

Abstract
Biological membranes and their diverse lipid constituents play key roles in a broad spectrum of cellular and physiological processes. Characterization of membrane-associated phenomena at a microscopic level is therefore essential to our fundamental understanding of such processes. Due to the semi-fluid and dynamic nature of lipid bilayers, and their complex compositions, detailed characterization of biological membranes at an atomic scale has been refractory to experimental approaches. Computational modeling and simulation offer a highly complementary toolset with sufficient spatial and temporal resolutions to fill this gap. Here, we review recent molecular dynamics studies focusing on the diversity of lipid composition of biological membranes, or aiming at the characterization of lipid-protein interaction, with the overall goal of dissecting how lipids impact biological roles of the cellular membranes.
Keywords: Molecular Dynamics, Lipid Bilayers, Biological Membranes, Molecular Simulation, Membrane Proteins, Peripheral Proteins
Introduction
Biological membranes define the physical boundaries of living cells and their internal compartments. Composed of a myriad of diverse molecular constituents, ranging from phospholipids and other small amphiphilic/hydrophobic molecules, to peptides and proteins, many components of biological membranes have been shown to directly participate in a multitude of cellular processes, e.g., signaling, transport, cell-cell communication, and chemical catalysis. Moreover, a considerable fraction of cellular processes take place at the membrane or its proximity, where the membrane serves as a platform for signaling partners to convene, assemble and function.
At least 1/3 of human genes encode membrane or secretory proteins, and approximately 1/2 of drug targets are membrane proteins. The function of these proteins depends not only on their localization in the membrane but also quite often regulated by specific interactions with certain lipid molecules. As a result, investigating lipids and characterizing their functional roles are now an integral part of modern structural and functional studies of membrane proteins. However, even with the significant progress made over the last decade in structural biology of membrane proteins, lipids and membranes continue to pose a major challenge to detailed experimental structural studies. This is mostly due to the highly dynamic nature of lipid bilayers, compounded by the diversity of the molecular constituents commonly found in biological membranes. Computational modeling methods, in particular molecular dynamics (MD) simulations, have served as a powerful complementary approach to experiments in this regard and have been used quite effectively to fill the gap in our structural description of biological membranes.
In this article, we review recent molecular simulation studies of biological membranes, where the focus has been on the characterization of functional roles of diverse membrane lipid constituents, most importantly in the context of structure, dynamics, and function of membrane-associated proteins.
Heterogeneous Lipid Composition of Biological Membranes
The majority of MD simulations of biological membranes and membrane proteins have been limited to homogeneous bilayers of a single lipid type, particularly glycerophospholipids such as POPC (most representative of eukaryotic cells), and POPE (for bacterial membranes). These bilayers, however, do not emulate realistic biological membranes, as other membrane components including sphingolipids, sterols, glycolipids (Fig. 1), play essential roles in not only maintaining membrane integrity and properties but also in its function. Several membrane building tools, e.g., CHARMM-GUI [1], LipidBuilder [2], insane [3], and MemProtMD [4]), paired with continuously improved contemporary lipid force fields [5], have now enabled the construction and simulation of heterogeneous membrane systems of diverse lipid compositions.
Figure 1:
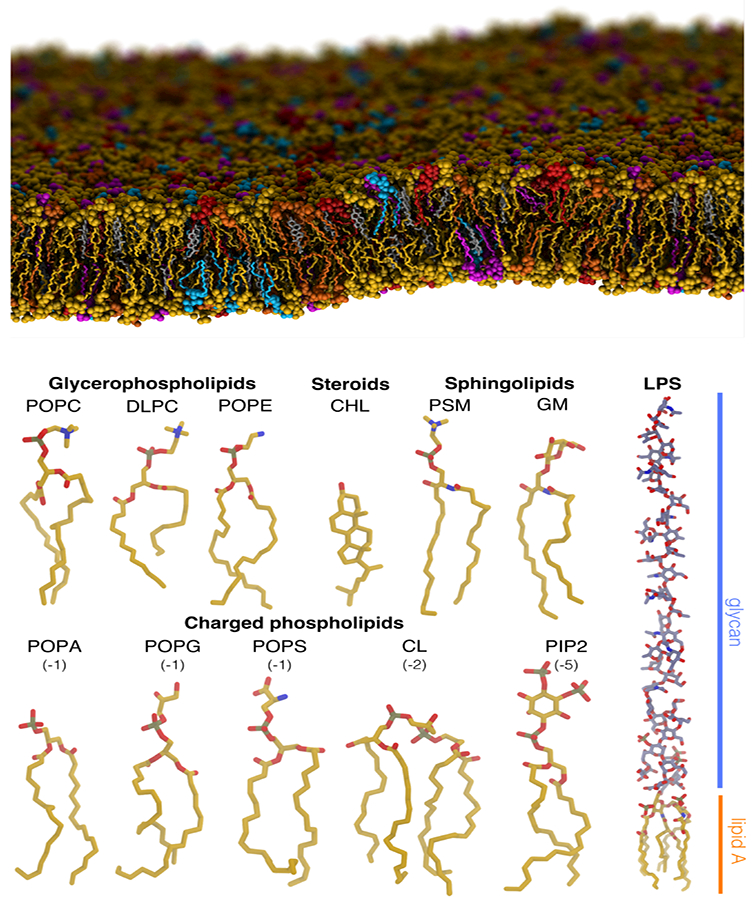
Heterogeneous lipid composition of biological membranes. (Top) An atomistic MD simulation system of a membrane with a complex, heterogeneous lipid composition. Seven different lipids, including cholesterol, cardiolipin, and a variety of phospholipids, are shown in different colors. Spontaneous curvature of the membrane arising from thermal fluctuations in the simulation can be observed. (Bottom) Structures of some exemplary lipids highlighting a variety of important features (e.g., head group charge and size, tail length and saturation, etc.) associated with lipids. Phospholipids and cholesterol (CHL) are major constituents of cellular membranes. Sphingolipids are common signaling lipids, cardiolipins (CL) are essential mitochondrial lipids, and lipopolysaccharides (LPS) are vital bacterial lipids of the outer membrane.
Cholesterol (CHL), a sterol, and sphingomyelin (SM), a sphingolipid, are major lipids, besides glycerophospholipids, in the cytoplasmic membranes of animal cells. These lipids not only have cellular regulatory functions but also modulate membrane structure and properties. Several extensive MD studies have shown that their presence affects the packing and thickness of the membrane, as well as its phase-transition temperature [6–9]. At certain concentrations, CHL and SM increase lipid ordering and decrease surface area, consequently increasing membrane condensation [10]. These effects were also found by MD studies to modulate transport properties, e.g., the permeability of small molecules, even nonpolar gases, through a lipid bilayer [11] and membrane dipole potentials [12]. Membrane lipid composition also plays a substantial role in the function of its occupants, and through various mechanisms (Fig. 2). For instance, an MD study reported composition-dependent partitioning of several key neurotransmitters that correlated with the membrane-relative position of their respective protein binding sites and the charge of lipid and neurotransmitter molecules [13].
Figure 2:
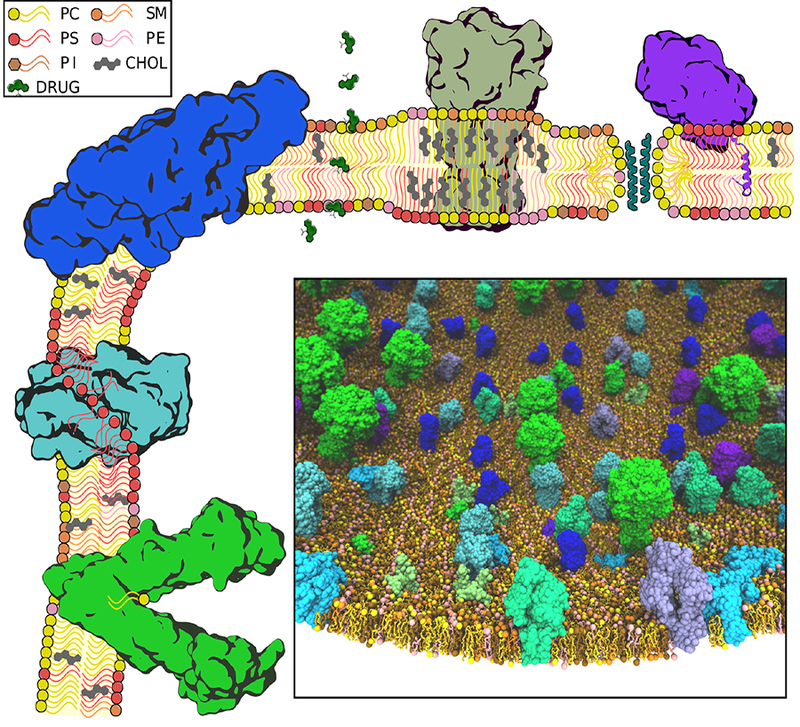
Diverse membrane-associated proteins and various modes of lipid interactions. Various lipid constituents and other small molecules (e.g., drugs) primarily partitioning in the membrane are drawn schematically (see inset key). Exemplary protein components with demonstrated significant interaction with lipids/membrane are drawn using a schematic-looking image based on their actual structure: green: an ABC transporter, bound to a lipid to be transported; cyan: scramblase mediating lipids traversing the two leaflets; blue: envelope and membrane proteins from dengue virus inducing a positive membrane curvature; olive: P2× receptor partitioned in a cholesterol-rich lipid raft, with an increased thickness and higher lipid order; teal: aggregation of antimicrobial peptides resulting in thoroidal pore formation; and, purple: cytochrome P450 anchored to the membrane through an inserted helix. (Inset) A fully atomistic model of a mesoscopic, heterogenous slice of the cellular membrane including various membrane-associated proteins. Several different types of proteins, including both peripheral and integral membrane proteins (shown in different colors) are included in the model. Construction of such complex molecular systems requires methodical placement of proteins and lipids and careful treatment of lipid-protein interfaces to ensure optimal packing.
Other important membrane lipids include, but are not limited to, lipopolysaccharides (LPS) and cardiolipins (CL). LPS, the most challenging lipid to model and simulate, is found in the outer leaflet of the outer membrane of Gram-negative bacteria, while CL is a major constituent of mitochondrial inner membranes of eukaryotes and the cytoplasmic membrane of prokaryotes. Due to the rise in antibiotic resistance, and the role of the outer membrane as a major permeation barrier, efforts have been made to understand the effects of LPS at molecular levels. An atomistic MD study showed that the crowding of LPS affects the diffusion of ions through OmpF porins [14]. A coarse-grained (CG) MD study of the antibiotic PMB1 correlated its antibacterial properties, at least in part, with the ability to alter the structure of the LPS-containing outer membrane [15]. A CG builder of bacterial membranes with LPS recently implemented in CHARMM-GUI has facilitated the modeling and simulation of the heterogeneous outer membrane systems [16]. A further study simulated CG models of a bacterial envelope comprising both outer and inner (cytoplasmic) membranes and associated membrane proteins, and revealed the influences of proteins on lipid distributions in the membrane and membrane morphology [17].
Coupling of Physical Properties of Membranes and Biological Function
The physical properties of membranes play vital roles in biological function, and computational techniques have begun to be used to illuminate the molecular bases of these roles. Most prominently, membrane curvature has been explored by MD in a variety of ways. MD can be used to determine the mechanism by which proteins induce curvature in membranes. For example, a recent study used CG MD to establish the structural basis by which the membrane proteins of dengue virus sculpt an otherwise spherical membrane into an icosahedral shape [18]. Another CG MD study demonstrated that ENTH domains, proteins involved in endocytosis, act cooperatively to transform thermal undulations of the membrane into larger scale, organized curvature [19]. MD can also be used to discover unexpected functionally relevant curvature. In an exemplary study, atomistic MD was used to suggest that polymyxin B, a ubiquitous antimicrobial peptide, disrupts the function of bacteria, in part, by inducing curvature in their outer membranes [20]. Finally, MD can be used to understand how lipids themselves stabilize curvature. For example, using CG MD, a recent study showed that a hemifusion diaphragm (HD), an intermediate state in the fundamental process of membrane fusion, relaxes to three distinct states (double bilayer, fusion pore, and metastable HD) depending on the proportion of lipids in the bilayer whose geometry favor negative curvature [21].
Beyond curvature, computational techniques can be used to explore the functional implications of other physical properties of the membrane, such as its thickness. For example, osmotic-gradient experiments in combination with atomistic MD have shown that the permeability of AQP4, the primary water channel in the mammalian brain, diminishes with decreasing bilayer thickness [22]. Atomistic MD simulations, in conjunction with in vivo and in vitro experiments, have also suggested that thicker membranes make oligomerization of Ire1, which plays an important role during viral infections and in certain types of cancer, more energetically favorable (Fig. 3) [23]. MD can be used to elucidate how drugs affect the thickness of the membrane. For instance, in conjunction with X-ray diffraction experiments, atomistic MD was used to show that caffeine, used medically as a drug adjuvant, increases the thickness of the membrane, perhaps explaining observed improved drug effectiveness when taken with caffeine [24]. MD is not the only computational technique that can be used to investigate membrane thickness. A recently developed continuum elastic model made predictions of the protein-induced changes in membrane thickness consistent with atomistic MD results at a fraction of the computational cost, paving the way for rapid characterization of membrane deformations at much larger scales [25]. Overall, computational techniques provide otherwise unobtainable insight into the fundamental molecular mechanisms derived from the membrane’s physical properties that generate basic biological function.
Figure 3:
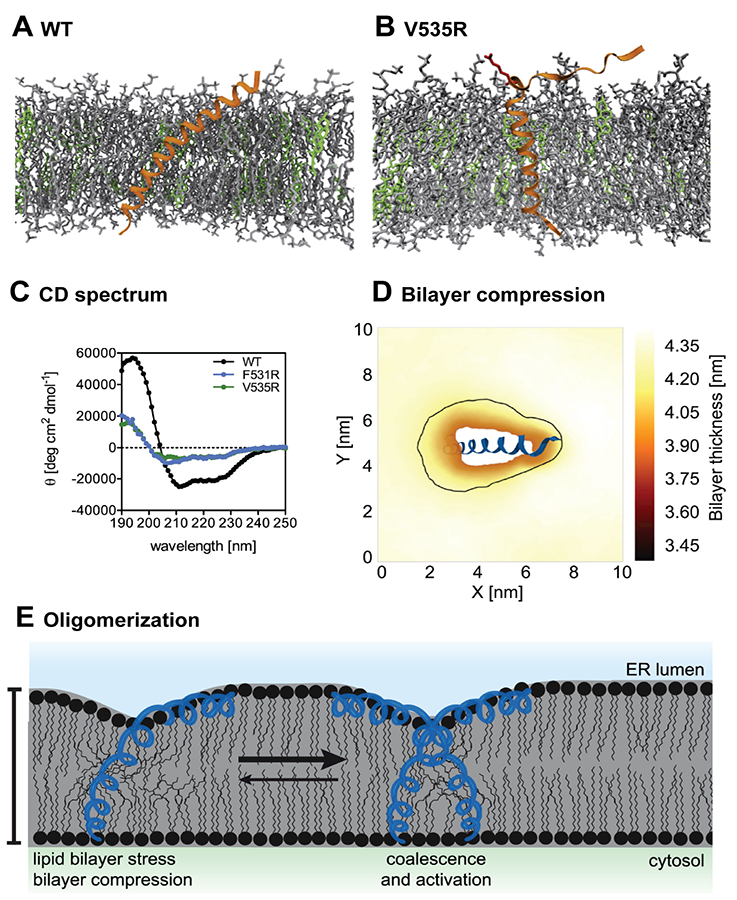
Cholesterol effect on the activation of unfolded protein response. The Ire1-derived sensor peptide and the associated V535R mutant are represented by orange helices in (A) and (B) with the amphipathic helix (AH) highlighted in red. Cholesterols and unsaturated phospholipids (POPC and DOPC) are respectively colored in green and gray. Helicity of the peptides was measured by circular dichroism (CD) spectroscopy (C) and together with MD simulations shows that a conserved structure of the AH is necessary for the sensor peptide to tilt. The presence of cholesterol thickens the membrane, and enhances the membrane compression upon the tilting of the sensor peptide (D). The enhanced bilayer stress induced upon the membrane compression facilitates the oligomerization of Ire1, and promotes the activation of unfolded protein response (E). The images are taken from Halbleib et al [23] with permission from Cell Press.
Membrane as a Platform for Protein Function
Interaction with the membrane is essential for peripheral membrane proteins (Fig. 2) to perform their biological functions and is facilitated by several elements, such as charge-charge interactions, protein structural features and hydrogen bonding effects.
Charged lipids and their counterions are often needed for the membrane binding of peripheral membrane proteins. For instance, Tim receptors are functionally associated with phosphatidylserine (PS) lipids. A combined MD and X-ray reflectivity study on one of its members, Tim1, demonstrated the role of Ca2+ ions in PS recognition and revealed two potential Ca2+-PS binding states, resulting in differential insertion depths of the protein [26]. The membrane binding of the blood coagulation Factor X is Ca2+-dependent, and a detailed MD analysis identified two potential sites of PS binding to the bound Ca2+ ions and the GLA domain [27].
In some cases, the membrane association of peripheral membrane proteins is lipid-specific, and often relies on the presence of multivalent anionic lipids, such as phosphatidylinositol bisphosphate (PIP2). Growth and proliferation factors, such as Brag2 and KRas4b, are among the proteins extensively examined by recent MD studies. CG simulations found that the pleckstrin homology and Sec7 domains of Brag2 prefer binding to PIP2 over monovalent lipids (e.g., PS) [28]. This specific binding potentially maintains the function of Brag2, which activates the ADP ribosylation factor GTPase. For KRas4b, a combined atomistic MD and fluorescence anisotropy study demonstrated its preferential binding to PIP2 over PS [29]. The long-lived salt bridges formed between KRas4b and PIP2 were suggested to provide an anchoring platform for the interactions between KRas4b and its signaling partners.
Peripheral membrane proteins often anchor to the membrane via specific structural elements providing lipid interactions. In cytochrome P450 3A4, a major metabolizing enzyme, an MD study showed that a positively charged motif of its catalytic domain binds favorably to anionic lipids, deepening its membrane immersion and potentially facilitating substrate delivery (Fig. 2) [30]. In KRas GTPases, which preferentially target anionic lipids, a simulation study showed that its membrane binding involves a polybasic motif, comprising six lysine residues, in addition to the farnesylated CAAX motif and helices of the catalytic domain [31]. A comprehensive MD study with replica-exchange umbrella sampling calculations was able to rationalize the difference in the experimentally observed binding rates of two synaptotagmin (Syt) vesicle-fusion proteins, Syt-1 and Syt-7 [32]. The presence of more basic residues in Syt-7 deepens its membrane insertion, which is in line with its slower dissociation relative to Syt-1.
Protein-membrane association can be mediated by water-mediated hydrogen bonding. A clear example is the membrane binding of the C1b domain of protein kinase C, in which a Markov state model analysis of atomistic MD trajectories revealed a shallower membrane insertion of the bryostatin-bound complex in contrast to the ligand-free one [33]. This binding state was found to be stabilized by extensive hydrogen bonding networks formed by structured water molecules bridging the protein and lipid molecules.
Lipid Modulation of Integral Membrane Proteins
Specific lipid-protein interactions are essential components in cellular signaling, metabolism, and trafficking. Several lipids, such as CHL, CL, PIP2, and glycolipids, are known to directly mediate activities of integral membrane proteins (Fig. 2) such as G-protein coupled receptors (GPCRs) and ion channels [34]. CHL modulates the structure and ligand binding properties of GPCRs. An extensive atomistic MD study of the β2 adrenergic receptor found that the incorporation of CHL into a lipid bilayer reduces the conformational flexibility of signaling helices, thereby hampering the transitions between the inactive and active states [35]. The observed diffusion of CHL into the ligand binding pocket in a recent MD study on the adenosine A2A receptor [36] indicates direct CHL actions on agonist and antagonist binding.
PIP2, an anionic lipid in the inner leaflet of mammalian plasma membranes, is required for the activation of Kir channels. Despite their strong binding, PIP2 does not seem to influence Kir2.2 clustering [37]. The binding energy of PIP2 to tetrameric Kir2.2 can be estimated with replica-exchange umbrella sampling [38]. As another example, PIP2 binding promotes the clustering and activation of EGFR, which is inhibited by ganglioside lipid GM3 from the outer leaflet of the membrane. The binding energies of these lipids to an EGFR dimer were also calculated using CG-based umbrella sampling [39]. Through extensive modeling and MD simulations, the PIP2 binding site was characterized in human dopamine transporter, where the binding induced the cytoplasmic opening of the transporter [40].
As a key mitochondrial lipid, the roles of CL on the respiratory chain have been extensively explored by CG simulations. The formation of a supercomplex composed of cytochrome bc1 and cytochrome c oxidase relies on bridging CL molecules that simultaneously bind both proteins [41], whereas the F0 domain of ATP synthase binds CL selectively but only intermittently, which possibly allows its rotation in the membrane during ATP synthesis [42]. The import of ADP and the export of ATP are facilitated by the adenine nucleotide translocase (ANT). Not only does ANT selectively bind CL [38, 43] but CL binding also initiates its oligomerization [43]. In addition to mitochondria, bidentate CL binding was found to be essential for the oligomerization of multiple bacterial membrane transporters [44].
Non-specific lipid binding can also directly control the function of a membrane protein. For example, ion permeation through a subfamily of K+ channels (K2P) could be blocked by a protruding phospholipid tail that accesses through an opening at the middle of the membrane, termed lateral fenestration [45–47]. Conversely, increased hydration brought about by lipid head groups could constitute a hydrophilic pathway, either allowing ions to enter the channel of the P2× receptor [48] (Fig. 4, left), or directly establishing a conducting pathway at the lateral surface of the channel/scramblase nhTMEM16 [49] (Fig. 4, right).
Figure 4:
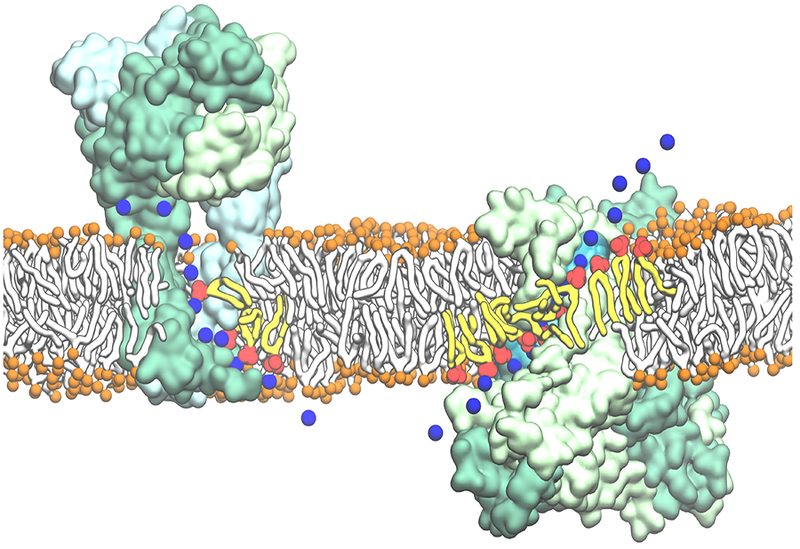
Examples of most direct involvement of phospholipids in biological membrane function, mediated by intimate lipid-protein interactions. Representative snapshots from MD simulations on human P2×3 receptor (Left) [48] and nhTMEM16 scramblase (Right) [49] demonstrating the direct involvement of lipids in ion translocation across the membrane. (Left) Lipids line the cytoplasmic fenestrations of the human P2×3 trimer to allow independent Na+ egress through the lateral cytoplasmic fenestrations during the simulation. (Right) Lipids lining the hydrophilic aqueduct on the surface of the nhTMEM16 scramblase play a structural role in forming a “proteolipidic” pore, which is likely to be used by ions to cross the membrane. The P2×3 trimer and nhTMEM16 dimer are shown in surface representations with each monomer colored in a different shade of green. The lipid headgroups interacting closely with the protein and coordinating permeating ions are shown in red with the tails drawn in yellow; bulk lipids are represented by orange spheres (phosphorus atoms) and white tails. The permeating Na+ ions are shown in time series snapshots (blue spheres). The hydration of the “proteolipidic” pore is illustrated by a transparent cyan surface.
Protein-Mediated Lipid Flip-Flop Across the Bilayer as a Major Signaling Mechanism
The transmembrane movement of lipids is critical to diverse cellular and physiological regulatory processes, including cell activation, blood coagulation, and apoptosis. Flip-flop rearrangement of phospholipids is an extremely slow process in intact lipid bilayers. The process, however, can be significantly accelerated through binding of molecular entities that deform the structure of the bilayer, a phenomenon captured in many simulation studies. Multiscale MD simulations have elucidated the role of local defects on spontaneous lipid flip-flop. For example, multi-μs MD simulations of antimicrobial peptide translocation revealed numerous lipid flip-flop events due to perturbation of the lipid headgroups and increased hydration in the hydrophobic core of membrane around the peptides, thus allowing lipid to more readily transverse (Fig. 2) [50]. Another CG study found that membrane partitioning of toxic anticoagulant brodifacoum causes bilayer thinning and permeabilization that promotes lipid flip-flop, which potentially plays a role in triggering cell death [51]. In addition, atomistic umbrella sampling simulations on the flip-flop of PS showed that membrane oxidation decreases the energy barrier for translocation [52]. In contrast, the presence of diacylglycerol increases the ordering of acyl chains and bilayer thickness, thereby increasing the energy barrier for lipid flip-flop [53]. Moreover, a simulation study found that positive transmembrane potential specifically reduces the free energy barrier of lipid flip-flop on the extracellular leaflet of the membrane, which may have substantial implications for biological activities that are associated with the disruption of cell membranes under physiological conditions [54]. Notably, by shifting the free energy profiles to account for membrane thickness differences among force fields, the free energy for membrane defect formation and lipid translocation can be reconciled among different force fields [55]. For fast-diffusing steroids, such as CHL, CG MD studies found that adding fullerene [56] or increasing the length of CHL aliphatic side chain [57] significantly reduced the flip-flop rate.
An extreme example of lipid translocation across perturbed lipid bilayer is the case of protein-mediated, physiologically relevant lipid transport catalyzed by effective machineries, such as scramblases, flippases, and floppases. Recent atomistic MD studies on nhTMEM16 scramblase (Fig. 4, right) [49, 58] and opsin GPCR [59] observed spontaneous diffusion of phospholipids between the two leaflets via a surface-exposed hydrophilic aqueduct provided by the proteins. Moreover, both MD simulations and continuum modeling on nhTMEM16 demonstrated a significant deformation of membrane structure induced by the protein, which decreases the effective membrane thickness near the aqueduct and greatly reduces the energy barrier against lipid translocation [49, 58]. In line with the observation on nhTMEM16 and opsin, simulations of bacteriorhodopsin [60] and P4-ATPase flippase [61] identified surface-exposed hydrated paths, potentially facilitating lipid translocation. With the advancements in non-equilibrium and enhanced sampling methods, atomistic descriptions of lipid translocation via active transporters are foreseeable in the near future.
Concluding Remarks and Perspective
A large body of evidence accumulated through numerous biochemical and biophysical studies has indisputably established considerable impacts of lipid constituents of the cellular membrane on its biological function. Lipids exert their effects either through modulating ensemble properties of the membrane which in turn can impact conformational dynamics and equilibria of membrane proteins, or via specific, direct interactions with membrane proteins. Specific lipid types have been shown to be directly involved in key signaling pathways, and the cell often relies on modulating their concentration and/or localization within the membrane to activate or shut down such pathways. The coagulation cascade and apoptosis are two major examples of direct roles of such “signaling lipids”. Biological membranes and their lipid constituents, therefore, can no longer be viewed as passive hydrophobic barriers, merely forming boundaries around the cell and its inner compartments.
Studying the role of lipids at a detailed level poses a major challenge, to low-resolution techniques, where lipids often manifest themselves as indistinct entities, collectively contributing to macroscopic bulk properties of the membrane. Characterizing the mechanisms by which lipids play their diverse roles requires sufficiently high resolutions, both spatially and temporally. While a number of experimental techniques, e.g., cryo-EM, X-ray crystallography, AFM, NMR, and EPR, have, in some cases, successfully provided information on the structure and specific interaction of lipids with membrane proteins, the dynamic nature of the lipid bilayers, and probably more importantly, the significant perturbation often imposed on the system under experimental conditions continue to limit the scope of such approaches. In this context, computational modeling and molecular simulation offer the required resolutions, thus providing us with a powerful, highly complementary approach to experiment. In particular, the technique of molecular dynamics (MD) has been widely used to gain information on lipid structure, dynamics, and interaction with membrane proteins.
Unraveling lipid-protein interactions and the mechanistic flavors through which such interactions impact biological function are now a major topic in membrane protein research, and we can only expect that this area continues to grow further in the future. Novel experimental techniques and computational algorithms, empowered by more precise measurements and more powerful hardware will allow us to gain a deeper and more complete understanding of dynamics of biological membranes, specific binding modes and sites for lipids in membrane proteins, and biological impacts of such phenomena.
On the computational side, a major recent shift has been employing mixed lipid bilayers, even taking into account the bilayer asymmetry, in MD studies, a trend that will soon become mainstream and provide increasingly more realistic representations of biological membranes, as the much needed experimental data on lipid composition of various cells and cellular compartments become available. With the increased speed in simulations, e.g., through GPU-enabled calculations, we should expect atomistic simulations to be able to capture longer time-scale phenomena, which are currently only available through CG simulations, e.g., meaningful lipid mixing, protein-induced aggregation of specific, charged lipids, and formation of microdomains. At the same time, the scope of CG simulations is expected to naturally expand into modeling larger, cell-scale membranes and their behavior.
Membranes and their interactions with other cellular elements are important factors in the morphology of cells, affecting aspects ranging from the overall shape of the cell, the organization of the cytoskeleton, to the shapes and locations of intracellular organelles. With access to more computing power and computational resources, we can expect to witness modeling of cellular events with large-scale modeling and simulations of membrane systems in the near future, for example, the budding and fusion of vesicles, the protrusion of filopodia, or the formation of irregularly shaped organelles, e.g., mitochondria and endoplasmic reticulum. Computational studies at these mesoscopic to macroscopic levels will rely on developments in several directions: (a) computational capability of handling massive simulation systems at millions to billions of atoms/particles; (b) tools to accurately model asymmetric, mixed lipid bilayers at physiological composition, even with heterogeneous local distributions mimicking microdomains; and (c) tools to easily generate structures of non-planar lipid bilayers, even to directly match the geometries of irregular organelle membranes that are observed experimentally. These advances will enable us to study function and regulation of constituents of biological membranes within more realistic conditions of a cell.
Acknowledgments
The authors would like to acknowledge support from the National Institute of General Medical Sciences of the National Institutes of Health under awards U54-GM087519, P41-GM104601, R01-GM101048, and R01-GM123455. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. N.T. would like to acknowledge support by the National Science Foundation Graduate Research Fellowship Program under Grant No. 1746047. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of the National Science Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Jo S, Cheng X, Lee J, Kim S, Park S-J, Patel DS, Beaven AH, Lee KI, Rui H, Parks S, Lee HS, Roux B, A. D. M. Jr, Klauda JB, Qi Y, Im W, CHARMM-GUI 10 years for biomolecular modeling and simulation, J. Comp. Chem 38 (2017) 1114–1124,• CHARMM-GUI is a swiss army knife for constructing membrane simulation systems, yet its functionality reaches far beyond membrane simulations. Some of the functional modules of CHARMM-GUI worth noting within the scope of this article include Membrane Builder, Glycolipid Modeler, LPS Modeler, and Martini Maker.
- [2].Bovigny C, Tamò G, Lemmin T, Maïno N, Dal Peraro M, LipidBuilder: a framework to build realistic models for biological membranes, J. Chem. Inf. Model 55 (2015) 2491–2499. [DOI] [PubMed] [Google Scholar]
- [3].Wassenaar TA, Ingólfsson HI, Böckmann RA, Tieleman DP, Marrink SJ, Computational lipidomics with insane: A versatile tool for generating custom membranes for molecular simulations, J. Chem. Theory Comput 11 (2015) 2144–2155,• The article describes a versatile method for building membranes, termed insane (INSert membrANE) that uses preset, CG lipid templates to build the membrane, also allowing on-the-fly generation of simple lipid types by specifying the headgroup, linker, and lipid tails, greatly improving our ability to create membranes of any lipid composition.
- [4].Stansfeld PJ, Goose JE, Caffrey M, Carpenter EP, Parker JL, Newstead S, Sansom MS, Mem-ProtMD: automated insertion of membrane protein structures into explicit lipid membranes, Structure 23 (2015) 1350–1361,• The article describes an automated protocol to model membrane proteins in explicit lipid bilayers, a valuable tool for setting up MD simulations of membrane proteins.
- [5].Lyubartsev AP, Rabinovich AL , Force field development for lipid membrane simulations, Biochim. Biophys. Acta Biomembr 1858 (2016) 2483–2497. [DOI] [PubMed] [Google Scholar]
- [6].Boughter CT, Monje-Galvan V, Im W, Klauda JB, Influence of cholesterol on phospholipid bilayer structure and dynamics, J. Phys. Chem. B 120 (2016) 11761–11772. [DOI] [PubMed] [Google Scholar]
- [7].Wang E, Klauda JB, Examination of mixtures containing sphingomyelin and cholesterol by molecular dynamics simulations, J. Phys. Chem. B 121 (2017) 4833–4844,• The article is among the pioneering studies in realistic representation of mammalian cellular membranes by explicitly including the effects of cholesterol and sphingomyelin, both representing major lipid constituents of the cellular membrane.
- [8].Adams M, Wang E, Zhuang X, Klauda JB, Simulations of simple Bovine and Homo sapiens outer cortex ocular lens membrane models, Biochim. Biophys. Acta Biomembr (2018) in press. [DOI] [PubMed] [Google Scholar]
- [9].Arnarez C, Webb A, Rouviere E, Lyman E, Hysteresis and the cholesterol dependent phase transition in binary lipid mixtures with the Martini model, J. Phys. Chem. B 120 (2017) 13086–13093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Bera I, Klauda JB, Molecular simulations of mixed lipid bilayers with sphingomyelin, glycerophos-pholipids, and cholesterol, J. Phys. Chem. B 121 (2017) 5197–5208. [DOI] [PubMed] [Google Scholar]
- [11].Dotson RJ, Smith CR, Bueche K, Angles G, Pias SC, Influences of cholesterol on the oxygen permeability of membranes: Insight from atomistic simulations, Biophys. J 112 (2017) 2336–2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Falkovich SG, Martinez-Seara H, Nesterenko AM, Vattulainen I, Gurtovenko AV, What can we learn about cholesterol’s transmembrane distribution based on cholesterol-induced changes in membrane dipole potential?, J. Phys. Chem. Lett 7 (2016) 4585–4590. [DOI] [PubMed] [Google Scholar]
- [13].Postila PA, Vattulainen I, Rog T, Selective effect of cell membrane on synaptic neurotransmission, Sci. Rep 6 (2016) 19345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Patel DS, Re S, Wu EL, Qi Y, Klebba PE, Widmalm G, Yeom MS, Sugita Y, Im W, Dynamics and interactions of OmpF and LPS: Influence on pore accessibility and ion permeability, Biophys. J 110 (2016) 930–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Jefferies D, Hsu P-C, Khalid S, Through the lipopolysaccharide glass: A potent antimicrobial peptide induces phase changes in membranes, Biochemistry 56 (2017) 1672–1679. [DOI] [PubMed] [Google Scholar]
- [16].Hsu P-C, Bruininks BMH, Jefferies D, Cesar P de Souza Telles, Lee J, Patel DS, Marrink SJ, Khalid S Qi Y, Im W, CHARMM-GUI Martini Maker for modeling and simulation of complex bacterial membranes with lipopolysaccharides, J. Comp. Chem 38 (2017) 2354–2363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Hsu P-C, Samsudin F, Shearer J, Khalid S, It is complicated: curvature, diffusion, and lipid sorting within the two membranes of Escherichia coli, J. Phys. Chem. Lett 8 (2017) 5513–5518. [DOI] [PubMed] [Google Scholar]
- [18].Marzinek JK, Holdbrook DA, Huber RG, Verma C, Bond PJ, Pushing the envelope: Dengue viral membrane coaxed into shape by molecular simulations, Structure 24 (2016) 1410–1420,• CG MD is used to determine the molecular basis by which an otherwise spherical membrane is sculpted into an icosahedral shape by the membrane proteins of dengue virus.
- [19].Bradley RP, Radhakrishnan R, Curvature-undulation coupling as a basis for curvature sensing and generation in bilayer membranes, Proc. Natl. Acad. Sci. USA 113 (2016) E5117–E5124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Santos DES, Pol-Fachin L, Lins RD, Soares TA, Polymyxin binding to the bacterial outer membrane reveals cation displacement and increasing membrane curvature in susceptible but not in resistant lipopolysaccharide chemotypes, J. Chem. Inf. Model 57 (2017) 2181–2193,• MD is used to demonstrate that polymyxin B induces curvature in membranes, suggesting a possible mechanism by which the antimicrobial peptide disrupts the function of bacteria.
- [21].Gardner JM, Abrams CF, Rate of hemifusion diaphragm dissipation and ability to form three-junction bound HD determined by lipid composition, J. Chem. Phys 147 (2017) 134903. [DOI] [PubMed] [Google Scholar]
- [22].Tong J, Wu Z, Briggs MM, Schulten K, Mclntosh TJ, The water permeability and pore entrance structure of aquaporin-4 channels depend on lipid bilayer thickness, Biophys. J 111 (2016) 90–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Halbleib K, Pesek K, Covino R, Hofbauer HF, Wunnicke D, Hanelt GHI, Ernst R, Activation of the unfolded protein response by lipid bilayer stress, Mol. Cell 67 (2017) 673–684,• A combined experimental and MD simulation study where it is shown how the increased thickness of the membrane caused by the presence of cholesterol molecules, together with the tendency of an anchoring helix to tilt in these domains can facilitate oligomerization of an integral membrane protein as part of its activation.
- [24].Khondker A, Dhaliwal A, Alsop RJ, Tang J, Backholm M, Shia A-C, Rheinstädter MC, Partitioning of caffeine in lipid bilayers reduces membrane fluidity and increases membrane thickness, Phys. Chem. Chem. Phys 19 (2017) 7101–7111,• MD and X-ray diffraction are used to show that caffeine increases the thickness of the membrane bilayer, an effect that perhaps explains how caffeine works as a medical adjuvant.
- [25].Argudo D, Bethel NP, Marcoline FV, Wolgemuth CW, Grabe M, New continuum approaches for determining protein-induced membrane deformations, Biophys. J 112 (2017) 2159–2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Tietjen GT, Baylon JL, Kerr D, Gong Z, Henderson JM, Heffern CTR, Meron M, Lin B, Schlossman ML, Adams EJ, Tajkhorshid E, Lee KYC, Coupling X-ray reflectivity and in silico binding to yield dynamics of membrane recognition by Tim1, Biophys. J 113 (2017) 1505–1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Muller MP, Wang Y, Morrissey JH, Tajkhorshid E, Lipid specificity of the membrane binding domain of coagulation factor X, J. Thromb. Haem 15 (2017) 2005–2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Karandur D, Nawrotek A, Kuriyan J, Cherfils J, Multiple interactions between an Arf/GEF complex and charged lipids determine activation kinetics on the membrane, Proc. Natl. Acad. Sci. USA 114 (2017) 11416–11421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Gregory MC, McLean MA, Sligar SG, Interaction of KRas4b with anionic membranes: A special role for PIP2, Biochem. Biophys. Res. Commun 487 (2017) 351–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Navrátilová V, Paloncýová M, Berka K, Otyepka M, Effect of lipid charge on membrane immersion of cytochrome P450 3A4, J. Phys. Chem. B 120 (2016) 11205–11213. [DOI] [PubMed] [Google Scholar]
- [31].Prakash P, Zhou Y, Liang H, Hancock JF, Gorfe AA, Oncogenic K-Ras binds to an anionic membrane in two distinct orientations: a molecular dynamics analysis, Biophys. J 110 (2016) 1125–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Vermaas JV, Tajkhorshid E, Differential membrane binding mechanics of synaptotagmin isoforms observed at atomic detail, Biochemistry 56 (2017) 281–293,• Using the accelerated HMMM membrane model, the simulations from this study revealed the key differences in the membrane binding of two synaptotagmin isoforms, with direct implications in their experimentally observed differential kinetics.
- [33].Ryckbosch SM, Wender PA, Pande VS, Molecular dynamics simulations reveal ligand-controlled positioning of a peripheral protein complex in membranes, Nat. Commun 8 (2017) 6,• All-atom MD simulations amounting collectively to ~0.5ms are used to show that different protein kinase C activators lead to distinct orientations of the complex due to different interactions with water molecules in the membrane inner leaflet.
- [34].Hedger G, Sansom MS, Lipid interaction sites on channels, transporters and receptors: Recent insights from molecular dynamics simulations, Biochim. Biophys. Acta Biomembr 1858 (2016) 2390–2400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Manna M, Niemelä M, Tynkkynen J, Javanainen M, Kulig W, Müller DJ, Rog T, Vattulainen I, Mechanism of allosteric regulation of β2-adrenergic receptor by cholesterol, eLife 5 (2016) 1–21,• All-atom MD simulations are used to demonstrate the structural roles of cholesterol in GPCR function.
- [36].Guixà-González R, Albasanz JL, Rodriguez-Espigares I, Pastor M, Sanz F, Martí-Solano M, Manna M, Martinez-Seara H, Hildebrand PW, Martin M, Selent J, Membrane cholesterol access into a G-protein-coupled receptor, Nat. Commun 8 (2017) 14505,• A combined study including experiments and all-atom MD simulations identifying direct regulatory actions of cholesterol on the binding of agonists or antagonists to GPCR.
- [37].Duncan AL, Reddy T, Koldso H, Helie J, Fowler PW, Chavent M, Sansom MSP, Protein crowding and lipid complexity influence the nanoscale dynamic organization of ion channels in cell membranes, Sci. Rep 7 (2017) 16647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Domanski J, Hedger G, Best RB, Stansfeld PJ, Sansom MSP, Convergence and sampling in determining free energy landscapes for membrane protein association, J. Phys. Chem. B 121 (2017) 3364–3375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Hedger G, Shorthouse D, Koldso H, Sansom MSP, Free energy landscape of lipid interactions with regulatory binding sites on the transmembrane domain of the EGF receptor, J. Phys. Chem. B 120 (2016) 8154–8163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Khelashvili G, Stanley N, Sahai MA, Medina J, LeVine MV, Shi L, De Fabritiis G, Weinstein H, Spontaneous inward opening of the dopamine transporter is triggered by PIP2-regulated dynamics of the N-terminus, ACS Chem. Neurosci 6 (2015) 1825–1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Arnarez C, Marrink SJ, Periole X, Molecular mechanism of cardiolipin-mediated assembly of respiratory chain supercomplexes, Chem. Sci 7 (2016) 4435–4443,• A self-assembly CG simulation demonstrates interactions involving cardiolipins in the oligomerization of critical bioenergetic proteins.
- [42].Duncan AL, Robinson AJ, Walker JE, Cardiolipin binds selectively but transiently to conserved lysine residues in the rotor of metazoan ATP synthases, Proc. Natl. Acad. Sci. USA 113 (2016) 8687–8692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Hedger G, Rouse SL, Domanski J, Chavent M, Koldso H, Sansom MSP, Lipid-loving ANTs: Molecular simulations of cardiolipin interactions and the organization of the adenne nucleotide translocase in model mitochondrial membranes, Biochemistry 55 (2016) 6238–6249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Gupta K, Donlan JAC, Hopper JTS, Uzdavinys P, Landreh M, Struwe WB, Drew D, Baldwin AJ, Stansfeld PJ, Robinson CV, The role of interfacial lipids in stabilizing membrane protein oligomers, Nature 541 (2017) 421–424,• Lipid contribution to oligomerization of membrane proteins is characterized with mass spectrometry, where MD simulations revealed that cardiolipin serves as a bidentate ligand stabilizing the multimeric interface.
- [45].Jorgensen C, Darre L, Oakes V, Torella R, Pryde D, Domene C, Lateral fenestrations in K+ channels explored using molecular dynamics simulations, Mol. Pharmacol 13 (2016) 2263–2273. [DOI] [PubMed] [Google Scholar]
- [46].Masetti M, Berti C, Ocello R, Di Martino GP, Recanatini M, Fiegna C, Cavalli A, Multiscale simulations of a two-pore potassium channel, J. Chem. Theory Comput 12 (2016) 5681–5687. [DOI] [PubMed] [Google Scholar]
- [47].Aryal P, Jarerattanachat V, Clausen MV, Schewe M, McClenaghan C, Argent L, Conrad LJ, Dong YY, Pike AC, Carpenter EP, Baukrowitz T, Sansom MS, Tucker SJ, Bilayer-mediated structural transitions control mechanosensitivity of the TREK-2 K2P channel, Structure 25 (2017) 708–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Mansoor SE, Lü W, Oosterheert W, Shekhar M, Tajkhorshid E, Gouaux E, X-ray structures define human P2×3 receptor gating cycle and antagonist action, Nature 538 (2016) 66–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Jiang T, Yu K, Hartzell HC, Tajkhorshid E, Lipids and ions traverse the membrane by the same physical pathway in the nhTMEM16 scramblase, eLife 6 (2017) e28671,• In a highly concerted study coupling experimental assays with MD simulations, this study revealed that the surface hydrophilic transmembrane cavity exposed to the lipid bilayer on a fungal scramblase serves as the pathway for both lipid translocation and ion conduction across the membrane.
- [50].Ulmschneider JP, Charged antimicrobial peptides can translocate across membranes without forming channel-like pores, Biophys. J 113 (2017) 73–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Ayee MAA, Roth CW, Akpa BS, Structural perturbation of a dipalmitoylphosphatidylcholine (DPPC) bilayer by warfarin and its bolaamphiphilic analogue: A molecular dynamics study, J. Coll. Interf. Sci 468 (2016) 227–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Razzokov J, Yusupov M, Vanuytsel S, Neyts EC, Bogaerts A, Phosphatidylserine flip-flop induced by oxidation of the plasma membrane: a better insight by atomic scale modeling, Plasma Processes Polym. 14 (2017) 1700013. [Google Scholar]
- [53].Alwarawrah M, Hussain F, Huang J, Alteration of lipid membrane structure and dynamics by diacylglycerols with unsaturated chains, Biochim. Biophys. Acta Biomembr 1858 (2016) 253–263. [DOI] [PubMed] [Google Scholar]
- [54].Lin J, Dargazany R, Alexander-Katz A, Lipid flip-flop and pore nucleation on zwitterionic bilayers are asymmetric under ionic imbalance, Small 13 (2017) 1603708. [DOI] [PubMed] [Google Scholar]
- [55].Bennett WFD, Hong CK, Wang Y, Tieleman DP, Antimicrobial peptide simulations and the influence of force field on the free energy for pore formation in lipid bilayers, J. Chem. Theory Comput 12 (2016) 4524–4533. [DOI] [PubMed] [Google Scholar]
- [56].Sastre J, Mannelli I, Reigada R, Effects of fullerene on lipid bilayers displaying different liquid ordering: a coarse-grained molecular dynamics study, Biochim. Biophys. Acta 1861 (2017) 2872–2882. [DOI] [PubMed] [Google Scholar]
- [57].Lin X, Zhang S, Ding H, Levental I, Gorfe AA, The aliphatic chain of cholesterol modulates bilayer interleaflet coupling and domain registration, FEBS Lett. 590 (2016) 3368–3374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Bethel NP, Grabe M, Atomistic insight into lipid translocation by a TMEM16 scramblase, Proc. Natl. Acad. Sci. USA 113 (2016) 14049–14054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Morra G, Razavi AM, Pandey K, Weinstein H, Menon AK, Khelashvili G, Mechanisms of lipid scrambling by the G protein-coupled receptor opsin, Structure 26 (2018) 356–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Verchere A, Ou W-L, Ploier B, Morizumi T, Goren MA, Biitikofer P, Ernst OP, Khelashvili G, Menon AK, Light-independent phospholipid scramblase activity of bacteriorhodopsin from halobacterium salinarum, Sci. Rep 7 (2017) 2045–2322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Andersen JP, Vestergaard AL, Mikkelsen SA, Mogensen LS, Chalat M, Molday RS, P4-atpases as phospholipid flippases-structure, function, and enigmas, Front. Physiol 7 (2016) 275. [DOI] [PMC free article] [PubMed] [Google Scholar]