

Abstract
Rationale:
Thrombotic thrombocytopenic purpura (TTP) is a rare, fatal disorder which could be caused by autoimmune diseases. However, TTP secondary to Sjögren syndrome (SS) is extremely rare.
Patient concerns:
A 47-year- old woman with an 8-year history of SS was admitted due to skin ecchymosis and bleeding gums. Then she gradually developed fever and headache.
Diagnoses:
Laboratory investigations suggested anemia, thrombocytopenia, increased lactic dehydrogenase, and a disintegrin-like metalloproteinase with thrombospondin motif type 1 member 13 (ADAMTS13) activity deficiency with high inhibitor titers. Acquired TTP was thus diagnosed.
Interventions:
Plasma exchange (PE) was the first choice for treatment, while glucocorticoid, cyclosporine A (CSA), rituximab, and intravenous immunoglobulin (IVIG) were used simultaneously. Bortezomib, a selective proteasome inhibitor and thereby inducing apoptosis in both B-cells and plasma cells, was added.
Outcomes:
She was discharged from the hospital and then treated with prednisone of 40 mg/d and hydroxychloroquine. The patient remained in full remission.
Lessons:
We conclude that bortezomib should be considered for patients with TTP refractory to PE, steroids, and rituximab due to its efficacy and relatively favorable side effect profile.
Keywords: ADAMTS13, bortezomib, Sjögren syndrome, thrombotic thrombocytopenic purpura
1. Introduction
As a critical disease with low incidence, thrombotic thrombocytopenic purpura (TTP) is commonly characterized by 5 typical symptoms, including fever, thrombocytopenia, microangiopathic hemolytic anemia, neurological symptoms, and renal damage.[1] Its pathogenesis is mainly due to deficiency of von Willebrand factor lyase (ADAMTS13) activity, which cannot cleave large von Willebrand factor (vWF) multimers in plasma, causing massive platelet (PLT) aggregation and extensive microvascular thrombosis. Plasma exchange (PE) is the most fundamental and optimal treatment for such a disorder, which can achieve an overall survival rate of 80% to 85%.[2] However, 40% of TTP patients still have early recurrence, and some refractory/recurrent TTP patients are facing death.[2] TTP can be secondary to a variety of autoimmune diseases. TTP secondary to Sjögren syndrome (SS) is not common, and refractory/recurrent manifestations are particularly rare. In the present study, we described and analyzed a case of refractory/recurrent TTP secondary to SS.
2. Case report
The study was approved by the Ethics Committee of the Soochow University. Informed written consent was obtained from the patient for publication of this case report and accompanying images. The medical records of the patient were anonymous. This was a 47-year-old woman with an 8-year history of SS. At diagnosis, she had a recurrent sensation of dry eyes, dry mouth, musculoskeletal pain, Raynaud phenomenon, and cough. The autoimmunity panel revealed the presence of nRNP, SSA/Ro, Ro52, and Ro60, and her antinuclear antibody titer reached 1/320. Ophthalmological examinations showed positive Bengal stain in each eye and shortened tear break-up time (both 2 s/5 min). Salivary gland biopsy indicated lymphocytic infiltration in salivary glands (a number of lymphocytic foci contained more than 50 lymphocytes) and fibrous tissue proliferation. She was treated with prednisone (30 mg/d) and hydroxychloroquine. Within her long-term follow-up, prednisone dose was gradually tapered at a dose of 5 mg/d, in addition to immunosuppressive mycophenolate mofetil of 1.5 g/d. After a 3-day treatment with cefixime and ibuprofen for toothache, she was admitted to our department on October 23, 2017 due to skin ecchymosis and bleeding gums. Her temperature and blood pressure were normal. She was further examined by laboratory tests. The counts of white blood cells were normal, while the PLT count was reduced to 2 × 109/L, and hemoglobin (HGB) content was 6.2 g/dL. Urinalysis revealed large amounts of urinary proteins and red blood cells. The titer of 24-hours urinary protein excretion was 0.60 g/d. The serum glutamic pyruvic transaminase, glutamic oxaloacetic transaminase, and total bilirubin levels were normal, whereas the lactic dehydrogenase (LDH) level was obviously increased to 624 U/L. The titer of C-reactive protein was 19.9 mg/L. Serum immunoglobulin G (IgG) and immunoglobulin A levels were slightly increased. Activated partial thromboplastin time was slightly prolonged. Prothrombin time, thrombin time, and fibrinogen remained unchanged. Moreover, results were negative for antidouble stranded DNA, antineutrophil antibody, anti-PLT antibody, and Comb test. The reticulocyte percentage was significantly increased. Bone marrow examination indicated iron-deficiency anemia, megakaryocyte dysmaturity, and thrombocytopenia.
The patient was immediately treated with methylprednisolone (MP) at a dose of 80 mg/d and intravenous immunoglobulin (IVIG) of 20 g/d for 5 consecutive days. The PLT count was decreased to 1 × 109/L. Therefore, MP pulse therapy (500 mg/d for 3 days) was given on hospital day 6. Intravenous steroids were also given (80 mg of MP daily except for the days of pulse therapy). Mycophenolate mofetil was changed to cyclosporine A (CSA) of 100 mg/d, in addition to IVIG (20 g/d for 5 days). The patient had a fever of 39°C and headache after 2 units of PLT transfusion. There were no obvious symptoms of infection. The head CT scan demonstrated lacunar infarction and right maxillary sinus inflammation. There was rare red cell debris in her peripheral blood smears. The diagnosis of TTP was confirmed based on a severe deficiency of ADAMTS13 activity as well as the presence of inhibitors. On hospital day 6, PE therapy (twice a day, each treatment contained 3000 mL plasma for a total of 17 times) and steroids were prescribed. The fever and headache were relieved significantly. PLT count was elevated from 90 to 180 × 109/L, and HGB was 98.0 g/L. LDH returned to normal range. However, her ADAMTS13 activity was 0%. The PLT function examination and lymphoma immunophenotyping were normal. The next-generation sequencing of the coagulation was used to detect heterozygous missense mutations in genes of ADAMTS13, vWF, and ITGA2B.
After another 55 times of PE, her PLT count still kept decreasing, finally reaching 21 × 109/L. On December 17, 2017, she received rituximab (375 mg/m2, once a week for 4 weeks), but she remained severely thrombocytopenic. On January 23, 2018, she received 2 circles of infusion of bortezomib (1.3 mg/m2, days 1, 4, 8, 11 on a 21-day cycle). She also received pulse MP (500 mg/d for 3 days and then reduced to 40 mg/d). On March 19, 2018, her ADAMTS13 activity returned to the normal level. The PLT count was increased to 66 × 109/L, HGB was increased to 91.0 g/L, and LDH returned to 384 U/L. She was discharged from the hospital and then treated with prednisone of 40 mg/d (Fig. 1) and hydroxychloroquine. The patient remained in full remission.
Figure 1.
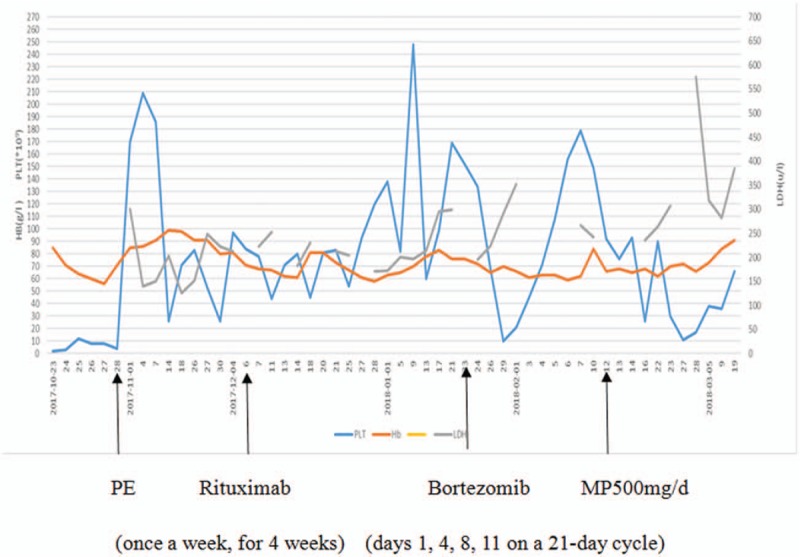
The changes of HGB, PLT and LDH, and treatment interventions during hospital course. HGB = hemoglobin, LDH = lactic dehydrogenase, MP = methylprednisolone, PE = plasma exchange, PLT = platelet.
3. Discussion
The cases of TTP associated with connective tissue disease are seldom reported. The spectrum of connective tissue disease causing acquired TTP includes systemic lupus erythematosus, mixed connective tissue disease, rheumatoid arthritis, systemic scleroderma, dermatomyositis, and antiphospholipid antibody syndrome. However, TTP secondary to SS is exceedingly rare. Only 11 cases of TTP complicating with SS have been reported in literatures in the past 40 years.[3] About half of these patients have not been diagnosed as SS previously. They also show variability in the severity of classic triad or pentad symptoms of TTP. Besides the damage to exocrine glands, SS can destroy the hematological system, with an incidence rate ranging from 34% to 44%. Its common clinical manifestations are thrombocytopenic purpura and immune-mediated hemolytic anemia. SS and TTP have nearly the same clinical manifestations in hematological system, leading to difficulty in distinguishing them. Our patient did not present with classic triad or pentad symptoms of TTP. There were no fragmented red cells in her peripheral blood smears. Her bone marrow examination indicated megakaryocyte dysmaturity and thrombocytopenia. The Coombs test was negative. All these information brought infrequent difficulties to differential diagnosis of anemia and thrombocytopenia. As our patient had a history of SS, we treated her with MP pulse. The immunosuppressive therapy was ineffective, and then the following PLT transfusion was administrated. Our SS patient complained of headache and fever. Finally, we confirmed the diagnosis of TTP according to decreased ADAMTS13 activity and positive anti-ADAMTS13 antibodies. As a result, the SS patients with hematological abnormalities, without classic TTP symptoms, are misdiagnosed and never diagnosed easily.
The pathogenesis of TTP secondary to SS still remains unclear. There are many autoantibodies in the serum of SS patients. Therefore, we speculated that the patient might have ADAMTS13 antibody in peripheral blood. Recent studies have shown that this antibody is a subtype of IgG. This anti-ADAMTS13 IgG usually inhibits the proteolytic activity of ADAMTS13 on VWF, resulting in the deposition of ULvWF on the vascular wall, injury of endothelium, and activation and aggregation of PLTs.[4]
According to the antibody of ADAMTS13, acquired TTP is divided into idiopathic TTP and secondary TTP. Acquired TTP is caused by autoimmune diseases, infections, tumors, drugs, pregnancy, and other factors.[5] In our case, drugs for relieving toothache could cause or trigger the TTP. The detection of decreased ADAMTS13 activity and positive expression of its inhibitor was helpful for the diagnostic progress of TTP and monitoring progression.[6] The persistently decreased ADAMTS13 activity and presence of inhibitor lead to a greater risk of relapse.[6] Our patient with severe ADAMTS13 deficiency (0%) became refractory and dependent on PE and steroids.
There is limited information or consensus on treatment of relapsed/refractory acquired TTP. In the management and treatment of refractory/relapsed TTP, PE (1.5 × plasma volume exchange for the first procedure) should be started at the early stage of TTP. PE can be performed even twice daily.[7] In general, standard-dose prednisone (1 mg/kg/d) can be used together with PE in patients with newly diagnosed TTP.[8] High-dose MP (10 mg/kg/d for 3 days) may be efficacious in refractory/relapsed TTP patients who do not respond to initial treatment of PE and low-dose steroids.[9] Other treatments include rituximab, vincristine, cyclophosphamide, CSA, and splenectomy.[10–13] Our case was treated with PE twice a day, 2 circles of high-dose MP pulse (500 mg/d for 3 days and then 80 mg/d), immunomodulators (CSA) and IVIG (20 g/d for 5 days), but the PLT count continued to fall below 30 × 109/L. Hemolysis did not cease such reduction, and severe ADAMTS13 deficiency (<10%) persisted.
Rituximab, a human-mouse chimeric monoclonal antibody binding to CD20, can kill normal B lymphocytes by complement-dependent cytotoxicity and antibody-dependent cytotoxicity, and deplete B-cell clones producing anti-ADAMTS13 antibodies. Therefore, rituximab has become a new and promising therapeutic approach for TTP,[14] which is currently recommended as a substitute of routine treatment for patients without significant effect of PE in the acute phase of TTP. In a multicenter, prospective, historical phase 2 clinical trial in the United Kingdom, rituximab in combination with PE is taken as a first-line treatment.[15] Bresin et al have reported that prophylactic infusion of rituximab given to patients at the remission phase with ADAMTS13 deficiency (activity <10%) can prevent the relapse of TTP.[16] Treating refractory/relapsed TTP with rituximab can improve remission rate of TTP, especially in patients with a severe ADAMTS13 deficiency and positive inhibitors.[17–19] A case meta-analysis has shown that the complete remission rate of rituximab in treating those refractory patients is up to 88%. In relapsed TTP, all patients achieve complete remission.[18] The optimal dose of rituximab has not been standardized. A recent article,[20] published in Hematology, has suggested that low-dose (100 mg, once a week for 4 weeks) of rituximab is equally effective as the standard-dose (375 mg/m2 once a week for 4 weeks) in TTP patients. Unfortunately, our patient did not respond to standard PE, steroids, CSA, IVIG, and rituximab. Though additional alternative therapies for refractory TTP, including cyclophosphamide, vincristine, and splenectomy, have been reported with variable success, considering the severity and specificity of this case, we selected bortezomib instead, a proteasome inhibitor.
The proteasome is an enzyme that degrades ubiquitous proteins in cells. Selective proteasome inhibitor bortezomib leads to cell cycle arrest and apoptosis. Bortezomib has been found to significantly reduce circulating autoantibody levels by inducing apoptosis of B and plasma cells,[21] which is of particular interest of autoimmune disease therapy. B cells form part of the physiopathology of SS. There have been reports of an increase in the number of plasma cells, which partly are Ro-52 or Ro-60 specific. Plasma cells without expression of CD20 are also important in the maintenance of the inflammatory response in SS. As an alternative treatment of SS, rituximab (anti-CD20 antibody) is not effective in targeting these plasma cells, which to a large extent may be responsible for the production of auto-antibodies.[22–27] However, the ability of bortezomib to target plasma cells has been well documented in plasma cell dyscrasia.[28] In patients with refractory liver and kidney transplant treated with rituximab, bortezomib has been shown to significantly reduce pathological antibody titers and promote long-term survival of organs.[15,29,30] It has been reported [31] that bortezomib can effectively ameliorate experimental SS in mice by inhibiting the Th17 response, which is helpful for the development of a novel therapeutic strategy for SS. Furthermore, Jakez-Ocampo et al[32] have reported a case of a severe SS patient refractory to conventional treatment in response to bortezomib. We believed that active treatment of the primary SS greatly contributed to improved curative effect of TTP.
Recently, bortezomib has been used to treat refractory/relapsed TTP,[33–41] which can eliminate residual anti-ADAMTS13 antibodies produced by plasma cells refractory to conventional immunosuppressive therapy.[34,35] There is an argument that auto-reactive B cells and plasma cells may produce ADAMTS13 antibodies, resulting in ongoing sequelae of PLT consumption in refractory TTP patients. Additional autoantibodies are thought to be synthesized from plasma cells. Bortezomib is administered to eradicate the autoantibody-producing plasma cells. In addition, bortezomib can induce apoptosis of immature dendritic cells, which is required for CD4+ T-cell activation and responsible for autoantibody production.[42] In 1 study, 5 relapsed TTP patients treated with bortezomib achieve complete remission, and 1 died of cardiac arrest with underlying cause.[39] In the past 10 years, 15 patients with relapsed/refractory acquired TTP have received bortezomib treatment. Moreover, 14 patients have survived and maintained remission during the acute attack (Table 1). Bortezomib directly exerts influence on plasma cells and significant inhibitory effect within 48 hours. This is why the refractory/relapsed TTP patients respond rapidly to bortezomib treatment.
Table 1.
Published cases with relapsed/refractory TTP who received bortezomib.

The source of IgG autoantibodies contains mature B cells and plasma cells. Rituximab, an effective treatment for TTP, can target against not CD20-negative plasma cells but CD20-positive B cells. Therefore, anti-ADAMTS13 autoantibody levels in TTP patients treated with standard rituximab may decrease, and the increase of ADAMTS13 activity due to continuous production of antibodies by plasma cells may delay. Our patient still showed severe deficiency of ADAMTS13 activity and presence of inhibitors after 4 circles of rituximab. Therefore, we assumed that the antibody response was mediated by long-lived plasma cells and but not inhibited by rituximab when the antibody titer was high and patients were continuously resistant to immunosuppressors.
Although a single case indicated that bortezomib was effective, we could not exclude the concomitant contribution of other immunosuppressive treatments and even omit the natural process of TTP. It was possible that combination therapy of rituximab and bortezomib might inhibit autoantibody production from CD20+ B cells and plasma cells. In many cases, prior to bortezomib administration, flow cytometry is used to confirm whether B cells are effectively depleted in peripheral blood serum. The combined effect depletes both specific mediated B cells and bortezomib-mediated plasma cells. Although the contribution of previous therapy cannot be ignored, the relationship between administration time and clinical improvement (almost 1 cycle) significantly contributes to dramatic bortezomib-specific therapeutic effects.[43–45] However, bortezomib-induced acquired TTP has been reported in the literature. Microvascular hemolytic anemia, thrombocytopenia, and neurologic symptoms have been documented after bortezomib administration. With drug withdrawal and PE therapy, the patient was clinically improved, PLT count was increased, red cell debris was removed, and neurological symptoms and hemolysis disappeared within 2 days. Therefore, prospective trials are needed to make a security evaluation of bortezomib.
4. Conclusions
We concluded that bortezomib was an option for the treatment of refractory/relapsed TTP since it showed a therapeutic effect and acceptable adverse effect. Considering other limited treatments and their significant side effects, such as cyclosporine, cyclophosphamide, vincristine and splenectomy, bortezomib was particularly effective for TTP compared with PE, steroids, and rituximab. However, prospective studies of bortezomib in TTP would be cumulatively difficult due to the low incidence of relapsed/refractory TTP. It is important to register and analyze a large-scale retrospective case review of TTP treated with bortezomib. In the next few years, the therapeutic field in TTP should pass the original treatment strategy derived from clinical experience and broaden the evaluation of currently administrated promising new drugs, including acetylcysteine, recombinant ADAMTS13, and caplacizumab (glycoprotein-Ib/IX-vWF axis inhibition agent) by large international clinical trials.
Author contributions
Conceptualization: Rurong Sun, Min Wu.
Data curation: Weiying Gu, Jing Wang.
Investigation: Yingchun Ma.
Methodology: Yingchun Ma.
Validation: Rurong Sun, Weiying Gu, Yingchun Ma, Jing Wang, Min Wu.
Writing – original draft: Rurong Sun.
Writing – review & editing: Min Wu.
Footnotes
Abbreviations: CSA = cyclosporine A, IVIG = intravenous immunoglobulin, PE = plasma exchange, SS = Sjögren syndrome, TTP = thrombotic thrombocytopenic purpura.
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
The authors have no conflicts of interest to disclose.
References
- [1].Zhou Z, Nguyen TC, Guchhait P, et al. Von Willebrand factor, ADAMTS-13, and thrombotic thrombocytopenic purpura. Semin Thromb Hemost 2010;36:71–81. [DOI] [PubMed] [Google Scholar]
- [2].Kremer Hovinga JA, Vesely SK, Terrell DR, et al. Survival and relapse in patients with thrombotic thrombocytopenic purpura. Blood 2010;115:1500–11. [DOI] [PubMed] [Google Scholar]
- [3].Xu X, Zhu T, Wu D, et al. Sjögren's syndrome initially presented as thrombotic thrombocytopenic purpura in a male patient: a case report and literature review. Clin Rheumatol 2017;37:1421–6. [DOI] [PubMed] [Google Scholar]
- [4].Verbij FC, Fijnheer R, Voorberg J, et al. Acquired TTP: ADAMTS13 meets the immune system. Blood Rev 2014;28:227–34. [DOI] [PubMed] [Google Scholar]
- [5].Mariotte E, Azoulay E, Galicier L, et al. French Reference Center for Thrombotic Microangiopathies. Epidemiology and pathophysiology of adulthood-onset thrombotic microangiopathy with severe ADAMTS13 deficiency (thrombotic thrombocytopenic purpura): a cross-sectional analysis of the French national registry for thrombotic microangiopathy. Lancet Haematol 2016;3:e237–45. [DOI] [PubMed] [Google Scholar]
- [6].Ferrafi S, Scheiflinger F, Rieger M, et al. Prognostic value of anti ADAMTS13 antibody features (Ig isotype, titer, and inhibitory effect) in a cohort of 35 adult French patients undergoing a first episode of thrombotic microangiopathy with undetectable ADAMTS13 activity. Blood 2007;109:2815–22. [DOI] [PubMed] [Google Scholar]
- [7].Nguyen L, Li X, Duvall D, et al. Twice-daily plasma exchange for patients with refractory thrombotic thrombocytopenic purpura: the experience of the Oklahoma Registry, 1989 through 2006. Transfusion 2008;48:349–57. [DOI] [PubMed] [Google Scholar]
- [8].Bell WR, Braine HG, Ness PM, et al. Improved survival in thrombotic thrombocytopenic purpura-hemolytic uremic syndrome. Clinical experience in 108 patients. N Engl J Med 1991;325:398–403. [DOI] [PubMed] [Google Scholar]
- [9].Balduini CL, Gugliotta L, Luppi M, et al. High versus standard dose methylprednisolone in the acute phase of idiopathic thrombotic thrombocytopenic purpura: a randomized study. Ann Hematol 2010;89:591–6. [DOI] [PubMed] [Google Scholar]
- [10].Lim W, Vesely SK, George JN. The role of rituximab in the management of patients with acquired thrombotic thrombocytopenic purpura. Blood 2015;125:1526–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Beloncle F, Buffet M, Coindre JP, et al. Splenectomy and/or cyclophosphamide as salvage therapies in thrombotic thrombocytopenic purpura: the French TMA reference center experience. Transfusion 2012;52:2436–44. [DOI] [PubMed] [Google Scholar]
- [12].Yilmaz M, Eskazan AE, Unsal A, et al. Cyclosporin A therapy on idiopathic thrombotic thrombocytopenic purpura in the relapse setting: two case reports and a review of the literature. Transfusion 2013;53:1586–93. [DOI] [PubMed] [Google Scholar]
- [13].Kappers-Klunne MC, Wijermans P, Fijnheer R, et al. Splenectomy for the treatment of thrombotic thrombocytopenic purpura. Br J Haematol 2005;130:768–76. [DOI] [PubMed] [Google Scholar]
- [14].Hie M, Gay J, Galicier L, et al. Preemptive Rituximab infusions after remission efficiently prevent relapses in acquired thrombotic thrombocytopenic purpura. Blood 2014;124:204–10. [DOI] [PubMed] [Google Scholar]
- [15].Scully M, McDonald V, Cavenagh J, et al. A phase 2 study of the safety and efficiency of rituximab with plasma exchange in acute acquired thrombotic thrombocytopenic purpura. Blood 2014;118:1746–53. [DOI] [PubMed] [Google Scholar]
- [16].Bresin E, Gastoldi S, Daina E, et al. Rituximab as preemptive treatment in patients with thrombotic thrombocytopenic purpura and evidence of anti-ADAMTS13 autoantibodies. Thrombo Haemost 2009;101:233–8. [PubMed] [Google Scholar]
- [17].Fakhouri F, Vemant JV, Veyradier A, et al. Efficiency of curative and prophylactic treatment with rituximab in ADAMTS-l3 deficient thrombotic thrombocytopenic purpura: a study of 11 cases. Blood 2005;106:1932–7. [DOI] [PubMed] [Google Scholar]
- [18].Caramazza D, Quintini G, Abbene I, et al. Rituximab for managing relapsing or refractory patients with idiopathic thrombotic thrombocytopenic purpura-hemolytic uraemic syndrome. Blood Transfus 2010;8:203–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Scully M, Cohen H, Cavenagh J, et al. Remission in acute refractory and relapsing thrombotic thrombocytopenic purpura following rituximab is associated with a reduction in IgG antibodies to ADAMTS-13. Br J Haematol 2007;136:45l–6. [DOI] [PubMed] [Google Scholar]
- [20].Vazquez-Mellado A, Pequeño-Luévano M, Cantu-Rodriguez OG, et al. More about low-dose Rituximab and plasma exchange as first-line therapy for patients with thrombotic thrombocytopenic purpura. Hematology 2006;21:311–6. [DOI] [PubMed] [Google Scholar]
- [21].Mulder A, Heidt S, Vergunst M, et al. Proteasome inhibition profoundly affects activated human B cells. Transplantation 2013;95:1331–7. [DOI] [PubMed] [Google Scholar]
- [22].Szyszko EA, Brun JG, Skarstein K, et al. Phenotypic diversity of peripheral blood plasma cells in primary Sjögren's syndrome. Scand J Immunol 2011;73:18–28. [DOI] [PubMed] [Google Scholar]
- [23].Aqrawi LA, Brokstad KA, Jakobsen K, et al. Low number of memory B cells in the salivary glands of patients with primary Sjögren's syndrome. Autoimmunity 2012;45:547–55. [DOI] [PubMed] [Google Scholar]
- [24].Szyszko EA, Brokstad KA, Oijordsbakken G, et al. Salivary glands of primary Sjögren's syndrome patients express factors vital for plasma cell survival. Arthritis Res Ther 2011;13:R2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Aqrawi LA, Skarstein K, Øijordsbakken G, et al. Ro52- and Ro60-specific B cell pattern in the salivary glands of patients with primary Sjögren's syndrome. Clin Exp Immunol 2013;172:228–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Aqrawi LA, Skarstein K, Bredholt G, et al. Autoantigen specific memory B cells in primary Sjogren's syndrome. Scand J Immunol 2012;75:61–8. [DOI] [PubMed] [Google Scholar]
- [27].Hiepe F, Dörner T, Hauser AE, et al. Long-lived autoreactive plasma cells drive persistent autoimmune inflammation. Nat Rev Rheumatol 2011;7:170–8. [DOI] [PubMed] [Google Scholar]
- [28].Painuly U, Kumar S. Efficacy of bortezomib as first-line treatment for patients with multiple myeloma. Clin Med Insights Oncol 2013;7:53–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Everly MJ, Everly JJ, Susskind B, et al. Bortezomib provides effective therapy for antibody- and cell-mediated acute rejection. Transplantation 2008;86:1754–61. [DOI] [PubMed] [Google Scholar]
- [30].Paterno F, Shiller M, Tillery G, et al. Bortezomib for acute antibody-mediated rejection in liver transplantation. Am J Transplant 2012;12:2526–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Xiao F, Lin X, Tian J, et al. Proteasome inhibition suppresses Th17 cell generation and ameliorates autoimmune development in experimental Sjögren's syndrome. Cell Mol Immunol 2017;Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Jakez-Ocampo J, Atisha-Fregoso Y, Llorente L. Refractory primary Sjögren syndrome successfully treated with bortezomib. J Clin Rheumatol 2015;21:31–2. [DOI] [PubMed] [Google Scholar]
- [33].Shortt J, Oh DH, Opat SS. ADAMTS13 antibody depletion by bortezomib in thrombotic thrombocytopenic purpura. N Engl J Med 2013;368:90–2. [DOI] [PubMed] [Google Scholar]
- [34].Yates S, Matevosyan K, Rutherford C, et al. Bortezomib for chronic relapsing thrombotic thrombocytopenic purpura: a case report. Transfusion 2014;54:2064–7. [DOI] [PubMed] [Google Scholar]
- [35].Mazepa MA, Raval JS, Moll S, et al. Bortezomib induces clinical remission and reduction of ADAMTS13 inhibitory antibodies in relapsed refractory idiopathic thrombotic thrombocytopenic purpura. Br J Haematol 2014;164:900–2. [DOI] [PubMed] [Google Scholar]
- [36].van Balen T, Schreuder MF, de Jong H, et al. Refractory thrombotic thrombocytopenic purpura in a 16-year-old girl: successful treatment with bortezomib. Eur J Haematol 2014;92:80–2. [DOI] [PubMed] [Google Scholar]
- [37].Patel PP, Becker J, Freyer C, et al. Rituximab-refractory thrombotic thrombocytopenic purpura responsive to intravenous but not subcutaneous bortezomib. Transfusion 2016;56:970–4. [DOI] [PubMed] [Google Scholar]
- [38].Acedillo RR, Govind M, Kashgary A, et al. Treatment of severe, refractory and rapidly evolving thrombotic thrombocytopenic purpura. BMJ Case Rep 2016;doi:10.1136/bcr-2016-215491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Patriquin CJ, Thomas MR, Dutt T, et al. Bortezomib in the treatment of refractory thrombotic thrombocytopenic purpura. Br J Haematol 2016;173:779–85. [DOI] [PubMed] [Google Scholar]
- [40].Ratnasingam S, Walker PA, Tran H, et al. Bortezomib-based antibody depletion for refractory autoimmune hematological diseases. Blood Adv 2016;1:31–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Pandey MR, Vachhani P, Ontiveros EP. Remission of severe, relapsed, and refractory TTP after multiple cycles of bortezomib. Case Rep Hematol 2017;2017:9681832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Park SJ, Cheong HI, Shin JI. Antibody depletion by bortezomib through blocking of antigen presentation. N Engl J Med 2013;368:1364–5. [DOI] [PubMed] [Google Scholar]
- [43].Morita R, Hashino S, Shirai S, et al. Thrombotic microangiopathy after treatment with bortezomib and dexamethasone in a patient with multiple myeloma. Int J Hematol 2008;88:248–50. [DOI] [PubMed] [Google Scholar]
- [44].Moore H, Romeril K. Multiple myeloma presenting with a fever of unknown origin and development of thrombotic thrombocytopenic purpura post-bortezomib. Intern Med J 2011;41:348–50. [DOI] [PubMed] [Google Scholar]
- [45].Mehta N, Saxena A, Niesvizky R. Bortezomib-induced thrombotic thrombocytopenic purpura. BMJ Case Rep 2012;doi:10.1136/bcr-2012-006461. [DOI] [PMC free article] [PubMed] [Google Scholar]