rojo AI & Cuadrado A (2016) Transcription factor NFE2L2/NRF2 is a regulator of macroautophagy genes. Autophagy 12, 1902–1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Komatsu M, Kurokawa H, Waguri S, Taguchi K, Kobayashi A, Ichimura Y, Sou YS, Ueno I, Sakamoto A, Tong KI et al (2010) The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol 12, 213–223. [DOI] [PubMed] [Google Scholar]
- 12. Wild AC, Moinova HR & Mulcahy RT (1999) Regulation of gamma‐glutamylcysteine synthetase subunit gene expression by the transcription factor Nrf2. J Biol Chem 274, 33627–33636. [DOI] [PubMed] [Google Scholar]
- 13. Moinova HR & Mulcahy RT (1999) Up‐regulation of the human gamma‐glutamylcysteine synthetase regulatory subunit gene involves binding of Nrf‐2 to an electrophile responsive element. Biochem Biophys Res Commun 261, 661–668. [DOI] [PubMed] [Google Scholar]
- 14. Thimmulappa RK, Mai KH, Srisuma S, Kensler TW, Yamamoto M & Biswal S (2002) Identification of Nrf2‐regulated genes induced by the chemopreventive agent sulforaphane by oligonucleotide microarray. Cancer Res 62, 5196–5203. [PubMed] [Google Scholar]
- 15. Lee JM, Calkins MJ, Chan K, Kan YW & Johnson JA (2003) Identification of the NF‐E2‐related factor‐2‐dependent genes conferring protection against oxidative stress in primary cortical astrocytes using oligonucleotide microarray analysis. J Biol Chem 278, 12029–12038. [DOI] [PubMed] [Google Scholar]
- 16. Wu KC, Cui JY & Klaassen CD (2011) Beneficial role of Nrf2 in regulating NADPH generation and consumption. Toxicol Sci 123, 590–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mitsuishi Y, Taguchi K, Kawatani Y, Shibata T, Nukiwa T, Aburatani H, Yamamoto M & Motohashi H (2012) Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell 22, 66–79. [DOI] [PubMed] [Google Scholar]
- 18. Holmstrom KM, Baird L, Zhang Y, Hargreaves I, Chalasani A, Land JM, Stanyer L, Yamamoto M, Dinkova‐Kostova AT & Abramov AY (2013) Nrf2 impacts cellular bioenergetics by controlling substrate availability for mitochondrial respiration. Biol Open 2, 761–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dinkova‐Kostova AT & Abramov AY (2015) The emerging role of Nrf2 in mitochondrial function. Free Radic Biol Med 88, 179–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. East DA, Fagiani F, Crosby J, Georgakopoulos ND, Bertrand H, Schaap M, Fowkes A, Wells G & Campanella M (2014) PMI: a DeltaPsi independent pharmacological regulator of mitophagy. Chem Biol 21, 1585–1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Georgakopoulos ND, Frison M, Alvarez MS, Bertrand H, Wells G & Campanella M (2017) Reversible Keap1 inhibitors are preferential pharmacological tools to modulate cellular mitophagy. Sci Rep 7, 10303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Knatko EV, Ibbotson SH, Zhang Y, Higgins M, Fahey JW, Talalay P, Dawe RS, Ferguson J, Huang JT, Clarke R et al (2015) Nrf2 activation protects against solar‐simulated ultraviolet radiation in mice and humans. Cancer Prev Res (Phila) 8, 475–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kobayashi EH, Suzuki T, Funayama R, Nagashima T, Hayashi M, Sekine H, Tanaka N, Moriguchi T, Motohashi H, Nakayama K et al (2016) Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat Commun 7, 11624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Johnson DA & Johnson JA (2015) Nrf2–a therapeutic target for the treatment of neurodegenerative diseases. Free Radic Biol Med 88, 253–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yamazaki H, Tanji K, Wakabayashi K, Matsuura S & Itoh K (2015) Role of the Keap1/Nrf2 pathway in neurodegenerative diseases. Pathol Int 65, 210–219. [DOI] [PubMed] [Google Scholar]
- 26. Kumar H, Lim HW, More SV, Kim BW, Koppula S, Kim IS & Choi DK (2012) The role of free radicals in the aging brain and Parkinson's Disease: convergence and parallelism. Int J Mol Sci 13, 10478–10504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wang Q, Li WX, Dai SX, Guo YC, Han FF, Zheng JJ, Li GH & Huang JF (2017) Meta‐analysis of Parkinson's disease and Alzheimer's disease revealed commonly impaired pathways and dysregulation of NRF2‐dependent genes. J Alzheimers Dis 56, 1525–1539. [DOI] [PubMed] [Google Scholar]
- 28. Mota SI, Costa RO, Ferreira IL, Santana I, Caldeira GL, Padovano C, Fonseca AC, Baldeiras I, Cunha C, Letra L et al (2015) Oxidative stress involving changes in Nrf2 and ER stress in early stages of Alzheimer's disease. Biochim Biophys Acta 1852, 1428–1441. [DOI] [PubMed] [Google Scholar]
- 29. Rojo AI, Pajares M, Rada P, Nunez A, Nevado‐Holgado AJ, Killik R, Van Leuven F, Ribe E, Lovestone S, Yamamoto M et al (2017) NRF2 deficiency replicates transcriptomic changes in Alzheimer's patients and worsens APP and TAU pathology. Redox Biol 13, 444–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Prasad KN (2016) Simultaneous activation of Nrf2 and elevation of antioxidant compounds for reducing oxidative stress and chronic inflammation in human Alzheimer's disease. Mech Ageing Dev 153, 41–47. [DOI] [PubMed] [Google Scholar]
- 31. Lipton SA, Rezaie T, Nutter A, Lopez KM, Parker J, Kosaka K, Satoh T, McKercher SR, Masliah E & Nakanishi N (2016) Therapeutic advantage of pro‐electrophilic drugs to activate the Nrf2/ARE pathway in Alzheimer's disease models. Cell Death Dis 7, e2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kwon SH, Ma SX, Hwang JY, Lee SY & Jang CG (2015) Involvement of the Nrf2/HO‐1 signaling pathway in sulfuretin‐induced protection against amyloid beta25‐35 neurotoxicity. Neuroscience 304, 14–28. [DOI] [PubMed] [Google Scholar]
- 33. Williamson TP, Johnson DA & Johnson JA (2012) Activation of the Nrf2‐ARE pathway by siRNA knockdown of Keap1 reduces oxidative stress and provides partial protection from MPTP‐mediated neurotoxicity. Neurotoxicology 33, 272–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chen PC, Vargas MR, Pani AK, Smeyne RJ, Johnson DA, Kan YW & Johnson JA (2009) Nrf2‐mediated neuroprotection in the MPTP mouse model of Parkinson's disease: critical role for the astrocyte. Proc Natl Acad Sci USA 106, 2933–2938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rojo AI, Innamorato NG, Martin‐Moreno AM, De Ceballos ML, Yamamoto M & Cuadrado A (2010) Nrf2 regulates microglial dynamics and neuroinflammation in experimental Parkinson's disease. Glia 58, 588–598. [DOI] [PubMed] [Google Scholar]
- 36. Jazwa A, Rojo AI, Innamorato NG, Hesse M, Fernandez‐Ruiz J & Cuadrado A (2011) Pharmacological targeting of the transcription factor Nrf2 at the basal ganglia provides disease modifying therapy for experimental parkinsonism. Antioxid Redox Signal 14, 2347–2360. [DOI] [PubMed] [Google Scholar]
- 37. Alarcon‐Aguilar A, Luna‐Lopez A, Ventura‐Gallegos JL, Lazzarini R, Galvan‐Arzate S, Gonzalez‐Puertos VY, Moran J, Santamaria A & Konigsberg M (2014) Primary cultured astrocytes from old rats are capable to activate the Nrf2 response against MPP+ toxicity after tBHQ pretreatment. Neurobiol Aging 35, 1901–1912. [DOI] [PubMed] [Google Scholar]
- 38. Jimenez‐Sanchez M, Licitra F, Underwood BR & Rubinsztein DC (2017) Huntington's disease: mechanisms of pathogenesis and therapeutic strategies. Cold Spring Harb Perspect Med 7, a024240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Prasad KN & Bondy SC (2016) Inhibition of early biochemical defects in prodromal Huntington's disease by simultaneous activation of Nrf2 and elevation of multiple micronutrients. Curr Aging Sci 9, 61–70. [DOI] [PubMed] [Google Scholar]
- 40. Esteras N, Dinkova‐Kostova AT & Abramov AY (2016) Nrf2 activation in the treatment of neurodegenerative diseases: a focus on its role in mitochondrial bioenergetics and function. Biol Chem 397, 383–400. [DOI] [PubMed] [Google Scholar]
- 41. Ludtmann MH, Angelova PR, Zhang Y, Abramov AY & Dinkova‐Kostova AT (2014) Nrf2 affects the efficiency of mitochondrial fatty acid oxidation. Biochem J 457, 415–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Greco T & Fiskum G (2010) Brain mitochondria from rats treated with sulforaphane are resistant to redox‐regulated permeability transition. J Bioenerg Biomembr 42, 491–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Greco T, Shafer J & Fiskum G (2011) Sulforaphane inhibits mitochondrial permeability transition and oxidative stress. Free Radic Biol Med 51, 2164–2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Piantadosi CA, Carraway MS, Babiker A & Suliman HB (2008) Heme oxygenase‐1 regulates cardiac mitochondrial biogenesis via Nrf2‐mediated transcriptional control of nuclear respiratory factor‐1. Circ Res 103, 1232–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sabouny R, Fraunberger E, Geoffrion M, Ng AC, Baird SD, Screaton RA, Milne R, McBride HM & Shutt TE (2017) The Keap1‐Nrf2 stress response pathway promotes mitochondrial hyperfusion through degradation of the mitochondrial fission protein Drp1. Antioxid Redox Signal 27, 1447–1459. [DOI] [PubMed] [Google Scholar]
- 46. Kandimalla R, Manczak M, Fry D, Suneetha Y, Sesaki H & Reddy PH (2016) Reduced dynamin‐related protein 1 protects against phosphorylated Tau‐induced mitochondrial dysfunction and synaptic damage in Alzheimer's disease. Hum Mol Genet 25, 4881–4897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Navarro E, Gonzalez‐Lafuente L, Perez‐Liebana I, Buendia I, Lopez‐Bernardo E, Sanchez‐Ramos C, Prieto I, Cuadrado A, Satrustegui J, Cadenas S et al (2017) Heme‐oxygenase I and PCG‐1alpha regulate mitochondrial biogenesis via microglial activation of alpha7 nicotinic acetylcholine receptors using PNU282987. Antioxid Redox Signal 27, 93–105. [DOI] [PubMed] [Google Scholar]
- 48. Tufi R, Gandhi S, de Castro IP, Lehmann S, Angelova PR, Dinsdale D, Deas E, Plun‐Favreau H, Nicotera P, Abramov AY et al (2014) Enhancing nucleotide metabolism protects against mitochondrial dysfunction and neurodegeneration in a PINK1 model of Parkinson's disease. Nat Cell Biol 16, 157–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Dinkova‐Kostova AT, Baird L, Holmstrom KM, Meyer CJ & Abramov AY (2015) The spatiotemporal regulation of the Keap1‐Nrf2 pathway and its importance in cellular bioenergetics. Biochem Soc Trans 43, 602–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Paupe V, Dassa EP, Goncalves S, Auchere F, Lonn M, Holmgren A & Rustin P (2009) Impaired nuclear Nrf2 translocation undermines the oxidative stress response in Friedreich ataxia. PLoS One 4, e4253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Shan Y, Schoenfeld RA, Hayashi G, Napoli E, Akiyama T, Iodi Carstens M, Carstens EE, Pook MA & Cortopassi GA (2013) Frataxin deficiency leads to defects in expression of antioxidants and Nrf2 expression in dorsal root ganglia of the Friedreich's ataxia YG8R mouse model. Antioxid Redox Signal 19, 1481–1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. D'Oria V, Petrini S, Travaglini L, Priori C, Piermarini E, Petrillo S, Carletti B, Bertini E & Piemonte F (2013) Frataxin deficiency leads to reduced expression and impaired translocation of NF‐E2‐related factor (Nrf2) in cultured motor neurons. Int J Mol Sci 14, 7853–7865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Abeti R, Uzun E, Renganathan I, Honda T, Pook MA & Giunti P (2015) Targeting lipid peroxidation and mitochondrial imbalance in Friedreich's ataxia. Pharmacol Res 99, 344–350. [DOI] [PubMed] [Google Scholar]
- 54. Sahdeo S, Scott BD, McMackin MZ, Jasoliya M, Brown B, Wulff H, Perlman SL, Pook MA & Cortopassi GA (2014) Dyclonine rescues frataxin deficiency in animal models and buccal cells of patients with Friedreich's ataxia. Hum Mol Genet 23, 6848–6862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Jarrott B & Williams SJ (2016) Chronic brain inflammation: the neurochemical basis for drugs to reduce inflammation. Neurochem Res 41, 523–533. [DOI] [PubMed] [Google Scholar]
- 56. Hoenen C, Gustin A, Birck C, Kirchmeyer M, Beaume N, Felten P, Grandbarbe L, Heuschling P & Heurtaux T (2016) Alpha‐synuclein proteins promote pro‐inflammatory cascades in microglia: stronger effects of the A53T mutant. PLoS One 11, e0162717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Quinti L, Dayalan Naidu S, Trager U, Chen X, Kegel‐Gleason K, Lleres D, Connolly C, Chopra V, Low C, Moniot S et al (2017) KEAP1‐modifying small molecule reveals muted NRF2 signaling responses in neural stem cells from Huntington's disease patients. Proc Natl Acad Sci USA 114, E4676–E4685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Townsend BE & Johnson RW (2016) Sulforaphane induces Nrf2 target genes and attenuates inflammatory gene expression in microglia from brain of young adult and aged mice. Exp Gerontol 73, 42–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Quinti L, Casale M, Moniot S, Pais TF, Van Kanegan MJ, Kaltenbach LS, Pallos J, Lim RG, Naidu SD, Runne H et al (2016) SIRT2‐ and NRF2‐targeting thiazole‐containing compound with therapeutic activity in Huntington's disease models. Cell Chem Biol 23, 849–861. [DOI] [PubMed] [Google Scholar]
- 60. Dinkova‐Kostova AT, Jenkins SN, Fahey JW, Ye L, Wehage SL, Liby KT, Stephenson KK, Wade KL & Talalay P (2006) Protection against UV‐light‐induced skin carcinogenesis in SKH‐1 high‐risk mice by sulforaphane‐containing broccoli sprout extracts. Cancer Lett 240, 243–252. [DOI] [PubMed] [Google Scholar]
- 61. Dinkova‐Kostova AT, Liby KT, Stephenson KK, Holtzclaw WD, Gao X, Suh N, Williams C, Risingsong R, Honda T, Gribble GW et al (2005) Extremely potent triterpenoid inducers of the phase 2 response: correlations of protection against oxidant and inflammatory stress. Proc Natl Acad Sci USA 102, 4584–4589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Liu H, Dinkova‐Kostova AT & Talalay P (2008) Coordinate regulation of enzyme markers for inflammation and for protection against oxidants and electrophiles. Proc Natl Acad Sci USA 105, 15926–15931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Fragoulis A, Siegl S, Fendt M, Jansen S, Soppa U, Brandenburg LO, Pufe T, Weis J & Wruck CJ (2017) Oral administration of methysticin improves cognitive deficits in a mouse model of Alzheimer's disease. Redox Biol 12, 843–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Serini S & Calviello G (2016) Reduction of oxidative/nitrosative stress in brain and its involvement in the neuroprotective effect of n‐3 PUFA in Alzheimer's disease. Curr Alzheimer Res 13, 123–134. [DOI] [PubMed] [Google Scholar]
- 65. Vargas MR, Pehar M, Cassina P, Beckman JS & Barbeito L (2006) Increased glutathione biosynthesis by Nrf2 activation in astrocytes prevents p75NTR‐dependent motor neuron apoptosis. J Neurochem 97, 687–696. [DOI] [PubMed] [Google Scholar]
- 66. Neymotin A, Calingasan NY, Wille E, Naseri N, Petri S, Damiano M, Liby KT, Risingsong R, Sporn M, Beal MF et al (2011) Neuroprotective effect of Nrf2/ARE activators, CDDO ethylamide and CDDO trifluoroethylamide, in a mouse model of amyotrophic lateral sclerosis. Free Radic Biol Med 51, 88–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Giordano S, Darley‐Usmar V & Zhang J (2014) Autophagy as an essential cellular antioxidant pathway in neurodegenerative disease. Redox Biol 2, 82–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Hensley K & Harris‐White ME (2015) Redox regulation of autophagy in healthy brain and neurodegeneration. Neurobiol Dis 84, 50–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Dayalan Naidu S, Dikovskaya D, Gaurilcikaite E, Knatko EV, Healy ZR, Mohan H, Koh G, Laurell A, Ball G, Olagnier D et al (2017) Transcription factors NRF2 and HSF1 have opposing functions in autophagy. Sci Rep 7, 11023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Olagnier D, Lababidi RR, Hadj SB, Sze A, Liu Y, Naidu SD, Ferrari M, Jiang Y, Chiang C, Beljanski V et al (2017) Activation of Nrf2 signaling augments vesicular stomatitis virus oncolysis via autophagy‐driven suppression of antiviral immunity. Mol Ther 25, 1900–1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Calap‐Quintana P, Soriano S, Llorens JV, Al‐Ramahi I, Botas J, Molto MD & Martinez‐Sebastian MJ (2015) TORC1 inhibition by rapamycin promotes antioxidant defences in a Drosophila model of Friedreich's ataxia. PLoS One 10, e0132376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Joshi G, Gan KA, Johnson DA & Johnson JA (2015) Increased Alzheimer's disease‐like pathology in the APP/PS1DeltaE9 mouse model lacking Nrf2 through modulation of autophagy. Neurobiol Aging 36, 664–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Clark LN, Wang Y, Karlins E, Saito L, Mejia‐Santana H, Harris J, Louis ED, Cote LJ, Andrews H, Fahn S et al (2006) Frequency of LRRK2 mutations in early‐ and late‐onset Parkinson disease. Neurology 67, 1786–1791. [DOI] [PubMed] [Google Scholar]
- 74. Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R et al (1997) Mutation in the alpha‐synuclein gene identified in families with Parkinson's disease. Science 276, 2045–2047. [DOI] [PubMed] [Google Scholar]
- 75. Skibinski G, Hwang V, Ando DM, Daub A, Lee AK, Ravisankar A, Modan S, Finucane MM, Shaby BA & Finkbeiner S (2017) Nrf2 mitigates LRRK2‐ and alpha‐synuclein‐induced neurodegeneration by modulating proteostasis. Proc Natl Acad Sci USA 114, 1165–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Gotzl JK, Lang CM, Haass C & Capell A (2016) Impaired protein degradation in FTLD and related disorders. Ageing Res Rev 32, 122–139. [DOI] [PubMed] [Google Scholar]
- 77. Rainero I, Rubino E, Michelerio A, D'Agata F, Gentile S & Pinessi L (2017) Recent advances in the molecular genetics of frontotemporal lobar degeneration. Funct Neurol 32, 7–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Luis E, Ortiz A, Eudave L, Ortega‐Cubero S, Borroni B, van der Zee J, Gazzina S, Caroppo P, Rubino E, D'Agata F et al (2016) Neuroimaging correlates of frontotemporal dementia associated with SQSTM1 mutations. J Alzheimers Dis 53, 303–313. [DOI] [PubMed] [Google Scholar]
- 79. Kovacs GG, van der Zee J, Hort J, Kristoferitsch W, Leitha T, Hoftberger R, Strobel T, Van Broeckhoven C & Matej R (2016) Clinicopathological description of two cases with SQSTM1 gene mutation associated with frontotemporal dementia. Neuropathology 36, 27–38. [DOI] [PubMed] [Google Scholar]
- 80. Goode A, Butler K, Long J, Cavey J, Scott D, Shaw B, Sollenberger J, Gell C, Johansen T, Oldham NJ et al (2016) Defective recognition of LC3B by mutant SQSTM1/p62 implicates impairment of autophagy as a pathogenic mechanism in ALS‐FTLD. Autophagy 12, 1094–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Kumar V, Islam A, Hassan MI & Ahmad F (2016) Delineating the relationship between amyotrophic lateral sclerosis and frontotemporal dementia: sequence and structure‐based predictions. Biochim Biophys Acta 1862, 1742–1754. [DOI] [PubMed] [Google Scholar]
- 82. Yang Y, Tang L, Zhang N, Pan L, Hadano S & Fan D (2015) Six SQSTM1 mutations in a Chinese amyotrophic lateral sclerosis cohort. Amyotroph Lateral Scler Frontotemporal Degener 16, 378–384. [DOI] [PubMed] [Google Scholar]
- 83. Goode A, Rea S, Sultana M, Shaw B, Searle MS & Layfield R (2016) ALS‐FTLD associated mutations of SQSTM1 impact on Keap1‐Nrf2 signalling. Mol Cell Neurosci 76, 52–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Bartolome F, Esteras N, Martin‐Requero A, Boutoleau‐Bretonniere C, Vercelletto M, Gabelle A, Le Ber I, Honda T, Dinkova‐Kostova AT, Hardy J et al (2017) Pathogenic p62/SQSTM1 mutations impair energy metabolism through limitation of mitochondrial substrates. Sci Rep 7, 1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Meng J, Lv Z, Qiao X, Li X, Li Y, Zhang Y & Chen C (2017) The decay of redox‐stress response capacity is a substantive characteristic of aging: revising the redox theory of aging. Redox Biol 11, 365–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Kubben N, Zhang W, Wang L, Voss TC, Yang J, Qu J, Liu GH & Misteli T (2016) Repression of the antioxidant NRF2 pathway in premature aging. Cell 165, 1361–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Corenblum MJ, Ray S, Remley QW, Long M, Harder B, Zhang DD, Barnes CA & Madhavan L (2016) Reduced Nrf2 expression mediates the decline in neural stem cell function during a critical middle‐age period. Aging Cell 15, 725–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Flowers A, Lee JY, Acosta S, Hudson C, Small B, Sanberg CD & Bickford PC (2015) NT‐020 treatment reduces inflammation and augments Nrf‐2 and Wnt signaling in aged rats. J Neuroinflammation 12, 174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Acosta S, Jernberg J, Sanberg CD, Sanberg PR, Small BJ, Gemma C & Bickford PC (2010) NT‐020, a natural therapeutic approach to optimize spatial memory performance and increase neural progenitor cell proliferation and decrease inflammation in the aged rat. Rejuvenation Res 13, 581–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Liddell JR (2017) Are astrocytes the predominant cell type for activation of Nrf2 in aging and neurodegeneration? Antioxidants (Basel) 6, 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Bell KF, Al‐Mubarak B, Martel MA, McKay S, Wheelan N, Hasel P, Markus NM, Baxter P, Deighton RF, Serio A et al (2015) Neuronal development is promoted by weakened intrinsic antioxidant defences due to epigenetic repression of Nrf2. Nat Commun 6, 7066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Vargas MR & Johnson JA (2009) The Nrf2‐ARE cytoprotective pathway in astrocytes. Expert Rev Mol Med 11, e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Bellaver B, Souza DG, Souza DO & Quincozes‐Santos A (2017) Hippocampal astrocyte cultures from adult and aged rats reproduce changes in glial functionality observed in the aging brain. Mol Neurobiol 54, 2969–2985. [DOI] [PubMed] [Google Scholar]
- 94. Baxter PS & Hardingham GE (2016) Adaptive regulation of the brain's antioxidant defences by neurons and astrocytes. Free Radic Biol Med 100, 147–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Vargas MR, Johnson DA, Sirkis DW, Messing A & Johnson JA (2008) Nrf2 activation in astrocytes protects against neurodegeneration in mouse models of familial amyotrophic lateral sclerosis. J Neurosci 28, 13574–13581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Morrison CD, Pistell PJ, Ingram DK, Johnson WD, Liu Y, Fernandez‐Kim SO, White CL, Purpera MN, Uranga RM, Bruce‐Keller AJ et al (2010) High fat diet increases hippocampal oxidative stress and cognitive impairment in aged mice: implications for decreased Nrf2 signaling. J Neurochem 114, 1581–1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Csiszar A, Gautam T, Sosnowska D, Tarantini S, Banki E, Tucsek Z, Toth P, Losonczy G, Koller A, Reglodi D et al (2014) Caloric restriction confers persistent anti‐oxidative, pro‐angiogenic, and anti‐inflammatory effects and promotes anti‐aging miRNA expression profile in cerebromicrovascular endothelial cells of aged rats. Am J Physiol Heart Circ Physiol 307, H292–H306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Hardie DG, Carling D & Gamblin SJ (2011) AMP‐activated protein kinase: also regulated by ADP? Trends Biochem Sci 36, 470–477. [DOI] [PubMed] [Google Scholar]
- 99. Csiszar A, Csiszar A, Pinto JT, Gautam T, Kleusch C, Hoffmann B, Tucsek Z, Toth P, Sonntag WE & Ungvari Z (2015) Resveratrol encapsulated in novel fusogenic liposomes activates Nrf2 and attenuates oxidative stress in cerebromicrovascular endothelial cells from aged rats. J Gerontol A Biol Sci Med Sci 70, 303–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Jang M, Cai L, Udeani GO, Slowing KV, Thomas CF, Beecher CW, Fong HH, Farnsworth NR, Kinghorn AD, Mehta RG et al (1997) Cancer chemopreventive activity of resveratrol, a natural product derived from grapes. Science 275, 218–220. [DOI] [PubMed] [Google Scholar]
- 101. FangFang, Li H, Qin T, Li M & Ma S (2017) Thymol improves high‐fat diet‐induced cognitive deficits in mice via ameliorating brain insulin resistance and upregulating NRF2/HO‐1 pathway. Metab Brain Dis 32, 385–393. [DOI] [PubMed] [Google Scholar]
- 102. Zhang GL, Wang W, Kang YX, Xue Y, Yang H, Zhou CM & Shi GM (2013) Chronic testosterone propionate supplement could activated the Nrf2‐ARE pathway in the brain and ameliorated the behaviors of aged rats. Behav Brain Res 252, 388–395. [DOI] [PubMed] [Google Scholar]
- 103. Linker RA, Lee DH, Ryan S, van Dam AM, Conrad R, Bista P, Zeng W, Hronowsky X, Buko A, Chollate S et al (2011) Fumaric acid esters exert neuroprotective effects in neuroinflammation via activation of the Nrf2 antioxidant pathway. Brain 134, 678–692. [DOI] [PubMed] [Google Scholar]
- 104. Dinkova‐Kostova AT, Kostov RV & Canning P (2017) Keap1, the cysteine‐based mammalian intracellular sensor for electrophiles and oxidants. Arch Biochem Biophys 617, 84–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Lastres‐Becker I, Garcia‐Yague AJ, Scannevin RH, Casarejos MJ, Kugler S, Rabano A & Cuadrado A (2016) Repurposing the NRF2 activator dimethyl fumarate as therapy against synucleinopathy in Parkinson's disease. Antioxid Redox Signal 25, 61–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Kerr F, Sofola‐Adesakin O, Ivanov DK, Gatliff J, Gomez Perez‐Nievas B, Bertrand HC, Martinez P, Callard R, Snoeren I, Cocheme HM et al (2017) Direct Keap1‐Nrf2 disruption as a potential therapeutic target for Alzheimer's disease. PLoS Genet 13, e1006593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Stack C, Ho D, Wille E, Calingasan NY, Williams C, Liby K, Sporn M, Dumont M & Beal MF (2010) Triterpenoids CDDO‐ethyl amide and CDDO‐trifluoroethyl amide improve the behavioral phenotype and brain pathology in a transgenic mouse model of Huntington's disease. Free Radic Biol Med 49, 147–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Bott LC, Badders NM, Chen KL, Harmison GG, Bautista E, Shih CC, Katsuno M, Sobue G, Taylor JP, Dantuma NP et al (2016) A small‐molecule Nrf1 and Nrf2 activator mitigates polyglutamine toxicity in spinal and bulbar muscular atrophy. Hum Mol Genet 25, 1979–1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Tian YP, Che FY, Su QP, Lu YC, You CP, Huang LM, Wang SG, Wang L & Yu JX (2017) Effects of mutant TDP‐43 on the Nrf2/ARE pathway and protein expression of MafK and JDP2 in NSC‐34 cells. Genet Mol Res 16, 1–13. [DOI] [PubMed] [Google Scholar]
- 110. Moujalled D, Grubman A, Acevedo K, Yang S, Ke YD, Moujalled DM, Duncan C, Caragounis A, Perera ND, Turner BJ et al (2017) TDP‐43 mutations causing amyotrophic lateral sclerosis are associated with altered expression of RNA‐binding protein hnRNP K and affect the Nrf2 antioxidant pathway. Hum Mol Genet 26, 1732–1746. [DOI] [PubMed] [Google Scholar]
- 111. Sheng XJ, Tu HJ, Chien WL, Kang KH, Lu DH, Liou HH, Lee MJ & Fu WM (2017) Antagonism of proteasome inhibitor‐induced heme oxygenase‐1 expression by PINK1 mutation. PLoS One 12, e0183076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Valente EM, Abou‐Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, Ali Z, Del Turco D, Bentivoglio AR, Healy DG et al (2004) Hereditary early‐onset Parkinson's disease caused by mutations in PINK1. Science 304, 1158–1160. [DOI] [PubMed] [Google Scholar]
- 113. Jin YN, Yu YV, Gundemir S, Jo C, Cui M, Tieu K & Johnson GV (2013) Impaired mitochondrial dynamics and Nrf2 signaling contribute to compromised responses to oxidative stress in striatal cells expressing full‐length mutant huntingtin. PLoS One 8, e57932. [DOI] [PMC free article] [PubMed] [Google Scholar]