

Abstract
Parkinson's disease motor symptoms are treated with levodopa, but long‐term treatment leads to disabling dyskinesia. Altered synaptic transmission and maladaptive plasticity of corticostriatal glutamatergic projections play a critical role in the pathophysiology of dyskinesia. Because the noble gas xenon inhibits excitatory glutamatergic signaling, primarily through allosteric antagonism of the N‐methyl‐d‐aspartate receptors, we aimed to test its putative antidyskinetic capabilities. We first studied the direct effect of xenon gas exposure on corticostriatal plasticity in a murine model of levodopa‐induced dyskinesia We then studied the impact of xenon inhalation on behavioral dyskinetic manifestations in the gold‐standard rat and primate models of PD and levodopa‐induced dyskinesia. Last, we studied the effect of xenon inhalation on axial gait and posture deficits in a primate model of PD with levodopa‐induced dyskinesia. This study shows that xenon gas exposure (1) normalized synaptic transmission and reversed maladaptive plasticity of corticostriatal glutamatergic projections associated with levodopa‐induced dyskinesia, (2) ameliorated dyskinesia in rat and nonhuman primate models of PD and dyskinesia, and (3) improved gait performance in a nonhuman primate model of PD. These results pave the way for clinical testing of this unconventional but safe approach. © 2018 The Authors. Movement Disorders published by Wiley Periodicals, Inc. on behalf of International Parkinson and Movement Disorder Society.
Keywords: rodent, primate, behavior, corticostriatal plasticity, NMDA
The reduced movement repertoire of Parkinson's disease (PD) is a result primarily of degeneration of nigrostriatal dopamine neurons. Levodopa (l‐dopa) restores dopamine‐mediated modulation, which can successfully relieve motor symptoms of PD. However, the repeated intake of l‐dopa leads to severe side effects, known as l‐dopa‐induced dyskinesia (LID).1
Several cellular and molecular mechanisms have been linked to the development and onset of LID in PD.1, 2 Among these mechanisms, impaired synaptic transmission and maladaptive plasticity of corticostriatal glutamatergic projections have been shown to play a critical role.3, 4 Clinical evidence also suggests involvement of l‐dopa‐induced disruption in the trafficking of N‐methyl‐d‐aspartate receptor (NMDA) receptor subunits associated with the development of LID. Indeed, an evidence‐based medicine review recommended the addition of amantadine, a multitarget drug that antagonizes the activity of NMDA to l‐dopa treatment to mitigate dyskinesia.5 Other drugs that target glutamatergic transmission with higher specificity have been evaluated in clinical tests, but they either failed to reduce LID or were not well tolerated.1
The noble gas xenon is capable of inhibiting excitatory glutamatergic signaling through antagonism of NMDA receptors.6 Here, we provide electrophysiological and behavioral evidence that supports the hypothesis that xenon gas exposure reverses LID‐inducing abnormal corticostriatal plasticity and ameliorates LID in gold‐standard rodent and primate models of PD and LID. Furthermore, we show that xenon inhalation can alleviate axial gait and posture deficits in a primate model of PD with LID. In contrast to other PD motor symptoms, l‐dopa treatment exerts little effect on, or even further exacerbates, these symptoms.7 These results establish a mechanistic framework to evaluate the ability of safe xenon inhalation to alleviate dyskinesia‐related symptoms in patients with PD and LID.
Methods and Materials
Detailed experimental procedures are provided in the Supplementary Methods.
Ex Vivo Electrophysiology in the 6‐OHDA‐Lesioned Mouse Model of PD
Corticostriatal plasticity was studied in l‐dopa‐treated 6‐hydroxydopamine (6‐OHDA)‐lesioned bacterial artificial chromosome transgenic mice expressing enhanced green fluorescent protein (EGFP) under the control of the promoter for the dopamine receptor type 1 (D1R) (Drd1a‐EGFP; 000297‐MU/H), prepared as previously described.8, 9, 10 abnormal involuntary movement (AIMs) were scored using a 0‐4 rating scale according to a validated rating system.8, 11 Electrophysiology on brain slices was conducted as described elsewhere.4, 12
l‐Dopa‐Induced AIMs in the 6‐OHDA‐Lesioned Rat Model
The AIM model was prepared in male Sprague‐Dawley rats (Charles River Laboratories) as previously described.13, 14, 15 The 4 AIM categories (axial, limb, and orolingual, hereafter referred to as ALO, and locomotive) were scored using a validated rating scale16, 25 by a trained investigator as previously described.13, 14, 15
l‐Dopa‐Induced Dyskinesia in the MPTP‐Treated Macaque Model
We conducted the 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine (MPTP) intoxication protocol, chronic l‐dopa treatment, and clinical assessments in 6 male macaques (Macaca mulatta, weight, 4.7 ± 0.3 kg; Xierxin, Beijing, PR of China), as previously published.8, 10, 13, 15, 17, 18, 19
Kinematics in the MPTP‐Treated Macaque Model
Gait pattern assessments were performed in 4 dyskinetic parkinsonian and 6 healthy able‐bodied macaques using a high‐speed motion capture system (Simi Reality Motion Systems, Germany), combining 4 or 6 video cameras (100 Hz) as previously described.20, 21, 22
Results
Xenon Exposure Normalizes Maladaptive Corticostriatal Plasticity Through NMDA Receptors
We first investigated the impact of xenon exposure on glutamatergic corticostriatal transmission and plasticity. We performed experiments in the 6‐OHDA‐lesioned mouse model treated with l‐dopa. Following behavioral evaluation, we sacrificed the mice to harvest acute corticostriatal slices4, 10, 23 (Fig. 1A and Supplementary Fig. S1). To compare the effects of xenon exposure on corticostriatal transmission between both groups of mice, we performed whole‐cell patch‐clamp recordings of spiny projection neurons (SPNs) in acute brain slices following 1‐hour exposure to xenon (5% CO2/21% O2/24% N2/50% Xe) or a vehicle gas (5% CO2/21% O2/74% N2); see Supplementary Figure S1. To quantify glutamatergic corticostriatal transmission, we measured the NMDA/α‐amino‐3‐hydroxy‐5‐methyl‐4‐isoxazolepropionic acid (AMPA) ratios of amplitudes of evoked excitatory postsynaptic currents (EPSCs) mediated by either receptor in SPNs.
Figure 1.
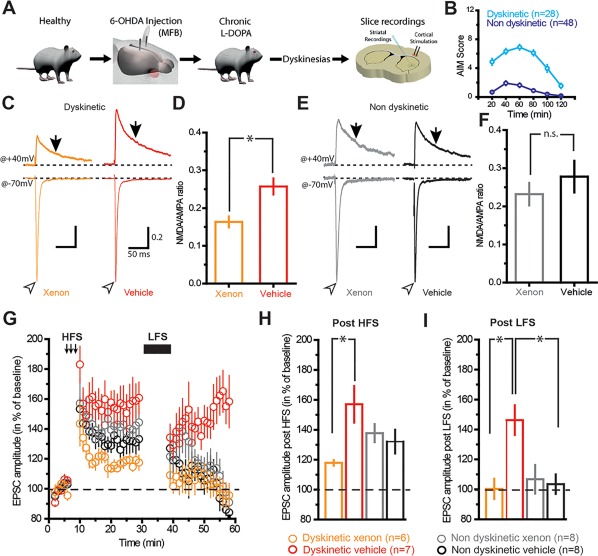
Corticostriatal plasticity in SPN is selectively restored by xenon exposure in dyskinetic mice. (A) Scheme depicting the experimental procedures for ex vivo electrophysiology. (B) Graph depicting abnormal voluntary movements (AIM) scored over a period of 2 hours in dyskinetic and nondyskinetic mice. (C) Sample traces of synaptic EPSC NMDA/AMPA ratios (determined by electrical stimulation of corticostriatal axons) recorded from SPNs in brain slices exposed for 1 hour to xenon or vehicle gas. Traces are normalized to the peak of AMPA currents. (D) Population data indicating the reduction of NMDA/AMPA ratio in SPN exposed to xenon. Open arrowheads and arrows indicate where AMPA and NMDA current amplitudes were measured, respectively (ratio xenon, 0.163 ± 0.014; n = 30 neurons; ratio vehicle, 0.257 ± 0.021; n = 34). (E, F) Same as C and D but in nondyskinetic mice. Note the absence of change in NMDA/AMPA ratios (ratio xenon, 0.231 ± 0.029; n = 15 neurons; ratio vehicle, 0.277 ± 0.041; n = 16 neurons). Stimulus artifacts have been deleted from sample traces for clarity. Scales in E are the same as C. *P < 0.05, unpaired t test. (G) Representative time course of peak EPSCs recorded in SPN before (left), after high‐frequency stimulation (middle), and after low‐frequency stimulation (right: post‐LFS) obtained from brain slices of dyskinetic mice exposed to xenon (orange trace) or vehicle gas (red trace) and from brain slices of nondyskinetic mice exposed to xenon (gray trace) or vehicle gas (black trace). (H) Bar graph shows that xenon reduces the magnitude of corticostriatal LTP compared with vehicle gas in dyskinetic mice (dyskinetic xenon, 117.9% ± 1.86%; n = 6 neurons; dyskinetic vehicle, 157% ± 12.25%; n = 7 neurons; nondyskinetic xenon, 140.77% ± 7.31%; n = 8 neurons; nondyskinetic vehicle, 132.11% ± 7.92%; n = 8 neurons; F 1,25 = 7.894, P = 0.0095, 2‐way ANOVA). (I) Bar graph showing that xenon selectively restores corticostriatal depotentiation compared with vehicle gas in dyskinetic mice (dyskinetic xenon, 100.28% ± 6.74%; n = 6 neurons; dyskinetic vehicle, 146.24% ± 9.95%, n = 7 neurons; nondyskinetic xenon, 106.75% ± 9.56%; n = 8 neurons; nondyskinetic vehicle, 103.46% ± 6.49%; n = 8 neurons; F 1,25 = 8.326; P = 0.0079, 2‐way ANOVA). *P < 0.05, 2‐way ANOVA, Bonferroni post hoc test. B, D, F‐I, values shown as mean ± standard error. [Color figure can be viewed at http://wileyonlinelibrary.com]
Xenon exposure mediated a significant decrease in NMDA/AMPA ratios in dyskinetic mice following xenon treatment (P < 0.05; Fig. 1C,D), whereas the NMDA/AMPA ratio did not change significantly in nondyskinetic mice (Fig. 1E‐F). This decrease was a result of a reduction of the NMDA component of EPSCs recorded at +40 mV. This xenon‐mediated reduction of the NMDA/AMPA ratio was observed both in the direct SPN pathway and in the indirect SPN pathway of dyskinetic mice (Supplementary Fig. S2), suggesting that xenon inhibits NMDA receptors in SPNs independently of their projection targets. The excitability of SPNs was not affected per se by xenon treatment (Supplementary Fig. S3).
We then investigated whether xenon‐mediated inhibition of NMDA receptors also affected corticostriatal synaptic plasticity. The synaptic hallmark of LID is the inability to depotentiate previously potentiated corticostriatal synapses.4, 10, 23 We potentiated synapses of SPNs with a high‐frequency stimulation train (HFS) and then delivered a low‐frequency stimulation train (LFS) to reverse this potentiation. As expected, LFS failed to reverse the depotentiation of corticostriatal synapses in dyskinetic mice exposed to vehicle gas (Fig. 1G‐I and Supplementary Fig. S3A‐C). However, exposure to xenon led to depotentiation of corticostriatal synapses (P < 0.05; Supplementary Fig. S4D‐F), similarly to the depotentiation observed in nondyskinetic mice (Fig. 1G‐I and Supplementary Fig. S4G‐I). Moreover, xenon exposure significantly reduced the magnitude of HFS‐induced long‐term potentiation (LTP) in dyskinetic mice (P < 0.05; Fig. 1G,H). In nondyskinetic mice, LFS reversed the depotentiation of corticostriatal synapses both in slices exposed to xenon and in vehicle (P < 0.05; Fig. 1G‐H and Supplementary Fig. S4G‐L), suggesting that corticostriatal transmission and plasticity were not altered in nondyskinetic mice.
Xenon Inhalation Reduces Abnormal Involuntary Movements in the 6‐OHDA Rat Model
We then evaluated the behavioral effects of xenon exposure on AIMs in the 6‐OHDA‐lesioned rat model of PD and dyskinesia (Fig. 2A).13, 14, 15, 17, 24 We assessed the effects of xenon inhalation (50% Xe/50% O2) for 30 minutes on the behavioral response to l‐dopa in the 6‐OHDA‐lesioned rats (Fig. 2B). The dose was chosen based on previous experiments in pain26 and the knowledge that the anesthetic dose in rats is 161%.27 This dose decreased axial, limb, and orofacial (ALO) AIMs, with a peak effect at 30 and 60 minutes (P < 0.01; Fig. 2C). There was also a significant therapeutic effect of xenon gas exposure over the entire 3‐hour observation period (P < 0.05; Fig. 2D), an efficacy similar to the amantadine benchmark treatment (Fig. 2C,D).
Figure 2.
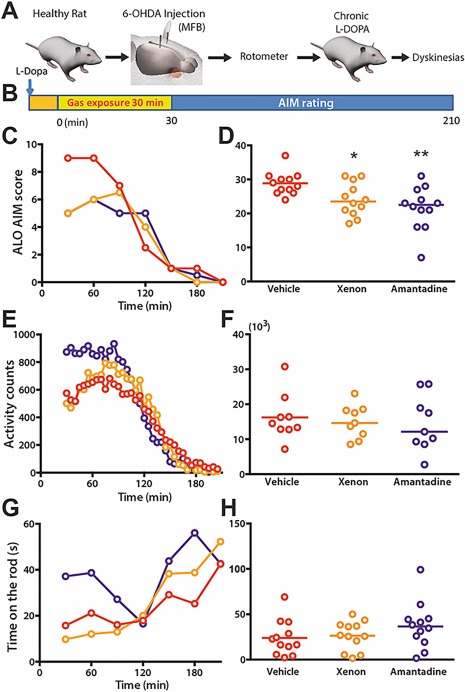
Xenon inhalation improves AIMs in dyskinetic rat during the first hour postexposure with similar amplitude as amantadine. (A) Schematic timeline for rodent behavioral experiments showing sequence of events of 6‐OHDA lesion and chronic l‐dopa treatment for inducing parkinsonism and AIMs, respectively. (B) Sequence of l‐dopa treatment followed by 30 minutes pf gas exposure for each rat before behavioral assessment. The orange square illustrates the time to obtain the selected gas concentration (wash‐in). (C, E, G) Plot of ALO scores (median), activity counts (mean), and time on the rod (mean) as a function of time (n = 12), respectively. (D) Sum of ALO scores over 3 hours' behavioral observation showed that xenon and amantadine treatment significantly ameliorate AIMs (Fr = 11.87, P < 0.05) *P < 0.05, **P < 0.01 cf vehicle. (F, H) There was no significant difference in motor skills between vehicle, xenon, and amantadine treatments, as measured by the sum of total activity counts (F 2,22 = 0.1586, P > 0.05) and mean time on the rod ([F 2,22 = 2.122, P > 0.05) over the 3‐hour behavioral observation period. *P < 0.05, **P < 0.01, t test. [Color figure can be viewed at http://wileyonlinelibrary.com]
At high concentrations, xenon mediates anesthetic effects. We thus verified the absence of sedation at the selected concentration (50% Xe/50% O2) by assessing subtle motor skills in 6‐OHDA‐lesioned rats. Compared with vehicle gas (50% N2/50% O2) or amantadine, the 30‐minute exposure to xenon did not alter the amount of locomotor activity (Fig. 2E,F) or the performance in the rotarod test (Fig. 2G,H), showing that such concentration was not sedatng.
Although xenon is the only noble gas that antagonizes NMDA receptors, we could not exclude an alternative mechanism of action that might be associated with a generic and inherent property of all noble gases. To refute this possibility, we evaluated the impact of inhaling krypton, argon, or neon noble gases on AIM severity in the l‐dopa‐treated 6‐OHDA‐lesioned rats (Supplementary Fig. S5A). We found that none of these additional gas treatments induced a significant positive effect on l‐dopa‐induced AIMs (Supplementary Fig. S5B), locomotor activity (Supplementary Fig. S5C), or rotarod performance (Supplementary Fig. S5D).
Xenon Inhalation Reduces Dyskinesia in the MPTP‐Treated Macaque Model of PD
Rodent models only partially reproduce the motor behaviors of LID observed in PD patients. Instead, MPTP‐treated macaque monkeys repeatedly exposed to l‐dopa develop LID that closely mimics the dyskinetic manifestations reported in patients with PD.1, 28 The majority of previously tested antidyskinetic strategies primarily reduced the severity of choreic dyskinesia, but had little impact on dystonic dyskinesia.1, 19 We therefore sought to validate the antidyskinetic effects of xenon inhalation in 6 macaque monkeys first rendered parkinsonian with MPTP (Fig. 3A) and second dyskinetic by chronic l‐dopa exposure (Fig. 3A,C,D and see the Materials and Methods section).8, 10, 13, 15, 17, 18
Figure 3.
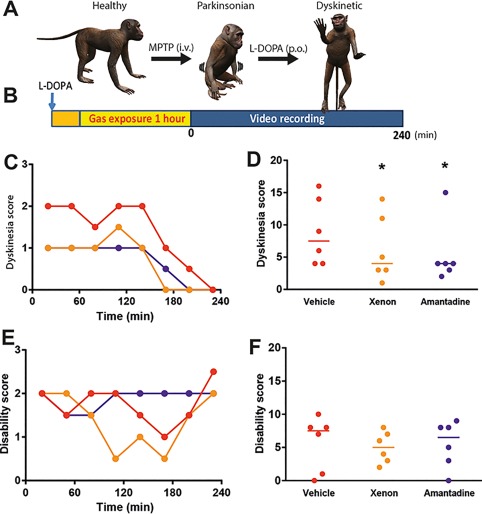
Xenon inhalation improves dyskinesia in dyskinetic nonhuman primates without interfering with antiparkinsonian action of l‐dopa, with amplitude similar to amantadine. (A) Schematic timeline for producing a dyskinetic macaque showing sequence of events from MPTP intoxication for inducing parkinsonism to chronic l‐dopa treatment to elicit dyskinesia. After 7 months, the macaques were ready for behavioral investigation. (B) Schematic of the behavior experiments used to evaluate the dyskinesia and disability scores. We treated the monkeys with l‐dopa, exposed them to the vehicle or xenon gas mixture for 1 hour, and then recorded videos of their behavior over the following 4 hours. The orange square illustrates the time to obtain the selected gas concentration (wash‐in). (C, E) Dyskinesia and disability scores (median) as a function of time (n = 6 macaques), respectively. (D, F) Sum of dyskinesia and disability scores noted over the first 2 hours of behavioral observation. Xenon and amantadine treatments ameliorated dyskinesia (Fr = 7.913, *P < 0.05) without worsening PD disability (Fr = 0.333, P > 0.05); n = 6 macaques; Friedman nonparametric ANOVA and Dunn multiple‐comparison post hoc analysis; *P < 0.05 compared with vehicle). [Color figure can be viewed at http://wileyonlinelibrary.com]
We conducted preliminary tests to calibrate different parameters of xenon gas exposure for the treatment of dyskinesia, which included (1) the effects of varied gas exposure times (15, 30, and 60 minutes) and (2) the effects of varied gas concentrations (50%, 35%, and 20% xenon mixed with 50%, 65 %, and 80 % O2, respectively) of acute xenon treatment, knowing that in the macaque monkey the anesthetic dose of xenon is 98%. To enhance the translational value of this calibration, gas delivery was performed using a human‐approved closed‐circuit respirator (FELIX DUAL, Air Liquide Healthcare, France). We found that 1‐hour exposure to the concentration of 50% Xe and 50% O2 was the most effective intervention for reducing dyskinesia.
Xenon exposure significantly decreased LID both at the l‐dopa peak effect (90‐180 minutes; P < 0.05; Fig. 3C) and over the 4‐hour period (P < 0.05; Fig. 3D), without deteriorating the antiparkinsonian effect of l‐dopa (Fig. 3E,F). Xenon mediated antidyskinetic effects whose amplitude matched the improvements mediated by amantadine (P < 0.01; Fig. 3C,D). Analysis of the subtype of dyskinesia revealed that xenon inhalation (1) significantly reduced the choreic form (Supplementary Fig. S6A) and (2) tended to reduce the dystonic form (Supplementary Fig. S6B).
Xenon Inhalation Preserves l‐Dopa Benefits and Improves Its Effect on Gait and Balance
The antidyskinetic effects of potential adjunct therapies can only be considered suitable if the antiakinetic effects of l‐dopa are preserved. In the 6 tested monkeys, we did not detect a difference in disability scores (Fig. 3E,F), in on‐time (Supplementary Fig. S6C), or in general motor activity following l‐dopa combined with xenon or vehicle gas treatment (Supplementary Fig. S6D). We even observed a trend toward an increase in good on‐time (Supplementary Fig. S6C), that is, more time spent without troublesome dyskinesia interfering with normal daily activities, suggestive of an increased quality of life and an overall reduction of dyskinesia impact on daily activities.
These results encouraged us to quantitatively study the effect of xenon alone or in combination with l‐dopa on gait and balance deficits. Both in patients with PD and non‐human primate (NHP) models of PD, these symptoms include freezing of gait, postural instability, and other axial impairments. l‐Dopa traditionally mediates variable effects on these deficits or even exacerbates them.7, 29, 30, 31, 32 We conducted detailed kinematic analyses of gait in 4 additional monkeys that underwent successive MPTP and l‐dopa treatments to develop PD symptoms and dyskinesia (Fig 3A) and in 6 healthy monkeys. We then trained these monkeys to walk along a straight corridor without any constraints (Fig. 4A).20, 21, 22 Video recordings (100 Hz) of markers painted onto hind‐limb and forelimb joints allowed 3‐D reconstruction of whole‐body kinematics (Fig. 4A).20, 21, 22
Figure 4.

l‐Dopa mediates subject‐specific improvements in gait and balance in the NHP model of PD. (A) Recordings of whole‐body kinematics during locomotion along a runway. Gait patterns are recorded using a high‐speed motion capture system. They are subsequently reconstructed in 3‐D, discretized per gait cycle, decomposed into numerous gait parameters, and analyzed using PC analysis. All gait cycles for 4 monkeys (S1‐S4) are shown in the space defined by PC1 and PC2, which also includes the mean gait cycles of 6 healthy monkeys for comparison (each symbol is an average of 10 to 20 gait cycles; black square, healthy; white circle, MPTP no l‐dopa; blur circle, MPTP 50% l‐dopa; purple circle, MPTP 100% l‐dopa). Plus signs mark the mean position of gait cycles for each condition. Gait of monkeys S1 and S2 became more similar (closer) to the gait of healthy monkeys following 100% l‐dopa treatment. Monkey S3 exhibited severe dyskinesia following 100% l‐dopa treatment, which was captured in the large displacement of gait cycles in the PC1‐2 space. Following the same treatment, the gait of monkey S4 moved farther away from those of healthy monkeys, indicating deterioration of gait performance. (B) Representative stick diagram decomposition of hind‐limb movements (left column) and traces of key gait variables (right column) for 3 l‐dopa doses (0%, 50%, and 100%) followed by 1 hour of vehicle gas inhalation (50% N2, 50% O2). (C) Bar plots showing changes in the mean Euclidean distance between all gait cycles in a given condition and the average gait cycle of all healthy monkeys. The other bar plots report average values for key parameters of gait (speed and stance duration) and posture (hip position) for the 3 experimental conditions. Data reported as mean. Error bars ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001; Wilcoxon rank sum test. [Color figure can be viewed at http://wileyonlinelibrary.com]
A total of 64 gait parameters were calculated from these kinematic recordings. We then used these parameters in a principal‐component (PC) analysis that allowed visualization of gait performance between experimental conditions.20 To quantify gait performance, we measured the Euclidean distance between MPTP monkeys and healthy monkeys in the space spanning all the computed gait parameters (Fig. 4A).
A full l‐dopa dose led to an immediate reduction of gait deficits in all 4 MPTP monkeys. However, l‐dopa produced variable effects on gait parameters affected by PD (Fig. 4A‐C). Two monkeys (S3 and S4) exhibited limited gait improvement. Specifically, S4 did not exhibit improvement of gait features following l‐dopa administration. Instead, exposure to 100% l‐dopa dose led to impaired balance and excessive excitation (Fig. 4C), despite the reduced disability. In S3, 50% l‐dopa treatment alleviated some of the gait deficits (namely, the hip forward position), but 100% l‐dopa treatment caused severe dyskinesia, which resulted in disturbed gait patterns. The other 2 monkeys (S1 and S2) displayed improvements in walking speed, step length, and posture (eg, maximum hip forward position) when treated with a 100% l‐dopa dose (Fig. 4C). These gait parameters capture the main spatiotemporal features of gait that are affected in individuals with PD.7 These results are in agreement with clinical observations that have shown that l‐dopa fails to consistently restore normal gait across individuals with PD.7
We then evaluated whether xenon could improve gait performance and reduce the detrimental effects of l‐dopa on locomotion in S3 and S4. We compared the effects of 1‐hour inhalation of xenon (50% Xe/50% O2) with vehicle gas (50% N2/50% O2) following administration of a suboptimal (50%) tailored dose of l‐dopa (Fig. 5). Both monkeys rapidly traversed the corridor to reach food reward. Inhaling xenon significantly improved gait performance, enabling both monkeys to produce locomotor movements that closely resembled those recorded in healthy monkeys (Fig. 5A). Even S3, who exhibited severe dyskinesia at the 100% l‐dopa dose (Fig. 5B), showed significant improvement in several clinically relevant gait variables (stepping speed and length and stance duration) when tested after xenon inhalation (Fig. 5B). For monkeys S1 and S2, which responded positively to l‐dopa, the addition of xenon to l‐dopa therapy did not substantially further normalize their gait (Supplementary Fig. S6).
Figure 5.
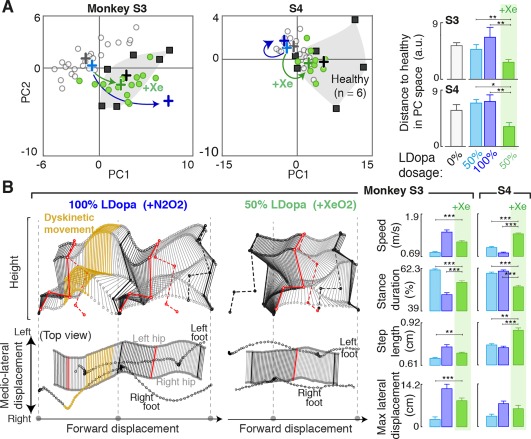
Xenon not only reduces the undesired effects of l‐dopa, but also alleviates locomotor deficits. (A) Plots showing all the gait cycles in the space defined by PC1 and PC2 (same projection as in Fig. 4A). Conventions are the same as in Figure 4. Colored arrows illustrate the changes of gait patterns observed following 100% l‐dopa and 50% l‐dopa + xenon treatments. Bar plots report mean Euclidean distance of all gait cycles compared with the average gait cycle of all healthy monkeys. Unlike the 50% or 100% l‐dopa treatments, which did not affect locomotion (S4) or cause further deterioration in gait performance (S3). The 50% l‐dopa treatment followed by xenon exposure enabled performance of gait patterns that resembled those of healthy monkeys. (B) Stick diagram decomposition of leg movements and pelvic movements viewed from the lateral aspect (top) and from above (bottom), respectively. The conditions 100% l‐dopa dose followed by vehicle inhalation (2 steps) and 50% l‐dopa dose followed by xenon inhalation (1 step) are shown for monkey S3. The steps in the 100% l‐dopa + vehicle condition showed a typical dyskinetic event (yellow sticks) and impaired balance, which is captured in the rightward offset of the foot positions compared with the hip. For the 50% l‐dopa + xenon condition, dyskinetic movements became rare or even absent. The feet were positioned under the hip throughout walking. Bar plots report changes in key gait parameters (stepping speed, step length, and stance duration) and the unbalanced hip displacement gait parameter designed to measure the frequency and severity of unbalanced movements for all tested conditions. Data are reported as means. Error bars ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001; Wilcoxon rank sum test. [Color figure can be viewed at http://wileyonlinelibrary.com]
Although it is difficult to dissociate dyskinetic and nondyskinetic gait deficits, these variables commonly employed to characterize parkinsonian gait (and often nonresponsive to l‐dopa therapies) can help to measure amelioration in gait patterns independent of dyskinetic symptoms. Conversely, dyskinetic gait changes characterized by uncontrollable movements of the legs or arms can disturb balance and mediolateral body stability. The stick diagrams in Figure 5B illustrate the difficulty of S3 maintaining balance, as its right and left hips consistently swung outside the stability support base between the feet. We quantified these excessive movements by measuring unbalanced hip displacement, the distance the hip midpoint swung beyond the area between the legs, in each step. The unbalanced hip displacement will be absent for animals that do not exhibit uncontrollable movements but will become substantial with the appearance of dyskinetic symptoms. Both S3 and S4 displayed substantial unbalanced hip displacement for the 100% l‐dopa condition, whereas S4 displayed some significant hip displacement, even for the 50% l‐dopa condition (Fig. 5B). In comparison, the 50% l‐dopa + xenon condition successfully alleviated parkinsonian gait deficits (Fig. 5A) while abolishing the dyskinetic unbalanced hip movements (Fig. 5B).
Discussion
Three important results emerged from these studies. First, xenon effectively targets NMDA receptors located on SPNs and normalizes the disrupted transmission of glutamatergic corticostriatal synapses in parkinsonian mice with AIMs. Second, xenon inhalation ameliorates LID in both rat and NHP models of PD and LID. Third, not only does xenon inhalation support the antiparkinsonian activity of l‐dopa, but it also mitigates the deleterious effects of l‐dopa on gait performance in NHP models of PD. These findings open a concrete and safe therapeutic strategy for addressing LID and parkinsonian locomotor symptoms. Here, we discuss the mechanisms underlying the effects of xenon and envisage the next steps to apply this therapy in clinical settings.
Dyskinetic motor abnormalities have been associated with an absence of bidirectional synaptic plasticity in striatal projection neurons.1, 3, 4, 33 This observation established the conceptual framework to guide the identification of possible synaptic mechanisms underlying LID. These symptoms emerge in response to chronic l‐dopa treatment. However, parkinsonian animals generally exhibit 2 types of responses to l‐dopa treatment. Animals that do not develop LID (20%‐30%) benefit from the antiparkinsonian effects of l‐dopa, which restores the physiological bidirectional synaptic plasticity in striatal projecting neurons, including long‐term depression (LTD), LTP, and depotentiation. In dyskinetic animals (70%‐80%), l‐dopa treatment also restores normal LTP and improves parkinsonian motor behaviors, but also triggers severe LID. Moreover, in these animals, striatal projection neurons fail to regain either LTD or depotentiation in response to low‐frequency stimulation (LFS) protocols following l‐dopa treatment.1, 3, 33 However, blockade of NMDA receptors33, 34 as well as D1 or M4 receptors10 reestablished these depotentiation capabilities, which effectively alleviated LID in rodent and primate models of LID. Yet the pharmacological agents used in these studies are not safe for clinical practice.
The noble gas xenon functionally antagonizes NMDA receptors containing either NR2A or NR2B subunits,6 the 2 major subunits of the corticostriatal glutamatergic receptors.35 This gas has a safety profile demonstrated by its utilization in clinical trials in anesthesia36 and in neonates37 that should enable repeated clinical applications at subanesthetic doses. The safety profile of xenon, both on vital cardiorespiratory and toxicity parameters, has allowed for its market authorization in anesthesia. This has been reinforced since then by demonstration of safety in clinical trials on anesthesia of fragile patients36 and in critical care on neonates.37 Despite the relatively rapid blood washout on termination of inhalation therapy, xenon might be effective under chronic repeated dosing through affinity for lipophilic tissues.38 It is thus a candidate for a safe and reliable intervention at subanesthetic dosages to reduce LID.
We found that xenon exposure not only restores depotentiation of striatal projecting neurons in dyskinetic mice, but also prevents increased potentiation by high‐frequency stimulation. Thus, xenon reestablishes physiological properties of striatal projecting neurons that do not differ from those of nondyskinetic animals. Because these effects were only observed in dyskinetic animals, we surmise that xenon specifically reverses pathological enhancement of corticostriatal transmission. This paradoxical effect was previously reported in dyskinetic compared with nondyskinetic animals. Time‐dependent measurements of monoamine levels revealed that this effect was essentially linked to elevated dopamine levels in the striatum following the last l‐dopa injection, as opposed to a fundamental change in plasticity signaling machinery.10 Several lines of evidence associated this aberrant potentiation of glutamatergic synapses from striatal projecting neurons with a dopamine D1 receptor‐dependent central feature of the LID pathophysiology.1 Drug‐naive and l‐dopa‐treated mice that do not develop dyskinesia differ from l‐dopa‐treated dyskinetic mice in the spatiotemporal distribution of D1 receptor stimulation. Whereas burst spiking of dopaminergic neurons only briefly stimulates nondyskinetic mice, D1 receptors exhibit prolonged stimulation in mice that develop LID. The amount of dopamine release also differs between nondyskinetic and dyskinetic animals. Indeed, the release of dopamine far exceeds normal levels in dyskinetic animals, which leads to a robust decrease in the firing discharge of the output of basal ganglia.39 Striatal D1 receptor stimulation is therefore spatially and temporally different between dyskinetic and nondyskinetic animals. The differential stimulation pattern of D1 receptors is responsible for the abnormal plasticity of striatal projecting neurons, as such corticostriatal plasticity is under the tight control of dopamine receptors. In turn, this property explains the restricted effects of gaseous or pharmacological manipulations of glutamatergic transmission in dyskinetic animals.
We suggest that alleviation of dyskinetic behaviors in both rat and primate models of LID following l‐dopa treatment and xenon inhalation is mediated through this xenon‐mediated improvement of glutamatergic transmission in striatal projecting neurons. Although the antidyskinetic effects were modest, the amplitude of these improvements matched those afforded by amantadine, whose efficacy has been demonstrated in patients with PD and LID.5 A series of parameters could enhance the ability of xenon inhalation to alleviate LID. These improvements may include optimizing the timing of exposure vis‐à‐vis l‐dopa administration and evaluating the impact of chronic versus acute exposure. For example, the identification of strategies to match the exposure and thus bioavailability of xenon to that of l‐dopa may effectively moderate the activation of D1 receptors during the on‐state, thus improving the behavioral effects of both drugs.
Importantly, xenon alleviated LID without compromising the symptomatic benefit of l‐dopa treatment, both in rat and NHP models. High‐resolution kinematic recordings combined with objective statistical analyses confirmed the beneficial effects of xenon on gait and balance deficits. As previously described in patients with PD, we found that l‐dopa mediated variable effects on gait performance and could even exacerbate freezing of gait, postural instability, and other axial impairments.7, 29, 30, 31, 32 Here, we obtained evidence that suggests that xenon may alleviate some of the gait and postural symptoms that are refractory to or even enhanced by l‐dopa. Previous experimental strategies to alleviate LID did not investigate potential effects on gait and posture. The lack of efficacy of dopaminergic treatments on these axial symptoms7, 40 suggests that the underlying lesions involve nondopamine systems and may thus involve lesions located outside the dopaminergic system.41 Several studies have shown that the cholinergic neurons of the pedunculopontine nucleus (PPN) undergo advanced degeneration in PD.42, 43 Growing evidence suggests that the PPN plays a crucial role in the development of gait and balance disorders.44 Indeed, the PPN is a surgical target to alleviate gait deficits with deep brain stimulation in patients with PD.45 Degeneration of cholinergic neurons in this dual cholinergic‐glutamatergic nucleus leads to increased glutamatergic tone and most likely abnormal plasticity,41, 44 as observed in striatal projecting neurons, Therefore, xenon may alleviate gait deficits through normalization of glutamatergic transmission and plasticity in the PPN, similarly to its action on corticostriatal synapses. Because the reported benefit varied from animal to animal, patients must be selected carefully for clinical studies. The presence of proven gait disorders is a prerequisite for such investigations.
A key unmet medical need of the PD community is a strategy for ameliorating LID. The hope is that a rational therapy will arise from the understanding of the mechanisms responsible for LID. Although various NMDA antagonists such as dextromethorphan, remacemide, milacemide, CP‐101,606, and memantine have been assessed for treating LID, their success has been limited.1 Only amantadine exhibited modest antidyskinetic effects in the gold‐standard NHP models of LID prior to evaluation in patients.1 At‐home distribution of a gaseous therapeutic is already taking place for several indications, with millions of patients receiving gas‐based home care throughout the world. Altogether, our results in the gold‐standard mouse, rat, and NHP models of LID provide a strong rationale to expedite clinical testing of this safe yet unconventional approach. Patients at an advanced stage with severe dyskinesia uncontrolled by treatment or severe gait disorders (skewness, freezing) who may need a “rescue therapy” could take advantage of such an inhaled therapy.
Author contributions
J.B., G.C., G.F., A.M., J.P., B.B., and E.B. designed the research and analyzed the data. J.B., M.C., S.M., and E.N. performed the ex vivo electrophysiology experiments, analyzed the data, and wrote the electrophysiology section. Q.L., W.K.D.K., S.McG., G.P., E.P., and E.B. performed the behavioral experiments in rodents and nonhuman primates, analyzed the data, and wrote the behavioral section. T.M., E.M.M., S.S., W.K.D.K., and G.C. performed the kinematics experiments, analyzed the data, and wrote the kinematics section. J.B., G.C., T.M., B.B., and E.B. wrote the article.
Financial disclosures
G.F., A.M., J.P., and B.B. are employees of Air Liquide Santé International. Q.L., W.K.D.K., S.McG., and E.P. are employees of Motac Neuroscience Ltd, a contract research organization.
E.B. has an equity stake in Motac holding Ltd. and receives consultancy payments from Motac Neuroscience Ltd. Current grant support includes Agence Nationale de la Recherche (E.B.), China Science Fund (E.B.), Michael J. Fox Foundation for Parkinson Research (E.B.), France Parkinson (E.B.), Fondation de France (E.B.), Medical Research Council (E.B.), Innovative Medicines Initiative (H2020, IMPRIND; E.B.), IDEX Bordeaux (E.B.), COEN (E.B.), MSA Coalition (E.B.), Rotary Foundation (E.B.), Nouvelle‐Aquitaine Regional Council (E.B.), and LABEX Brain (E.B.). J.B.'s current grant support includes Agence Nationale de la Recherche and Fondation de France. G.C. is a shareholder of G‐Therapeutics SA, a company developing neurotechnologies not directly related to this work.
Acknowledgments
We are grateful to Y.J.Z., L.H., C.Y.L., and X.R.L. for the care of the nonhuman primates and to F. Roguet, M. Dehors, A. Le Bris, and G. Dabée from the in vivo platform (PIV) for the nursing of rodents included in this study. We thank A. S. Zana, F. Michel, and Zhang Huiliang for their help in tracking the monkey kinematics. We thank Francois Gilles and Christian Daviet for their technical assistance regarding gas administration.
Jérôme Baufreton, Tomislav Milekovic, and Qin Li PhD are co–first authors.
Funding agencies: This work was supported by Air Liquide Santé International, in part with a grant received from the Michael J. Fox Foundation. M.C. was supported by LABEX BRAIN grant (ANR‐10‐LABX‐43).
References
- 1. Bastide MF, Meissner WG, Picconi B, et al. Pathophysiology of L‐dopa‐induced motor and non‐motor complications in Parkinson's disease. Prog Neurobiol 2015;132:96‐168. [DOI] [PubMed] [Google Scholar]
- 2. Bezard E, Brotchie JM, Gross CE. Pathophysiology of levodopa‐induced dyskinesia: Potential for new therapies. Nat Rev Neurosci 2001;2:577‐588. [DOI] [PubMed] [Google Scholar]
- 3. Calabresi P, Di Filippo M, Ghiglieri V, Tambasco N, Picconi B. Levodopa‐induced dyskinesias in patients with Parkinson's disease: filling the bench‐to‐bedside gap. Lancet Neurol 2010;9(11):1106‐1117. [DOI] [PubMed] [Google Scholar]
- 4. Picconi B, Centonze D, Hakansson K, et al. Loss of bidirectional striatal synaptic plasticity in L‐DOPA‐induced dyskinesia. Nat Neurosci 2003;6(5):501‐506. [DOI] [PubMed] [Google Scholar]
- 5. Fox SH, Katzenschlager R, Lim SY, et al. The Movement Disorder Society Evidence‐Based Medicine Review Update: treatments for the motor symptoms of Parkinson's disease. Mov Disord 2011;26(Suppl 3):S2‐S41. [DOI] [PubMed] [Google Scholar]
- 6. Haseneder R, Kratzer S, Kochs E, et al. The xenon‐mediated antagonism against the NMDA receptor is non‐selective for receptors containing either NR2A or NR2B subunits in the mouse amygdala. Eur J Pharmacol 2009;619(1‐3):33‐37. [DOI] [PubMed] [Google Scholar]
- 7. Boonstra TA, van der Kooij H, Munneke M, Bloem BR. Gait disorders and balance disturbances in Parkinson's disease: clinical update and pathophysiology. Curr Opin Neurol 2008;21(4):461‐471. [DOI] [PubMed] [Google Scholar]
- 8. Fasano S, Bezard E, D'Antoni A, et al. Inhibition of Ras‐guanine nucleotide‐releasing factor 1 (Ras‐GRF1) signaling in the striatum reverts motor symptoms associated with L‐dopa‐induced dyskinesia. Proc Natl Acad Sci U S A 2010;107(50):21824‐21829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Berthet A, Bezard E, Porras G, et al. L‐DOPA impairs proteasome activity in parkinsonism through D1 dopamine receptor. J Neurosci 2012;32(2):681‐691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shen W, Plotkin JL, Francardo V, et al. M4 Muscarinic Receptor signaling ameliorates striatal plasticity deficits in models of L‐dopa‐induced dyskinesia. Neuron 2015;88(4):762‐773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lundblad M, Picconi B, Lindgren H, Cenci MA. A model of L‐DOPA‐induced dyskinesia in 6‐hydroxydopamine lesioned mice: relation to motor and cellular parameters of nigrostriatal function. Neurobiol Dis 2004;16(1):110‐123. [DOI] [PubMed] [Google Scholar]
- 12. Miguelez C, Morin S, Martinez A, et al. Altered pallido‐pallidal synaptic transmission leads to aberrant firing of globus pallidus neurons in a rat model of Parkinson's disease. J Physiol 2012;590(22):5861‐5875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Porras G, Berthet A, Dehay B, et al. PSD‐95 expression controls l‐DOPA dyskinesia through dopamine D1 receptor trafficking. J Clin Invest 2012;122(11):3977‐3989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Schuster S, Nadjar A, Guo JT, et al. The 3‐hydroxy‐3‐methylglutaryl‐CoA reductase inhibitor lovastatin reduces severity of L‐DOPA‐induced abnormal involuntary movements in experimental Parkinson's disease. J Neurosci 2008;28(17):4311‐4316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Urs NM, Bido S, Peterson SM, et al. Targeting beta‐arrestin2 in the treatment of L‐DOPA‐induced dyskinesia in Parkinson's disease. Proc Natl Acad Sci U S A 2015;112(19):E2517‐2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cenci MA, Lee CS, Bjorklund A. L‐DOPA‐induced dyskinesia in the rat is associated with striatal overexpression of prodynorphin‐ and glutamic acid decarboxylase mRNA. Eur J Neurosci 1998;10(8):2694‐2706. [PubMed] [Google Scholar]
- 17. Ahmed M, Berthet A, Bychkov E, et al. Lentiviral overexpression of GRK6 alleviates L‐dopa‐induced dyskinesia in experimental Parkinson's disease. Sci Transl Med 2010;2(28):28ra28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bezard E, Ferry S, Mach U, et al. Attenuation of levodopa‐induced dyskinesia by normalizing dopamine D3 receptor function. Nat Med 2003;9(6):762‐767. [DOI] [PubMed] [Google Scholar]
- 19. Bezard E, Pioli EY, Li Q, et al. The mGluR5 negative allosteric modulator dipraglurant reduces dyskinesia in the MPTP macaque model. Mov Disord 2014;29(8):1074‐1079. [DOI] [PubMed] [Google Scholar]
- 20. Capogrosso M, Milekovic T, Borton D, et al. A brain‐spine interface alleviating gait deficits after spinal cord injury in primates. Nature 2016;539(7628):284‐288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Friedli L, Rosenzweig ES, Barraud Q, et al. Pronounced species divergence in corticospinal tract reorganization and functional recovery after lateralized spinal cord injury favors primates. Sci Transl Med 2015;7(302):302ra134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yin M, Borton DA, Komar J, et al. Wireless Neurosensor for Full‐Spectrum Electrophysiology Recordings during Free Behavior. Neuron 2014;84(6):1170‐1182. [DOI] [PubMed] [Google Scholar]
- 23. Calabresi P, Pisani A, Rothwell J, Ghiglieri V, Obeso JA, Picconi B. Hyperkinetic disorders and loss of synaptic downscaling. Nat Neurosci 2016;19(7):868‐875. [DOI] [PubMed] [Google Scholar]
- 24. Schuster S, Doudnikoff E, Rylander D, et al. Antagonizing L‐type Ca2 + channel reduces development of abnormal involuntary movement in the rat model of L‐3,4‐dihydroxyphenylalanine‐induced dyskinesia. Biol Psychiatry 2009;65(6):518‐526. [DOI] [PubMed] [Google Scholar]
- 25. Lundblad M, Andersson M, Winkler C, Kirik D, Wierup N, Cenci MA. Pharmacological validation of behavioral measures of akinesia and dyskinesia in a rat model of Parkinson's disease. Eur J Neurosci 2002;15(1):120‐132. [DOI] [PubMed] [Google Scholar]
- 26. Bessiere B, Laboureyras E, Laulin JP, Simonnet G. Xenon prevents inflammation‐induced delayed pain hypersensitivity in rats. Neuroreport 2010;21(18):1167‐1171. [DOI] [PubMed] [Google Scholar]
- 27. Koblin DD, Fang Z, Eger EI 2nd, et al. Minimum alveolar concentrations of noble gases, nitrogen, and sulfur hexafluoride in rats: helium and neon as nonimmobilizers (nonanesthetics). Anesth Analg 1998;87(2):419‐424. [DOI] [PubMed] [Google Scholar]
- 28. Fox SH, Johnston TH, Li Q, Brotchie J, Bezard E. A critique of available scales and presentation of the Non‐Human Primate Dyskinesia Rating Scale. Mov Disord 2012;27(11):1373‐1378. [DOI] [PubMed] [Google Scholar]
- 29. Giladi N, Nieuwboer A. Understanding and treating freezing of gait in parkinsonism, proposed working definition, and setting the stage. Mov Disord 2008;23(Suppl 2):S423‐S425. [DOI] [PubMed] [Google Scholar]
- 30. Nonnekes J, Snijders AH, Nutt JG, Deuschl G, Giladi N, Bloem BR. Freezing of gait: a practical approach to management. Lancet Neurol 2015;14(7):768‐778. [DOI] [PubMed] [Google Scholar]
- 31. Nantel J, McDonald JC, Bronte‐Stewart H. Effect of medication and STN‐DBS on postural control in subjects with Parkinson's disease. Parkinsonism Relat Disord 2012;18(3):285‐289. [DOI] [PubMed] [Google Scholar]
- 32. Bloem BR. Postural instability in Parkinson's disease. Clin Neurol Neurosurg 1992;94(Suppl):S41‐S45. [DOI] [PubMed] [Google Scholar]
- 33. Thiele SL, Chen B, Lo C, et al. Selective loss of bi‐directional synaptic plasticity in the direct and indirect striatal output pathways accompanies generation of parkinsonism and l‐DOPA induced dyskinesia in mouse models. Neurobiol Dis 2014;71:334‐344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ghiglieri V, Bagetta V, Pendolino V, Picconi B, Calabresi P. Corticostriatal plastic changes in experimental L‐DOPA‐induced dyskinesia. Parkinsons Dis 2012;2012:358176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hallett PJ, Dunah AW, Ravenscroft P, et al. Alterations of striatal NMDA receptor subunits associated with the development of dyskinesia in the MPTP‐lesioned primate model of Parkinson's disease. Neuropharmacology 2005;48(4):503‐516. [DOI] [PubMed] [Google Scholar]
- 36. Law LS, Lo EA, Gan TJ. Xenon anesthesia: a systematic review and meta‐analysis of randomized controlled trials. Anesth Analg 2016;122(3):678‐697. [DOI] [PubMed] [Google Scholar]
- 37. Azzopardi D, Robertson NJ, Bainbridge A, et al. Moderate hypothermia within 6 h of birth plus inhaled xenon versus moderate hypothermia alone after birth asphyxia (TOBY‐Xe): a proof‐of‐concept, open‐label, randomised controlled trial. Lancet Neurol 2016;15(2):145‐153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Katz I, Murdock J, Palgen M, Pype J, Caillibotte G. Pharmacokinetic analysis of the chronic administration of the inert gases Xe and Ar using a physiological based model. Med Gas Res 2015;5:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Meissner W, Ravenscroft P, Reese R, et al. Increased slow oscillatory activity in substantia nigra pars reticulata triggers abnormal involuntary movements in the 6‐OHDA‐lesioned rat in the presence of excessive extracellular striatal dopamine. Neurobiol Dis 2006;22(3):586‐598. [DOI] [PubMed] [Google Scholar]
- 40. Bloem BR, Hausdorff JM, Visser JE, Giladi N. Falls and freezing of gait in Parkinson's disease: a review of two interconnected, episodic phenomena. Mov Disord 2004;19(8):871‐884. [DOI] [PubMed] [Google Scholar]
- 41. Grabli D, Karachi C, Folgoas E, et al. Gait disorders in parkinsonian monkeys with pedunculopontine nucleus lesions: a tale of two systems. J Neurosci 2013;33(29):11986‐11993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hirsch EC, Graybiel AM, Duyckaerts C, Javoy‐Agid F. Neuronal loss in the pedunculopontine tegmental nucleus in Parkinson disease and in progressive supranuclear palsy. Proc Natl Acad Sci U S A 1987;84(16):5976‐5980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Jellinger K. The pedunculopontine nucleus in Parkinson's disease, progressive supranuclear palsy and Alzheimer's disease. J Neurol Neurosurg Psychiatry 1988;51(4):540‐543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Nutt JG, Bloem BR, Giladi N, Hallett M, Horak FB, Nieuwboer A. Freezing of gait: moving forward on a mysterious clinical phenomenon. Lancet Neurol 2011;10(8):734‐744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Thevathasan W, Debu B, Aziz T, et al. Pedunculopontine nucleus deep brain stimulation in Parkinson's disease: A clinical review. Mov Disord 2018;33(1):10‐20. [DOI] [PubMed] [Google Scholar]