

Abstract
Catalytic hydrogenation of cyclic carbonates to diols and methanol was achieved using a molecular catalyst based on earth‐abundant manganese. The complex [Mn(CO)2(Br)[HN(C2H4PiPr2)2] 1 comprising commercially available MACHO ligand is an effective pre‐catalyst operating under relatively mild conditions (T=120 °C, p(H2)=30–60 bar). Upon activation with NaOtBu, the formation of coordinatively unsaturated complex [Mn(CO)2[N(C2H4PiPr2)2)] 5 was spectroscopically verified, which confirmed a kinetically competent intermediate. With the pre‐activated complex, turnover numbers up to 620 and 400 were achieved for the formation of the diol and methanol, respectively. Stoichiometric reactions under catalytically relevant conditions provide insight into the stepwise reduction form the CO2 level in carbonates to methanol as final product.
Keywords: carbon dioxide, carbonates, hydrogenation, manganese, methanol
Currently, there is a strong interest in the hydrogenation of CO2 to methanol for the chemical supply chain and within the context of novel energy carriers.1, 2 The synthesis of methanol by hydrogenation of CO2 using molecular catalyst systems has been successfully addressed by various working groups in recent years. The field was pioneered in 2011 by Milstein and co‐workers, who demonstrated the possibility to reduce CO2 indirectly via hydrogenation of isolated derivatives such as cabonates, carbamates, and formates using Ru pincer complex under mild conditions (Scheme 1).3 Seminal contributions in this field were made by the groups of Sanford,4 Prakash, and Olah,5 and Klankermayer and Leitner.6 The latter work demonstrated that there are also direct pathways that do not require a stable organic intermediate.6a While these studies used noble metals as active catalytic centers, few examples employing non‐noble metal catalyst systems have also been described. The group of Beller reported a catalytic system based on Cobalt reaching turnover numbers (TONs) of up to 78.7 Recently, Prakash et al. reported a Manganese catalyzed sequential hydrogenation of CO2 to methanol via a pre‐formed formamide reaching TONs up to 36.8
Scheme 1.
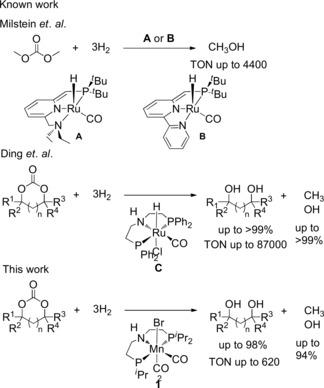
Molecular homogeneous catalysts for the hydrogenation of carbonates (n=0, 1; R1‐R4=H or alkyl).
Cyclic carbonates are particularly attractive as intermediate CO2 derivatives in this context, as they can be formed readily from the reaction of CO2 with the corresponding epoxides or oxetanes. In 2012, Ding et al. reported a highly efficient catalytic system for the hydrogenation of cyclic carbonates using Ru‐MACHO pincer catalyst with TONs up to 87 000 (Scheme 1).9 The diols formed as stoichiometric co‐product are valuable products, making this transformation in particular also as an attractive synthetic approach to these compounds.10 In fact, the carbonate group has been suggested as valuable protecting group for diols in organic synthesis.11
The potential of hydrogenation catalysts based on abundant and cheap third‐row metal Manganese and its pincer complexes12 has been pointed out by the groups of Milstein13 and Beller14 in 2016. In particular, complexes of this type have been successfully employed in the hydrogenation of CO2 to formic acid derivatives.5, 15 Encouraged by these reports, we were able to develop a catalytic procedure for catalytic hydrogenation of cyclic carbonates to diols and methanol using manganese complex 1 comprising the well‐known and commercially available PNP pincer ligand MACHO. While this manuscript was in preparation, we became aware of two independent parallel studies employing newly synthesized PNN‐pincer ligands.16, 17
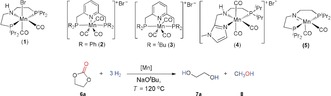
Initial studies were conducted using ethylene carbonate 6 a as a benchmark substrate (Table 1). Reactions were carried out with 1 mol % of catalyst in presence of NaOtBu as basic co‐catalyst (2 equiv) in THF (0.7 mL) at 120 °C under 30 bar of H2. While complex 2–4 showed only low activity, the Mn‐MACHO complex 1 gave 86 % conversion already under screening conditions. Ethylene glycol (7 a) was detected by standard analytical techniques in quantities corresponding to nearly perfect selectivity, while the yield of methanol was consistently somewhat lower. Formate esters were observed as side products in small amounts to account for the difference at least partly. Complex 1 was still quite active at 100 °C, reaction giving 74 % conversion with 71 % yield to 7 a and 62 % yield into methanol.
Table 1.
Manganese catalyzed hydrogenation of 6 a: Influence of catalyst precursors and reaction conditions.[a,b]
| Entry | [Mn] No. | [Mn] [mol %] | NaOtBu [mol %] | H2 [bar] | t [h] | X [%] | 7 a (%) [TON] | 8 (%) [TON] |
|---|---|---|---|---|---|---|---|---|
| 1 | 1 | 1 | 2 | 30 | 26 | 86 | 86 (86) | 62 (62) |
| 2 | 2 | 1 | 2 | 30 | 26 | 23 | 10 (10) | 4 (4) |
| 3 | 3 | 1 | 2 | 30 | 26 | 23 | 14 (14) | 3 (3) |
| 4 | 4 | 1 | 2 | 30 | 26 | 21 | 11 (11) | 5 (5) |
| 5[c] | 1 | 1 | 2 | 30 | 26 | 74 | 71 (71) | 62 (62) |
| 6 | 1 | 2 | 3 | 30 | 26 | 98 | 90 (45) | 75 (38) |
| 7 | 1 | 0.5 | 1 | 30 | 26 | 70 | 66 (132) | 46 (92) |
| 8[d] | 1 | 0.2 | 0.5 | 30 | 40 | 56 | 48 (240) | 35 (175) |
| 9[d] | 1 | 0.2 | 0.5 | 60 | 40 | 60 | 52 (260) | 30 (147) |
| 10[d] | 5[e] | 0.2 | – | 60 | 14 | 38 | 37 (187) | 26 (129) |
| 11[d] | 5[e] | 0.2 | – | 60 | 40 | 70 | 67 (336) | 44 (220) |
| 12[d] | 5[e] | 0.1 | – | 60 | 40 | 66 | 62 (620) | 40 (400) |
[a] Conditions: 6 a (44 mg, 0.5 mmol), H2, Mn complex, NaOtBu, temp. (120 °C), THF (0.7 mL). [b] Yield was calculated using gas chromatography, ethyl heptanoate (25 μL, 0.15 mmol) was used an internal standard. [c] T=100 °C. [d] 6 a (440 mg, 5 mmol), H2, Mn complex, NaOtBu, temp. (120 °C), THF (2 mL). [e] Pre‐catalyst 1 was treated with NaOtBu in THF for 30 min at RT; reaction mixture was passed through celite, after removal of all volatiles, the formation of 5 was verified by 31P{1H} NMR spectroscopy and it was used directly without further work‐up.
Next, we investigated the influence of catalyst and co‐catalyst loading on catalyst performance with the most promising pre‐catalyst 1 (Table 1, entries 6–9). Increasing the loading of 1 to 2 mol % lead to almost full conversion of 6 a and 90 % yield of 7 a. Reducing the Mn loading to 0.5 mol % lead to 70 % conversion and 66 % yield of 7 a corresponding to a TON of 132. At a loading as low as 0.2 mol % for complex 1 and 0.5 mol % for NaOtBu and extended reaction time of 40 h, TONs of 240 for 7 a and 175 for methanol were obtained, respectively.
Interestingly, the reaction showed significantly higher TONs to the corresponding products, when complex 1 was pre‐activated with NaOtBu in THF for 30 min at room temperature. After removal of all volatiles, the nearly quantitative formation of the unsaturated complex 5 was verified by 1H and 31P{1H} NMR spectroscopy (Scheme 2).18 The remaining solid was re‐dissolved in THF and used directly without further purification. At a 0.2 mol % of manganese loading, the use of 5 at 60 bar pressure of hydrogen after 14 h gave 38 % conversion with 187 TONs into diol and 129 TONs to MeOH (Table 1, entry 10). Gratifyingly, when the reaction time was increased to 40 h, 70 % conversion was achieved with 336 TONs into diol and 220 TONs into methanol (Table 1, entry 11). The best results were found with 0.1 mol % manganese, leading to 66 % conversion corresponding to TONs of 620 for 7 a and 400 for methanol, respectively (Table 1, entry 12).
Scheme 2.
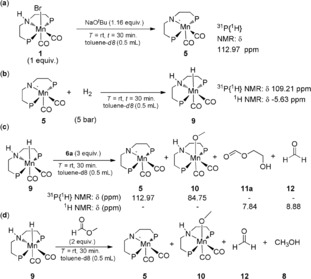
Formation of presumed MnI intermediates and their reactivity towards organic substrates; P=PiPr2.
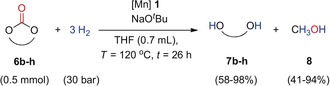
The substrate scope of the reaction was assessed for complex 1 using the standard procedure without pre‐activation for the hydrogenation of cyclic 5‐membered and 6‐membered carbonates. Reaction of 5‐membered cyclic carbonates in THF (0.7 mL) at 120 °C under 30 bar of H2 using 1 (1–2 mol %) and NaOtBu (1.1 equiv) revealed conversion up to >99 % with yield up to 97 % for diol and 74 % into methanol (Table 2, entries 1–7). The activity of complex 1 for the hydrogenation of six‐membered ring carbonates was equally high and even exceeded that observed for the five‐membered ring substrates under the same conditions. Hydrogenation of six‐membered cyclic carbonates under standard conditions showed conversion up to >99 % with yield up to 98 % for diol and 94 % for methanol (Table 2, entries 8–11).
Table 2.
Hydrogenation of cyclic carbonates using Mn complex 1 as a pre‐catalyst.[a,b]
| Entry | Carbonate | [Mn] [mol %] |
Conv. [%] |
Diol [7, %] |
Methanol [8, %] |
|
|---|---|---|---|---|---|---|
| 1 |
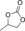
|
6 b | 1 | 89 | 75 | 66 |
| 2 | 2 | >99 | 82 | 75 | ||
| 3 |
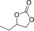
|
6 c | 1 | 86 | 82 | 65 |
| 4 | 2 | >99 | 97 | 74 | ||
| 5 |
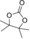
|
6 d | 1 | 82 | 76 | 66 |
| 6[c] |
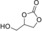
|
6 e | 1 | 67 | 59 | 43 |
| 7[c] | 2 | 77 | 71 | 60 | ||
| 8 |
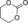
|
6 f | 1 | 79 | 58 | 41 |
| 9 | 2 | 97 | 80 | 75 | ||
| 10 |
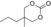
|
6 g | 1 | >99 | 98 | 94 |
| 11 |
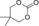
|
6 h | 1 | 80 | 73 | 57 |
[a] Conditions: 6 (0.5 mmol), H2 (30 bar), complex 1, NaOtBu (1.1 equiv with respect to 1), THF (0.7 mL), temp. (120 °C), 26 h. [b] Yield was calculated using gas chromatography, ethyl heptanoate (25 μL, 0.15 mmol) was used as an internal standard. [c] 1,3‐Propanediol (25 μL, 0.34 mmol) was used as an internal standard.
A series of control reactions were performed to get insight into possible intermediates resulting from pre‐catalyst 1 and their reactivity towards the organic substrates. Reaction of complex 1 with NaOtBu leads to the formation of complex 5 (Scheme 2, entry a), which confirmed a kinetically competent intermediate in the catalytic experiments summarized in Table 1. In presence of hydrogen (5 bar), the Mn‐monohydride complex 9 is formed within 30 minutes in [D8]toluene as confirmed by 1H NMR and 31P NMR spectrum (1H NMR: δ −5.63 ppm (t, J=51 ppm, Mn‐H); 31P{1H} NMR: δ 109.21 ppm, Scheme 2, entry b). Adding 3 equiv of ethylene carbonate 6 a to this solution regenerated the unsaturated Mn complex 5 according to 31P{1H} NMR spectra together with the smaller amounts of the methanol coordinated manganese complex 10 (Supporting Information, Figure S2).8 The formate ester of ethylene glycol 11 a and free formaldehyde 12 were observed as organic products in the 1H NMR spectrum (11 a:12=75:25) (Supporting Information, Figure S1; Scheme 2, entry c). Treating complex 9 with methyl formate lead also to formation of manganese complex 5 and 10 together with free formaldehyde and methanol (Supporting Information, Figure S3, S4, and S5; Scheme 2, entry d). These results confirm the high reactivity of the Mn‐MACHO framework for the sequential transfer of H2 formally as H− and H+ to the C=O units of CO2 derivatives along the reduction path to methanol.
The observed species can be related directly to a plausible mechanism for the catalytic cycle in line with previous reports for other metals (Scheme 3). Activation of complex 1 leads to the catalyst species I, which corresponds to the experimentally observed complex 5. Heterolytic cleavage of the H2 molecule across the Mn=N double bond leads to Intermediate II detected as the mono‐hydride complex 9. The carbonyl group of the substrate interacts with the protic N−H hydrogen activating the C=O unit for nucleophilic attack by Mn‐bound hydride. This leads to an ortho‐ester as first organic reduction product. Cyclic ortho‐esters are typically more stable in the isomeric open form of the formate ester and hydroxyl functionality, in line with the detection of this species in the control reactions. This principle of H−H addition to the C=O unit can be re‐iterated until the methanol stage is reached. Coordination of CH3OH to species I would explain the presence of complex 10.
Scheme 3.
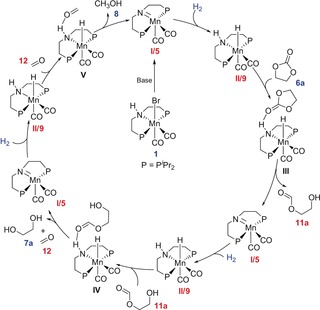
Possible mechanism for catalytic hydrogenation of cyclic carbonates using the Mn‐MACHO complex 1 as pre‐catalyst; starting compounds and products are shown in blue, experimentally detected complexes and intermediates are highlighted in red.19
The catalytic cycle and the observed intermediates are in accord with the DFT calculations reported by the Rüping group for their PNN‐ligand system. Their data associated the highest activation barrier with the initial hydride transfer to 6 a.17 This in line with the observation of organic products at all reduction levels down to methanol upon reaction with the hydride complex 9. However, a concerted H2 activation and transfer mechanism19 as suggested for the ligand framework introduced by Milstein cannot be excluded.16 The two pathways follow basically the same bond breaking and bond formation trajectory with similar energy spans, differentiated by the existence of structures akin to 9 as intermediates or off‐loop species. Further studies are required to elucidate these alternatives for manganese catalyzed CO2 hydrogenation, which may well depend on the individual ligand framework given their close similarity.
In conclusion, the manganese complex [Mn(CO)2(Br)[HN(C2H4PiPr2)2] 1 was identified as efficient catalyst for the hydrogenation of cyclic carbonates. The complex is readily accessible from commercially available starting materials comprising the PNP pincer ligand MACHO. High yields for the diols and for methanol were achieved under relatively mild conditions, corresponding to up to 620 TON and 400 TON, respectively. These data match or even surpass the performance of the PNN‐ligand based catalysts developed in parallel studies.16, 17 The formation of unsaturated complex 5 and the hydride complex 9 was validated under relevant reaction conditions, corresponding to two crucial intermediates of a potential catalytic cycle for this reaction. In particular, their reactivity towards H2 and the organic substrates substantiated the pivotal function of the M=N/M−N unit as a formal hydride/proton relay in the stepwise reduction of the C=O unit. Further studies are necessary to distinguish between a stepwise or concerted mechanism for this heterolytic H2 transfer. Overall, these results substantiate the great potential of manganese complexes for hydrogenation of CO2‐derived functional groups, opening the path for catalytic conversion of carbon dioxide into methanol using earth abundant, cheap, and benign manganese as metal component.
Experimental Section
Standard procedure for the catalytic hydrogenation of cyclic carbonates to diols and methanol using Mn catalyst 1: The catalytic reactions were carried out in externally heated 20 mL stainless‐steel reactors equipped with a glass inlet and a magnetic stir bar. Mn‐complex 1 and NaOtBu were weighed into the glass inlet inside a glovebox. The glass inlet was closed with a septum and transferred into the bottom part of the steel autoclave, where it was opened under a stream of argon. After sealing, the autoclave was purged with argon three times. The required amounts of carbonates 6 and THF were added by syringe through a needle valve under argon flow at room temperature. The autoclave was sealed and pressurized with hydrogen gas and heated to the reaction temperature. After the given reaction time, the autoclave was cooled to room temperature and carefully vented under continuous stirring. Ethyl heptanoate was added as an internal standard and the resulting solution was analyzed by gas chromatography.
Procedure with pre‐activated complex 5: A solution of complex 1 (0.1 mmol) and NaOtBu (0.12 mmol) in THF (0.5 mL) was stirred for 30 min at RT and the formation of 5 was checked by 31P{1H} NMR spectroscopy. After 30 minutes, the reaction mixture was passed through a short pad of celite. After thorough removal of all volatiles under vacuum, the solid was re‐dissolved in THF. This solution was used for catalysis following the standard procedure.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was carried out as part of our activities in the Cluster of Excellence “Tailor‐Made Fuels from Biomass” funded by the Excellence Initiative by the German federal and state governments to promote science and research at German universities (EXC 236) and within the Kopernikus Project “P2X: Flexible use of renewable resources—exploration, validation and implementation of ”Power‐to‐X“ concepts” supported by the German Federal Ministry of Education and Research (BMBF) (Grant Number 03SFK2A). A.K. thanks the Erasmus Mundus Action 1 Programme (FPA2013‐0037) “SINCHEM” for a stipend. We thank Prof. Dr. Magnus Rüping for sharing the results of his group prior to publication.
A. Kaithal, M. Hölscher, W. Leitner, Angew. Chem. Int. Ed. 2018, 57, 13449.
References
- 1.
- 1a. Olah G. A., Alain G., Prakash G. K. Surya, Beyond Oil and Gas: The Methanol Economy, Wiley-VCH, Weinheim, 2009; [Google Scholar]
- 1b. Methanol: The Basic Chemical and Energy Feedstock of the Future (Eds.: M. Bertau, H. Offermanns, L. Plass, F. Schmidt, H.-J. Wernicke), Springer, Heidelberg, 2013; [Google Scholar]
- 1c. Chemical Energy Storage (Ed.: R. Schlögl), deGruyter, Berlin, 2013. [Google Scholar]
- 2.
- 2a. Centi G., Quadrelli E. A., Perathoner S., Energy Environ. Sci. 2013, 6, 1711–1731; [Google Scholar]
- 2b. Aresta M., Dibenedetto A., Angelini A., Chem. Rev. 2014, 114, 1709–1742; [DOI] [PubMed] [Google Scholar]
- 2c. Alberico E., Nielsen M., Chem. Commun. 2015, 51, 6714–6725; [DOI] [PubMed] [Google Scholar]
- 2d. Klankermayer J., Wesselbaum S., Beydoun K., Leitner W., Angew. Chem. Int. Ed. 2016, 55, 7296–7343; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 7416–7467; [Google Scholar]
- 2e. Artz J., Müller T. E., Thenert K., Kleinekorte J., Meys R., Sternberg A., Bardow A., Leitner W., Chem. Rev. 2018, 118, 434–504. [DOI] [PubMed] [Google Scholar]
- 3. Balaraman E., Gunanathan C., Zhang J., Shimon L. J., Milstein D., Nat. Chem. 2011, 3, 609–614. [DOI] [PubMed] [Google Scholar]
- 4. Rezayee N. M., Huff C. A., Sanford M. S., J. Am. Chem. Soc. 2015, 137, 1028–1031. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Kar S., Sen R., Goeppert A., Prakash G. K. S., J. Am. Chem. Soc. 2018, 140, 1580–1583; [DOI] [PubMed] [Google Scholar]
- 5b. Kothandaraman J., Goeppert A., Czaun M., Olah G. A., Prakash G. K. S., J. Am. Chem. Soc. 2016, 138, 778–781. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Wesselbaum S., vom Stein T., Klankermayer J., Leitner W., Angew. Chem. Int. Ed. 2012, 51, 7499–7502; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 7617–7620; [Google Scholar]
- 6b. Wesselbaum S., Moha V., Meuresch M., Brosinski S., Thenert K. M., Kothe J., vom Stein T., Englert U., Hölscher M., Klankermayer J., Leitner W., Chem. Sci. 2015, 6, 693–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Schneidewind J., Adam R., Baumann W., Jackstell R., Beller M., Angew. Chem. Int. Ed. 2017, 56, 1890–1893; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 1916–1919. [Google Scholar]
- 8. Kar S., Goeppert A., Kothandaraman J., Prakash G. K. S., ACS Catal. 2017, 7, 6347–6351. [Google Scholar]
- 9. Han Z., Rong L., Wu J., Zhang L., Wang Z., Ding K., Angew. Chem. Int. Ed. 2012, 51, 13041–13045; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 13218–13222. [Google Scholar]
- 10.
- 10a. Tamura M., Ito K., Honda M., Nakagawa Y., Sugimoto H., Tomishige K., Sci. Rep. 2016, 6, 24038; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10b. Arnaud S. P., Wu L., Wong Chang M.-A., Comerford J. W., Farmer T. J., Schmid M., Chang F., Li Z., Mascal M., Faraday Discuss. 2017, 202, 61–77; [DOI] [PubMed] [Google Scholar]
- 10c. Saxena R. K., Anand P., Saran S., Isar J., Agarwal L., Indian J. Microbiol. 2010, 50, 2–11; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10d. Bucher B., Curran D. P., Tetrahedron Lett. 2000, 41, 9617–9621. [Google Scholar]
- 11.
- 11a. Laserna V., Fiorani G., Whiteoak C. J., Martin E., Escudero-Adán E., Kleij A. W., Angew. Chem. Int. Ed. 2014, 53, 10416–10419; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 10584–10587; [Google Scholar]
- 11b. Crich D., Vinod A. U., Picione J., J. Org. Chem. 2003, 68, 8453–8458. [DOI] [PubMed] [Google Scholar]
- 12.For recent reviews, see:
- 12a. Garbe M., Junge K., Beller M., Eur. J. Org. Chem. 2017, 4344–4362; [Google Scholar]
- 12b. Kallmeier F., Kempe R., Angew. Chem. Int. Ed. 2018, 57, 46–60; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 48–63; [Google Scholar]
- 12c. Filonenko G. A., van Putten R., Hensen E. J. M., Pidko E. A., Chem. Soc. Rev. 2018, 47, 1459–1483; [DOI] [PubMed] [Google Scholar]
- 12d. Maji B., Barman M., Synthesis 2017, 49, 3377–3393. [Google Scholar]
- 13. Mukherjee A., Nerush A., Leitus G., Shimon L. J. W., Ben David Y., Espinosa Jalapa N. A., Milstein D., J. Am. Chem. Soc. 2016, 138, 4298–4301. [DOI] [PubMed] [Google Scholar]
- 14. Elangovan S., Topf C., Fischer S., Jiao H., Spannenberg A., Baumann W., Ludwig R., Junge K., Beller M., J. Am. Chem. Soc. 2016, 138, 8809–8814. [DOI] [PubMed] [Google Scholar]
- 15.
- 15a. Dubey A., Nencini L., Fayzullin R. R., Nervi C., Khusnutdinova J. R., ACS Catal. 2017, 7, 3864–3868; [Google Scholar]
- 15b. Bertini F., Glatz M., Gorgas N., Stöger B., Peruzzini M., Veiros L. F., Kirchner K., Gonsalvi L., Chem. Sci. 2017, 8, 5024–5029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kumar A., Janes T., Espinosa-Jalapa N. A., Milstein D., Angew. Chem. Int. Ed. 2018, 57, 12076–12080; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 12252–12256. [Google Scholar]
- 17. Zubar V., Lebedev Y., Azofra L. M., Cavallo L., El-Sepelgy O., Rueping M., Angew. Chem. Int. Ed. 2018, 10.1002/anie.201805630; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 10.1002/ange.201805630. [DOI] [Google Scholar]
- 18. Fu S., Shao Z., Wang Y., Liu Q., J. Am. Chem. Soc. 2017, 139, 11941–11948. [DOI] [PubMed] [Google Scholar]
- 19.
- 19a. Gusev D. G., ACS Catal. 2016, 6, 6967–6981; [Google Scholar]
- 19b. Dub P. A., Henson N. J., Martin R. L., Gordon J. C., J. Am. Chem. Soc. 2014, 136, 3505–3521; [DOI] [PubMed] [Google Scholar]
- 19c. Hasanayn F., Baroudi A., Bengali A. A., Goldman A. S., Organometallics 2013, 32, 6969–6985; [Google Scholar]
- 19d. Hasanayn F., Morris R. H., Inorg. Chem. 2012, 51, 10808–10818. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary