

Abstract
In this review, we discuss the poorly explored role of calcium/calmodulin‐dependent protein kinase II (CaMKII) in memory maintenance, and its influence on memory destabilization. After a brief review on CaMKII and memory destabilization, we present critical pieces of evidence suggesting that CaMKII activity increases retrieval‐induced memory destabilization. We then proceed to propose two potential molecular pathways to explain the association between CaMKII activation and increased memory destabilization. This review will pinpoint gaps in our knowledge and discuss some ‘controversial’ observations, establishing the basis for new experiments on the role of CaMKII in memory reconsolidation. The role of CaMKII in memory destabilization is of great clinical relevance. Still, because of the lack of scientific literature on the subject, more basic science research is necessary to pursue this pathway as a clinical tool.
Keywords: calcium/calmodulin‐dependent kinase II, reconsolidation, synaptic signaling
Abbreviations used
- AIP
autocamtide‐2‐related inhibitory peptide
- AMPAR
α‐amino‐3‐hydroxy‐5‐methyl‐4‐isoxazolepropionic acid receptor
- CaM
calcium/calmodulin
- CaMKII
calcium/calmodulin‐dependent protein kinase II
- CaMKIV
CaM‐dependent protein kinase type IV
- CaMKV
CaM kinase like vesicle associated
- CFC
contextual fear condition
- CYLD
cylindromatosis
- LTD
long‐term depression
- LTP
long‐term potentiation
- LVGCCs
L‐type voltage‐gated calcium channels
- MWM
Morris water maze
- NMDAR
N‐methyl‐d‐aspartate receptor
- PKC
protein kinase C
- PKD1
protein kinase D1
- PSD
post‐synaptic density
- RRID
Research Resource Identifier
- S‐site
substrate binding site
- T286
threonine 286
- T305
threonine 305
- T306
threonine 306
- tat
trans‐acting activator of transcription
- UPS
ubiquitin proteasome system
CaMKII
Calcium/calmodulin‐dependent kinase II (CaMKII) is the major post‐synaptic density (PSD) protein in the brain, accounting for 1–2% of total proteins (Erondu and Kennedy 1985; Peng et al. 2004; Cheng et al. 2006). CaMKII is a serine/threonine kinase composed of an auto‐inhibitory regulatory domain, an N‐terminal kinase domain and a C‐terminal self‐association domain (Chao et al. 2011; Hell 2014; Myers et al. 2017). During resting state, this enzyme is inactive because of blocking of the substrate binding site (S‐site) and the catalytic domain, both found in the kinase domain, by the pseudosubstrate segment in the regulatory domain (Braun and Schulman 1995b; Hell 2014). Binding with the calcium/calmodulin (CaM) complex causes a conformational change in CaMKII that unblocks the kinase domain from the inhibitory domain, activating the enzyme (Colbran et al. 1989; Meyer et al. 1992; Grant et al. 2008).
CaMKII has a wide range of substrates and is involved in many aspects of cellular function, such as the regulation of ion channel function, neurotransmitter release, gene transcription, cytoskeleton organization and intracellular calcium homeostasis (Erondu and Kennedy 1985; Tobimatsu and Fujisawa 1989; Hudmon and Schulman 2002; Lisman et al. 2002, 2012; Lucchesi et al. 2011). In mammals, four different isoforms of this enzyme are expressed: α, β, γ and δ isoforms (Tobimatsu and Fujisawa 1989; Gaertner et al. 2004). The most abundant isoforms in the brain are α and β CaMKII (Bennett et al. 1983; Tobimatsu and Fujisawa 1989; Peng et al. 2004). These isoforms are usually associated with each other, creating a holoenzyme composed of 12 CaMKII subunits organized into two hexameric rings (Kolodziej et al. 2000; Hoelz et al. 2003; Rosenberg et al. 2005). The 12 subunits are primarily composed of α and β CaMKII heteromers, but homomers consisting of only αCaMKII have been observed (Bronstein et al. 1988).
The enzyme is organized into a complex of subunits, thereby facilitating the occurrence of autophosphorylation. Examples of autophosphorylation sites of CaMKII are threonine 305 (T305) and threonine 306 (T306). Phosphorylation of these sites are believed to be inhibitory because of blocking of the CaM binding site, causing CaMKII to translocate out of the PSD area and decreasing long‐term potentiation (LTP) and learning (Hanson and Schulman 1992; Shen et al. 2000; Elgersma et al. 2002). The most studied autophosphorylation site of CaMKII isoforms is threonine 286 (T286) for αCaMKII and threonine 287 (T287) for βCaMKII.
Phosphorylation at the T286/287 sites occurs between neighboring subunits within the same holoenzyme, and requires binding of CaM to both of the subunits involved (Hanson et al. 1994; Mukherji and Soderling 1994; Rich and Schulman 1998). Phosphorylation at T286/287 allows CaMKII to remain in an active state, even in the absence of CaM, serving as an example of a CaM‐independent state of activation (Miller et al. 1988; Hanson et al. 1994; Irvine et al. 2006). Autophosphorylation at T286 also enhances CaM complex's binding affinity for the enzyme by 1000‐fold, with an increase in release time from less than a second to hundreds of seconds (Meyer et al. 1992; Tzortzopoulos and Torok 2004; Tzortzopoulos et al. 2004). T286/287 autophosphorylation also changes CaMKII binding affinity for other molecules. For example, it increases the holoenzyme's binding affinity to the N‐methyl‐d‐aspartate receptor (NMDAR) (Bayer et al. 2001).
Because of its ability to switch from a CaM‐dependent to a CaM‐independent state of activation by T286/287 autophosphorylation (bistability), CaMKII has been suggested to act as a memory molecule, preserving ‘memories’ of strong calcium signals (Lisman 1994). T286A‐mutant mice lack the ability to autophosphorylate at the T286 site, and have one of the most severe spatial learning deficits described in a mutant mouse (Giese et al. 1998; Need and Giese 2003). T286A mutation also blocks the induction of NMDAR‐dependent LTP at excitatory hippocampal CA1 synapses (Giese et al. 1998; Cooke et al. 2006).
Indeed, the role of CaMKII in learning is widely accepted; however, its role as a memory molecule is still a matter of debate (Lucchesi et al. 2011; Coultrap and Bayer 2012; Sanhueza and Lisman 2013). Buard et al. (2010) has shown that blocking CaMKII activity via systemic injection of the CaMKII inhibitor, tatCN21, prior to performing a contextual fear long‐term memory test, but after conditioning, had no effect on memory storage. Even T286A‐mutant mice can learn and maintain contextual and cued fear memory, if they were conditioned using extended protocols (Irvine et al. 2005, 2011). Although αCaMKII T286 autophosphorylation is required for LTP induction in pyramidal CA1 neurons (Giese et al. 1998; Cooke et al. 2006), it can also induce long‐term depression in the same cells (Marsden et al. 2007; Mockett et al. 2011) and it is not necessary for LTP induction in dentate gyrus granule cells (Cooke et al. 2006; Wu et al. 2006). Moreover, various authors have observed that LTP induction results in a transient increase in CaMKII autonomous activity, lasting for only a few minutes (Lengyel et al. 2004; Lee et al. 2009; Fujii et al. 2013).
Nonetheless, CaMKII has been shown to be important for memory extinction. Prolonged and repetitive re‐exposure to the conditioned stimulus without the unconditioned stimulus leads to a gradual weakening of the conditioned response, called memory extinction. Memory extinction is the learning of new environmental conditions that suppresses the previously learned conditioned response (Pavlov 1927; Eisenberg et al. 2003; Pedreira and Maldonado 2003; Myers and Davis 2007; Quirk and Mueller 2008; Pape and Pare 2010). The partial reduction of CaMKII autophosphorylation in heterozygous T286A mutants impairs extinction of contextual fear memory (Kimura et al. 2008). Furthermore, blocking of hippocampal CaMKII kinase activity impairs memory extinction (Szapiro et al. 2003). Inhibition of CaMKII activity by intrahippocampal injection of autocamtide‐2‐related inhibitory peptide (AIP) blocks the facilitation of memory extinction, which results from exposure to a novel stimulus (de Carvalho Myskiw et al. 2014). Therefore, CaMKII may play a role in memory maintenance as a biological substrate of memory extinction.
Here, we propose a different, novel and unexplored role for CaMKII in memory. We will avoid the ‘traditional’ discussion of CaMKII as a learning or memory molecule, in addition to its role in memory extinction. Instead, we will explore a different role for CaMKII in memory maintenance. CaMKII's role in memory destabilization, an important step of retrieval‐induced memory reconsolidation.
Memory reconsolidation: destabilization and restabilization
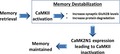
The first evidence of memory reconsolidation was presented in Misanin et al. (1968), where an amnesic effect was induced by electroconvulsive shock 24 h after fear conditioning training. Such amnesic effect could only be achieved if the electroconvulsive shock was presented after re‐exposure to the conditioned stimulus. In other words, the associative memory between a neutral conditioned stimulus and an unconditioned stimulus was lost after electroconvulsive shock, only if the memory was retrieved (Misanin et al. 1968). This observation challenged the long prevailing theory that memories once consolidated would no longer be labile. Recently, memory reconsolidation has been shown to be an important process for the maintenance and further strengthening of a memory (Lee 2008; Fukushima et al. 2014). Retrieval‐induced reconsolidation can destabilize a memory, which involves proteasome‐dependent degradation of synaptic proteins, followed by restabilization of the memory, a protein synthesis‐dependent process (Fig. 1) (Kelly et al. 2003; Nader 2003; Lee et al. 2004, 2008; Lee 2008).
Figure 1.
Schematic representation of how memory reconsolidation works. Retrieval of a memory by the presentation of the conditioned and/or unconditioned stimulus initiates the reconsolidation process. During reconsolidation, memory‐related proteins are degraded, which is called memory destabilization (Lee et al. 2004, 2008). Concurrent with reconsolidation, memory is restabilized by protein synthesis (Nader et al. 2000). Although it remains clear that protein synthesis is necessary to compensate for protein degradation, it is unclear if one precedes the other or if memory destabilization and restabilization happen simultaneously. Nonetheless, the result is maintenance of the memory with the possibility of alterations in the memory during the reconsolidation process (Lee 2008). Although the NMDAR is involved in memory destabilization and restabilization, it has been reported that isoform specificity can be found for these two different steps of memory reconsolidation (Milton et al. 2013).
A previously consolidated memory is impaired by pharmacological blocking of protein synthesis after the retrieval process (Nader et al. 2000). Blocking the proteasome system with clasto‐lactacystin‐β‐lactone, a specific, cell permeable and irreversible inhibitor of the catalytic proteasome subunit 20S, reverts the memory impairment effect elicited by blocking protein synthesis (Lee et al. 2008). Henceforth, protein synthesis in memory reconsolidation is important to revert protein degradation‐dependent memory destabilization. The ubiquitin proteasome system (UPS) is the main mechanism for protein catabolism in mammalian cells and works by targeting proteins via ubiquitination with posterior degradation by the 26S proteasome enzyme (Varshavsky et al. 2000; Leestemaker and Ovaa 2017). Whether or not memory destabilization is necessary for memory maintenance after retrieval is still a matter of debate. Pharmacological blocking of the catalytic subunit 20S, immediately after retrieval, has been shown to impair memory maintenance (Artinian et al. 2008). However, as discussed by Artinian et al. (2008), it is unclear if protein degradation is solely involved in the destabilization process. The UPS might be working in memory reconsolidation by degradation of memory suppressor proteins, thereby facilitating memory restabilization. However, Lee et al. (2008) have reported that retrieval‐induced protein degradation by the UPS system is in fact related to memory destabilization. These authors have observed no effect on memory maintenance by pharmacological inhibition of the UPS system, after retrieval. Furthermore, Lee et al. (2008) have also shown that inhibition of protein degradation increases memory maintenance by inhibiting memory extinction. The apparent contradiction between the observations of Artinian et al. (2008) and Lee et al. (2008) could be a consequence of the different experimental designs. Artinian et al. (2008) and Lee et al. (2008) studies differ in terms of the hippocampal area where protein degradation was inhibited; the CA3 region was targeted in the former and CA1 region was targeted the latter. The CA1 and CA3 areas might have a different dependence for protein degradation after retrieval. Both areas have been shown to play distinct roles in memory maintenance (Ji and Maren 2008; Langston et al. 2010) and have different proteomic profiles (Gozal et al. 2002). Both articles also present results from different behavioral paradigms. While Artinian et al. (2008) used the Morris water maze (MWM), Lee et al. (2008) utilized the contextual fear conditioning paradigm. The two behavioral paradigms are known to produce different phenotypes with animals harboring the same genetic mutation (Sterneck et al. 1998), or even in animals exposed to the same pharmacological intervention (Shuman et al. 2009). The repetitive training spread throughout many days, which is required by the MWM, might create, for example, a more flexible memory that becomes more sensible to changes in the UPS function because of the continuous processes of destabilization–restabilization during training.
Nevertheless, the necessity for and the roles played by destabilization in memory maintenance after retrieval are still questions to be answered. For example, the identification of proteins targeted to degradation during the destabilization process is still poorly studied. First efforts have identified proteins involved in translational control, like MOV10 (Jarome et al. 2011), and synaptic structure, like Shank (Lee et al. 2008; Jarome et al. 2011). It is possible that memory destabilization plays different roles depending on the area of the brain being studied, and protein degradation could be relevant for both memory destabilization and restabilization.
Evidences for CaMKII regulation of memory destabilization
One of the strongest pieces of evidence for the role of CaMKII in memory destabilization are the observations of Cao et al. (2008). By over‐expressing a transgenic form of αCaMKII that has a different ATP‐binding site structure, referred to as the αCaMKII‐F89G transgene, Cao et al. (2008) could increase CaMKII levels and activity, as well as specifically block αCaMKII‐F89G activity. The authors observed that if αCaMKII activity was increased at the time of retrieval of cued or contextual fear memory, the memory was specifically erased. There was no spontaneous recovery, indicating that this was a true memory erasure and not an enhancement of extinction. Cao et al. (2008) suggested that the memory erasure phenotype could be related to an increase in reconsolidation‐induced protein degradation, citing Lee et al. (2008) published in the same year.
A more direct link between CaMKII and UPS protein degradation during destabilization was the observation made in Jarome et al. (2016). The authors reported that administration of AIP, an inhibitor of CaMKII, in the amygdala did not affect fear memory, but it rescued retrieval‐dependent memory impairment, which was induced by blocking protein synthesis. This is a characteristic phenotype observed after blocking memory destabilization, supporting the role of CaMKII in memory destabilization. Additionally, AIP treatment stopped the retrieval‐induced proteasome activity, in vitro and in vivo, and impaired retrieval‐induced phosphorylation of the proteasome subunit Rpt6 on serine 120 in synaptosomes (Jarome et al. 2016). This suggested that CaMKII activity, at the time of retrieval, regulates protein degradation at the synapse.
A less substantial piece of evidence for the role of CaMKII activation inducing memory destabilization can be conjectured from the results reported by Rossetti et al. (2017). Rossetti et al. (2017) showed that viral‐induced expression of αCaMKII T286D/T305A/T306A gene (a hyperactive form of αCaMKII) in the hippocampus, blocks previously learned place‐avoidance behavior. It is plausible to propose that the expression of a highly active form of CaMKII increases memory destabilization, resulting in the memory impairment phenotype observed in Rossetti et al. (2017), which is in agreement with Cao et al. 2008;. Since the viral vector was injected 3 days after the first memory test, any CaMKII‐induced memory destabilization enhancement likely occurred during the second memory test when the phenotype was observed. However, a clear link between CaMKII activation and retrieval‐induced memory destabilization is difficult to establish because of the behavioral protocol used by Rossetti et al. (2017). The conditioned place‐avoidance task used demands repetitive trials to be conducted both during training and memory test, prior to viral injection. In each of these trials, memory was retrieved, which probably initiated memory destabilization/reconsolidation throughout the training and memory test sessions. Furthermore, in this behavioral paradigm, the unconditioned stimulus (shock) is present during the memory test, allowing for continued conditioning of the animal. Henceforth, this model might be too complex to deduce whether a memory impairment is retrieval dependent or independent. Rossetti et al. (2017) presents another plausible interpretation of the memory impairment resulting from the αCaMKII T286D/T305A/T306A mutation. Based on the memory engram theory (Tonegawa et al. 2015; Josselyn et al. 2017), the authors proposed that excessive CaMKII activity from αCaMKII T286D/T305A/T306A mutation resulted in association of the conditioned stimulus to multiple and unspecific synaptic/neuronal pathways. Consequently, the memory was lost since the memory engram was also lost. It is our opinion that the data collected by Rossetti et al. (2017) does not enable one to definitively determine the memory processes affected by the mutation. Even though an increase in memory destabilization or an impairment of the memory engram are possible explanations, with the data available it is impossible to determine the cause of the memory impairment.
We have recently observed that dorso‐hippocampal knockdown of CaMKII endogenous inhibitor, CaMK2N1, results in a retrieval‐dependent memory impairment (Vigil et al. 2017). CaMK2N1 is a specific endogenous inhibitor of CaMKII kinase activity (Chang et al. 1998). In our experiments, CaMK2N1 knockdown animals presented normal freezing scores in a first contextual fear memory test, but lower freezing scores in a subsequent test. This retrieval‐induced memory impairment can be interpreted as an increase in memory destabilization. We have also observed that 2 h after contextual fear memory retrieval, there was a decrease in αCaMKII T286 phosphorylation and such decrease was dependent on CaMK2N1 expression. Additionally, contextual fear memory retrieval promotes CaMK2N1 expression in dorsal hippocampi (Vigil et al. 2017). If CaMKII activation induces memory destabilization, CaMK2N1 expression could be induced by memory retrieval to control the destabilization process. This could explain why knockdown of CaMK2N1 results in retrieval‐dependent memory impairment. It is unlikely that this memory impairment was the result of an enhancement in memory extinction, as no extinction was observed in the control group. Extinction and reconsolidation seem to be exclusive processes (Merlo et al. 2014). It is important to notice that although CaMK2N1 was knocked‐down before conditioning, a memory impairment was observed only in the second memory test. This is different from Cao et al. (2008), who found that increased CaMKII activation impairs memory, even during the first test. This comparison leads to two important conclusions. First, memory reconsolidation is a process that extends beyond the retrieval session. Second, under physiological conditions, CaMK2N1 is important when it comes to reducing CaMKII‐induced memory destabilization after, but not during, memory retrieval. Therefore, CaMKII‐induced memory destabilization likely starts during memory retrieval, as observed by Cao et al. (2008), and needs to be controlled by CaMK2N1 after the memory is retrieved (Vigil et al. 2017).
Corroborating with a role for CaMKII in memory destabilization, Rich et al. (2016) performed a phosphoproteomic study of basolateral amygdala samples from rats subjected to either extinction or reconsolidation of previously learned cocaine seeking behavior. αCaMKII phosphorylation at S331, a largely understudied site, was shown to decrease when memory was retrieved and increase after extinction. In the same article, Rich et al. (2016) showed that S331 phosphorylation reduces αCaMKII kinase activity. Thus, reduction in S331 phosphorylation after memory retrieval is thought to increase CaMKII activity, possibly initiating CaMKII‐induced memory destabilization. If this were the case, memory retrieval would first reduce CaMKII S331 phosphorylation, resulting in memory destabilization followed by a later increase in CaMK2N1 expression in order to stop the destabilization process. Unfortunately, the authors did not test behavioral phenotypes induced by specific manipulation of S331 phosphorylation in vivo. Consequently, the role of CaMKII S331 phosphorylation in memory destabilization is still a hypothesis lacking substantial evidence.
The memory phenotypes of the articles cited in this section are possible observations of changes in memory destabilization by manipulation of CaMKII, and are summarized in Table 1. Based on these observations, we can raise the hypothesis that CaMKII activation during and after memory retrieval induces memory destabilization. But, it remains unclear which molecular pathways are involved in CaMKII‐induced memory destabilization. Here, we propose two possible mechanisms by which CaMKII can regulate memory destabilization.
Table 1.
This table summarizes evidences that CaMKII activity regulates memory destabilization
| Authors (year) | Journal | Brain region | CaMKII manipulation | Behavioral phenotype |
|---|---|---|---|---|
| Cao et al. (2008) | Neuron | Forebrain | Over‐expression of αCaMKII‐F86G transgene and specific inhibition of αCaMKII‐F86G kinase activity | Retrieval‐dependent erasure of tone and contextual fear memory because of over‐expression; Phenotype reversed by blocking αCaMKII‐F86G ATP‐binding site |
| Jarome et al. (2016) | Neurobiol. Learn. Mem. | Amygdala | Pharmacological blocking of CaMKII with AIP | No effect on contextual fear memory alone; Rescued retrieval‐dependent fear memory impairment induced by protein synthesis blocking, suggesting blocking of memory destabilization by CaMKII blocking |
| Rossetti et al. (2017) | Neuron | Hippocampi | Viral‐induced expression of αCaMKII T286D/T305A/T306A | Impairment of previously learned place‐avoidance memory |
| Vigil et al. (2017) | Sci. Rep. | Dorsal‐hippocampi | Viral‐induced knock‐down of CaMKII endogenous inhibitor, CaMK2N1 | Retrieval‐dependent impairment of contextual fear memory |
CaMKII, memory destabilization and GluN2B
Memory destabilization was first associated with NMDAR activity by Ben Mamou et al. (2006). The authors show that pharmacological inhibition of NMDAR with intra‐basolateral amygdala injection of ifenprodil or AP5 prevented the retrieval‐dependent cued fear memory impairment that was induced by protein synthesis inhibition. That is, NMDAR inhibition prior to the memory retrieval session eliminated the necessity for memory restabilization, as memory destabilization was diminished.
Using more specific pharmacological tools, Milton et al. (2013) have suggested that within the basolateral amygdala, the regulation of destabilization and restabilization are dissociated. While memory destabilization is regulated by activation of NMDAR subunit GluN2B, memory restabilization is regulated by NMDAR subunit GluN2A. Milton et al. (2013) did not observe any change in auditory fear memory after specific pharmacological inhibition of GluN2B activity, but GluN2B inhibition prevented the memory impairment induced by blocking protein synthesis after memory retrieval. Hence, GluN2B inhibition impairs fear memory destabilization. In contrast, injection of a GluN2A‐prefferring antagonist, NVP‐AAM077, reduces freezing behavior after reactivation much like protein synthesis inhibition.
A GluN2B‐induced memory destabilization was also later described by Crestani et al. (2015) and Ferrer Monti et al. (2016). Both used a distractor stimulus to erase memory in a memory retrieval‐dependent matter. Crestani et al. (2015) used an air puff during retrieval as a distractor to induce contextual fear memory impairment. This impairment could be blocked by intra‐CA1 injection of the GluN2B antagonist ifenprodil, prior to retrieval session. Ferrer Monti et al. (2016) used sucrose solution after memory retrieval to erase contextual fear memory. Memory erasure was blocked by injection of ifenprodil in the basolateral amygdala. Interestingly, Ferrer Monti et al. (2016) confirmed that his behavioral protocol induced memory reconsolidation by using i.p. injections of midazolam, a fast‐acting enhancer of GABA‐A receptor activation that has previously been shown to disrupt memory reconsolidation (Bustos et al. 2006; Robinson and Franklin 2010; De Oliveira Alvares et al. 2013; Pineyro et al. 2013). Crestani et al. (2015), on the other hand, tested if the exposure of a rat to a different environment would induce memory erasure, and it did not, confirming the specificity and necessity of memory retrieval for memory erasure. Therefore, both articles further support the existence of a GluN2B‐induced memory destabilization mechanism.
CaMKII is known to bind to the GluN2B subunit (Strack and Colbran 1998) in a process that regulates synaptic plasticity (Barria and Malinow 2005; Zhou et al. 2007) and is necessary for memory formation (Zhou et al. 2007; Halt et al. 2012; Stein et al. 2014). CaMKII binding to NMDAR increases CaMKII activity by facilitating CaMKII T286/T287 autophosphorylation and inhibiting its dephosphorylation (Lisman and Raghavachari 2015). CaMKII complex can bind in to two different sites of GluN2B. One site is dependent on CaMKII's association with CaM (within residues 1120–1480), and the second binding site depends on CaMKII T286 phosphorylation (residues 839–1120) (Bayer et al. 2001, 2006). Moreover, the inhibition of CaMKII kinase activity by AIP treatment of hippocampal‐neuronal culture and hippocampal slices reduces GluN2B colocalization with PSD‐95 within the synapses (Gardoni et al. 2009). Hence, it is possible that CaMKII activity increases the synaptic levels of GluN2B, increasing memory destabilization after memory retrieval.
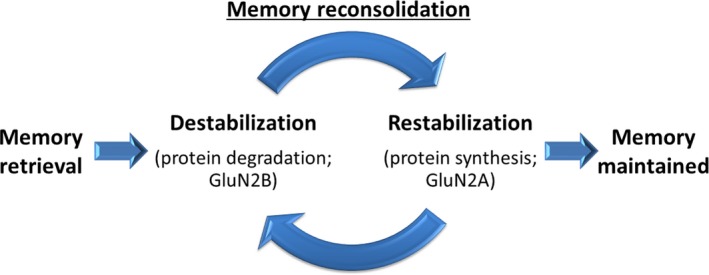
If CaMKII activity induces memory destabilization, one could predict that its inhibition is necessary for the maintenance of the memory after retrieval. In other words, excessive CaMKII activation might result in memory impairment because of excessive destabilization. Corroborating with this hypothesis, we have recently reported a contextual fear memory retrieval‐induced hippocampal expression of CaMK2N1, and this expression was necessary for memory maintenance after retrieval (Vigil et al. 2017). CaMK2N1 is known to block CaMKII binding to GluN2B (Vest et al. 2007). Thus, retrieval‐induced expression of CaMK2N1 could stop memory destabilization by blocking CaMKII interaction with GluN2B. Figure 2 is a schematic view of how CaMKII activity might induce memory destabilization via regulation of GluN2B synaptic levels and how this process would be stopped by retrieval‐induced CaMK2N1 expression. To further test this hypothesis and to understand how GluN2B activity regulates memory destabilization, more experiments are necessary.
Figure 2.
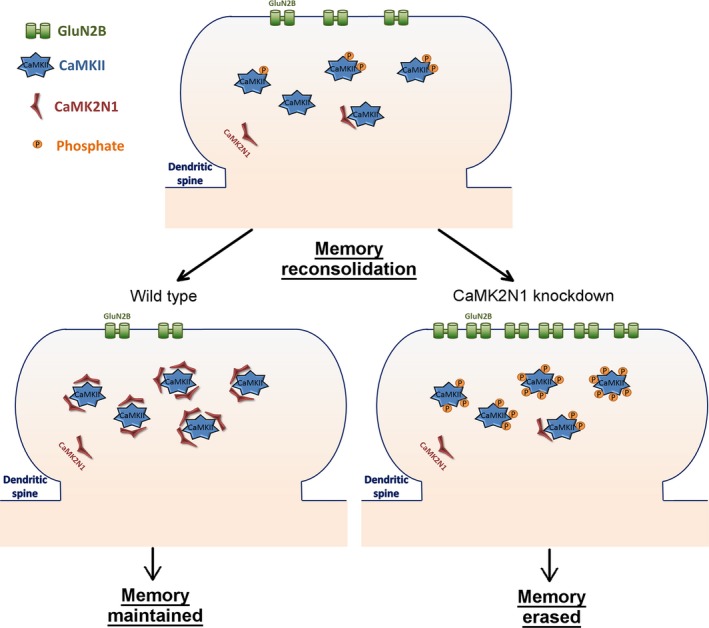
This schematic representation shows that CaMKII regulates the levels of GluN2B‐containing NMDAR in the synapse after retrieval, affecting the maintenance of a memory. This hypothesis could explain the observations of Vigil et al. (2017). Once memory is retrieved, CaMKII increases GluN2B localization within the synapse, starting a memory destabilization process. In normal (wild‐type) animals, this process is stopped by expression of CaMK2N1, which inhibits CaMKII and blocks the GluN2B‐induced memory destabilization. On the other hand, in Vigil et al. (2017), CaMK2N1 knockdown caused memory erasure because of excessive GluN2B‐induced memory destabilization resulting from the uncontrolled CaMKII activity and consequent increase in synaptic levels of GluN2B. GluN2B increase could be related to anchoring of extrasynaptic GluN2B in the post‐synaptic density (PSD) and/or decrease in GluN2B degradation. The mechanism is still unknown.
CaMKII, memory destabilization and protein degradation
Another possible mechanism of how CaMKII might play a role in memory destabilization is via regulation of UPS‐dependent protein degradation. It has been observed that post‐retrieval inhibition of CaMKII stops retrieval‐induced protein degradation and rescues memory impairment resulting from protein synthesis inhibition (Jarome et al. 2016). Autophosphorylation of T286 increases αCaMKII's affinity for the proteasome and promotes proteasome recruitment to the PSD (Bingol et al. 2010). CaMKII can also phosphorylate serine 120 of proteasome subunit Rpt6 and increase its activity (Djakovic et al. 2009; Jarome et al. 2013). The phosphorylation of Rpt6 seems to decrease synaptic strength by impairing miniature excitatory post‐synaptic current (Djakovic et al. 2012). CaMKII also phosphorylates the protein cylindromatosis. Once phosphorylated, cylindromatosis is activated and facilitates proteasomal degradation of proteins by removing K63‐linked polyubiquitins from targeted proteins (Thein et al. 2014). Thus, CaMKII activation increases protein degradation by incrementing proteasome activity, by anchoring it in the PSD area and by facilitating access to target proteins. This regulation of protein degradation by CaMKII is probable related to the retrieval‐induced CaMKII‐dependent proteasome activation reported by Jarome et al. (2016) (Figure 3). Furthermore, the retrieval‐dependent memory impairment observed after transgenic CaMKII over‐expression in Cao et al. (2008) experiments, could also be explained by uncontrolled activation of protein degradation. Nonetheless, a direct link between the memory maintenance impairment induced by αCaMKII‐F89G over‐expression and UPS protein degradation is still to be shown.
Figure 3.
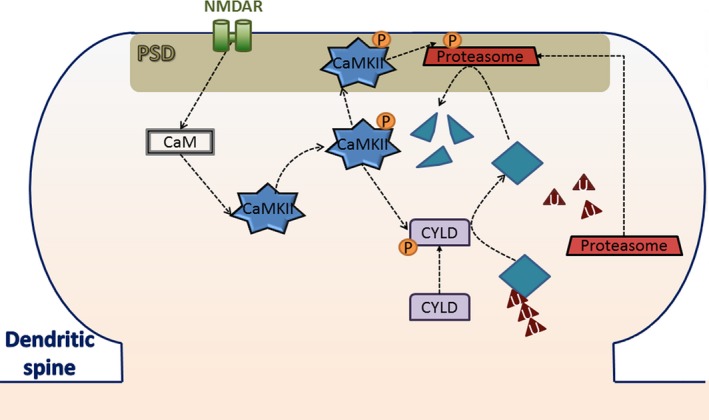
Schematic representation of how CaMKII can regulate ubiquitin proteasome system (UPS) activity and increase memory destabilization by increasing protein degradation. Calcium coming from open NMDAR binds with calmodulin creating the CaM complex that activates CaMKII. Once active, CaMKII autophosphorylates threonine 286, further increasing its activity. Active CaMKII phosphorylates cylindromatosis (CYLD), which activates this enzyme. Active CYLD removes K63‐linked polyubiquitins from proteins, targeting them for degradation. CaMKII also phosphorylates proteasome subunit Rpt6 on serine 120, increasing its activation. Active CaMKII also increases proteasome localization in the PSD. Such CaMKII/UPS pathway can help explain the behavioral phenotypes observed by Jarome et al. (2016) and Cao et al. (2008). Additionally, it is in accordance with the retrieval‐induced decrease in Shank levels, observed in Lee et al. (2008), and GluA1 (Vigil et al. 2017) levels in the post‐synaptic density.
Although the results of Jarome et al. (2016) clearly support the existence of a role of CaMKII in memory destabilization by regulation of UPS protein degradation, it does not establish a definitive role for CaMKII in memory destabilization. AIP, the inhibitor of CaMKII used, belongs to a family of CaMKII inhibitory peptides designed based on T286 autophosphorylation site of αCaMKII. These peptides are fragments of the T286 area, but with a substitution of the threonine to alanine (Hanson et al. 1989; Braun and Schulman 1995a; Ishida et al. 1995; Pellicena and Schulman 2014). The specificity of such substrate‐based inhibitors of CaMKII is still a matter of debate. They have also been reported to inhibit protein kinase D1 (PKD1) (Backs et al. 2009) and protein kinase C (Smith et al. 1990; Hvalby et al. 1994).
A more specific inhibitor of CaMKII has been used by Naskar et al. (2014), the inhibitory peptide CaMKIINtide. By inhibiting CaMKII with CaMKIINtide treatment in Lymnaea stagnalis snail model, Naskar et al. (2014) described a memory consolidation impairment that could be rescued by proteasome inhibition. CaMKIINtide treatment also inhibited αCaMKII T305 autophosphorylation and decreased the levels of the α‐amino‐3‐hydroxy‐5‐methyl‐4‐isoxazolepropionic acid receptor subunit GluA1. The GluA1 decrease was rescued by proteasome inhibition (Naskar et al. 2014). CaMKIINtide is derived from the endogenous inhibitor CaMK2N1, and so far, it has been shown to block CaMKII kinase activity specifically (Chang et al. 1998; Vest et al. 2007). CaMK2N1‐derived peptides have also been reported to reduce levels of CaMKII at the synapse (Sanhueza et al. 2011), inhibit T305 autophosphorylation (Vest et al. 2007), block binding to Densin (Jiao et al. 2011) and decrease clustering of CaMKII in the dendrites (Tao‐Cheng et al. 2013). Although the study in Naskar et al. (2014) has used a more specific tool, they tested the role of CaMKII regulation for protein degradation in memory consolidation. Nevertheless, like reconsolidation, consolidation also induces a wave of UPS‐dependent protein degradation (Lopez‐Salon et al. 2001; Artinian et al. 2008; Jarome et al. 2011; Jarome and Helmstetter 2014). Similar mechanisms might be used in both consolidation‐ and reconsolidation‐induced protein degradation.
Conclusion
The role of CaMKII in memory maintenance has always been a matter of debate (Lisman 1994; Irvine et al. 2005; Buard et al. 2010; Lucchesi et al. 2011; Sanhueza and Lisman 2013; Rossetti et al. 2017). Here, we have gathered pieces of evidence suggesting that CaMKII may play a role in reconsolidation‐induced memory destabilization, where CaMKII activation facilitates memory destabilization after retrieval. Cao et al. (2008) presents the first evidence for CaMKII‐induced memory destabilization. Jarome et al. (2016) established the most direct link between CaMKII and memory destabilization, identifying protein degradation as a molecular pathway involved. Vigil et al. (2017) supports the role of CaMK2N1 as a physiological mechanism by which CaMKII‐induced memory destabilization can be controlled. Additionally, reduction in CaMKII S331 phosphorylation could be responsible for initiating CaMKII‐induced memory destabilization (Rich et al. 2016). Finally, Rossetti et al. (2017) also observed that an increase in hippocampal CaMKII activity could lead to memory impairment. Although these observations suggest that CaMKII activation induces memory destabilization, none of these observations provides definitive evidence. Jarome et al. (2016) uses a pharmacological tool that is limited by its unspecific activity. Vigil et al. (2017) and Cao et al. (2008) report the occurrence of a retrieval‐induced memory erasure, but lack the direct link with a biological marker of memory destabilization. Example of these markers would be changes in S120 Rpt6 proteasome phosphorylation (Djakovic et al. 2009, 2012; Jarome et al. 2013, 2016) and decrease in the levels of MOV10 (Jarome et al. 2011) or in the synaptic levels of Shank (Lee 2008; Jarome et al. 2011). Rich et al. (2016) failed to study any behavioral phenotype resulting from specific manipulation of S331 phosphorylation. The retrieval dependence of the behavioral phenotype observed by Rossetti et al. (2017) was not tested. Consequently, experiments employing refined specific tools to manipulate and quantify memory destabilization and CaMKII activity, levels and localization are necessary.
Here, we propose two possible mechanisms by which CaMKII may regulate memory destabilization. It is possible that CaMKII controls memory destabilization via regulation of synaptic levels of GluN2B and/or via the regulation of protein degradation in the synapse. These two mechanisms can also be linked or interact with one another. The UPS activity pathway is a more direct link between CaMKII and memory destabilization, and has a larger body of evidence supporting it. The CaMKII/GluN2B pathway proposed here has never been tested and lacks the essential understanding of how GluN2B regulates memory destabilization. Aside from the involvement of Ca2+ influx, which is mediated by L‐type voltage‐gated calcium channels (Crestani et al. 2015), not much is known about the mechanism.
The hypothesis that memory destabilization is induced via a CaMKII‐dependent mechanism is not without apparent controversy. Da Silva et al. (2013) advocates a role for CaMKII in reconsolidation, more specifically, in the restabilization process. Da Silva et al. (2013) observed that hippocampal CaMKII inhibition by AIP after spatial memory retrieval induces memory impairment, which was rescued by inhibiting protein degradation. This memory impairment phenotype was time‐dependent, not present 24 h after AIP treatment but present 5 days after. So, the phenotype observed by Da Silva et al. (2013) was interpreted as an indication that CaMKII activity is necessary for memory restabilization. The AIP‐induced behavioral phenotype reported by Da Silva et al. (2013) and Jarome et al. (2016) are quite different from each other. However, if we consider that CaMKII might be important in both memory destabilization and restabilization, we eliminate the controversy between these two different observations. While Jarome's manipulation of CaMKII could have affected memory destabilization, Da Silva's might have changed memory restabilization. It is also important to consider that Da Silva et al. (2013) uses a different behavioral paradigm, the MWM. The MWM paradigm is not the most conventional paradigm used to test memory reconsolidation, as memory formation requires several training trials spread throughout various days of training. Therefore, memory destabilization and restabilization will occur during training, making it impossible to confirm the retrieval dependence of the phenotype by using a memory retrieval free group. Still, the MWM is a very useful and important paradigm to test different aspects of memory and learning.
Similar to Da Silva et al. (2013), Rich et al. (2016) also observed that intra‐basolateral amygdala inhibition of CaMKII, after memory reactivation, impaired cocaine cued memory reconsolidation. CaMKII inhibition reduced the cocaine‐seeking behavior induced by presentation of previously paired stimulus (3 tone‐light stimulation). Nevertheless, for inhibition of CaMKII, Rich et al. (2016) applied bilateral‐injections of KN‐93 or KN‐62, which have been shown to be non‐specific inhibitors of CaMKII. For instance, they also inhibit the kinase activity of CaM‐dependent protein kinase IV (Redondo et al. 2010), ‘calmodulin kinase‐like vesicle‐associated’ (Mochizuki et al. 1993) and others (Wayman et al. 2008).
It is our opinion that CaMKII likely plays a role in memory destabilization. Consequently, CaMKII plays a role in memory maintenance, not as a ‘memory molecule’, but rather as biological substrate of memory reconsolidation. If this is the case, understanding how CaMKII regulates retrieval‐induced memory destabilization could have an enormous impact on the treatment of post‐traumatic stress disorder and addiction. We also do not refute or discard the notion that CaMKII may play a role in memory maintenance via other mechanisms like extinction (Szapiro et al. 2003; Kimura et al. 2008; de Carvalho Myskiw et al. 2014), memory restabilization (Da Silva et al. 2013) or even as a memory molecule (Rossetti et al. 2017).
More studies are necessary to properly dissect the roles of CaMKII on memory maintenance. It is of paramount importance to include a retrieval free group in order to test the dependence of any memory phenotype to memory retrieval. The use of gene therapy treatments to knockdown, knockin or knockout specific genes can yield rich observations on the functions of CaMKII in memory maintenance. Pharmacological tools derived from the endogenous inhibitor CaMK2N1, like CaMKIINtide (Chang et al. 1998), are preferable because of specificity. The CaMKIINtide has been fused to the trans‐acting activator of transcription (tat) domain, increasing cell penetration and creating the 21‐amino acid peptide, tatCN21 (Vest et al. 2007; Buard et al. 2010). One can also find shorter versions like CN19 (Coultrap and Bayer 2011) and the 17‐amino acid CN17 (Gomez‐Monterrey et al. 2013), which have been shown to work as effectively as CN21. Transgenic animals like the T286A mutant have always been and continue to be important models for studying the roles of CaMKII in memory (Giese et al. 1998; Rossetti et al. 2017). Still, the use of inducible mutations needs to be explored further, as it avoids long‐term plasticity compensations that might bias observations. Finally, investigating the role of CaMKII in different brain areas, as well as the effect of CaMKII manipulation at different time points after the process of learning and retrieval will require a collaborative, long and challenging effort from many researchers.
Acknowledgments and conflict of interest disclosure
We thank the Brazilian National Council for Scientific and Technological Development (CNPq), as well as the British MRC for their generous support. We also thank Liliana Espinoza for reviewing this scientific paper. The authors declare that they have no conflict of interests.
References
- Artinian J., McGauran A. M., De Jaeger X., Mouledous L., Frances B. and Roullet P. (2008) Protein degradation, as with protein synthesis, is required during not only long‐term spatial memory consolidation but also reconsolidation. Eur. J. Neurosci. 27, 3009–3019. [DOI] [PubMed] [Google Scholar]
- Backs J., Backs T., Neef S. et al (2009) The delta isoform of CaM kinase II is required for pathological cardiac hypertrophy and remodeling after pressure overload. Proc. Natl Acad. Sci. USA 106, 2342–2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barria A. and Malinow R. (2005) NMDA receptor subunit composition controls synaptic plasticity by regulating binding to CaMKII. Neuron 48, 289–301. [DOI] [PubMed] [Google Scholar]
- Bayer K. U., De Koninck P., Leonard A. S., Hell J. W. and Schulman H. (2001) Interaction with the NMDA receptor locks CaMKII in an active conformation. Nature 411, 801–805. [DOI] [PubMed] [Google Scholar]
- Bayer K. U., LeBel E., McDonald G. L., O'Leary H., Schulman H. and De Koninck P. (2006) Transition from reversible to persistent binding of CaMKII to postsynaptic sites and NR2B. J. Neurosci. 26, 1164–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben Mamou C., Gamache K. and Nader K. (2006) NMDA receptors are critical for unleashing consolidated auditory fear memories. Nat. Neurosci. 9, 1237–1239. [DOI] [PubMed] [Google Scholar]
- Bennett M. K., Erondu N. E. and Kennedy M. B. (1983) Purification and characterization of a calmodulin‐dependent protein kinase that is highly concentrated in brain. J. Biol. Chem. 258, 12735–12744. [PubMed] [Google Scholar]
- Bingol B., Wang C. F., Arnott D., Cheng D., Peng J. and Sheng M. (2010) Autophosphorylated CaMKIIalpha acts as a scaffold to recruit proteasomes to dendritic spines. Cell 140, 567–578. [DOI] [PubMed] [Google Scholar]
- Braun A. P. and Schulman H. (1995a) The multifunctional calcium/calmodulin‐dependent protein kinase: from form to function. Annu. Rev. Physiol. 57, 417–445. [DOI] [PubMed] [Google Scholar]
- Braun A. P. and Schulman H. (1995b) A non‐selective cation current activated via the multifunctional Ca(2 + )‐calmodulin‐dependent protein kinase in human epithelial cells. J. Physiol. 488(Pt 1), 37–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronstein J. M., Wasterlain C. G. and Farber D. B. (1988) A retinal calmodulin‐dependent kinase: calcium/calmodulin‐stimulated and ‐inhibited states. J. Neurochem. 50, 1438–1446. [DOI] [PubMed] [Google Scholar]
- Buard I., Coultrap S. J., Freund R. K., Lee Y. S., Dell'Acqua M. L., Silva A. J. and Bayer K. U. (2010) CaMKII “autonomy” is required for initiating but not for maintaining neuronal long‐term information storage. J. Neurosci. 30, 8214–8220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bustos S. G., Maldonado H. and Molina V. A. (2006) Midazolam disrupts fear memory reconsolidation. Neuroscience 139, 831–842. [DOI] [PubMed] [Google Scholar]
- Cao X., Wang H., Mei B., An S., Yin L., Wang L. P. and Tsien J. Z. (2008) Inducible and selective erasure of memories in the mouse brain via chemical‐genetic manipulation. Neuron 60, 353–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Carvalho Myskiw J., Furini C. R., Benetti F. and Izquierdo I. (2014) Hippocampal molecular mechanisms involved in the enhancement of fear extinction caused by exposure to novelty. Proc. Natl Acad. Sci. USA 111, 4572–4577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang B. H., Mukherji S. and Soderling T. R. (1998) Characterization of a calmodulin kinase II inhibitor protein in brain. Proc. Natl Acad. Sci. USA 95, 10890–10895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao L. H., Stratton M. M., Lee I. H. et al (2011) A mechanism for tunable autoinhibition in the structure of a human Ca2 + /calmodulin‐ dependent kinase II holoenzyme. Cell 146, 732–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng D., Hoogenraad C. C., Rush J. et al (2006) Relative and absolute quantification of postsynaptic density proteome isolated from rat forebrain and cerebellum. Mol. Cell Proteomics 5, 1158–1170. [DOI] [PubMed] [Google Scholar]
- Colbran R. J., Smith M. K., Schworer C. M., Fong Y. L. and Soderling T. R. (1989) Regulatory domain of calcium/calmodulin‐dependent protein kinase II. Mechanism of inhibition and regulation by phosphorylation. J. Biol. Chem. 264, 4800–4804. [PubMed] [Google Scholar]
- Cooke S. F., Wu J., Plattner F. et al (2006) Autophosphorylation of alphaCaMKII is not a general requirement for NMDA receptor‐dependent LTP in the adult mouse. J. Physiol. 574, 805–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coultrap S. J. and Bayer K. U. (2011) Improving a natural CaMKII inhibitor by random and rational design. PLoS ONE 6, e25245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coultrap S. J. and Bayer K. U. (2012) CaMKII regulation in information processing and storage. Trends Neurosci. 35, 607–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crestani A. P., Zacouteguy Boos F., Haubrich J., Ordonez Sierra R., Santana F., Molina J. M., Cassini Lde F., Alvares Lde O. and Quillfeldt J. A. (2015) Memory reconsolidation may be disrupted by a distractor stimulus presented during reactivation. Sci. Rep. 5, 13633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Da Silva W. C., Cardoso G., Bonini J. S., Benetti F. and Izquierdo I. (2013) Memory reconsolidation and its maintenance depend on L‐voltage‐dependent calcium channels and CaMKII functions regulating protein turnover in the hippocampus. Proc. Natl Acad. Sci. USA 110, 6566–6570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Oliveira Alvares L., Crestani A. P., Cassini L. F., Haubrich J., Santana F. and Quillfeldt J. A. (2013) Reactivation enables memory updating, precision‐keeping and strengthening: exploring the possible biological roles of reconsolidation. Neuroscience 244, 42–48. [DOI] [PubMed] [Google Scholar]
- Djakovic S. N., Schwarz L. A., Barylko B., DeMartino G. N. and Patrick G. N. (2009) Regulation of the proteasome by neuronal activity and calcium/calmodulin‐dependent protein kinase II. J. Biol. Chem. 284, 26655–26665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djakovic S. N., Marquez‐Lona E. M., Jakawich S. K., Wright R., Chu C., Sutton M. A. and Patrick G. N. (2012) Phosphorylation of Rpt6 regulates synaptic strength in hippocampal neurons. J. Neurosci. 32, 5126–5131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberg M., Kobilo T., Berman D. E. and Dudai Y. (2003) Stability of retrieved memory: inverse correlation with trace dominance. Science 301, 1102–1104. [DOI] [PubMed] [Google Scholar]
- Elgersma Y., Fedorov N. B., Ikonen S., Choi E. S., Elgersma M., Carvalho O. M., Giese K. P. and Silva A. J. (2002) Inhibitory autophosphorylation of CaMKII controls PSD association, plasticity, and learning. Neuron 36, 493–505. [DOI] [PubMed] [Google Scholar]
- Erondu N. E. and Kennedy M. B. (1985) Regional distribution of type II Ca2 + /calmodulin‐dependent protein kinase in rat brain. J. Neurosci. 5, 3270–3277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer Monti R. I., Giachero M., Alfei J. M., Bueno A. M., Cuadra G. and Molina V. A. (2016) An appetitive experience after fear memory destabilization attenuates fear retention: involvement GluN2B‐NMDA receptors in the Basolateral Amygdala Complex. Learn Mem 23, 465–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii H., Inoue M., Okuno H., Sano Y., Takemoto‐Kimura S., Kitamura K., Kano M. and Bito H. (2013) Nonlinear decoding and asymmetric representation of neuronal input information by CaMKIIalpha and calcineurin. Cell Rep. 3, 978–987. [DOI] [PubMed] [Google Scholar]
- Fukushima H., Zhang Y., Archbold G., Ishikawa R., Nader K. and Kida S. (2014) Enhancement of fear memory by retrieval through reconsolidation. Elife 3, e02736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaertner T. R., Kolodziej S. J., Wang D., Kobayashi R., Koomen J. M., Stoops J. K. and Waxham M. N. (2004) Comparative analyses of the three‐dimensional structures and enzymatic properties of alpha, beta, gamma and delta isoforms of Ca2 + ‐calmodulin‐dependent protein kinase II. J. Biol. Chem. 279, 12484–12494. [DOI] [PubMed] [Google Scholar]
- Gardoni F., Mauceri D., Malinverno M. et al (2009) Decreased NR2B subunit synaptic levels cause impaired long‐term potentiation but not long‐term depression. J. Neurosci. 29, 669–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giese K. P., Fedorov N. B., Filipkowski R. K. and Silva A. J. (1998) Autophosphorylation at Thr286 of the alpha calcium‐calmodulin kinase II in LTP and learning. Science 279, 870–873. [DOI] [PubMed] [Google Scholar]
- Gomez‐Monterrey I., Sala M., Rusciano M. R. et al (2013) Characterization of a selective CaMKII peptide inhibitor. Eur. J. Med. Chem. 62, 425–434. [DOI] [PubMed] [Google Scholar]
- Gozal E., Gozal D., Pierce W. M. et al (2002) Proteomic analysis of CA1 and CA3 regions of rat hippocampus and differential susceptibility to intermittent hypoxia. J. Neurochem. 83, 331–345. [DOI] [PubMed] [Google Scholar]
- Grant P. A., Best S. L., Sanmugalingam N., Alessio R., Jama A. M. and Torok K. (2008) A two‐state model for Ca2 + /CaM‐dependent protein kinase II (alphaCaMKII) in response to persistent Ca2 + stimulation in hippocampal neurons. Cell Calcium 44, 465–478. [DOI] [PubMed] [Google Scholar]
- Halt A. R., Dallapiazza R. F., Zhou Y. et al (2012) CaMKII binding to GluN2B is critical during memory consolidation. EMBO J. 31, 1203–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson P. I. and Schulman H. (1992) Inhibitory autophosphorylation of multifunctional Ca2 + /calmodulin‐dependent protein kinase analyzed by site‐directed mutagenesis. J. Biol. Chem. 267, 17216–17224. [PubMed] [Google Scholar]
- Hanson P. I., Kapiloff M. S., Lou L. L., Rosenfeld M. G. and Schulman H. (1989) Expression of a multifunctional Ca2 + /calmodulin‐dependent protein kinase and mutational analysis of its autoregulation. Neuron 3, 59–70. [DOI] [PubMed] [Google Scholar]
- Hanson P. I., Meyer T., Stryer L. and Schulman H. (1994) Dual role of calmodulin in autophosphorylation of multifunctional CaM kinase may underlie decoding of calcium signals. Neuron 12, 943–956. [DOI] [PubMed] [Google Scholar]
- Hell J. W. (2014) CaMKII: claiming center stage in postsynaptic function and organization. Neuron 81, 249–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoelz A., Nairn A. C. and Kuriyan J. (2003) Crystal structure of a tetradecameric assembly of the association domain of Ca2 + /calmodulin‐dependent kinase II. Mol. Cell 11, 1241–1251. [DOI] [PubMed] [Google Scholar]
- Hudmon A. and Schulman H. (2002) Neuronal CA2 + /calmodulin‐dependent protein kinase II: the role of structure and autoregulation in cellular function. Annu. Rev. Biochem. 71, 473–510. [DOI] [PubMed] [Google Scholar]
- Hvalby O., Hemmings H. C., Jr , Paulsen O. et al (1994) Specificity of protein kinase inhibitor peptides and induction of long‐term potentiation. Proc. Natl Acad. Sci. USA 91, 4761–4765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irvine E. E., Vernon J. and Giese K. P. (2005) AlphaCaMKII autophosphorylation contributes to rapid learning but is not necessary for memory. Nat. Neurosci. 8, 411–412. [DOI] [PubMed] [Google Scholar]
- Irvine E. E., von Hertzen L. S., Plattner F. and Giese K. P. (2006) alphaCaMKII autophosphorylation: a fast track to memory. Trends Neurosci. 29, 459–465. [DOI] [PubMed] [Google Scholar]
- Irvine E. E., Danhiez A., Radwanska K., Nassim C., Lucchesi W., Godaux E., Ris L. and Giese K. P. (2011) Properties of contextual memory formed in the absence of alphaCaMKII autophosphorylation. Mol. Brain 4, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida A., Kameshita I., Okuno S., Kitani T. and Fujisawa H. (1995) A novel highly specific and potent inhibitor of calmodulin‐dependent protein kinase II. Biochem. Biophys. Res. Commun. 212, 806–812. [DOI] [PubMed] [Google Scholar]
- Jarome T. J. and Helmstetter F. J. (2014) Protein degradation and protein synthesis in long‐term memory formation. Front Mol. Neurosci. 7, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarome T. J., Werner C. T., Kwapis J. L. and Helmstetter F. J. (2011) Activity dependent protein degradation is critical for the formation and stability of fear memory in the amygdala. PLoS ONE 6, e24349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarome T. J., Kwapis J. L., Ruenzel W. L. and Helmstetter F. J. (2013) CaMKII, but not protein kinase A, regulates Rpt6 phosphorylation and proteasome activity during the formation of long‐term memories. Front. Behav. Neurosci. 7, 115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarome T. J., Ferrara N. C., Kwapis J. L. and Helmstetter F. J. (2016) CaMKII regulates proteasome phosphorylation and activity and promotes memory destabilization following retrieval. Neurobiol. Learn. Mem. 128, 103–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji J. and Maren S. (2008) Differential roles for hippocampal areas CA1 and CA3 in the contextual encoding and retrieval of extinguished fear. Learn Mem 15, 244–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao Y., Jalan‐Sakrikar N., Robison A. J., Baucum A. J., 2nd , Bass M. A. and Colbran R. J. (2011) Characterization of a central Ca2 + /calmodulin‐dependent protein kinase IIalpha/beta binding domain in densin that selectively modulates glutamate receptor subunit phosphorylation. J. Biol. Chem. 286, 24806–24818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josselyn S. A., Kohler S. and Frankland P. W. (2017) Heroes of the engram. J. Neurosci. 37, 4647–4657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly A., Laroche S. and Davis S. (2003) Activation of mitogen‐activated protein kinase/extracellular signal‐regulated kinase in hippocampal circuitry is required for consolidation and reconsolidation of recognition memory. J. Neurosci. 23, 5354–5360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura R., Silva A. J. and Ohno M. (2008) Autophosphorylation of alphaCaMKII is differentially involved in new learning and unlearning mechanisms of memory extinction. Learn Mem 15, 837–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolodziej S. J., Hudmon A., Waxham M. N. and Stoops J. K. (2000) Three‐dimensional reconstructions of calcium/calmodulin‐dependent (CaM) kinase IIalpha and truncated CaM kinase IIalpha reveal a unique organization for its structural core and functional domains. J. Biol. Chem. 275, 14354–14359. [DOI] [PubMed] [Google Scholar]
- Langston R. F., Stevenson C. H., Wilson C. L., Saunders I. and Wood E. R. (2010) The role of hippocampal subregions in memory for stimulus associations. Behav. Brain Res. 215, 275–291. [DOI] [PubMed] [Google Scholar]
- Lee J. L. (2008) Memory reconsolidation mediates the strengthening of memories by additional learning. Nat. Neurosci. 11, 1264–1266. [DOI] [PubMed] [Google Scholar]
- Lee J. L., Everitt B. J. and Thomas K. L. (2004) Independent cellular processes for hippocampal memory consolidation and reconsolidation. Science 304, 839–843. [DOI] [PubMed] [Google Scholar]
- Lee S. H., Choi J. H., Lee N., Lee H. R., Kim J. I., Yu N. K., Choi S. L., Kim H. and Kaang B. K. (2008) Synaptic protein degradation underlies destabilization of retrieved fear memory. Science 319, 1253–1256. [DOI] [PubMed] [Google Scholar]
- Lee S. J., Escobedo‐Lozoya Y., Szatmari E. M. and Yasuda R. (2009) Activation of CaMKII in single dendritic spines during long‐term potentiation. Nature 458, 299–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leestemaker Y. and Ovaa H. (2017) Tools to investigate the ubiquitin proteasome system. Drug Discov Today Technol. 26, 25–31. [DOI] [PubMed] [Google Scholar]
- Lengyel I., Voss K., Cammarota M., Bradshaw K., Brent V., Murphy K. P., Giese K. P., Rostas J. A. and Bliss T. V. (2004) Autonomous activity of CaMKII is only transiently increased following the induction of long‐term potentiation in the rat hippocampus. Eur. J. Neurosci. 20, 3063–3072. [DOI] [PubMed] [Google Scholar]
- Lisman J. (1994) The CaM kinase II hypothesis for the storage of synaptic memory. Trends Neurosci. 17, 406–412. [DOI] [PubMed] [Google Scholar]
- Lisman J. and Raghavachari S. (2015) Biochemical principles underlying the stable maintenance of LTP by the CaMKII/NMDAR complex. Brain Res. 1621, 51–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisman J., Schulman H. and Cline H. (2002) The molecular basis of CaMKII function in synaptic and behavioural memory. Nat. Rev. Neurosci. 3, 175–190. [DOI] [PubMed] [Google Scholar]
- Lisman J., Yasuda R. and Raghavachari S. (2012) Mechanisms of CaMKII action in long‐term potentiation. Nat. Rev. Neurosci. 13, 169–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez‐Salon M., Alonso M., Vianna M. R., Viola H., Mello e Souza T., Izquierdo I., Pasquini J. M. and Medina J. H. (2001) The ubiquitin‐proteasome cascade is required for mammalian long‐term memory formation. Eur. J. Neurosci. 14, 1820–1826. [DOI] [PubMed] [Google Scholar]
- Lucchesi W., Mizuno K. and Giese K. P. (2011) Novel insights into CaMKII function and regulation during memory formation. Brain Res. Bull. 85, 2–8. [DOI] [PubMed] [Google Scholar]
- Marsden K. C., Beattie J. B., Friedenthal J. and Carroll R. C. (2007) NMDA receptor activation potentiates inhibitory transmission through GABA receptor‐associated protein‐dependent exocytosis of GABA(A) receptors. J. Neurosci. 27, 14326–14337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merlo E., Milton A. L., Goozee Z. Y., Theobald D. E. and Everitt B. J. (2014) Reconsolidation and extinction are dissociable and mutually exclusive processes: behavioral and molecular evidence. J. Neurosci. 34, 2422–2431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer T., Hanson P. I., Stryer L. and Schulman H. (1992) Calmodulin trapping by calcium‐calmodulin‐dependent protein kinase. Science 256, 1199–1202. [DOI] [PubMed] [Google Scholar]
- Miller S. G., Patton B. L. and Kennedy M. B. (1988) Sequences of autophosphorylation sites in neuronal type II CaM kinase that control Ca2(+)‐independent activity. Neuron 1, 593–604. [DOI] [PubMed] [Google Scholar]
- Milton A. L., Merlo E., Ratano P., Gregory B. L., Dumbreck J. K. and Everitt B. J. (2013) Double dissociation of the requirement for GluN2B‐ and GluN2A‐containing NMDA receptors in the destabilization and restabilization of a reconsolidating memory. J. Neurosci. 33, 1109–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misanin J. R., Miller R. R. and Lewis D. J. (1968) Retrograde amnesia produced by electroconvulsive shock after reactivation of a consolidated memory trace. Science 160, 554–555. [DOI] [PubMed] [Google Scholar]
- Mochizuki H., Ito T. and Hidaka H. (1993) Purification and characterization of Ca2 + /calmodulin‐dependent protein kinase V from rat cerebrum. J. Biol. Chem. 268, 9143–9147. [PubMed] [Google Scholar]
- Mockett B. G., Guevremont D., Wutte M., Hulme S. R., Williams J. M. and Abraham W. C. (2011) Calcium/calmodulin‐dependent protein kinase II mediates group I metabotropic glutamate receptor‐dependent protein synthesis and long‐term depression in rat hippocampus. J. Neurosci. 31, 7380–7391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherji S. and Soderling T. R. (1994) Regulation of Ca2 + /calmodulin‐dependent protein kinase II by inter‐ and intrasubunit‐catalyzed autophosphorylations. J. Biol. Chem. 269, 13744–13747. [PubMed] [Google Scholar]
- Myers K. M. and Davis M. (2007) Mechanisms of fear extinction. Mol. Psychiatry 12, 120–150. [DOI] [PubMed] [Google Scholar]
- Myers J. B., Zaegel V., Coultrap S. J., Miller A. P., Bayer K. U. and Reichow S. L. (2017) The CaMKII holoenzyme structure in activation‐competent conformations. Nat. Commun. 8, 15742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nader K. (2003) Memory traces unbound. Trends Neurosci. 26, 65–72. [DOI] [PubMed] [Google Scholar]
- Nader K., Schafe G. E. and Le Doux J. E. (2000) Fear memories require protein synthesis in the amygdala for reconsolidation after retrieval. Nature 406, 722–726. [DOI] [PubMed] [Google Scholar]
- Naskar S., Wan H. and Kemenes G. (2014) pT305‐CaMKII stabilizes a learning‐induced increase in AMPA receptors for ongoing memory consolidation after classical conditioning. Nat. Commun. 5, 3967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Need A. C. and Giese K. P. (2003) Handling and environmental enrichment do not rescue learning and memory impairments in alphaCamKII(T286A) mutant mice. Genes Brain Behav. 2, 132–139. [DOI] [PubMed] [Google Scholar]
- Pape H. C. and Pare D. (2010) Plastic synaptic networks of the amygdala for the acquisition, expression, and extinction of conditioned fear. Physiol. Rev. 90, 419–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlov I. P. (1927) Conditioned Reflexes: an Investigation of the Physiological Activity of the Cerebral Cortex. Oxford University Press, London. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedreira M. E. and Maldonado H. (2003) Protein synthesis subserves reconsolidation or extinction depending on reminder duration. Neuron 38, 863–869. [DOI] [PubMed] [Google Scholar]
- Pellicena P. and Schulman H. (2014) CaMKII inhibitors: from research tools to therapeutic agents. Front Pharmacol. 5, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng J., Kim M. J., Cheng D., Duong D. M., Gygi S. P. and Sheng M. (2004) Semiquantitative proteomic analysis of rat forebrain postsynaptic density fractions by mass spectrometry. J. Biol. Chem. 279, 21003–21011. [DOI] [PubMed] [Google Scholar]
- Pineyro M. E., Ferrer Monti R. I., Alfei J. M., Bueno A. M. and Urcelay G. P. (2013) Memory destabilization is critical for the success of the reactivation‐extinction procedure. Learn Mem 21, 46–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quirk G. J. and Mueller D. (2008) Neural mechanisms of extinction learning and retrieval. Neuropsychopharmacology 33, 56–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redondo R. L., Okuno H., Spooner P. A., Frenguelli B. G., Bito H. and Morris R. G. (2010) Synaptic tagging and capture: differential role of distinct calcium/calmodulin kinases in protein synthesis‐dependent long‐term potentiation. J. Neurosci. 30, 4981–4989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rich R. C. and Schulman H. (1998) Substrate‐directed function of calmodulin in autophosphorylation of Ca2 + /calmodulin‐dependent protein kinase II. J. Biol. Chem. 273, 28424–28429. [DOI] [PubMed] [Google Scholar]
- Rich M. T., Abbott T. B., Chung L. et al (2016) Phosphoproteomic analysis reveals a novel mechanism of CaMKIIalpha regulation Inversely induced by cocaine memory extinction versus reconsolidation. J. Neurosci. 36, 7613–7627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson M. J. and Franklin K. B. (2010) Reconsolidation of a morphine place preference: impact of the strength and age of memory on disruption by propranolol and midazolam. Behav. Brain Res. 213, 201–207. [DOI] [PubMed] [Google Scholar]
- Rosenberg O. S., Deindl S., Sung R. J., Nairn A. C. and Kuriyan J. (2005) Structure of the autoinhibited kinase domain of CaMKII and SAXS analysis of the holoenzyme. Cell 123, 849–860. [DOI] [PubMed] [Google Scholar]
- Rossetti T., Banerjee S., Kim C., Leubner M., Lamar C., Gupta P., Lee B., Neve R. and Lisman J. (2017) Memory erasure experiments indicate a critical role of CaMKII in memory storage. Neuron 96(207–216), e202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanhueza M. and Lisman J. (2013) The CaMKII/NMDAR complex as a molecular memory. Mol. Brain 6, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanhueza M., Fernandez‐Villalobos G., Stein I. S., Kasumova G., Zhang P., Bayer K. U., Otmakhov N., Hell J. W. and Lisman J. (2011) Role of the CaMKII/NMDA receptor complex in the maintenance of synaptic strength. J. Neurosci. 31, 9170–9178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen K., Teruel M. N., Connor J. H., Shenolikar S. and Meyer T. (2000) Molecular memory by reversible translocation of calcium/calmodulin‐dependent protein kinase II. Nat. Neurosci. 3, 881–886. [DOI] [PubMed] [Google Scholar]
- Shuman T., Wood S. C. and Anagnostaras S. G. (2009) Modafinil and memory: effects of modafinil on Morris water maze learning and Pavlovian fear conditioning. Behav. Neurosci. 123, 257–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith M. K., Colbran R. J. and Soderling T. R. (1990) Specificities of autoinhibitory domain peptides for four protein kinases. Implications for intact cell studies of protein kinase function. J. Biol. Chem. 265, 1837–1840. [PubMed] [Google Scholar]
- Stein I. S., Donaldson M. S. and Hell J. W. (2014) CaMKII binding to GluN2B is important for massed spatial learning in the Morris water maze. F1000Res 3, 193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterneck E., Paylor R., Jackson‐Lewis V., Libbey M., Przedborski S., Tessarollo L., Crawley J. N. and Johnson P. F. (1998) Selectively enhanced contextual fear conditioning in mice lacking the transcriptional regulator CCAAT/enhancer binding protein delta. Proc. Natl Acad. Sci. USA 95, 10908–10913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strack S. and Colbran R. J. (1998) Autophosphorylation‐dependent targeting of calcium/calmodulin‐dependent protein kinase II by the NR2B subunit of the N‐methyl‐ D‐aspartate receptor. J. Biol. Chem. 273, 20689–20692. [DOI] [PubMed] [Google Scholar]
- Szapiro G., Vianna M. R., McGaugh J. L., Medina J. H. and Izquierdo I. (2003) The role of NMDA glutamate receptors, PKA, MAPK, and CAMKII in the hippocampus in extinction of conditioned fear. Hippocampus 13, 53–58. [DOI] [PubMed] [Google Scholar]
- Tao‐Cheng J. H., Yang Y., Bayer K. U., Reese T. S. and Dosemeci A. (2013) Effects of CaMKII inhibitor tatCN21 on activity‐dependent redistribution of CaMKII in hippocampal neurons. Neuroscience 244, 188–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thein S., Tao‐Cheng J. H., Li Y., Bayer K. U., Reese T. S. and Dosemeci A. (2014) CaMKII Mediates Recruitment and Activation of the Deubiquitinase CYLD at the Postsynaptic Density. PLoS ONE 9, e91312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobimatsu T. and Fujisawa H. (1989) Tissue‐specific expression of four types of rat calmodulin‐dependent protein kinase II mRNAs. J. Biol. Chem. 264, 17907–17912. [PubMed] [Google Scholar]
- Tonegawa S., Liu X., Ramirez S. and Redondo R. (2015) Memory engram cells have come of age. Neuron 87, 918–931. [DOI] [PubMed] [Google Scholar]
- Tzortzopoulos A. and Torok K. (2004) Mechanism of the T286A‐mutant alphaCaMKII interactions with Ca2 + /calmodulin and ATP. Biochemistry 43, 6404–6414. [DOI] [PubMed] [Google Scholar]
- Tzortzopoulos A., Best S. L., Kalamida D. and Torok K. (2004) Ca2 + /calmodulin‐dependent activation and inactivation mechanisms of alphaCaMKII and phospho‐Thr286‐alphaCaMKII. Biochemistry 43, 6270–6280. [DOI] [PubMed] [Google Scholar]
- Varshavsky A., Turner G., Du F. and Xie Y. (2000) Felix Hoppe‐Seyler lecture 2000. The ubiquitin system and the N‐end rule pathway. Biol. Chem. 381, 779–789. [DOI] [PubMed] [Google Scholar]
- Vest R. S., Davies K. D., O'Leary H., Port J. D. and Bayer K. U. (2007) Dual mechanism of a natural CaMKII inhibitor. Mol. Biol. Cell 18, 5024–5033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigil F. A., Mizuno K., Lucchesi W., Valls‐Comamala V. and Giese K. P. (2017) Prevention of long‐term memory loss after retrieval by an endogenous CaMKII inhibitor. Sci. Rep. 7, 4040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wayman G. A., Lee Y. S., Tokumitsu H., Silva A. J. and Soderling T. R. (2008) Calmodulin‐kinases: modulators of neuronal development and plasticity. Neuron 59, 914–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J., Rowan M. J. and Anwyl R. (2006) Long‐term potentiation is mediated by multiple kinase cascades involving CaMKII or either PKA or p42/44 MAPK in the adult rat dentate gyrus in vitro. J. Neurophysiol. 95, 3519–3527. [DOI] [PubMed] [Google Scholar]
- Zhou Y., Takahashi E., Li W. et al (2007) Interactions between the NR2B receptor and CaMKII modulate synaptic plasticity and spatial learning. J. Neurosci. 27, 13843–13853. [DOI] [PMC free article] [PubMed] [Google Scholar]