

Abstract
Rationale:
X-linked lymphoproliferative syndromes (XLPs) are rare, yet often fatal primary immunodeficiency diseases, which rarely manifest as Langerhans cell histiocytosis (LCH) complicated with hemophagocytic lymphohistiocytosis (HLH). Clinical data of a case of XLP-2 manifesting as LCH complicated with HLH was retrospectively analyzed to determine the etiology and causal gene.
Patient concerns and diagnosis:
The diagnosis of multisystem LCH was confirmed by skin biopsy and other examinations in a 13-month boy with recurrent ear discharge, fever and hemorrhagic papules for 3 months. A good therapeutic response to LCH-III protocol-directed induction chemotherapy was achieved but unremitting HLH developed 6 weeks after the initiation of induction chemotherapy. To identify possible underlying genetic causes, gene mutation analysis was undertaken. A novel XIAP gene mutation (c.99delT, p.F33fsX37) was documented.
Interventions:
After the diagnosis of HLH had been confirmed, HLH-2004-directed chemotherapy was instituted.
Outcomes:
The clinical condition of the patient had become progressively deteriorating after 8-week chemotherapy of HLH-2004 protocol, requiring frequent infusions of RBC suspensions and apheresis platelets. His parents decided to receive no further therapy, and the patient died soon after discharge.
Lessons:
Meticulous laboratory investigations including genetic studies should be undertaken in young children with LCH complicated with HLH and poor therapeutic response.
Keywords: hemophagocytic lymphohistiocytosis, Langerhans cell histiocytosis, x-linked lymphoproliferative disease
1. Introduction
X-linked lymphoproliferative syndrome (XLP) is a rare form of primary immunodeficiency caused either by SH2D1A or XIAP gene mutation and known as XLP-1 and XLP-2 respectively.[1] Hemophagocytic lymphohistiocytosis (HLH), a group of macrophage-related histiocytic disorders, occurs frequently in the setting of XLP and is most commonly triggered by Epstein-Barr virus (EBV) infection. Langerhans cell histiocytosis (LCH), another group of dendritic cell-related histiocytic disorders, is most prevalent in childhood.[2,3] We report here for the first time a boy with biopsy-proven LCH and HLH secondary to XLP-2 with underlying XIAP gene mutation.
2. Case report
A 13-month-old boy with recurrent ear discharge, fever, and hemorrhagic papules for 3 months was admitted to our hospital. The boy looked pale. Hemorrhagic papules over his abdomen and white mucoid secretion in bilateral external auditory canals were documented. No palpable skull mass and defect were found. The liver and spleen were palpable at 4 cm and 5 cm below the costal margin, respectively. Complete blood count revealed that Hb was 65 g/L, WBC 2.4 × 109/L, absolute neutrophil count 0.77 × 109/L, and platelet 41 × 109/L. The liver function, total plasma triglycerides, serum ferritin, and coagulation screening tests were normal. Serological investigations, including cytomegalovirus (CMV), hepatitis B, hepatitis C, Mycoplasma, Rubella, EBV, Salmonella, and HIV were all negative. Skin biopsy confirmed the diagnosis of LCH, with positive expression of CD1a, S100, and CD207 (Langerin). Bone marrow aspiration showed no hemophagocytosis. Diffuse interstitial infiltration in the lungs, bilateral pleural effusion, hepatosplenomegaly, and peritoneal effusion were documented by CT scanning. Accordingly, the diagnosis of multisystem LCH was made. The clinical condition improved remarkably 1 month after the initiation of LCH-III protocol-based induction chemotherapy, with almost complete disappearance of skin rashes and remarkable retraction of enlarged liver and spleen. Nevertheless, he suffered from high fever (over 39°C), with concomitant reappearance of hemorrhagic skin eruptions spread over the trunk and foot base, and even enlarged liver and spleen in the 6th week of induction chemotherapy. Laboratory investigations revealed progressive panctocytopenia and hyperferritinemia (serum ferritin 959.9ng/mL), and conspicuous hemophagocytosis. EBV-IgM, TORCH, blood culture were negative. In order to elucidate the underlying etiology of HLH, genetic investigation was carried out in the 3rd months of disease onset. A novel mutation (c.99delT, p.F33fsX70) in exon 2 of the XIAP gene was documented (Fig. 1). Therefore, the diagnosis of genetic HLH secondary to XLP-2 was unequivocally proven. Nevertheless, the same mutation was not detected in his mother and old brother, suggesting a de novo XIAP mutation. Accordingly, HLH-2004-directed chemotherapy was instituted. However, the clinical condition became progressively deteriorating after 8-week chemotherapy, requiring frequent infusions of RBC suspensions and apheresis platelets. His parents decided to receive no further therapy, and he died soon after discharge.
Figure 1.
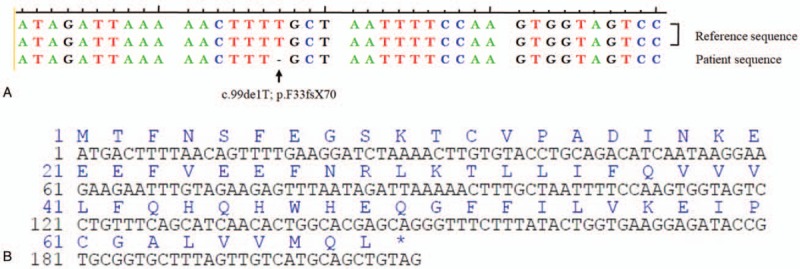
Result of XIAP sequence analysis in the patient. A. Deletion of the 99th base, T, in exon 2 of XIAP resulted in a frame-shift mutation. B. The partial coding and amino acid sequences of XIAP in the patient indicating the frame-shift mutation.
3. Discussion
XLP is a rare form of primary immunodeficiency, with X-linked mode of inheritance. Collectively, the annual incidence of XLP is approximately 2 to 3 per million. XLP-1 caused by SH2D1A mutation accounts for the majority of cases (60%–97%), while XLP-2 due to XIAP mutation comprises just about 10% of cases.[1,4]
Although XIAP gene was discovered early in 1996, the direct link between XIAP mutation and XLP was definitely proven in 2006 when XIAP mutation was detected in a patient cohort with XLP-1 phenotype without SH2D1A mutation.[5] Since then, XLP-2 due to XIAP mutation was recognized as an independent disease entity distinct from XLP-1.
Clinically, XLP -2 is exceedingly rare, with less than 100 cases reported to date. XLP most often presents within the first year of life, with a variety of clinical manifestations and is characterized by frequent infections, hypogammaglobulinemia and increased risk of lymphoma. As compared to XLP-1, HLH develops in up to 90% of patients with XLP-2 and is most commonly triggered by EBV infection (30%–70% of cases) or CMV infection (20% of cases).[1,6] In addition, HLH associated with XLP-2 tends to be recurrent, but with relatively mild disease severity.[6] This could be explained by relatively preserved NK and T cell-mediated cytotoxic response in XLP-2 patients as against to XLP-1 counterparts.
As stated previously, both LCH and HLH belong to histiocytic disorders but are dendritic cell-related and macrophage-related respectively. Numerous researches in the last decade have shown that LCH is a clonal myeloid neoplasm with over 50% of cases harboring BRAF V600E point mutation.[7,8] HLH secondary to LCH has been reported sparsely and is primarily found in young children with multisystem LCH, which could be triggered by a variety of infectious and non-infectious factors. Nevertheless, the majority of studies were case reports or small series studies.[9–11] So far, the study with largest patient cohort regarding the interrelationship between childhood LCH and HLH showed that a tentative diagnosis of HLH could be made in 12 out of 30 cases (40%) of childhood LCH, with 7 cases (23.3%) fully fulfilling the clinical diagnostic criteria of HLH. The authors speculated that cytokines produced by activated cytotoxic T lymphocytes or CD1a positive histiocytes might be the underlying mechanism of macrophage activation and resultant HLH.[12] Whether HLH in patients with LCH is just triggered by viral infections or has an underlying genetic basis remains elusive.
To our best knowledge, there has been just 1 case report of LCH in patients with XLP-1, but none in patients with XLP-2.[13] Here, we reported for the first time a boy with XLP-2-associated genetic HLH and coexisting biopsy-proven LCH. Genetic analysis revealed a novel c.99delT in exon 2 of the XIAP gene, resulting in a frame-shift mutation and premature termination of XIAP synthesis (p.F33fsX70). The majority of HLH in patients with LCH are multisystem disorders with extensive infiltration of organ-system by CD1a positive pathologic Langerhans cells.[12] Although without documented EBV infection, we speculate that HLH development might be triggered by other unidentified viral infection in our index case that is immunocompromised. Alternatively, HLH might be more likely triggered by marked cytokine release from CD1a positive pathologic Langerhans cells and sustained macrophage activation possibly related to LCH-directed chemotherapy, as evidenced by HLH onset in the 6th week of induction therapy.[10]
Of particular clinical importance is that great efforts should be made to identify triggering factors or underlying genetic defects in young children with LCH characterized by poor response to HLH-direct chemotherapy or unremitting and deteriorating clinical course. Further studies are needed to elucidate the possible link between HLH and LCH.
Author contributions
Data curation: Xia Guo.
Formal analysis: Xia Guo, Qiang Li, Ju Gao.
Project administration: Xia Guo.
Writing – original draft: Xia Guo.
Writing – review & editing: Xia Guo, Qiang Li, Ju Gao.
Footnotes
Abbreviations: CMV = cytomegalovirus, EBV = Epstein-Barr virus, HLH = hemophagocytic lymphohistiocytosis, LCH = Langerhans cell histiocytosis, XLP = X-linked lymphoproliferative syndrome.
This research was supported by the research fund from Sichuan Provincial Health Bureau (No. 15012) and Applied Basic Research Project of Science and Technology, Sichuan Province, China (No. 2015JY0044).
An oral consent via telephone had been given by the parents who lived in a remote village in Yun Nan province. Because it was not a clinical trial and no off-label drugs were used, the ethical approval is not necessary for this case report.
QL and JG contributed equally to the work and should be listed as the co-responding authors
The authors have no conflicts of interest to disclose.
References
- [1].Filipovich AH, Zhang K, Snow AL, et al. X-linked lymphoproliferative syndromes: brothers or distant cousins? Blood 2010;116:3398–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Erker C, Harker-Murray P, Talano JA. Usual and unusual manifestations of familial hemophagocytic lymphohistiocytosis and langerhans cell histiocytosis. Pediatr Clin North Am 2017;64:91–109. [DOI] [PubMed] [Google Scholar]
- [3].Ranganathan S. Histiocytic proliferations. Semin Diagn Pathol 2016;33:396–409. [DOI] [PubMed] [Google Scholar]
- [4].Sumegi J, Huang D, Lanyi A, et al. Correlation of mutations of the SH2D1A gene and epstein-barr virus infection with clinical phenotype and outcome in X-linked lymphoproliferative disease. Blood 2000;96:3118–25. [PubMed] [Google Scholar]
- [5].Rigaud S, Fondaneche MC, Lambert N, et al. XIAP deficiency in humans causes an X-linked lymphoproliferative syndrome. Nature 2006;444:110–4. [DOI] [PubMed] [Google Scholar]
- [6].Marsh RA, Madden L, Kitchen BJ, et al. XIAP deficiency: a unique primary immunodeficiency best classified as X-linked familial hemophagocytic lymphohistiocytosis and not as X-linked lymphoproliferative disease. Blood 2010;116:1079–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Berres ML, Merad M, Allen CE. Progress in understanding the pathogenesis of Langerhans cell histiocytosis: back to histiocytosis X. Br J Haematol 2015;169:3–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Delprat C, Arico M. Blood spotlight on Langerhans cell histiocytosis. Blood 2014;124:867–72. [DOI] [PubMed] [Google Scholar]
- [9].Washio K, Muraoka M, Kanamitsu K, et al. A case of refractory Langerhans cell histiocytosis complicated with hemophagocytic lymphohistiocytosis rescued by cord blood transplantation with reduced-intensity conditioning. Acta Med Okayama 2017;71:249–54. [DOI] [PubMed] [Google Scholar]
- [10].Dokmanovic L, Krstovski N, Jankovic S, et al. Hemophagocytic lymphohistiocytosis arising in a child with Langerhans cell histiocytosis. Turkish J Pediatr 2014;56:452–7. [PubMed] [Google Scholar]
- [11].Povoas MI, Luis PP, Esteves I, et al. Severe Langerhans cell histiocytosis in an infant: haemophagocytic syndrome association. BMJ Case Rep 2014;pii: bcr2014206983.doi:10.1136/bcr-2014-206983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Favara BE, Jaffe R, Egeler RM. Macrophage activation and hemophagocytic syndrome in langerhans cell histiocytosis: report of 30 cases. Pediatr Dev Pathol 2002;5:130–40. [DOI] [PubMed] [Google Scholar]
- [13].Zhang X, Zhu D, Lan H, et al. Langerhans cell histiocytosis, a new clinical phenotype of x-linked lymphoproliferative disease. Eur J Med Genet 2011;54:165–8. [DOI] [PubMed] [Google Scholar]