

Ponatinib, a cancer drug, inhibits occurrence and growth of cerebral cavernous malformation in mouse models.
Abstract
Cerebral cavernous malformation (CCM) is a common cerebrovascular disease that can occur sporadically or be inherited. They are major causes of stroke, cerebral hemorrhage, and neurological deficits in the younger population. Loss-of-function mutations in three genes, CCM1, CCM2, and CCM3, have been identified as the cause of human CCMs. Currently, no drug is available to treat CCM disease. Hyperactive mitogen-activated protein kinase kinase Kinase 3 (MEKK3) kinase signaling as a consequence of loss of CCM genes is an underlying cause of CCM lesion development. Using a U.S. Food and Drug Administration–approved kinase inhibitor library combined with virtual modeling and biochemical and cellular assays, we have identified a clinically approved small compound, ponatinib, that is capable of inhibiting MEKK3 activity and normalizing expression of downstream kruppel-like factor (KLF) target genes. Treatment with this compound in neonatal mouse models of CCM can prevent the formation of new CCM lesions and reduce the growth of already formed lesions. At the ultracellular level, ponatinib can normalize the flattening and disorganization of the endothelium caused by CCM deficiency. Collectively, our study demonstrates ponatinib as a novel compound that may prevent CCM initiation and progression in mouse models through inhibition of MEKK3-KLF signaling.
INTRODUCTION
Cerebral cavernous malformations (CCMs) are common vascular malformations in the brain, with a prevalence of ~0.5% in the human population (1, 2). CCMs manifest as clusters of thin-walled dilated vessels in the venous vascular beds of the central nervous system. CCM lesions can lead to hemorrhagic rupture that cause stroke, or their growth places pressure on the surrounding neuronal tissues and result in neurological deficits. Currently, there is no available drug treatment for CCM disease, and high-risk surgical resection remains the only option for patients with this life-threatening condition.
Loss-of-function mutations in one of three genes, CCM1 (also known as KRIT1), CCM2, and CCM3 (also known as PDCD10), cause human familial CCM (2). CCM genes encode nonhomologous cytoplasmic proteins, forming a single signaling complex (3–5). Recent studies reveal that this CCM protein complex acts as a suppressor of the MEKK3 [also known as MAP3K3 (mitogen-activated protein kinase kinase kinase 3)] kinase cascade. Deletions of CCM genes lead to activation of the MEKK3 kinase cascade and increased expression of downstream targets such as KLF2/4 and Adamts4/5 (6, 7). Through developmental studies using mouse and zebrafish models, we have previously found a novel CCM-MEKK3-KLF-Adamts signaling axis in endothelial cells that controls cardiovascular development (7). Further studies in mouse models of CCM diseases confirmed that this signaling pathway is conserved in CCM pathogenesis. Genetic ablation of MEKK3-KLF signaling can prevent CCM lesion formation in mouse models (6, 8). These results suggest that pharmacological inhibition of the MEKK3-KLF signaling pathway may be a promising approach to treat CCM disease.
In this study, we have identified ponatinib (AP24534), a U.S. Food and Drug Administration (FDA)–approved kinase inhibitor currently used to treat cancer, as an effective inhibitor of MEKK3-KLF signaling. Ponatinib can prevent CCM lesion initiation and progression in mouse models of CCM.
RESULTS
Ponatinib directly inhibits MEKK3 kinase activity
Loss of CCM genes increases the expression KLF2/4 and their target genes as a result of increased MEKK3 activity (6, 7, 9, 10). This suggests that hyperactive MEKK3 is a key component in the pathway leading to CCM lesion development. MEKK3 belongs to the Map3k family and shares the highest similarity with MEKK2 [94% conservation in their catalytic domains (11)]. Both are MAPK kinases that activate the extracellular signal–regulated kinase 5 (ERK5) pathway by phosphorylating and activating the MAPK kinase MEK5 (12, 13). Compounds that can inhibit MEKK2 at the nanomolar scale have recently been reported (14, 15).
To test whether these inhibitors can also repress MEKK3 kinase activity, we performed compound-protein modeling based on their identified crystal structures, as well as in vitro kinase assays using purified MEKK3 and its interacting partner and kinase substrate, MEK5. Ponatinib is a type II kinase inhibitor that binds kinases in their ASP-Phe-Gly (DFG)–out conformation (16). On the basis of the previously solved crystal structures of the ponatinib–fibroblast growth factor receptor 4 (FGFR4) complex (17), the ponatinib-Abl complex (16), and a structurally solved DFG-out kinase P21-activated kinase 1 (PAK1) (18), which is highly homologous to the MEKK3 kinase domain, computer modeling revealed that ponatinib can bind to the MEKK3 kinase domain in the adenosine 5′-triphosphate (ATP) binding pocket with hydrogen bonds and π-π interaction (Fig. 1, A to C, and figs. S1 and S2).
Fig. 1. Ponatinib directly inhibits MEKK3 kinase activity.
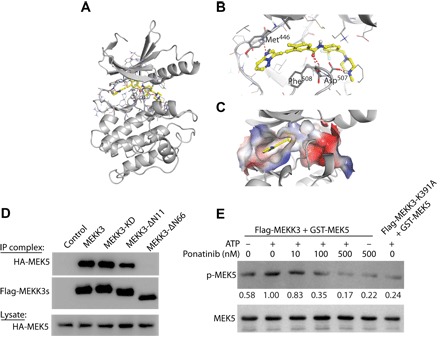
(A to C) Computer modeling demonstrating that ponatinib can bind with the MEKK3 kinase domain in the ATP-binding pocket with hydrogen-bonds and π-π interaction (color) as demonstrated by a ribbon plot with full-length proteins (A) and the ribbon plot (B) and surface plot (C) of the binding pocket. (D) Immunoprecipitation (IP) demonstrating robust MEKK3 and MEK5 interactions. Deletion of 66 amino acids at the N terminus (MEKK3-ΔN66) abolishes the interaction. Deletion of 11 amino acids at the N terminus (MEKK3-ΔN11) or kinase-dead MEKK3 (MEKK3-KD) do not affect MEKK3-MEK5 interaction. (E) In vitro kinase assays showing changes in MEK5 phosphorylation following treatment with ponatinib. MEKK3-K391A, a kinase-dead mutant, was included as a negative control. The quantification of p-MEK5/MEK5 density ratios was shown between the blots. Results are representative of three independent experiments.
Our immunoprecipitation studies confirmed direct interaction between MEKK3 and MEK5 (Fig. 1D). Deletion of the N-terminal 11–amino acids (MEKK3-ΔN11) or kinase-dead MEKK3 (MEKK3-KD) had no effect on MEKK3-MEK5 binding. In contrast, deletion of MEKK3’s N-terminal 66 amino acids (MEKK3-ΔN66) completely abolished the MEKK3-MEK5 interaction. These results agree with previous reports that binding between MEKK3 and MEK5 is dependent on interactions between their respective PB-1 domains (Fig. 1D) (12). To determine whether ponatinib inhibits MEKK3 kinase activity by inhibiting MEK5 phosphorylation, we performed in vitro kinase assays using purified flag-MEKK3 and glutathione S-transferase (GST)–MEK5. Addition of ATP (1 mM) was able to increase p-MEK5 levels, while treatment with ponatinib dose-dependently reduced this increase in MEK5 phosphorylation. Notably, ponatinib treatment (500 nM) decreased p-MEK5 levels to baseline, similar to that in the reaction with the kinase-dead mutant of MEKK3 (flag-MEKK3-K391A) (Fig. 1E). These results indicate that ponatinib can directly inhibit MEKK3 kinase activity, as reflected by the inhibition of MEK5 phosphorylation.
Ponatinib inhibits MEKK3 signaling activation conferred by CCM deficiency
Loss of CCM genes in endothelial cells lead to CCM formation through elevated MEKK3 signaling (7). Whether ponatinib can rescue the CCM phenotype by inhibiting aberrant MEKK3 signaling is currently unknown. We knocked down CCM1 in human umbilical vein endothelial cells (HUVECs) using small interfering RNA (siRNA) (fig. S3A), and in agreement with our published data, loss of CCM1 increased the expression levels of MEKK3 downstream targets (Fig. 2, A to F) (7). Ponatinib had no effect on KLF2, eNOS, and ADAMTS1 expression levels in HUVECs transfected with an siRNA control (Scr), while high concentrations of ponatinib decreased AQP1, ADAMTS4, and ADAMTS9 levels compared with vehicle (Fig. 2, A to F). Ponatinib treatment was able to reduce the elevated expression of these genes in HUVECs transfected with si-CCM1 to similar levels of the siRNA controls (Fig. 2, A to F). Western blots further confirmed the normalization of KLF2, eNOS, ADAMTS1, and ADAMTS4 expressions in si-CCM1-HUVECs following ponatinib treatment (Fig. 2, G and H). Elevated MEKK3 signaling leads to increased phosphorylation of MEK5 that, in turn, results in hyperphosphorylation of ERK5 (12). Phosphorylated and total ERK5 levels were up-regulated in the si-CCM1-HUVECs compared with those in si-Scr controls, and this increase in expression was reduced to baseline levels following ponatinib treatment (Fig. 2I). p38 MAPK is another downstream target of MEKK3 (19); however, ponatinib had no effect on p38 MAPK phosphorylation or expression (fig. S3B). These results indicate that ponatinib is a potent inhibitor of MEKK3 signaling in endothelial cells and acts to block overactive MEKK3-induced downstream signaling.
Fig. 2. Ponatinib blocks MEKK3-induced signaling in endothelial cells.
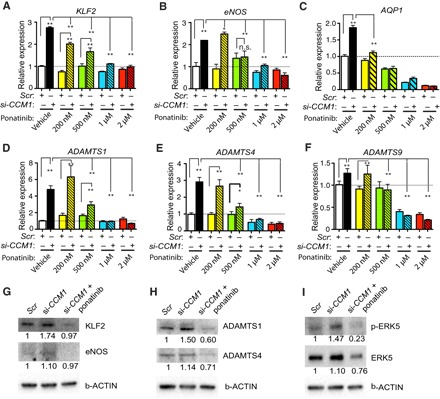
(A to F) Gene expression analysis of ponatinib-treated HUVECs with siRNA-induced CCM1 (si-CCM1) gene knockdown. Ponatinib treatment in HUVECs normalized the increased expression of KLF2 (A), eNOS (B), and AQP1 (C), as well as ADAMTS1 (D), ADAMTS4 (E), and ADAMTS9 (F), following CCM1 knockdown. (G to H) Western blotting analysis showing that ponatinib treatment decreased KLF2, eNOS, ADAMTS1, and ADAMTS4 expression. (I) Ponatinib treatment decreased the expression levels of ERK5 and p-ERK5. Error bars shown as SEM and significance determined by one-way analysis of variance (ANOVA) for multiple comparisons (n = 4). “*” indicates P < 0.05, “**” indicates P < 0.001, and n.s. indicates P > 0.05. Western blotting images are representatives of three independent experiments. The quantification of relative density ratios is shown underneath each blot.
Ponatinib is an FDA-approved compound, designed to target BCR-ABL tyrosine kinase to treat acute myeloid leukemia (20). A recent report also identified ponatinib as an inhibitor of MEKK2 activity (14). We now demonstrate that ponatinib is also a potent inhibitor of the MEKK3 signaling cascade (Fig. 2). To determine whether loss of BCR-ABL and MEKK2 can also regulate downstream genes of CCM signaling, we used siRNA to knock down the expression of MEKK2 or ABL in si-CCM1–knocked down HUVECs. si-MEKK3–knocked down si-CCM1 HUVECs were included as controls. We demonstrated efficient and specific knockdown of CCM1, MEKK2, MEKK3, and ABL in HUVECs (fig. S4, F to I). Combined knockdown of CCM1 and MEKK2 can partially rescue the elevated KLF2 and KLF4 gene expressions observed in si-CCM1–knocked down HUVECs, as well as its downstream targets eNOS and ADAMTS1 (fig. S4, A to D). Notably, loss of CCM1 and ABL did not reduce expression of KLF2/4 or its target genes; rather, we observed an increase in the expression of these genes (fig. S4, A to E). The most significant rescue in gene expression was observed with simultaneous knockdown of CCM1 and MEKK3, where KLF2, KLF4, eNOS, and ADAMTS1 expression was similar to the Scr control HUVECs (fig. S4, A to E). Therefore, these results suggest that the ponatinib’s ability to normalize abnormal gene expression following loss of CCM1 is through MEKK3 signaling and, to a lesser extent, MEKK2. ABL is not a target of ponatinib in endothelial cells that contributes to CCM signaling.
The hyperactive MEKK-KLF-ADAMTS signaling cascade observed in endothelial cells during CCM pathogenesis is also conserved during cardiovascular development (7). To further confirm that MEKK2 is not the dominant MEKK family member involved in CCM pathology, we knocked down ccm2 using morpholino in the Tg(cmcl2:EGFP) zebrafish model and visualized cardiac development in vivo. Consistent with previous studies, knockdown of ccm2 induced a dilated heart phenotype (fig. S5) (5, 21). While mekk3 knockdown can rescue the dilated cardiac defects in the ccm2 morphants (7), reduction in mekk2 had no effect on dilated cardiac phenotype (fig. S5). Together, these data suggest that mekk2 is not involved in CCM signaling during cardiac development in vivo. In addition, these results further confirm that a MEKK3-mediated signaling cascade underlies CCM lesion formation.
In addition to MEKK2/3 and ABL, ponatinib also targets several other kinases, including platelet-derived growth factor receptor α/β (PDGFRα/β), FGFRs, vascular endothelial growth factor receptor 2 (VEGFR2), Fms-related tyrosine kinase 3 (FLT3), and SRC (22). To determine whether some of these targets are also involved in CCM signaling, we treated si-CCM1–knocked down HUVECs with selective inhibitors against these targets. Among the five inhibitors tested, PD173074, a selective inhibitor for FGFR1 and VEGFR2, had no effect on si-CCM1 CCM1-induced KLF2 expression (fig. S6). In contrast, crenolanib (inhibitor for PDGFRα/β), masitinib (inhibitor for PDGFRα/β, ABL, KIT, and LYN), dovitinib (inhibitor against FGFR1/2, FLT3, FLT4, and c-KIT), and KX2-391 (SRC inhibitor) all partially reduced elevated KLF2 and its target gene expression in si-CCM1–treated HUVECs (fig. S6). These data suggest that in the absence of CCM1, and in addition to MEKK3, PDGFR and SRC signaling pathways may also play a role in CCM pathology by up-regulating KLF2. Further in vivo studies are needed to confirm the role of these signaling cascades in CCM pathogenesis.
Ponatinib inhibits CCM lesion initiation and growth
Our in vitro data thus far demonstrate that ponatinib can rescue aberrant signaling associated with CCM pathogenesis in CCM1–knocked down endothelial cultures (Fig. 2 and fig. S4). To address whether ponatinib can alleviate CCM lesion formation in vivo, we delivered ponatinib (10 mg/kg) intragastrically to mice with induced endothelial cell–specific deletion of Ccm1 (Cdh5-CreERT2;Ccm1fl/fl, denoted the Ccm1iECKO mice) (Fig. 3A). Ccm1 gene deletion was induced by 4-hydroxytamoxifen (4-HT) injection at postnatal day 1 (P1) (fig. S7A). The first CCM lesions were detected at P6, and at P13, numerous vascular malformations were observed in the sham-treated Ccm1iECKO pups, as visualized by micro-CT (Fig. 3, B to D). In contrast, a single dose of ponatinib at P6 in the Ccm1iECKO mice significantly reduced the number of developed lesions detected in the hindbrains (Fig. 3, E to G). CCM lesion burden was reduced by 72% (Fig. 3H) and total lesion number by 35% in ponatinib-injected Ccm1iECKO pups compared with sham-treated Ccm1iECKO mice (Fig. 3I). Histological analysis of P13 brains corroborated these micro-CT findings (fig. S8). We further classified lesion number according to their size and observed significantly reduced number of medium-sized (106 to 107 μm3 in volume) and large-sized (> 107 μm3) lesions in the ponatinib-treated Ccm1iECKO mice compared with the sham-treated Ccm1iECKO group. The number of small-sized lesions (< 106 μm3) was similar between the treatment groups (Fig. 3I). The reductions in total lesion volume observed in the Ccm1iECKO mice treated with ponatinib were due to the significantly decreased total volume of large lesions (Fig. 3J).
Fig. 3. Ponatinib inhibits CCM lesion formation and progression in the CCM1-deficient CCM model.
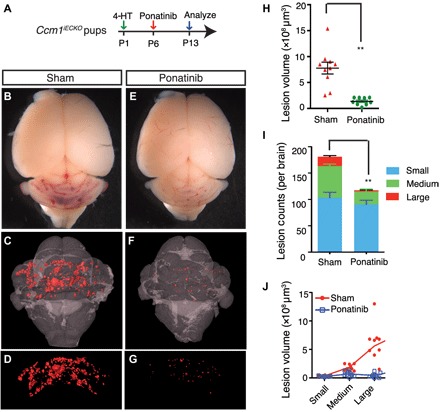
(A) Schematic of experimental design. Neonatal pups at P1 were induced with 4-HT and treated with ponatinib at P6. Brains were collected at P13 for micro–computed tomography (CT) analysis. (B to G) Micro-CT imaging of CCM lesions in Ccm1iECKO with (E to G) or without (B to D) ponatinib treatment. (H to J) Quantification of micro-CT analysis shows that ponatinib treatment in the Ccm1iECKO reduced CCM lesion burden by 72% (H) and total lesions number by 35% (I) compared with that of sham-treated controls. CCM lesions distribution analysis showing decreased number (I) and total volume (J) of medium and large lesions in ponatinib-treated Ccm1iECKO mice, but the number and collective volume of small lesions did not change. Error bars are shown as SEM, and significance was determined by Student’s t test. “**” indicates P < 0.001. n = 10 for the sham group, n = 9 for the ponatinib treatment group.
In the CCM1-CCM2-CCM3 protein complex, the CCM2 protein directly interacts with MEKK3 (23), and loss of Ccm2 also leads to vascular malformations as a result of increased endothelial Mekk3 signaling (6, 7). Ccm2 gene deletions were again induced by 4-HT injection at P1 (fig. S7B). Similar to the results from the Ccm1iECKO mice, a single dose of ponatinib in the endothelial cell–specific Ccm2 knockout (Ccm2iECKO) mice was also able to decrease CCM lesion volume by 85% and CCM lesion count by 36% compared with sham-treated Ccm2iECKOmice (Fig. 4, A to J). The decrease in total lesion counts indicates that ponatinib treatment limits lesion initiation. In addition, the more marked reduction of CCM lesion volume suggests that ponatinib is highly effective in limiting CCM lesion growth. Together, these data demonstrate that ponatinib can efficiently limit the development of vascular malformations.
Fig. 4. Ponatinib inhibits CCM lesion formation and progression in the CCM2-deficient CCM model.
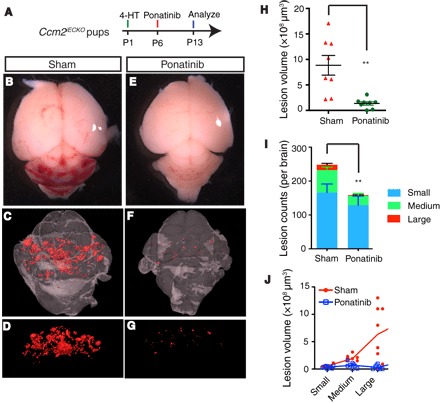
(A) Schematic of experimental design. (B to G) Micro-CT imaging of CCM lesions in the CCM2iECKO with (E to G) or without (B to D) ponatinib treatment. (H and I) Quantification of micro-CT analysis shows that ponatinib treatment reduced CCM lesion burden by 85% (H) and total lesions number by 36% (I) in the Ccm2iECKO mice compared with that of sham controls. (J) CCM lesions distribution analysis showing that the decreased lesion burden was mainly due to the reduction in the number of large lesions. Error bars are shown as SEM, and significance was determined by Student’s t test. “**” indicates P < 0.001; n = 8 for both sham and ponatinib treatment groups.
Ponatinib prevents progression of late-stage CCM lesions
In the neonatal CCM models, CCM lesions start to appear around P6 and then undergo rapid growth and formation such that numerous well-established lesions are formed by P11. Thereafter, lesions will continue to grow, albeit at a relatively slower pace. This lesion development pattern in mice closely recapitulates human CCM lesion growth as detected using magnetic resonance imaging scans. Treatment of endothelial-specific Ccm knockout mice with ponatinib at the time of lesion growth significantly reduced the number of lesions formed and the total lesion volume, indicating that ponatinib can effectively protect against lesion initiation and progression (Figs. 3 and 4). To determine whether ponatinib can reduce lesion growth or rescue already formed lesions, we again injected Ccm1iECKO pups with 4-HT at P1 to induce lesion formation, followed by a single dose of ponatinib at P11, and lesion burden was analyzed at P22 (Fig. 5A). Ponatinib treatment resulted in significantly reduced CCM lesion burden at P22 compared with the sham-treated Ccm1iECKO mice (Fig. 5, B to G, and fig. S9). Quantitative analyses of micro-CT imaging data demonstrate that ponatinib treatment can decrease the lesion volume by > 70% relative to sham-treated Ccm1iECKO controls (Fig. 5H). Treatment with ponatinib had no effect on the total number of developed CCM lesions at P22, but rather, it significantly decreased the presence of large- and medium-sized lesions with a concomitant increased number of small-sized lesions (Fig. 5, I and J).
Fig. 5. Ponatinib impairs the growth of established CCM lesions in mouse model.
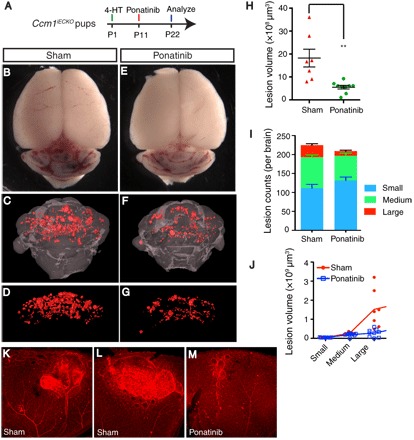
(A) Schematic of experimental design. Neonatal pups at P1 is induced with 4-HT and treated with ponatinib at P11. Brains and eyes were collected at P22 for micro-CT analysis and retina staining. (B to G) Micro-CT imaging of CCM lesions in brain in Ccm1iECKO with (E to G) or without (B to D) ponatinib treatment. (H to J) Quantification of micro-CT analysis shows that ponatinib treatment reduced CCM lesion volume burden by 70% (H) without changes to the total lesion number (I) in Ccm1iECKO mice compared with that of sham controls. CCM lesion distribution analysis shows decreased number of medium and large lesions in ponatinib-treated Ccm1iECKO mice, but the number of small lesions increased (I). Collective volume of small and medium lesions did not change, but the collective volume of large lesions is decreased (J). (K to M) Isolectin staining of retinal vasculature demonstrates smaller CCM lesions in the ponatinib-treated retina (M) compared with that of sham treatment (K and L). Error bars are shown as SEM, and significance was determined by Student’s t test. “**” indicates P < 0.001; n = 7 for the sham group, n = 9 for the ponatinib treatment group.
In addition to lesion development in the cerebellum, CCM model mice also develop retinal lesions. Large vascular malformations at the periphery of the retinal vascular plexus formed in the sham-treated Ccm1iECKO mice (Fig. 5, K and L). Treatment with ponatinib had a similar inhibitory effect in reducing retinal lesion development in the Ccm1iECKO mice as it did in reducing cerebellum lesion formation (Fig. 5M). These data demonstrate that ponatinib can effectively reduce the growth of already established CCM lesions to limit the transition from small lesions to large lesions.
Ponatinib normalizes aberrant endothelial genes expression and morphology in CCM-deficient mice
To determine whether ponatinib inhibits CCM lesion formation and progression through targeting of KLF2/4 and its downstream targets, we isolated murine brain endothelial cells and analyzed gene expressions by quantitative polymerase chain reaction (qPCR). Brain endothelial cells were isolated from the P8 Ccm1iECKO pups, in which gene deletion was induced at P1, and ponatinib was administered at P6. Consistent with the data from HUVECs (Fig. 2), qPCR results revealed increased expression of Klf2, Klf4, eNos, and Id1 mRNA compared with that of cells from Ccm1fl/fl littermate controls. A single treatment with ponatinib in Ccm1iECKO mice was sufficient to normalize the elevated gene expression levels in brain endothelial cells to that of the littermate controls (Fig. 6, A to D). Normalization of Klf4 protein levels in fresh isolated brain endothelial cells seen by Western analysis further confirmed the qPCR data (Fig. 6E).
Fig. 6. Ponatinib normalized MEKK3-induced signaling and endothelium ultrastructure in CCM mouse models.
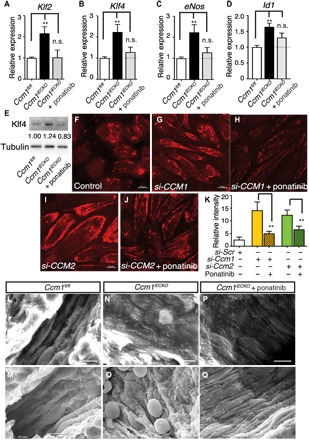
(A to D) Gene expression analysis of ponatinib-treated Ccm1iECKO mice. Ponatinib treatment at P6 normalized the increased expression of Klf2, Klf4, eNos, and Id1 in the freshly isolated brain endothelial cells from Ccm1iECKO mice as analyzed at P8. Error bars are shown as SEM, and significance was determined by one-way ANOVA, n = 6. “**” indicates P < 0.001; (E) Western blotting analysis of Klf4 protein level in freshly isolated brain endothelial cells. Band density ratios are shown underneath the Klf4 blot. (F to J) Immunocytochemistry analysis shows increased pMLC2 level in the si-CCM1 (G) and si-CCM2 (I) endothelial cells compared with control (F), while treatment with ponatinib returned pMLC2 levels to near baseline (H and J). (K) Quantification of pML2 relative fluorescence intensity. (L to Q) Scanning electron microscope images of murine brain microvessels in P30 mouse brains. In microvessels without CCM lesions (L and M), endothelium ridges are present and aligned in the direction of blood flow. In CCM lesions (N and O), endothelium is flattened and disorganized. CCM lesions treated with ponatinib (P and Q) show partially normalized ridge structures of endothelium and alignment in the direction of blood flow. Results are representative of three independent experiments.
It has been shown that loss of CCM genes in cultured human endothelial cells and endothelial cells isolated from a mouse brain leads to activation of Rho signaling (5, 6, 24). To determine whether ponatinib can reverse the aberrant activation of Rho signaling conferred by CCM deficiency, we treated cultured HUVECs with siRNA against CCM1 or CCM2, followed by ponatinib treatment. Immunocytochemistry analysis revealed a marked increase of pMLC2 level in cells transfected with si-CCM1 (Fig. 6G) or si-CCM2 (Fig. 6I) compared with the control group (Fig. 6F). Ponatinib treatment reversed the aberrant increases in pMLC2 levels in both si-CCM1–treated (Fig. 6H) and si-CCM2–treated (Fig. 6J) cells. Quantification of relative fluorescence intensity is shown in Fig. 6K.
The major pathological hallmarks of CCM lesions are flattening of endothelial cells and disruptions to the endothelial cell–endothelial cell and endothelial cell–pericyte interactions. Scanning electron microscopy was used to determine whether ponatinib can normalize the structural changes in the endothelium with lesions. Under normal conditions, the endothelium ridges are well organized and align in the direction of blood flow (Fig. 6, L to M). In contrast, the endothelium within the CCM lesions was flattened and disorganized (Fig. 6, N and O). Ponatinib treatment can partially rebuild the ridge structures of the endothelium and return endothelial cell alignment along the direction of blood flow (Fig. 6, P and Q). These results demonstrate that ponatinib can normalize the underlying dysfunctional endothelial structural phenotype in brain microvessels that contain CCM lesions and represent a potential therapy for CCM disease.
DISCUSSION
Recent genetic studies reveal that elevated MEKK3 signaling is a causal factor for CCM lesion formation (6). In the present study, we have identified an FDA-approved kinase inhibitor, ponatinib, originally developed as a tyrosine kinases inhibitor to treat chronic myeloid leukemia (16), as a potent inhibitor of hyperactive endothelial MEKK3 signaling, with potential as a novel therapy to treat CCM disease. Ponatinib is capable of robustly inhibiting CCM lesion initiation and progression in both Ccm1- and Ccm2-deficient mouse models. These preclinical data provide the rationale for repurposing the use of ponatinib or design of new ponatinib derivatives for treating patients with CCM.
The FDA has approved ponatinib for clinical use, but its use has been reported to cause significant adverse effects in the cardiovascular system (20). The exact cellular and molecular targets leading to these detrimental effects remain unknown. A kinase inhibition spectrum assay has revealed that ponatinib can target a dozen kinases with high efficiency (25). Our kinase assays and gene knockdown studies demonstrate that ponatinib can efficiently target MEKK3. A previous study has also established ponatinib as a potent inhibitor of MEKK2 (14). Our in vitro data indicated that MEKK2 could also mediate CCM signaling but to a lesser extent when compared with MEKK3. In vivo studies using zebrafish models suggest that MEKK2 is not involved in CCM signaling (fig. S5), but it is necessary to confirm whether MEKK2 is involved in CCM pathogenesis in in vivo mouse CCM models. Ponatinib was designed to target the tyrosine protein kinase ABL1, but we did not find any role of ABL in CCM signaling (fig. S4). Whether other potent targets of ponatinib in addition to MEKK3 also play a potential role in CCM pathogenesis will require further investigation.
MEKK3 is an upstream activator of the MEK5-ERK5 signaling cascade (12, 26). Our previous study has shown that MEK5 inhibition can reduce elevated KLF2/4 expression that is induced with CCM deficiency (7). In this study, we found that ponatinib can block MEK5 phosphorylation in in vitro kinase assays. Furthermore, we demonstrate that ponatinib can inhibit the elevated expression of MEKK3 downstream genes such as KLF, eNOS, and Adamts genes that are up-regulated in CCM-deficient cells. While ponatinib can efficiently rescue the elevated p-ERK5 level in CCM-deficient endothelial cells, it had no effect on p38 MAPK signaling (fig. S3B), although p38 MAPK is another pathway downstream of MEKK3. These results indicate that in endothelial cells, ponatinib’s mode of action is through the MEKK3/ERK5 signaling cascade.
In the current study, we could inhibit new CCM lesion formations (below the level of detection) and the growth of already developed CCM lesions with high efficacy. These results were achieved with only a single intragastric administration of the compound in mice. How ponatinib achieves this high efficiency remains to be fully elucidated. It could be due to several reasons: (i) In the neonatal murine model of CCM disease, CCM lesions only develop when CCM genes are deleted between P1 and P3. Deletion of CCM genes outside this critical window leads to minimal or even lack of formed CCM lesions. This temporal dependence suggests that the downstream signaling may be a critical component in lesion development only within the specific time window, and inhibition of signaling within this time window may be sufficient to block lesion formation. (ii) CCM lesion progression may require continuous up-regulation of KLFs. Transient KLF down-regulation by ponatinib through blocking of MEKK3 signaling may have allowed endothelial cells to adapt to elevated KLFs, and therefore, they do not develop lesions anymore. (iii) A clinical trial study reported that ponatinib is readily absorbed in humans with a median time to maximal plasma radioactivity of 5 hours and mean terminal elimination half-life of radioactivity of 66.4 hours (27). This study suggests that even with a single dose, ponatinib can be readily absorbed and active for a long period of time and may contribute to its effectiveness against CCM lesions.
Ponatinib is recommended for use at a starting dosage of 30 mg/kg for patients with cancer (16). In our study, ponatinib (10 mg/kg) can potently block MEKK3 activity and reduce CCM lesion burden. Given our results that ponatinib targets MEKK3, together with previous genetic data demonstrating that deleting just one allele of Mekk3 is sufficient to block lesion formation (6, 8), this may account for the potent effect of ponatinib in reducing CCM pathology. The ability to prevent lesion formation using a lower dose of ponatinib is an important factor, as ponatinib has been associated with adverse cardiovascular events and that usage at a lower dosage may mitigate these effects. Further investigations are required to translate these results from mouse models to the clinic. The neonatal CCM model used in our study is the best available model to mimic human CCM disease. However, it still does not fully recapitulate human conditions. For example, in humans, CCM mostly manifests during adulthood (30s to 40s) and can be detected in various areas throughout the brain. In the neonatal murine model, CCM gene deletions must be induced in the first few days after birth for lesions to form, and these lesions are mostly limited to the cerebellum. Because of these differences, the dosage concentration, timing of administration, and potential adverse effect will have to be carefully assessed in human clinical trials to determine the beneficial effects of ponatinib as a therapy for CCM. It is likely that treatment and management of CCM lesions in humans require long-term drug therapy, and whether any unintended effects on other kinases will require further investigation. Chemical modifications to improve the target selectivity of ponatinib will be necessary for the development of a potent yet safe drug of CCM treatment. A similar design has been achieved to redirect the selectivity of ponatinib derivative toward PIPK1 (25).
Various agents based on different targeting mechanisms have been tested in preclinical studies to treat CCM. Among them, fasudil, which targets Rho signaling, has been shown to be effective in reducing lesion burden in CCM1 or CCM2 heterozygous models enhanced by Msh2 or p53 deficiency (28). Sulindac sulfide and its analogs have been shown to effectively impair lesion formation in the CCM3 knockout mice (29), and Tak242, a Toll-like receptor 4 signaling blocker, can reduce CCM burden in both CCM1-and CCM2-deficient mice (6). Statins have been tested in clinical setting without positive effects, while ROCK inhibitors are currently in preclinical and clinical trials. Our previous studies have shown that Rho signaling lies downstream of MEKK3 during CCM lesion development (6), and that ponatinib is effective at reducing elevated Rho signaling in CCM-deficient endothelial cells (Fig. 6). Whether a combination of drugs such as ponatinib with fasudil or other ROCK inhibitors that target CCM signaling at multiple nodes will be more effective in preventing CCM lesion formation and development will need to be investigated.
In conclusion, we have identified an FDA-approved drug, ponatinib, which can markedly reduce CCM lesion burden by inhibiting MEKK3-KLF activity. As a currently used clinical drug, it can be rapidly applied as a therapeutic treatment for patients with CCM for which there are no current available treatments. Ponatinib’s adverse effects observed from clinical use in patients with cancer indicate that it may have other unintended kinase targets and suggest that improved derivatives of ponatinib with improved specificity may be required for use in both patients with cancer and patients with CCM.
MATERIALS AND METHODS
Study design
This study was designed to specifically identify therapeutic compounds that can effectively prevent CCM lesion formation and progression through inhibition of the MEKK3-KLF-ADAMTS signaling axis. The strategy used an FDA-approved kinase screen to identify compounds that inhibited KLF2/4 gene expression levels in CCM1/2-deficient endothelial cells. Ponatinib was found to be the most promising candidate following in vitro studies and testing in in vivo mouse models of CCM lesion development. The endothelial-specific Ccm1 or Ccm2 knockout (Ccm1iECKO or Ccm2iECKO) mice on C57Bl/6 background were treated with sham or ponatinib. Animals were randomly assigned to treatment groups from litters with at least four pups. Only data from animal studies with the appropriate littermate controls (Cre-negative controls, Ccm1iECKO or Ccm2iECKO pups treated with sham, and Ccm1iECKO or Ccm2iECKO treated with ponatinib) were included in the analysis. Therapeutic potential of ponatinib was examined by micro-CT for reductions in lesion number, size, and volume. Changes in gene expression were measured by qPCR. Restoration of misaligned endothelial lining was observed by scanning electron microscopy.
Mouse models
The Cdh5-CreERT2 PAC transgenic mice (iECre), and Ccm1fl/fl and Ccm2fl/fl animals have been previously described (7, 30, 31). All experimental animals were maintained on a C57BL/6J genetic background. The Sydney Local Health District Animal Welfare Committee and Institutional Animal Care and Use Committee of Tianjin Medical University approved all animal ethics and protocols. All experiments were conducted under the guidelines/regulations of Centenary Institute, The University of Sydney, and Tianjin Medical University.
CCM lesion induction and ponatinib administration in mouse model
The Ccm1iECKO mice and the Ccm2iECKO mice were generated by crossing the Cdh5-CreERT2;Ccm1fl/fl or Cdh5-CreERT2;Ccm2fl/fl mice with the Ccm1fl/fl or Ccm2fl/fl mice, respectively. To induce Ccm gene deletion, 50 μl of 4-HT (0.5 mg/ml; H7904, Sigma-Aldrich) was given intragastrically to neonatal pups at P1. Ponatinib [Selleck, 10 mg/kg made in 25 mM citrate buffer (pH 2.75)] was administrated to the neonatal CCM disease model via intragastric administration at either P6 or P11. Control animals were similarly injected with 4-HT and administered with vehicle [25 mM citrate buffer (pH 2.75)].
Immunoprecipitation analysis and in vitro kinase assay
Cultured human embryonic kidney 293 cells were transfected with flag-MEKK3 and HA-MEK5 plasmids, and cells were lysed with radioimmunoprecipitation assay buffer 48 hours after transfection. The lysates were subject to pulldown with anti-flag antibody affinity gel (Sigma-Aldrich). After washing, protein bound to the beads were eluted and subjected to Western blot analysis.
For in vitro kinase assays, purified GST-MEK5 was used as the substrate. Purified flag-MEKK3 and GST-MEK5 were incubated in the kinase reaction buffer system [20 mM Hepes/KOH (pH 7.5), 5 mM MgCl2, 2 mM dithiothreitol, 1 mM ATP] for 4 hours at 30°C. The reaction mixtures were then denatured with Laemmli loading buffer, and Western blots were performed using a standard protocol. Phosphorylated MEK5 was detected with the p-MEK5 antibody from Novus.
Ponatinib treatment and gene expression analysis in cultured endothelial cells
HUVECs were purchased from Lonza or freshly isolated from umbilical vessels, as previously described (32), and siRNA-mediated gene knockdown in these cells was performed, as previously described (31). Briefly, HUVECs plated on six-well plates at 80% confluence were transfected with 10 nM siRNA using Lipofectamine RNAiMAX transfection reagents (Thermo Fisher Scientific). Transfected cells were treated with ponatinib (0.2 to 2 μM) or vehicle 24 hours after transfection, and RNA or protein was harvested 48 hours after initial transfection. siRNAs directed against CCM1, MEKK2, MEKK3, and ABL1 were used for the knockdown experiments. After siRNA knockdown of CCM1, HUVECs were treated with ponatinib (0.2 to 2 μM) for 24 hours, RNA was collected using an RNeasy micro kit (Qiagen 74004), and complementary DNA (cDNA) was synthesized using the the SuperScript VILO cDNA Synthesis Kit and Master Mix (Thermo Fisher Scientific). PCR reactions were set up in triplicate, and data shown were from at least three independent repeats. Real-time PCR was performed with the PowerUp SYBR Green PCR Master Mix (Thermo Fisher Scientific) using the following human primers: GAPDH, 5′-GAGTCAACGGATTTGGTCGT-3′ (forward) and 5′-GATCTCGCTCCTGGAAGATG-3′ (reverse); KLF2, 5′-CTACACCAAGAGTTCGCATCTG-3′ (forward) and 5′-CCGTGTGCTTTCGGTAGTG-3′ (reverse); KLF4, 5′-GGCGGGCTGATGGGCAAGTT-3′ (forward) and 5′-TGCCGTCAGGGCTGCCTTTG-3′ (reverse); eNOS, 5′-GGGTCCTGTGTATGGATGAGT-3′ (forward) and 5′-ATGCTGTTGAAGCGGATCTTA-3′ (reverse); AQP1, 5′-TCATGTACATCATCGCCCAGT-3′ (forward) and 5′-GGTAGTAGCCAGCACGCATAG-3′ (reverse); ADAMTS1, 5′-GTAGGACAGCCCACAGGA-ACT-3′ (forward) and 5′-GTAGGACAGCCCACAGGAACT-3′ (reverse); ADAMTS4, 5′-AACATGCTCCATGACAACTCC-3′ (forward) and 5′-AATGGAGCCTCTGGTTTGTCT-3′ (reverse); ADAMTS9, 5′-CCGATCGTGAGCAGTGTAAC-3′ (forward) and 5′-TTGTTTATGCCCTCGACCAC-3′ (reverse); CCM1, 5′-AATGGCAGAGAAGCATGAGCAGTG-3′ (forward) and 5′-ATTTCAGCATGTCCTCCTCCAGCA-3′ (reverse); MEKK2, 5′-AGAATCCTTCAGTTCCCCAGA-3′ (forward) and 5′-TACTACGATCCAGCAGTTCCA-3′ (reverse); MEKK3, 5′-AGATGTGGAGCACAAGGTGAC-3′ (forward) and 5′-ATTTATATCCCCTGCGGACTG-3′ (reverse); and ABL1, 5′-GTTCCATCTCCCACTTGTCGT-3′ (forward) and 5′-TGAGATACGAAGGGAGGGTGT-3′ (reverse).
Isolation of cerebellar endothelial cells and gene expression analysis
Cerebellar endothelial cells were isolated through enzymatic digestion, followed by separation using magnetic-activated cell sorting by anti-CD31–conjugated magnetic beads (MACS MS system, Miltenyi Biotec), as previously described (6). Isolated cerebellar endothelial cells were pelleted, and total RNA was extracted using the RNeasy micro kit (Qiagen 74004). PCR reactions were set up in triplicate, and data shown are from at least three independent repeats. Changes in gene expression were detected using the following mouse primers: Gapdh, 5′-GTCCCGTAGACAAAATGGTGA-3′ (forward) and 5′-TTTGATGTTAGTGGGGTCTCG-3′ (reverse); Klf2, 5′-CGCCTCGGGTTCATTTC-3′ (forward) and 5′-AGCCTATCTTGCCGTCCTTT-3′ (reverse); Klf4, 5′-GTGCCCCGACTAACCGTTG-3′ (forward) and 5′-GTCGTTGAACTCCTCGGTCT-3′ (reverse); eNos, 5′-CCTGTGCATGGATGAGTATGA-3′ (forward) and 5′-TGAGCAGGAGACACTGTTGAA-3′ (reverse); and Id1, 5′-ATCCTGCAGCATGTAATCGAC-3′ (forward) and 5′-GAGTCCATCTGGTCCCTCAGT-3′ (reverse).
Immunocytochemistry analysis
HUVECs were plated on coverslips in six-well plates and transfected with si-CCM1 or si-CCM2 for 24 hours. Cells were treated with ponatinib (1 μM) for a further 24 hours before fixation and immunocytochemistry staining with p-MLC2 antibody (Cell Signaling Technology). Images were taken with a Zeiss LSM800 confocal microscope.
Histology
Tissue samples were fixed in 4% formaldehyde overnight and embedded in paraffin. Five-micrometer-thick sections were stained for hematoxylin and eosin using a standard protocol and observed under a light microscope.
Isolectin staining of retina vasculature
For flat-mount retina staining, fixed retinas were blocked overnight in 4°C with 1% bovine serum albumin and 0.3% Triton X-100 in phosphate-buffered saline (PBS). Retinas were conditioned with PBLEC buffer [1% Triton X-100, 1 mM CaCl2, 1 mM MgCl2, and 0.1 mM MnCl2 in PBS (pH 6.8)] before they were incubated overnight with a 1:25 Daylight 594–conjugated isolectin B4 (Vector Laboratories, Burlingame, CA, USA) in a PBLEC buffer at 4°C to stain for retinal vessels. After washing, the stained retinas were postfixed with 4% paraformaldehyde (PFA) for 5 min and washed again with PBS. Images of retinal vessels were taken with a confocal sSP5 microscope (Leica Microsystems, Germany).
Micro-CT scan and analysis
CCM lesion burden was analyzed using micro-CT imaging techniques, as previously described (33).
Computer modeling
Homology models of MEKK3 and MEKK2 were built on the basis of the crystal structure of the serine/threonine protein kinase PAK1 (Protein Data Bank ID: 4ZLO) (18) as the template by Prime (Schrödinger, LLC) (34). Protein minimization was then carried out using Prime. Ponatinib was prepared (35) and docked into the models, respectively, at the XP precision using Glide (Schrödinger, LLC) (36). The docked ligand-protein complex in three dimensions was presented using PyMOL (37). The two-dimensional ligand-protein interaction diagrams were presented using Maestro (Schrödinger, LLC) (38).
Brain microvascular analysis by scanning electron microscopy
Mice were perfused with 10 ml of scanning electron microscopy fixative [2.5% electron microscopy grade glutaraldehyde, 2% formaldehyde, 2 mM calcium chloride, 2% sucrose, and 0.1 M cacodylate buffer (pH 7.4)]. Hindbrain was dissected and fixed in the scanning electron microscopy fixative overnight. Samples were further prepared for scanning electron microscopy, as previous described (39). Brain microvasculature was examined with a JEOL Neoscope Tabletop scanning electron microscope at up to ×5000 magnification.
Zebrafish studies
Zebrafish strain including AB and Tg(cmlc2:GFP) transgenic lines were raised and maintained at 28.5°C in system water and staged as previously described (40). This study was approved by the Ethical Review Committee of Institute of Zoology, Chinese Academy of Sciences, China. Two antisense morpholino oligonucleotides (MOs) were used: ccm2 MO (GAAGCTGAGTAATACCTTAACTTCC) and mekk2 MO (GGCTGAAAAATGCTGCTCACCTTGC).
Morpholinos were injected at the one-cell stage. Tg(cmlc2:GFP) embryos at 3 days after fertilization were mounted in 1% low–melting point agarose. Images were acquired with an Andor dragonfly spinning disk confocal microscope (Dragonfly 505).
Statistical analysis
All the statistical analysis in the study was done using t tests and one-way ANOVA in GraphPad Prism statistical software. Values are presented as means ± SE. Statistical significance was considered when P ≤ 0.05.
Supplementary Material
Acknowledgments
We acknowledge the facilities and the technical assistance of the Sydney Microscopy & Microanalysis (SMM), the Australian Centre for Microscopy & Microanalysis (ACMM) at the University of Sydney, and the micro-CT facility at the Institute of Zoology, Chinese Academy of Sciences. We thank D. Li for providing Ccm1fl/fl mouse line as a gift. We thank C. Kirkpatrick of Monash Institute of Pharmaceutical Sciences for help in assessing the pharmacokinetic/pharmacodynamic (PK/PD) of ponatinib. We thank J. Li of the Centenary Institute for assistance in HUVEC study. Funding: This study was supported by Australian National Health and Medical Research Council (NHMRC) project grant APP1124011, Australian Brain Foundation grant 2016, National Natural Science Foundation of China (NSFC grant no. 81771240) (X.Z.), and the Future Leader Fellowship (Award ID 101856) from the National Heart Foundation of Australia (R.L.). Author contributions: J.P.C., R.W., and X.Z. designed and performed most of the experiments. X.Y. and X.W. performed animal experiments and histological analysis. L.W. and F.L. performed zebrafish studies. K.K.T. performed retinal analysis. R.L. performed Western blots and analysis. M.F. and Z.H. provided technical and facility support for micro-CT analysis. V.C. contributed to scanning electron microscopy analysis. Z.Y. and J.B. performed computer modeling analyses. J.P.C., R.W., R.L., and X.Z. analyzed the data and wrote the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/11/eaau0731/DC1
Fig. S1. Three-dimensional structural comparison of the binding between ponatinib and Abl, FGFR4, and MEKK3.
Fig. S2. Two-dimensional mapping of ponatinib interaction with Abl, FGFR4, and MEKK3.
Fig. S3. CCM1, p38, and MEKK3 expression in cultured HUVECs.
Fig. S4. MEKK3 is the dominant regulator of KLF signaling.
Fig. S5. Confocal images of Tg(cmlc2:EGFP) embryos injected with control, ccm2, or ccm2 + mekk2 morpholinos.
Fig. S6. Effects of ponatinib and other kinase inhibitors on KLF2 expression.
Fig. S7. qPCR quantification of CCM gene expression in brain endothelial cells isolated from 4-HT–induced p6 pups.
Fig. S8. Hematoxylin and eosin staining of CCM lesions at P13.
Fig. S9. Hematoxylin and eosin staining of CCM lesions at P30.
REFERENCES AND NOTES
- 1.Vernooij M. W., Ikram M. A., Tanghe H. L., Vincent A. J., Hofman A., Krestin G. P., Niessen W. J., Breteler M. M., van der Lugt A., Incidental findings on brain MRI in the general population. N. Engl. J. Med. 357, 1821–1828 (2007). [DOI] [PubMed] [Google Scholar]
- 2.Fischer A., Zalvide J., Faurobert E., Albiges-Rizo C., Tournier-Lasserve E., Cerebral cavernous malformations: From CCM genes to endothelial cell homeostasis. Trends Mol. Med. 19, 302–308 (2013). [DOI] [PubMed] [Google Scholar]
- 3.Stalheim L., Uhlik M. T., Abell A. N., Ancrile B. B., Johnson G. L., Marchuk D. A., CCM1 and CCM2 protein interactions in cell signaling: Implications for cerebral cavernous malformations pathogenesis. Hum. Mol. Genet. 14, 2521–2531 (2005). [DOI] [PubMed] [Google Scholar]
- 4.Kleaveland B., Zheng X., Liu J. J., Blum Y., Tung J. J., Zou Z., Sweeney S. M., Chen M., Guo L., Lu M. M., Zhou D., Kitajewski J., Affolter M., Ginsberg M. H., Kahn M. L., Regulation of cardiovascular development and integrity by the heart of glass-cerebral cavernous malformation protein pathway. Nat. Med. 15, 169–176 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zheng X., Xu C., Di Lorenzo A., Kleaveland B., Zou Z., Seiler C., Chen M., Cheng L., Xiao J., He J., Pack M. A., Sessa W. C., Kahn M. L., CCM3 signaling through sterile 20-like kinases plays an essential role during zebrafish cardiovascular development and cerebral cavernous malformations. J. Clin. Invest. 120, 2795–2804 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhou Z., Tang A. T., Wong W. Y., Bamezai S., Goddard L. M., Shenkar R., Zhou S., Yang J., Wright A. C., Foley M., Arthur J. S., Whitehead K. J., Awad I. A., Li D. Y., Zheng X., Kahn M. L., Cerebral cavernous malformations arise from endothelial gain of MEKK3-KLF2/4 signalling. Nature 532, 122–126 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rawnsley D. R., Goddard L. M., Pan W., Cao X. J., Jakus Z., Zheng H., Yang J., Arthur J. S., Whitehead K. J., Li D., Zhou B., Garcia B. A., Zheng X., Kahn M. L., The cerebral cavernous malformation pathway controls cardiac development via regulation of endocardial MEKK3 signaling and KLF expression. Dev. Cell 32, 168–180 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Choi J. P., Foley M., Zhou Z., Wong W. Y., Gokoolparsadh N., Arthur J. S., Li D. Y., Zheng X., Micro-CT imaging reveals Mekk3 heterozygosity prevents cerebral cavernous malformations in Ccm2-deficient mice. PLOS ONE 11, e0160833 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cuttano R., Rudini N., Bravi L., Corada M., Giampietro C., Papa E., Morini M. F., Maddaluno L., Baeyens N., Adams R. H., Jain M. K., Owens G. K., Schwartz M., Lampugnani M. G., Dejana E., KLF4 is a key determinant in the development and progression of cerebral cavernous malformations. EMBO Mol. Med. 8, 6–24 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Otten C., Faurobert E., Rudolph F., Zhu Y., Boulday G., Duchene J., Mickoleit M., Dietrich A. C., Ramspacher C., Steed E., Manet-Dupé S., Benz A., Hassel D., Vermot J., Huisken J., Tournier-Lasserve E., Felbor U., Sure U., Albiges-Rizo C., Abdelilah-Seyfried S., Regulation of β1 integrin-Klf2-mediated angiogenesis by CCM proteins. Dev. Cell 32, 181–190 (2015). [DOI] [PubMed] [Google Scholar]
- 11.Gerwins P., Blank J. L., Johnson G. L., Cloning of a novel mitogen-activated protein kinase kinase kinase, MEKK4, that selectively regulates the c-Jun amino terminal kinase pathway. J. Biol. Chem. 272, 8288–8295 (1997). [DOI] [PubMed] [Google Scholar]
- 12.Nakamura K., Johnson G. L., PB1 domains of MEKK2 and MEKK3 interact with the MEK5 PB1 domain for activation of the ERK5 pathway. J. Biol. Chem. 278, 36989–36992 (2003). [DOI] [PubMed] [Google Scholar]
- 13.Zhang D., Facchinetti V., Wang X., Huang Q., Qin J., Su B., Identification of MEKK2/3 serine phosphorylation site targeted by the Toll-like receptor and stress pathways. EMBO J. 25, 97–107 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ahmad S., Johnson G. L., Scott J. E., Identification of ponatinib and other known kinase inhibitors with potent MEKK2 inhibitory activity. Biochem. Biophys. Res. Commun. 463, 888–893 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ahmad S., Hughes M. A., Johnson G. L., Scott J. E., Development and validation of a high-throughput intrinsic ATPase activity assay for the discovery of MEKK2 inhibitors. J. Biomol. Screen. 18, 388–399 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Metcalf C. A., Sundaramoorthi R., Wang Y., Zou D., Thomas R. M., Zhu X., Cai L., Wen D., Liu S., Romero J., Qi J., Chen I., Banda G., Lentini S. P., Das S., Xu Q., Keats J., Wang F., Wardwell S., Ning Y., Snodgrass J. T., Broudy M. I., Russian K., Zhou T., Commodore L., Narasimhan N. I., Mohemmad Q. K., Iuliucci J., Rivera V. M., Dalgarno D. C., Sawyer T. K., Clackson T., Shakespeare W. C., Discovery of 3-[2-(imidazo[1,2-b]pyridazin-3-yl)ethynyl]-4-methyl-N-{4-[(4-methylpiperazin-1-yl)methyl]-3-(trifluoromethyl)phenyl}benzamide (AP24534), a potent, orally active pan-inhibitor of breakpoint cluster region-abelson (BCR-ABL) kinase including the T315I gatekeeper mutant. J. Med. Chem. 53, 4701–4719 (2010). [DOI] [PubMed] [Google Scholar]
- 17.Tucker J. A., Klein T., Breed J., Breeze A. L., Overman R., Phillips C., Norman R. A., Structural insights into FGFR kinase isoform selectivity: Diverse binding modes of AZD4547 and ponatinib in complex with FGFR1 and FGFR4. Structure 22, 1764–1774 (2014). [DOI] [PubMed] [Google Scholar]
- 18.Karpov A. S., Amiri P., Bellamacina C., Bellance M. H., Breitenstein W., Daniel D., Denay R., Fabbro D., Fernandez C., Galuba I., Guerro-Lagasse S., Gutmann S., Hinh L., Jahnke W., Klopp J., Lai A., Lindvall M. K., Ma S., Möbitz H., Pecchi S., Rummel G., Shoemaker K., Trappe J., Voliva C., Cowan-Jacob S. W., Marzinzik A. L., Optimization of a dibenzodiazepine hit to a potent and selective allosteric PAK1 inhibitor. ACS Med. Chem. Lett. 6, 776–781 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Uhlik M. T., Abell A. N., Johnson N. L., Sun W., Cuevas B. D., Lobel-Rice K. E., Horne E. A., Dell’Acqua M. L., Johnson G. L., Rac-MEKK3-MKK3 scaffolding for p38 MAPK activation during hyperosmotic shock. Nat. Cell Biol. 5, 1104–1110 (2003). [DOI] [PubMed] [Google Scholar]
- 20.Poch Martell M., Sibai H., Deotare U., Lipton J. H., Ponatinib in the therapy of chronic myeloid leukemia. Expert Rev. Hematol. 9, 923–932 (2016). [DOI] [PubMed] [Google Scholar]
- 21.Mably J. D., Chuang L. P., Serluca F. C., Mohideen M.-A. P. K., Chen J.-N., Fishman M. C., santa and valentine pattern concentric growth of cardiac myocardium in the zebrafish. Development 133, 3139–3146 (2006). [DOI] [PubMed] [Google Scholar]
- 22.Anastassiadis T., Deacon S. W., Devarajan K., Ma H., Peterson J. R., Comprehensive assay of kinase catalytic activity reveals features of kinase inhibitor selectivity. Nat. Biotechnol. 29, 1039–1045 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fisher O. S., Deng H., Liu D., Zhang Y., Wei R., Deng Y., Zhang F., Louvi A., Turk B. E., Boggon T. J., Su B., Structure and vascular function of MEKK3–cerebral cavernous malformations 2 complex. Nat. Commun. 6, 7937 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Whitehead K. J., Chan A. C., Navankasattusas S., Koh W., London N. R., Ling J., Mayo A. H., Drakos S. G., Jones C. A., Zhu W., Marchuk D. A., Davis G. E., Li D. Y., The cerebral cavernous malformation signaling pathway promotes vascular integrity via Rho GTPases. Nat. Med. 15, 177–184 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Najjar M., Suebsuwong C., Ray S. S., Thapa R. J., Maki J. L., Nogusa S., Shah S., Saleh D., Gough P. J., Bertin J., Yuan J., Balachandran S., Cuny G. D., Degterev A., Structure guided design of potent and selective ponatinib-based hybrid inhibitors for RIPK1. Cell Rep. 10, 1850–1860 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Clark P. R., Jensen T., Kluger M. S., Morelock M., Hanidu A., Qi Z., Tatake R. J., Pober J. S., MEK5 is activated by shear stress, activates ERK5 and induces KLF4 to modulate TNF responses in human dermal microvascular endothelial cells. Microcirculation 18, 102–117 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ye Y. E., Woodward C. N., Narasimhan N. I., Absorption, metabolism, and excretion of [14C]ponatinib after a single oral dose in humans. Cancer Chemother. Pharmacol. 79, 507–518 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shenkar R., Shi C., Austin C., Moore T., Lightle R., Cao Y., Zhang L., Wu M., Zeineddine H. A., Girard R., McDonald D. A., Rorrer A., Gallione C., Pytel P., Liao J. K., Marchuk D. A., Awad I. A., RhoA kinase inhibition with fasudil versus simvastatin in murine models of cerebral cavernous malformations. Stroke 48, 187–194 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bravi L., Rudini N., Cuttano R., Giampietro C., Maddaluno L., Ferrarini L., Adams R. H., Corada M., Boulday G., Tournier-Lasserve E., Dejana E., Lampugnani M. G., Sulindac metabolites decrease cerebrovascular malformations in CCM3-knockout mice. Proc. Natl. Acad. Sci. U.S.A. 112, 8421–8426 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang Y., Nakayama M., Pitulescu M. E., Schmidt T. S., Bochenek M. L., Sakakibara A., Adams S., Davy A., Deutsch U., Lüthi U., Barberis A., Benjamin L. E., Mäkinen T., Nobes C. D., Adams R. H., Ephrin-B2 controls VEGF-induced angiogenesis and lymphangiogenesis. Nature 465, 483–486 (2010). [DOI] [PubMed] [Google Scholar]
- 31.Zheng X., Xu C., Smith A. O., Stratman A. N., Zou Z., Kleaveland B., Yuan L., Didiku C., Sen A., Liu X., Skuli N., Zaslavsky A., Chen M., Cheng L., Davis G. E., Kahn M. L., Dynamic regulation of the cerebral cavernous malformation pathway controls vascular stability and growth. Dev. Cell 23, 342–355 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Young J. A., Ting K. K., Li J., Moller T., Dunn L., Lu Y., Lay A. J., Moses J., Prado-Lourenço L., Khachigian L. M., Ng M., Gregory P. A., Goodall G. J., Tsykin A., Lichtenstein I., Hahn C. N., Tran N., Shackel N., Kench J. G., McCaughan G., Vadas M. A., Gamble J. R., Regulation of vascular leak and recovery from ischemic injury by general and VE-cadherin-restricted miRNA antagonists of miR-27. Blood 122, 2911–2919 (2013). [DOI] [PubMed] [Google Scholar]
- 33.Choi J. P., Yang X., Foley M., Wang X., Zheng X., Induction and micro-CT imaging of cerebral cavernous malformations in mouse model. J. Vis. Exp. e56476 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schrödinger Release 2017-4: Prime (Schrödinger, LLC, 2017).
- 35.Schrödinger Release 2017-4: LigPrep (Schrödinger, LLC, 2017).
- 36.Schrödinger Release 2017-4: Glide (Schrödinger, LLC, 2017).
- 37.Herbert S. P., Stainier D. Y. R., Molecular control of endothelial cell behaviour during blood vessel morphogenesis. Nat. Rev. Mol. Cell Biol. 12, 551–564 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schrödinger Release 2017-4: Maestro (Schrödinger, LLC, 2017).
- 39.Cogger V. C., O’Reilly J. N., Warren A., Le Couteur D. G.. A standardized method for the analysis of liver sinusoidal endothelial cells and their fenestrations by scanning electron microscopy. J. Vis. Exp. e52698 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kimmel C. B., Ballard W. W., Kimmel S. R., Ullmann B., Schilling T. F.. Stages of embryonic development of the zebrafish. Dev. Dynam. 203, 253–310 (1995). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/11/eaau0731/DC1
Fig. S1. Three-dimensional structural comparison of the binding between ponatinib and Abl, FGFR4, and MEKK3.
Fig. S2. Two-dimensional mapping of ponatinib interaction with Abl, FGFR4, and MEKK3.
Fig. S3. CCM1, p38, and MEKK3 expression in cultured HUVECs.
Fig. S4. MEKK3 is the dominant regulator of KLF signaling.
Fig. S5. Confocal images of Tg(cmlc2:EGFP) embryos injected with control, ccm2, or ccm2 + mekk2 morpholinos.
Fig. S6. Effects of ponatinib and other kinase inhibitors on KLF2 expression.
Fig. S7. qPCR quantification of CCM gene expression in brain endothelial cells isolated from 4-HT–induced p6 pups.
Fig. S8. Hematoxylin and eosin staining of CCM lesions at P13.
Fig. S9. Hematoxylin and eosin staining of CCM lesions at P30.