

Abstract
BACKGROUND
Juvéderm Vollure XC (VYC-17.5L) belongs to a family of nonanimal hyaluronic acid (HA) gels based on the Vycross technology platform.
OBJECTIVE
To evaluate the safety and effectiveness of VYC-17.5L for correction of moderate to severe nasolabial folds (NLFs) compared with a control HA dermal filler.
METHODS
In this double-blind study, 123 adults with 2 moderate or severe NLFs as measured on the 5-point photonumeric NLF severity scale (NLFSS) were randomized to VYC-17.5L in 1 NLF and control in the contralateral NLF. The coprimary effectiveness end points at Month 6 were difference in improvement in mean NLFSS score for VYC-17.5L versus control and NLFSS responder rate (≥1-point improvement) for VYC-17.5L.
RESULTS
The coprimary effectiveness end points were met. NLFSS scores improved by 1.4 with VYC-17.5L and by 1.3 with control; NLFSS responder rate with VYC-17.5L was 93.2%. More than three-quarters of subjects (82%) treated with VYC-17.5L were very satisfied at Month 6. Investigators reported that VYC-17.5L was smoother and more natural looking and easier to inject and mold than control. VYC-17.5L resulted in significantly fewer severe injection site responses than control.
CONCLUSION
VYC-17.5L was safe and effective for correcting moderate to severe NLFs, with results lasting through 6 months in 93% of subjects.
Nasolabial folds (NLFs) are caused by loss of deep fat and subsequent loss of muscle contour in the midface, leading to sagging and formation of wrinkles and folds.1–3 Nasolabial folds can be treated successfully with hyaluronic acid (HA) dermal gels, which provide volume to the targeted area, reduce the appearance of folds, and restore the natural 3-dimensional contour of the treated region.4,5
Juvéderm Vollure XC (VYC-17.5L; Allergan plc, Dublin, Ireland) belongs to a family of nonanimal HA versatile, highly moldable gels based on the Vycross technology platform (Allergan plc), which combines low– and high–molecular-weight HA to improve the cross-linking efficiency of the HA chains. The tightly cross-linked HA network yields a higher viscosity gel with greater lift capacity and improved response durability. These features result in a filler that achieves a natural look using a lower HA concentration while being less hygroscopic.6 VYC-17.5L contains lidocaine to make the injection process more comfortable for the patient and reduce the need for conventional anesthetics.7,8 The safety and effectiveness of Juvéderm products for treating moderate to severe facial wrinkles and folds such as NLFs have been shown in multiple clinical studies.9–14
The present study evaluated the safety and effectiveness of VYC-17.5L for correction of moderate to severe NLFs compared with a control HA dermal filler through 6 months after treatment.
Methods
Study Design
This prospective, multicenter, randomized, within-subject controlled study was conducted at 6 sites in the United States; each site had an unblinded treating investigator (TI) along with a blinded evaluating investigator (EI) who performed all safety and effectiveness assessments. The total study duration was up to 20 months (1 month for treatment and up to 19 months of follow-up). This report presents data from a preplanned interim analysis, with results through 6 months for all subjects.
Eligible subjects were randomized to treatment with VYC-17.5L in either the right or left NLF and control in the contralateral NLF; the order of the injections (left or right side) was also randomized. Randomization was based on a central randomization schedule stratified by investigational site using an automated interactive voice/web response system accessed by the TI or study coordinator. Subjects received initial treatment according to the randomization schedule and, if judged necessary by the TI, an optional touch-up treatment could be administered 30 days later. Each product was administered via injection using a 30-gauge half-inch needle in accordance with each product's respective directions for use. The TI determined injection volumes based on clinical experience, with the total volume not to exceed 4 mL in each NLF for the initial and touch-up treatments combined. Subjects and EIs remained blinded to the treatment assignment for each NLF throughout the study duration.
The study was conducted in compliance with Good Clinical Practice guidelines and was registered at Clinicaltrials.gov (identifier: NCT01976663). A central institutional review board covering all investigational sites approved the protocol before any subjects were enrolled, and all subjects provided written informed consent before participation.
Subjects
Adult subjects (aged ≥18 years) with 2 fully visible NLFs were eligible if they had severity scores of 2 (moderate) for both NLFs or scores of 3 (severe) for both NLFs on the validated, 5-point photonumeric NLF Severity Scale (NLFSS) as assessed by the EI, and agreed to refrain from treatment with other antiwrinkle/volumizing agents in facial regions below the orbital rim for the study duration. Subjects were excluded if they had undergone facial tissue augmentation in the lower two-thirds of the face with dermal fillers within the previous 12 months or with fat or botulinum toxin injections within the previous 6 months; had undergone cosmetic facial procedures in the face or neck within the previous 6 months; or had received semipermanent fillers or permanent facial implants in the lower face. Subjects with an uncontrolled disease, active inflammation, infection, or lesions in the NLF area, or who had a tendency for developing hypertrophic scarring were also excluded. Females who were pregnant or nursing were ineligible to participate. Drugs known to increase coagulation time were withdrawn for 10 days prior to and 3 days after study treatment.
Assessments
Prior to treatment, EIs evaluated the severity of NLFs using the NLFSS (0 = none; 1 = mild; 2 = moderate; 3 = severe; 4 = extreme). Facial digital photography was performed before and at 60 minutes after each treatment. Treating investigators evaluated the ease of injection and the moldability of each product on 3-point scales (left side easier, both sides the same, right side easier). The subjects rated their procedural pain for each NLF (pain during injection) on an 11-point scale (0 = no pain; 10 = worst pain imaginable) and completed a 30-day safety diary after each treatment reporting any injection site responses (ISRs). Subjects recorded the severity of ISRs in the safety diary as mild, moderate, or severe.
Subjects returned at 3 and 14 days after treatment to undergo facial digital photography, to rate satisfaction and NLF preference, and to complete safety assessments. On Day 3, subjects completed the items on the Recovery Early Symptoms scale of the FACE-Q questionnaire, and EIs compared the smoothness of each NLF region using a 3-point scale (left side felt smoother; both sides felt equally smooth; right side felt smoother) and natural look of each NLF region using a 3-point scale (left side looked more natural; both sides looked equally natural; right side looked more natural). Assessments of subject satisfaction and NLF preference were repeated if optional touch-up treatment was administered on Day 30.
Routine follow-up visits for assessment of safety and effectiveness occurred at 1, 3, and 6 months after the last treatment (initial or touch-up). At each visit, EIs evaluated NLF severity and the natural look of NLF regions, and facial digital photography was performed. Subjects rated satisfaction with treatment for each NLF using an 11-point scale (0 = completely dissatisfied; 10 = completely satisfied) and rated NLF preference. In addition, subjects completed the Appraisal of Nasolabial Folds scale of the FACE-Q questionnaire for each NLF at screening and at Months 3 and 6.
Statistics
Effectiveness analyses were conducted on the modified intent-to-treat population, which included all randomized subjects who received study treatment. The coprimary effectiveness end points were the difference in mean improvement from baseline in EI-assessed NLFSS scores for VYC-17.5L versus control at Month 6, and the observed responder rate for VYC-17.5L at Month 6, defined as the proportion of subjects with at least a 1-point improvement from baseline on the NLFSS. The primary effectiveness analysis determined whether VYC-17.5L was noninferior to control in terms of improvement in mean NLFSS scores at Month 6, wherein 0.5 points was the prespecified margin of noninferiority, and whether the responder rate for VYC-17.5L was statistically significantly greater than 50% at Month 6. Because each subject received both products (1 in each NLF), statistical comparisons were made using paired data. A 1-sided 95% Wald confidence interval (CI) for the mean difference in improvement in NLFSS scores between VYC-17.5L versus control was constructed to test for noninferiority; a p-value was determined using the Wilcoxon signed-rank test. A 1-sided exact binomial test was used to evaluate whether the responder rate for VYC-17.5L at 6 months was significantly greater than 50% and to compare injection characteristics between the 2 products. The Benjamini–Hochberg method was used to correct for statistical multiplicity. Other effectiveness end points and safety parameters were analyzed descriptively.
Results
Subject Disposition
Of the 126 subjects enrolled, 123 were randomized and treated. Sixty-three subjects (51.2%) received optional touch-up treatment. For the Month 6 visit (primary time point), 117 subjects (95.1%) completed the visit within the analysis window.
Subject Characteristics and Treatment Administration
The study cohort was primarily female (95.1%) and white (74.0%), with a median age of 54 (Table 1). All Fitzpatrick skin phototypes were represented in the study, although Phototypes II and III were the most common (Table 1). Mean baseline NLFSS score was 2.6, consistent with the enrollment criteria of moderate or severe NLFs. Median daily sunlight exposure was 1 hour, and most subjects (69.9%) had never used tobacco products.
TABLE 1.
Baseline Characteristics

In preparation for initial treatment, one or more types of anesthesia were administered to most subjects (82.1%; 101/123), most commonly a topical anesthetic (82.2%; 83/101) for a mean of 41 minutes; nerve block was administered to 17.8% (18/101) of subjects. During touch-up, all treated NLFs received a topical anesthetic for a mean of 32 minutes for VYC-17.5L and 31 minutes for control. The median volume injected for the combined initial treatment and touch-up was 1.7 mL for each product (Table 2). A serial puncture technique was used in both NLFs in over 90% of subjects, with tunneling used in 50% of subjects, fanning in over 30% of subjects, and cross-hatching in 16% of subjects.
TABLE 2.
Treatment Administration

At the initial treatment, the TI reported that VYC-17.5L and control had the same ease of injection and moldability in just 8.1% of subjects (10/123). Of the remaining 113 subjects, VYC-17.5L was significantly easier to inject and easier to mold (p < .001) in 73.5% of subjects (83/113).
Effectiveness
The coprimary effectiveness end points were met at Month 6. Improvement in NLF severity was seen with VYC-17.5L and control: mean NLFSS scores improved by 1.4 in the NLFs treated with VYC-17.5L and by 1.3 in the NLFs treated with control (p = .097). The difference between treatments in mean NLFSS scores was 0.1, and the lower 95% CI limit for this difference was −0.02, which was greater than the prespecified margin of −0.5. The NLFSS responder rate at Month 6 with VYC-17.5L was 93.2% (109/117), which was statistically greater than 50% on the 1-sided exact binomial test (p < .001). Figure 1 shows photographs representative of the treatment effect, with severe NLFs at baseline and improvements evident at Month 6.
Figure 1.
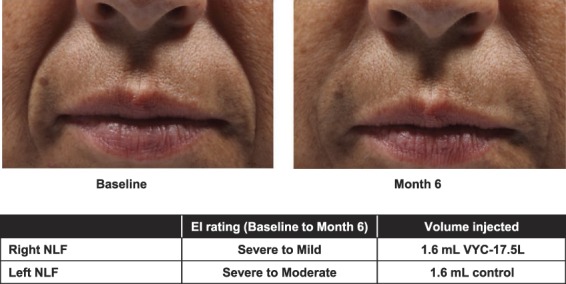
Representative photographs of a subject's NLFs at baseline and Month 6. EI, evaluating investigator; NLFs, nasolabial folds.
When assessed by time point after the last treatment, NLFSS responder rates were numerically higher with VYC-17.5L than with control at Months 1 and 6 (Figure 2). For the FACE-Q Appraisal of Nasolabial Folds, the mean score increased from 32 at baseline to 73 at Month 6 in subjects treated with VYC17.5L (Figure 3). At Month 6, the mean (SD) improvement from baseline in FACE-Q score was 40.6 (23.8) with VYC-17.5L and 37.8 (23.6) with control. On the FACE-Q question of how much subjects were bothered by the depth of their NLFs, there was a dramatic reduction from 87.0% (107/123 subjects) of subjects reporting moderately or extremely bothered at baseline to 13.7% (16/117 subjects) at Month 6 after VYC-17.5L treatment.
Figure 2.
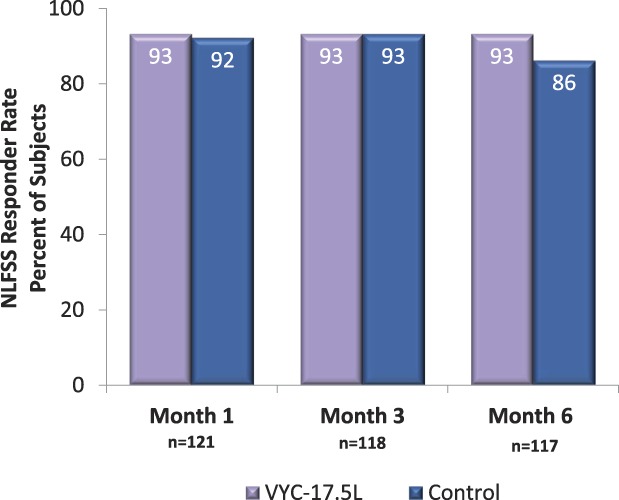
Nasolabial Fold Severity Scale responder rates based on EI assessment after treatment with VYC-17.5L and control by study visit. EI, evaluating investigator; NLFSS, Nasolabial Fold Severity Scale.
Figure 3.
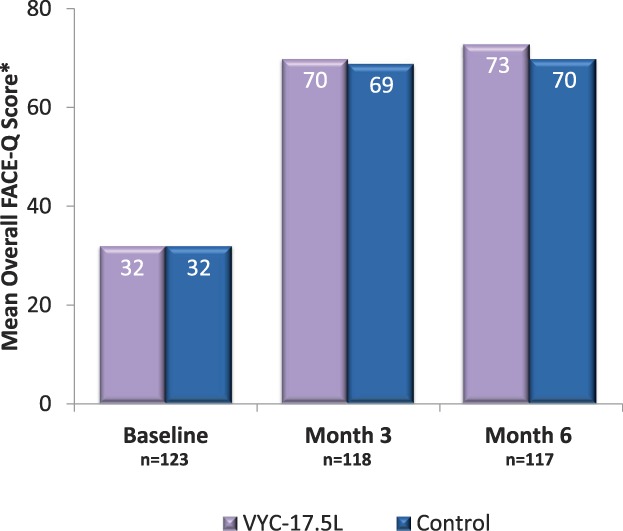
Overall appraisal of NLF FACE-Q score after treatment with VYC-17.5L and control by study visit. *Subject responses to the 5 FACE-Q questions were combined into an overall score for the NLF ranging from 0 (subject is extremely bothered by appearance of the NLF) to 100 (subject is not at all bothered by the appearance of the NLF) using the scale developers' scoring algorithm. NLF, nasolabial fold.
The EI evaluated the smoothness of each NLF at Day 3 after initial treatment: a difference was found in 91 of 121 subjects (75.2%), with VYC-17.5L rated smoother in 65 subjects (71.4%) and control rated smoother in 26 subjects (28.6%). From Day 3 to Month 6, a difference in the natural look of each NLF region was reported in 77%–85% of subjects, with VYC-17.5L providing a more natural look in twice as many subjects as control at all time points through Month 6 (Figure 4).
Figure 4.
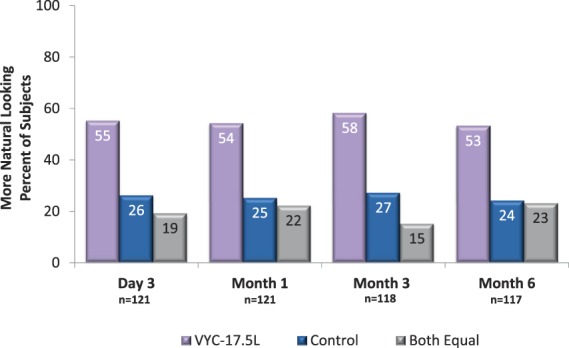
Evaluating investigator assessments of natural look, by visit.
Subjects reported a high level of satisfaction with VYC-17.5L through Month 6 (Figure 5). When subjects evaluated NLF preference for overall treatment outcome, 70.2% (85/121) reported a preference at Day 3, and 59.8% (70/117) reported a preference at Month 6. Among the subjects who expressed a preference, a numerical preference for VYC-17.5L over control was evident at Day 3 (70.6% vs 29.4%) and remained evident at Month 6 (62.9% vs 37.1%).
Figure 5.
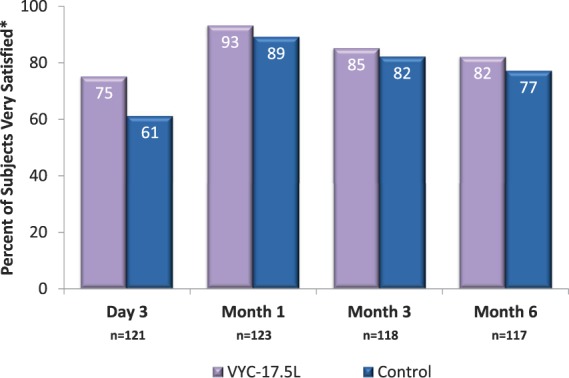
Subject satisfaction with treatment. *Shown are the percentage of subjects with scores of 7 through 10 on an 11-point scale (0 = completely dissatisfied; 10 = completely satisfied).
The primary effectiveness end point of NLFSS mean change from baseline was also evaluated for subgroups with Fitzpatrick skin Phototypes I and II, III and IV, and V and VI. Results of a 1-way ANOVA test of improvement in NLF severity by skin phototype at Month 6 showed that skin phototype had no impact on the effectiveness of VYC-17.5L or control.
Safety
Subjects assessed their pain immediately after completion of initial and touch-up injections of each NLF using an 11-point scale (0 = no pain; 10 = worst pain imaginable). Mean scores for procedural pain were 2.3 for VYC-17.5L and control on initial treatment, and 2.2 for VYC-17.5L and 2.3 for control on touch-up treatment.
Most subjects reported ISRs after initial treatment (Table 3). Rates were lower in all ISR categories after treatment with VYC-17.5L than with control, with rates at least 5 percentage points lower for swelling, tenderness, lumps/bumps, redness, pain after injection, and itching. The incidence of ISRs after touch-up treatment was lower than after initial treatment, and again was lower with VYC-17.5L than with control. The severity of ISRs was notably lower with VYC-17.5L compared with control. The incidence of severe ISRs was lower after initial treatment with VYC-17.5L (31.0%; 95% CI: 22.8%–40.3%) than with control (58.3%; 95% CI: 49.0%–67.3%); the nonoverlapping CIs indicate that the difference between treatments was significant. Severe ISRs were less common with VYC-17.5L than control in all categories except for itching, with a clear separation evident for the most common categories of firmness, swelling, tenderness to touch, and lumps/bumps. Severe ISRs were also less common after touch-up with VYC-17.5L than with control (20.0% vs 35.8%). Most ISRs resolved within 2 weeks of initial and touch-up treatments with each product. Injection site responses that were ongoing at the end of the diary entries were automatically considered to be adverse events (AEs).
TABLE 3.
Incidence of Injection Site Responses After Initial Treatment

Adverse events were reported by the EI for 29 NLFs (23.6%) with VYC-17.5L and for 27 NLFs (22.0%) with control. The most common AEs for both products were injection site induration (diary term firmness, 10.6% and 8.9%, respectively), injection site mass (diary term lumps/bumps, 7.3% and 7.3%), and injection site swelling (7.3% and 7.3%). All other AEs occurred at rates of less than 5%. Most AEs at both NLFs resolved within 60 days, and few required any treatment. Most AEs were mild or moderate; severe AEs were reported for 6 NLFs (4.9%) treated with VYC-17.5L and 10 NLFs (8.1%) treated with control. None of the severe AEs required treatment or were considered serious by the EI. No serious AEs or deaths related to treatment were reported. Adverse events that did not occur at NLFs were typically events common in the general population, such as nasopharyngitis or headache, and were generally considered unrelated to study treatment.
Subjects completed the FACE-Q Recovery Early Symptoms scale on Day 3 after treatment. After initial treatment, the majority of subjects reported feeling not at all bothered or a little bothered by the 17 symptoms listed on the FACE-Q scale. The proportion of subjects who reported feeling not at all bothered or a little bothered was 15 or more percentage points higher for VYC-17.5L compared with control on 4 questions: discomfort (90.1% vs 74.4%), tenderness (88.4% vs 69.4%), feeling sore (89.3% vs 71.1%), and swelling (84.9% vs 60.0%). Subjects reported feeling moderately bothered approximately 3 times less often after VYC-17.5L than with control on 6 questions: discomfort (5.8% vs 20.7%), tenderness (7.4% vs 23.1%), feeling sore (5.8% vs 22.3%), feeling bruised (5.8% vs 15.3%), swelling (10.1% vs 30.8%), and feeling that face is tight (5.0% vs 15.7%).
A subgroup analysis by Fitzpatrick skin phototype showed some small differences between subgroups in ISRs and AEs, but the overall results suggest that skin phototype has no effect on the safety of the products.
Discussion
The results of this study show that VYC-17.5L was safe and effective for correction of NLFs. This study met both coprimary end points, indicating that the effects of VYC-17.5L were noninferior to those of control and produced an NLFSS responder rate that was significantly greater than 50% at Month 6 (p < .001). The data for subject preference for overall treatment outcomes and for subject satisfaction with treatment provide further support of the effectiveness of VYC-17.5L for correction of moderate to severe NLFs.
Treating investigators felt that VYC-17.5L was easier to inject and mold compared with control. Furthermore, advantages in both smoother and more natural looking results were noted for VYC-17.5L versus control. These results suggest that VYC-17.5L may provide technological and aesthetic benefits.
The safety assessments demonstrated that both VYC-17.5L and control were safe and tolerable. Procedural pain was generally minimal with both products. Common ISRs, such as firmness, swelling, and tenderness to touch, were reported at lower rates with VYC-17.5L than with control. Moreover, there were significantly fewer subject-rated severe ISRs reported with VYC-17.5L, particularly firmness, swelling, tenderness to touch, and lumps/bumps. Adverse events at the NLFs were reported at similar rates with both treatments, and were most commonly injection site induration, mass, and swelling (corresponding to diary reports of ISRs of firmness, lumps/bumps, and swelling). Adverse events at other sites were generally unrelated to treatment and consistent with those expected in a general population. Results from the FACE-Q Early Symptoms scale showed that subjects reported fewer bothersome symptoms, such as tenderness, feeling sore, and swelling, after treatment with VYC-17.5L compared with control, suggesting that subjects may have recovered more quickly after VYC-17.5L.
The results reported herein are consistent with previous studies of the safety and effectiveness of other Juvéderm products for correction of NLFs.9–13 The current data are from a 6-month analysis of a longer-duration study. In studies presenting 6- or 7-month data, improvements from baseline in NLF severity either were similar for the HA filler in question and the control filler15–17 or showed greater effectiveness versus the control filler.18 A more complete understanding of the duration of effect of VYC-17.5L will be possible when the final results are available. Additionally, although EIs were blinded to treatment, the TIs were not blinded. The possibility that this may have influenced the way in which they treated each NLF or their assessments of product characteristics cannot be determined.
In conclusion, the results of this study demonstrate that VYC-17.5L is safe and effective for correcting moderate to severe NLFs, with treatment benefits lasting at least 6 months in 93% of subjects.
Acknowledgments
The authors express their appreciation to all treating and evaluating investigators who participated in this study and to Shraddha Mehta, PhD of Allergan plc for conducting the statistical analyses. Treating investigators were K.B. (West Palm Beach, FL); Fredric Brandt, MD and Joely Kauffman-Janette, MD (Coral Gables, FL); Steven H. Dayan, MD (Chicago, IL); P.E.G. (Los Angeles, CA); G.M. (Birmingham, AL); and Corey Maas, MD, FACS (San Francisco, CA). Evaluating investigators were Richard Schwartz, MD (West Palm Beach, FL); Christopher O'Connell, MD (Coral Gables, FL); Otto Placik, MD (Chicago, IL); Rajesh Chopra, MD, FACS (Los Angeles, CA); Melanie L. Appell, MD (Birmingham, AL); and Vic A. Narurkar, MD (San Francisco, CA).
Footnotes
This study was funded by Allergan plc, Dublin, Ireland. Editorial support for this article was provided by Peloton Advantage, Parsippany, New Jersey, and was funded by Allergan plc.
G. Monheit is an investigator for Allergan plc, Galderma, Alphaeon, and Teoxane and is a consultant for Allergan plc, Galderma, Suneva, and Merz. K. Beer is a clinical trial investigator, consultant, and speaker for Allergan plc, Galderma, and Merz. He is a shareholder in Anterios and a partner in The Cosmetic Bootcamp and Theraplex LLC. P.E. Grimes is an investigator for Allergan plc, Suneva, and Alphaeon, and is a consultant for Procter & Gamble. B. Hardas, V. Lin, and D.K. Murphy were employees and stockholders of Allergan plc at the time of the study. B.M. Weichman provided medical writing assistance at the request of the authors, which was funded by Allergan plc.
References
- 1.Gierloff M, Stohring C, Buder T, Gassling V, et al. Aging changes of the midfacial fat compartments: a computed tomographic study. Plast Reconstr Surg 2012;129:263–73. [DOI] [PubMed] [Google Scholar]
- 2.Le Louarn C, Buthiau D, Buis J. Structural aging: the facial recurve concept. Aesthet Plast Surg 2007;31:213–8. [DOI] [PubMed] [Google Scholar]
- 3.Ezure T, Amano S. Involvement of upper cheek sagging in nasolabial fold formation. Skin Res Technol 2012;18:259–64. [DOI] [PubMed] [Google Scholar]
- 4.Beasley KL, Weiss MA, Weiss RA. Hyaluronic acid fillers: a comprehensive review. Facial Plast Surg 2009;25:86–94. [DOI] [PubMed] [Google Scholar]
- 5.Fitzgerald R, Graivier MH, Kane M, Lorenc ZP, et al. Appropriate selection and application of nonsurgical facial rejuvenation agents and procedures: panel consensus recommendations. Aesthet Surg J 2010;30(Suppl1):36S–45S. [DOI] [PubMed] [Google Scholar]
- 6.Eccleston D, Murphy DK. Juvéderm (R) Volbella in the perioral area: a 12-month prospective, multicenter, open-label study. Clin Cosmet Investig Dermatol 2012;5:167–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Raspaldo H, De Boulle K, Levy PM. Longevity of effects of hyaluronic acid plus lidocaine facial filler. J Cosmet Dermatol 2010;9:11–5. [DOI] [PubMed] [Google Scholar]
- 8.Gassia V, Raspaldo H, Niforos FR, Michaud T. Global 3-dimensional approach to natural rejuvenation: recommendations for perioral, nose, and ear rejuvenation. J Cosmet Dermatol 2013;12:123–36. [DOI] [PubMed] [Google Scholar]
- 9.Baumann LS, Shamban AT, Lupo MP, Monheit GD, et al. Comparison of smooth-gel hyaluronic acid dermal fillers with cross-linked bovine collagen: a multicenter, double-masked, randomized, within-subject study. Dermatol Surg 2007;33(Suppl 2):S128–35. [DOI] [PubMed] [Google Scholar]
- 10.Pinsky MA, Thomas JA, Murphy DK, Walker PS. Juvéderm injectable gel: a multicenter, double-blind, randomized study of safety and effectiveness. Aesthet Surg J 2008;28:17–23. [DOI] [PubMed] [Google Scholar]
- 11.Lupo MP, Smith SR, Thomas JA, Murphy DK, et al. Effectiveness of Juvederm Ultra Plus dermal filler in the treatment of severe nasolabial folds. Plast Reconstr Surg 2008;121:289–97. [DOI] [PubMed] [Google Scholar]
- 12.Grimes PE, Thomas JA, Murphy DK. Safety and effectiveness of hyaluronic acid fillers in skin of color. J Cosmet Dermatol 2009;8:162–8. [DOI] [PubMed] [Google Scholar]
- 13.Smith SR, Jones D, Thomas JA, Murphy DK, et al. Duration of wrinkle correction following repeat treatment with Juvederm hyaluronic acid fillers. Arch Dermatol Res 2010;302:757–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weinkle SH, Bank DE, Boyd CM, Gold MH, et al. A multi-center, double-blind, randomized controlled study of the safety and effectiveness of Juvéderm injectable gel with and without lidocaine. J Cosmet Dermatol 2009;8:205–10. [DOI] [PubMed] [Google Scholar]
- 15.Nast A, Reytan N, Hartmann V, Pathirana D, et al. Efficacy and durability of two hyaluronic acid-based fillers in the correction of nasolabial folds: results of a prospective, randomized, double-blind, actively controlled clinical pilot study. Dermatol Surg 2011;37:768–75. [DOI] [PubMed] [Google Scholar]
- 16.Rzany B, Bayerl C, Bodokh I, Boineau D, et al. Efficacy and safety of a new hyaluronic acid dermal filler in the treatment of moderate nasolabial folds: 6-month interim results of a randomized, evaluator-blinded, intra-individual comparison study. J Cosmet Laser Ther 2011;13:107–12. [DOI] [PubMed] [Google Scholar]
- 17.Wu Y, Xu J, Jia Y, Murphy DK. Safety and effectiveness of hyaluronic acid injectable gel in correcting moderate nasolabial folds in Chinese subjects. J Drugs Dermatol 2016;15:70–6. [PubMed] [Google Scholar]
- 18.Ascher B, Bayerl C, Brun P, Kestemont P, et al. Efficacy and safety of a new hyaluronic acid dermal filler in the treatment of severe nasolabial lines - 6-month interim results of a randomized, evaluator-blinded, intra-individual comparison study. J Cosmet Dermatol 2011;10:94–8. [DOI] [PubMed] [Google Scholar]