

Abstract
The structural analysis of carbohydrates remains challenging mainly due to the lack of rapid analytical methods able to determine and quantitate glycosidic linkages between the diverse monosaccharides found in natural oligosaccharides and polysaccharides. In this research, we present the first LC-MS/MS-based method for the rapid and simultaneous relative quantitation of glycosidic linkages for oligosaccharide and polysaccharide characterization. The method developed employs ultra-high performance liquid chromatography coupled with triple quadrupole mass spectrometry (UHPLC/QqQ-MS) analysis performed in multiple reaction monitoring (MRM) mode. A library of 22 glycosidic linkages was built using commercial oligosaccharide standards. Permethylation and hydrolysis conditions along with LC-MS/MS parameters were optimized resulting in a workflow requiring only 50 μg of substrate for the analysis. Samples were homogenized, permethylated, hydrolyzed, and then derivatized with 1-phenyl-3-methyl-5-pyrazolone (PMP) prior to analysis by UHPLC/MRM-MS. Separation by C18 reverse-phase UHPLC along with the simultaneous monitoring of derivatized terminal, linear, bisecting, and trisecting monosaccharide linkages by mass spectrometry is achieved within a 15-minute run-time. Reproducibility, efficacy, and robustness of the method was demonstrated with galactan (Lupin) and polysaccharides within food such as whole carrots. The speed and specificity of the method enables its application toward the rapid glycosidic linkage analysis of oligosaccharides and polysaccharides.
Keywords: Glycosidic Linkage Analysis, Mass Spectrometry, Carbohydrates, Multiple Reaction Monitoring, Permethylation, 1-phenyl-3-methyl-5-pyrazolone, LC-MS, Tandem MS
Graphical Abstract
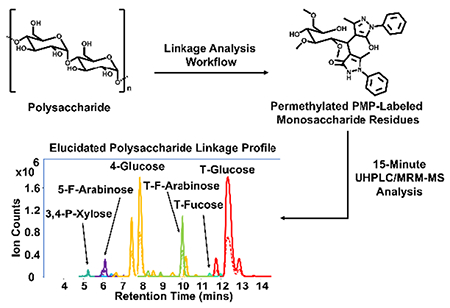
INTRODUCTION
Carbohydrates are the most abundant bio-molecules in nature and are composed of monosaccharides, oligosaccharides, and polysaccharides. Carbohydrates are of fundamental biological importance: both as a source of energy and as the main structural and functional components of plant, fungi cell walls, and animal cell membranes1–4. Structures of carbohydrates also display a wide range of bioactive properties in nature. For example, oligosaccharides can act as chemical messenger molecules and regulate physiological processes within plants5. Similarly, free oligosaccharides present in human milk are known to promote the growth of beneficial gut bacteria within infants6–8. Protein-bound oligosaccharides have been shown to be important for protein folding, biological recognition, and molecular trafficking9–11. In bacterial chemotaxis, monosaccharides such as galactose are known to be attractant for Escherichia coli12. As food, plant carbohydrates have unique nutritional digestibility. For example, amylopectin, a digestible polysaccharide component of starch, is composed of a α(1→4) glucose backbone with occasional α(1→4,6) glucose branching, while cellulose, a β(1→4) linear glucose polysaccharide, is non-digestible13 illustrating that glycosidic linkages along with the anomeric character play a key role in the bioactivity of carbohydrates.
Monosaccharide and oligosaccharide analysis have been the focus of several major efforts in our research and elsewhere14–19. We have recently developed a monosaccharide analysis method based on LC-MS with greater sensitivity and speed than previous methods15. Oligosaccharide and glycan analysis have significantly advanced and are now commonly applied in a rapid throughput manner20–25. However, methods for the rapid linkage analysis of plant polysaccharides remains elusive. Thus, our knowledge of food polysaccharides and their structures can be best described, with few exceptions, as rudimentary.
The common platform for the structural analysis of polysaccharides, involves gas chromatography coupled to mass spectrometry (GC-MS)26–29. However, analysis by GC-MS requires that the carbohydrates are volatile, which involves multiple derivatization steps and requires a high amount of material for analysis due to lack of sensitivity30. Other methods including high pH anion-exchange chromatography with pulsed amperometric detection (HPAEC-PAD), capillary electrophoresis (CE), high-performance liquid chromatography (HPLC) separation coupled with UV detection are used for monosaccharide profiling31–36. However, these methods are either non-structure informative, require long run-times to achieve isomer separation, or not amenable to rapid throughput analysis. Recent developments on the implementation of LC-MS-based techniques for carbohydrate analysis has greatly improved the sensitivity and specificity for the detection of monosaccharides15, 37–38. Furthermore, the use of multiple reaction monitoring (MRM) has been shown as an effective method for the quantification of glycoproteins39–40, metabolites41–42, oligosaccharides43–44, and monosaccharides15, 45–47.
The most common method for the glycosidic linkage analysis of oligosaccharides and polysaccharides typically involves permethylation of free hydroxyl groups using the Hakomori approach28. Next, the permethylated monosaccharides are released by acid hydrolysis resulting in residues that will only contain free hydroxyl groups in specific positions that indicate the locations of previous glycosidic bonds. Furthermore, alpha/beta stereochemical information is lost during this process. Whether the monosaccharides exist in either the cyclic or open-chain form, reduction of the anomeric center is often necessary as multiple peaks in the analysis can arise from the presence of anomers30. The remaining free hydroxyls located on the released residues can be further derivatized by either acetylation or silylation48–49. The derivatized compounds are usually prepared in volatile organic solvents prior to injection and analysis by GC-MS, making evaporation of samples a considerable concern and rendering the platform not suitable for large sample sets26–29, 50–52.
Derivatization of the reducing end of monosaccharides with 1-phenyl-3-methyl-5-pyrazolone (PMP), developed by Honda et al., has been commonly used for the quantitation of monosaccharides by liquid chromatography coupled with UV detection53–57. The labeling reaction involves the addition of two hydrophobic PMP molecules to the reducing end of a monosaccharide by Michael addition. The increased hydrophobicity of the derivatized monosaccharides facilitates improved separation by reversed-phase C18 liquid chromatography58–59. However, use of UV detection for accurate quantitation of PMP-labeled monosaccharides is non-structure selective, thus, relies heavily on chromatographic separation. Recently, an LC-MS-based method from this laboratory has been reported for the simultaneous quantitation of PMP-labeled monosaccharides15.
In this study, we developed a methodology which utilizes permethylation in combination with PMP derivatization followed by UHPLC/QqQ-MS analysis operated in the MRM mode for the elucidation and relative quantitation of glycosidic linkage residues derived from oligosaccharides and polysaccharides. We illustrate the broad scope of this method for the simultaneous analysis of 22 unique glycosidic linkages representing terminal, linear, branching bisecting and trisecting species within oligosaccharides and both soluble and insoluble polysaccharides. The method employs permethylation with iodomethane followed by acid hydrolysis using trifluoroacetic acid (TFA) and derivatization with PMP. Optimized UHPLC conditions offer efficient chromatographic separation and analysis of isomeric linkage residues within a 15-minute run. Validation of this approach was performed using polysaccharides from whole food such as whole carrots. Furthermore, the method’s speed and sensitivity make it ideal for the analysis of oligosaccharides and polysaccharides derived from foods and biological samples.
EXPERIMENTAL PROCEDURES
Samples and materials.
Ammonium acetate (NH4Ac), TFA, iodomethane (contains copper stabilizer, 99.5 %), sodium hydroxide pellets (NaOH) (semi-conductor grade, 99.99% trace metals basis), ammonium hydroxide solution (NH4OH) (28-30%, NH3 basis), PMP, dichloromethane (DCM), methanol (MeOH) (HPLC grade), stachyose, and anhydrous dimethyl sulfoxide (DMSO) were purchased from Sigma-Aldrich (St. Louis, MO). Acetonitrile (ACN) (HPLC grade) was purchased from Honeywell (Muskegon, MI). 2-O-(a-D-mannopyranosyl)-D-mannopyranose, 1,4-D-xylobiose, 1,5-a-L-arabinotriose, 1,3-a-1,6-a-D-mannotriose, isomaltotriose, 4-O-(b-D-galactopyranosyl)-D-galactopyranose, lactose, 2’-fucosyllactose (synthetic), nigerose, 3-O-(b-D-galactopyranosyl)-D-galactopyranose, 3-O-(a-D-mannopyranosyl)-D-mannopyranose, 1,4-b-D-mannotriose, maltohexaose, 1,6-a-D-mannotriose, and amylopectin were obtained from Carbosynth (Compton, UK). Galactan (Lupin), 33-α-L-arabinofuranosyl-xylotetraose and sophorose were acquired from Megazyme (Chicago, IL). Whole carrots were purchased from Whole Foods (Davis, CA). Nanopure water was used for all experiments.
Optimization of permethylation reaction.
Samples containing 50 μg maltohexaose oligosaccharide were permethylated using iodomethane in a solution of DMSO containing concentrated NaOH and reacted on a shaker at room temperature for 0, 10, 30, 50, and 70 minutes, respectively. The reaction was quenched at each time-point by the addition of ice-cold water. The addition of ice-cold water prevents a rise in temperature and degradation caused by the “peeling” reaction60. A liquid-liquid extraction using DCM and subsequent washes with ice-cold water was performed to remove excess NaOH and DMSO. The upper aqueous layer was discarded and the remaining bottom organic layer containing permethylated products was collected and dried to completion by vacuum centrifugation. Samples were subjected to acid hydrolysis with TFA followed by vacuum centrifugation. The released permethylated monosaccharide residues were derivatized with PMP following a previously established procedure by Xu et al15. Samples were subjected to analysis by ultra-high pressure liquid chromatography/multiple reaction monitoring mass spectrometry (UHPLC/MRM-MS).
Preparation of polysaccharide standards and whole carrot samples for derivatization and analysis.
Stock solutions of amylopectin and galactan (Lupin) polysaccharide standards were prepared in water. A whole carrot root was chopped, lyophilized, ground to a fine powder using a mortar and pestle, then a stock solution of this processed material was prepared in water. Further homogenization of sample was accomplished by bead blasting with 1.4 mm stainless steel magnetic beads for 3 minutes using a Next Advance Bullet Blender Storm 24 (Next Advance, Troy, NY).
Acid hydrolysis reaction optimization.
Aliquots containing 50 μg of amylopectin were permethylated using the optimal time of 50 minutes. Samples were subjected to acid hydrolysis for 30, 60, 90, 120, 150, and 180 minutes. Three experimental replicates for each time-point were performed. The resulting samples were subjected to PMP derivatization. Samples were enriched by liquid-liquid extraction with DCM and water, and analyzed by UHPLC/MRM-MS.
Ultra-high pressure liquid chromatography/triple quadrupole mass spectrometry (UHPLC/QqQ-MS) analysis.
Separation and analysis of the permethylated PMP-labeled monosaccharides was carried out on an Agilent 1290 Infinity II UHPLC system coupled to an Agilent 6495A triple quadrupole (QqQ) mass spectrometer (Agilent Technologies, Santa Clara, CA). For analysis, 1 μL of sample was injected onto an Agilent ZORBAX RRHD Eclipse Plus C18 column (2.1 mm × 150 mm i.d., 1.8 μm particle size) equipped with an Agilent ZORBAX Eclipse Plus C18 guard cartridge (2.1 mm × 5 mm i.d., 1.8 μm particle size) and separated using a 15-minute binary gradient with a constant flow rate of 0.22 mL/min. Mobile phase A: 25 mM NH4Ac in 5 % ACN/water (v/v), adjusted to pH 8.2 using NH4OH; Mobile phase B: 95 % ACN/water (v/v). The following binary gradient was used: 0.00-5.00 mins, 21.00 % B; 5.00-9.00 mins, 21.00-22.00 % B; 9.00-11.00 mins, 22.00 % B; 11.00-13.60 mins, 22.00-24.50 % B; 13.60-13.61 mins, 24.50-99.00% B; 13.61-13.80 mins, 99.00 % B; 13.80-13.81 mins, 99.00-21.00 % B; 13.81-15.00 mins, 21.00 % B.
Samples were introduced into the mass spectrometer using an electrospray ionization (ESI) source operated in the positive ion mode. Nitrogen drying and sheath gas temperatures were set at 290 °C and 300 °C, respectively. Drying and sheath gas flow rates were set at 11 L/min and 12 L/min, respectively. The nebulizer pressure was set to 30 psi. Capillary and fragmentor voltages were set at 1800 V and 380 V, respectively. To execute collision induced dissociation (CID), the collision energy was set to a constant 35 V. Data acquired from the UHPLC/QqQ-MS was collected using Agilent MassHunter Workstation Data Acquisition version B.06.01. Data analysis was performed using Agilent MassHunter Quantitative Analysis software version B.06.00.
RESULTS AND DISCUSSION
The method developed employs a UHPLC/MRM-MS platform for the glycosidic linkage analysis of oligosaccharides and polysaccharides using permethylation in combination with PMP derivatization. First, permethylation of free hydroxyls on the polysaccharide is performed using the Hakomori method28. Next, the monosaccharides are released at their respective glycosidic bonds by acid hydrolysis with TFA at an elevated temperature. Upon completion of the hydrolysis, positions of the hydrolyzed glycosidic bonds are retained as free hydroxyl groups. Lastly, the released monomers undergo derivatization at the reducing end with PMP and are analyzed by UHPLC/MRM-MS employing C18 as the stationary phase.
Construction of the MRM transitions
Depending on the degree of branching present within a given oligosaccharide or polysaccharide structure, the resulting hydrosylates would contain a unique degree of permethylation (DoPe) value. For example, Scheme S-1 displays the structure of derivatized galactose monosaccharide residues linked at either the (1→2)- or (1→4)-hydroxyl positions. These residues have a DoPe value of 3, which is equal to the number of methylation sites.
To determine transitions for the MRM method, fragment ions from the derivatized structures were scanned using the QqQ operated in product ion mode. The protonated precursor ion of each derivatized monosaccharide was subjected to CID. The protonated precursor ion mass is equal to the sum of the initial monosaccharide mass, the mass of a methyl group multiplied by the monosaccharide DoPe value, and the mass of two PMP residues ([M + 14*DoPe value + 330 + H]+). Three major fragment ions were observed by MS/MS and used for MRM transitions. The most abundant fragment ion, m/z 175.1, was chosen as the quantifier ion and is the result of fragmentation between PMP and C1 of the derivatized monosaccharide (Figure 1A-B, Scheme S-1).
Figure 1.
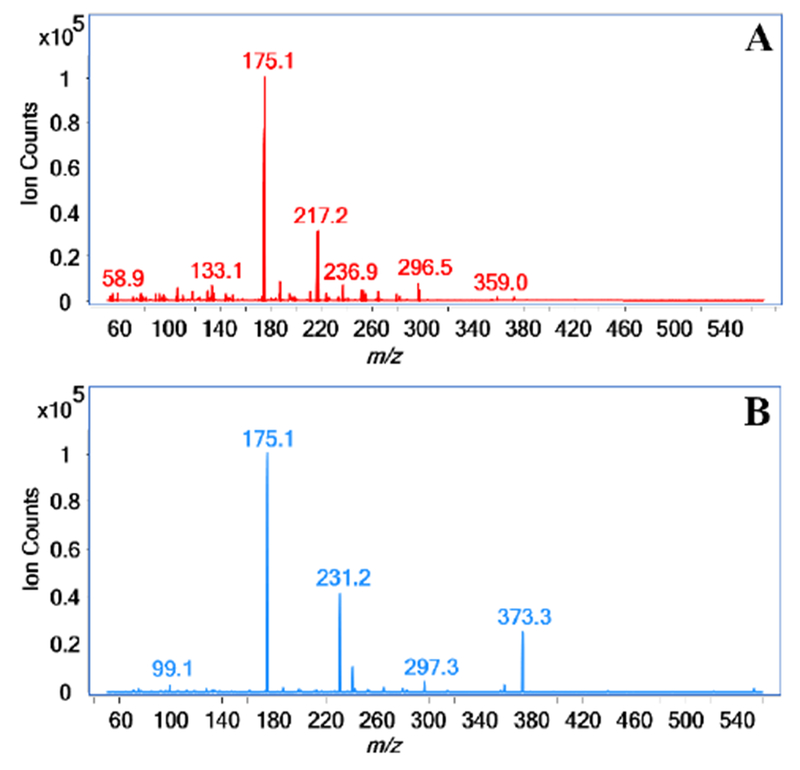
MS/MS spectra of the methylated PMP-labeled protonated precursor ion m/z 553.3 for (A) 2-galactose and (B) 4-galactose. Fragmentation by CID of 2-galactose results in signals m/z 175.1 and m/z 217.2 as the two most abundant fragment ions while the fragmentation of 4-galactose produces signals m/z 175.1 and m/z 231.2 as the two most abundant fragment ions.
The second most abundant fragment ion is the result of fragmentation between C2-C3 of the monosaccharide and one C1-PMP bond. If the monosaccharide residue is linked at the (1→2)-position, a signal at m/z 217.2 will be observed as the second most abundant fragment ion (Figure 1A, Scheme S-1). However, monosaccharide residues linked at positions other than the (1→2)-position yield m/z 231.2 as the second most abundant fragment ion (Figure 1B, Scheme S-1). The two fragment ions, m/z 217.2 and m/z 231.2 differentiate between monosaccharide residues linked at the (1→2)-position and those not linked at this position, respectively. Because terminal (T) linkage residues are only linked at the anomeric hydroxyl position, only fragment ions of m/z 175.1 and m/z 231.2 are produced upon CID.
To elaborate further, the following linear hexose monosaccharide residues will produce fragment ions of m/z 175.1 and m/z 231.2 upon CID: (1→3)-, (1→4)-, or (1→6)-hydroxyl positions, where (1)- denotes linkage at the anomeric position. Similarly, deoxyhexose residues produce these ions when linked in the (1→3)- or (1→4)-hydroxyl positions. Pentose monosaccharides, such as arabinose in the furanose form, will produce these ions when linked at either the (1→3)- or (1→5)-positions. Furthermore, pentoses such as xylose existing in the pyranose form, produce these ions when linked at either the (1→3)- or (1→4)-positions. However, linear hexoses, deoxyhexoses, and pentoses, in either the pyranose or furanose form, will produce fragment ions of m/z 175.1 and m/z 217.2 when linked only at the (1→2)-positions.
For branched bisecting hexose monosaccharide residues, the following pairs of linkages will produce fragment ions of m/z 175.1 and m/z 231.2: (1→3,4)-, (1→3,6)-, or (1→4,6)-hydroxyl positions. Deoxyhexoses, along with pentoses in the pyranose form, also produce these fragment ions when linked at the (1→3,4)-positions. Pentoses in the furanose form produce these fragment ions when linked at the (1→3,5)-positions. Bisecting hexose residues produce fragment ions of m/z 175.1 and m/z 217.2 when linked at the (1→2,3)-, (1→2,4)-, or (1→2,6)-positions. Similarly, deoxyhexoses and pentoses in the pyranose form produce these fragment ions when linked at the (1→2,3)- or (1→2,4)-positions and (1→2,3)- and (1→2,5)-positions for pentoses in the furanose form.
Trisecting hexose monosaccharide residues produce fragment ions corresponding to m/z 175.1 and m/z 231.2 when linked at the (1→3,4,6)-positions. However, fragment ions m/z 175.1 and m/z 217.2 are produced for trisecting hexoses linked at the (1→2,3,4)-, (1→2,3,6)-, or (1→2,4,6)-positions. Scheme S-1 provides the proposed structures of the fragment ions produced by CID of protonated precursor ions of monosaccharides with linkage combinations with or without linkage at the (1→2)-positions.
Because monosaccharides at the non-reducing end are always linked at the anomeric hydroxyl position, annotation for the 1-hydroxyl position can be dropped. A general and more simplified short-hand annotation system is further described. Linear monosaccharide residues are given the annotation of “X-” followed by the corresponding monosaccharide, where “X” represents a linkage position other than the anomeric 1-hydroxyl position. Bisecting and trisecting monosaccharide linkage residues are simplified with an annotation of “X,X-” and “X,X,X-”, respectively, followed by the corresponding monosaccharide. For example, instead of using (1→2,3,6)-glucose to describe a trisecting glucose residue, the short-hand annotation of 2,3,6-glucose is used. This short-hand annotation system is used to annotate glycosidic linkages throughout the remainder of this paper.
Permethylation reaction
A time-point study using a maltohexaose oligosaccharide standard was performed at 0, 10, 30, 50, and 70 minutes, in triplicate, to optimize the permethylation reaction for 50 μg of starting material. The optimal permethylation time was determined by monitoring the absolute abundances of the expected T-glucose and 4-glucose linkage residues at each time-point. A plot displaying the release of the linkage residues from maltohexaose at each of the different time-points is shown in Figure S-1. Both 4-glucose and T-glucose increased with the 4-glucose reaching maximum at 50 minutes. Released T-glucose residues reached maximum at approximately the same reaction period. The decline in abundance of the 4-glucose residue at the 70-minute time-point may be attributed to degradation by the peeling reaction. Based on these results, a reaction time of 50 minutes was used for all subsequent permethylation reactions.
Hydrolysis reaction
A crucial step in obtaining the most comprehensive profile of monosaccharide linkages within the analysis involves use of effective acid hydrolysis conditions. While the acid lability of monosaccharides in oligosaccharides and polysaccharides vary depending on monosaccharide structure and linkage composition61–62, a general acid hydrolysis method was developed for the analysis of glycosidic linkages within food and purified polysaccharides. Amylopectin was used as a model polysaccharide to determine the optimal hydrolysis conditions needed to release permethylated monosaccharides.
The optimal hydrolysis reaction time was determined by monitoring the release of permethylated monosaccharide residues at different reaction times after labeling with PMP. The time of acid hydrolysis was optimized to maximize the release of linkage residues by altering the reaction time from 30 to 180 minutes in 30-minute increments. Absolute peak areas using ion abundances for each identified linkage residue were compared at each time point to determine the optimal hydrolysis condition. The optimal hydrolysis condition for amylopectin was determined to be 120 minutes (Figure S-2). Degradation of all permethylated monosaccharide residues from amylopectin was observed at hydrolysis times > 150 minutes. Terminal and 4-glucose residues were the most abundant species observed as expected. Minor abundances corresponding to two branched bisecting hexose species were observed. While it is expected for amylopectin to contain 4,6-glucose residues, an additional unexpected branched species of different linkage was also observed. Neither species could be distinguished, therefore, were both given the designation of X,X-hexose (I) or X,X-hexose (II) to describe the unknown linkages.
Construction of the glycosidic linkage library
Commercial standards with known structures were used to create a library of various monosaccharide linkages. Standards that were utilized to construct the library include 2-O-(a-D-mannopyranosyl)-D-mannopyranose, 1,4-D-xylobiose, 1,5-a-L-arabinotriose, 1,3-a-1,6-a-D-mannotriose, isomaltotriose, 4-O-(b-D-galactopyranosyl)-D-galactopyranose, lactose, 2’-fucosyllactose, nigerose, 3-O-(b-D-galactopyranosyl)-D-galactopyranose, 1,6-a-D-mannotriose, 33-α-L-arabinofuranosyl-xylotetraose, sophorose, 3-O-(a-D-mannopyranosyl)-D-mannopyranose, 1,4-b-D-mannotriose, and stachyose. For the analysis, permethylated and PMP-labeled monosaccharide residues were prepared and analyzed by UHPLC/MRM-MS
To demonstrate the behavior of the standards when placed through the linkage analysis workflow, 4-O-(b-D-galactopyranosyl)-D-galactopyranose and 2’-fucosyllactose are described in greater detail. The standard 4-O-(b-D-galactopyranosyl)-D-galactopyranose is a disaccharide that contains one 4-galactose and one T-galactose. After work-up, the corresponding residue for a 4-galactose contains three methoxy groups (DoPe 3) and two PMP derivatives. Terminal galactoses, however, will contain four methoxy groups (DoPe 4) and two PMP derivatives. The resulting protonated precursor ion masses for 4-galactose and T-galactose are m/z 553.3 and m/z 567.6, respectively. Transitions of m/z 553.3 → m/z 175.1 and m/z 553.3 → m/z 231.2 for 4-galactose and m/z 567.6 → m/z 175.1 and m/z 567.6 → m/z 231.2 for T-galactose were monitored and observed (Figure S-3A).
The standard 2’-fucosyllactose is a trisaccharide that contains one 4-glucose, one 2-galactose, and one T-fucose. After putting 2’-fucosyllactose through the same reaction workflow, each of these expected linkage residues will contain three methoxy groups (DoPe 3) and two PMP derivatives, resulting in a protonated precursor ion mass of m/z 553.3, m/z 553.3, and m/z 537.2, respectively. Expected transitions of m/z 553.3 → m/z 175.1 and m/z 553.3 → m/z 231.2 for 4-glucose along with m/z 537.2 → m/z 175.1 and m/z 537.2→ m/z 231.2 for T-fucose were monitored and observed (Figure S-3B). Because the galactose was linked at the 2-position, transitions of m/z 553.3 → m/z 175.1 and m/z 553.3 → m/z 217.2 were also monitored and observed (Figure S-3B).
For 4-O-(b-D-galactopyranosyl)-D-galactopyranose, linkage residues corresponding to 4-galactose and T-galactose were observed at 7.9 and 12.3 minutes, respectively. The calculated ratio of linkage residues based on relative peak areas was found to be 0.76 4-galactose:1.00 T-galactose yielded, nearly to the expected 1.00 4-galactose:1.00 T-galactose ratio. For 2’-fucosyllactose, elution times of 8.3, 8.7, and 12.0 minutes were observed for 4-glucose, 2-galactose, and T-fucose, respectively. The calculated relative peak area ratio of 0.81 4-glucose:2.73 2-galactose:1.00 T-fucose differs from the expected 1.00 4-glucose:1.00 2-galactose:1.00 T-fucose ratio. This suggests either that the instrumental response varies for each of the linkage residues or that different acid hydrolysis conditions were required. While the calculated ratios slightly differ from the expected values for both 4-O-(b-D-galactopyranosyl)-D-galactopyranose and 2’-fucosyllactose, the method was sufficient to obtain retention time values necessary to construct the linkage library.
Based on 16 oligosaccharide standards, a library of 22 unique glycosidic linkage residues encompassing bisecting, linear, and terminal species for xylose, arabinose, fucose, mannose, galactose, and glucose was developed using the above approach. Bisecting residues obtained include 3,4-P-xylose (pyranose form) and 3,6-mannose. Linear residues obtained include 4-P-xylose, 5-F-arabinose (furanose form), 2-mannose, 3-mannose, 4-mannose, 6-mannose, 2-galactose, 3-galactose, 4-galactose, 6-galactose, 2-glucose, 3-glucose, 4-glucose, and 6-glucose. Terminal residues obtained include T-P-xylose, T-F-arabinose, T-fucose, T-mannose, T-galactose, and T-glucose.
The retention times, DoPe values, precursor ion masses, and product ion masses for the obtained glycosidic linkages are summarized in Table 1. Several of the standards used contained identical linkages, thus, were used to further validate retention times and identity of the linkage residues. Figure 2A shows an MRM chromatogram of pooled oligosaccharide standards placed through the workflow to demonstrate the chromatographic separation and analysis for a mixture of linkage residues.
Table 1.
Library containing 22 unique glycosidic linkages built using oligosaccharide standards.
| Linkage Residue | RT (mins) | DoPe Value | Pre-Cursor Ion (m/z) | Product Ions (m/z) |
|---|---|---|---|---|
| 2-Mannose | 4.88 | 3 | 553.3 | 175.1, 217.1 |
| 3,4-P-Xylose | 5.35 | 1 | 495.2 | 175.1, 231.2 |
| 4-P-Xylose | 5.98 | 2 | 509.2 | 175.1, 231.2 |
| 5-F-Arabinose | 6.12 | 2 | 509.2 | 175.1, 231.2 |
| 3,6-Mannose | 6.29 | 2 | 539.2 | 175.1, 231.2 |
| 6-Galactose | 6.76 | 3 | 553.3 | 175.1, 231.2 |
| 6-Glucose | 6.76 | 3 | 553.3 | 175.1, 231.2 |
| 6-Mannose | 7.52 | 3 | 553.3 | 175.1, 231.2 |
| 4-Galactose | 7.52 | 3 | 553.3 | 175.1, 231.2 |
| 4-Glucose | 7.92 | 3 | 553.3 | 175.1, 231.2 |
| 2-Galactose | 8.20 | 3 | 553.3 | 175.1, 217.1 |
| T-P-Xylose | 8.25 | 3 | 523.2 | 175.1, 231.2 |
| 3-Glucose | 8.52 | 3 | 553.3 | 175.1, 231.2 |
| 3-Galactose | 8.68 | 3 | 553.3 | 175.1, 231.2 |
| 2-Glucose | 9.39 | 3 | 553.3 | 175.1, 217.1 |
| T-F-Arabinose | 9.93 | 3 | 523.2 | 175.1, 231.2 |
| 3-Mannose | 10.23 | 3 | 553.3 | 175.1, 231.2 |
| 4-Mannose | 10.59 | 3 | 553.3 | 175.1, 231.2 |
| T-Fucose | 11.24 | 3 | 537.2 | 175.1, 231.2 |
| T-Galactose | 11.57 | 4 | 567.6 | 175.1, 231.2 |
| T-Glucose | 12.15 | 4 | 567.6 | 175.1, 231.2 |
| T-Mannose | 12.50 | 4 | 567.6 | 175.1, 231.2 |
Figure 2.
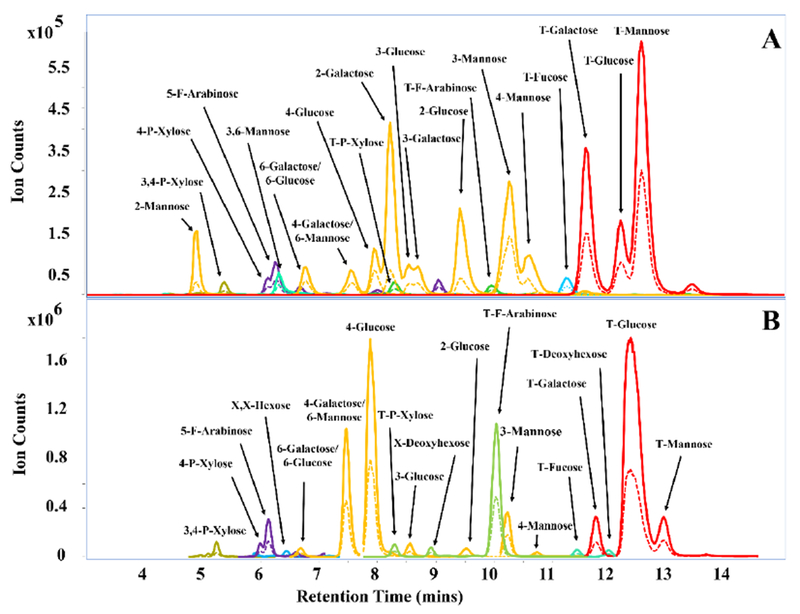
(A) MRM chromatogram of 22 methylated PMP-labeled monosaccharides obtained from pooled commercially available oligosaccharides. (B) - MRM chromatogram for whole carrots. Solid and dashed traces represent quantifying and qualifying transitions, respectively.
In general, linkage residues of different monosaccharide classes (for example, deoxyhexose versus hexose or pentose versus hexose) along with their corresponding DoPe values are resolved by mass. However, there are two pairs of isomeric compounds that are not readily chromatographically resolved such as: 6-galactose and 6-glucose; 6-mannose and 4-galactose. This ambiguity can be resolved based on a separate monosaccharide analysis, which can be performed complimentary to the linkage analysis. The standards developed in Table 1 were used to determine linkages in more complicated polysaccharides. Although the developed library was used for the positive identification of glycosidic linkages, MRM transitions for trisecting, bisecting, linear, and terminal species for each class of monosaccharide were additionally monitored.
Linkage analysis of polysaccharides
To demonstrate the method’s ability to accurately identify the linkage components of a polysaccharide, the approach was applied to the commercially available polysaccharide standard, galactan (Lupin). The specified structure of galactan contains a linear backbone of repeating β(1,4)-galactose monosaccharides. However, the standard is reported to contain other monosaccharides such as arabinose, rhamnose, and xylose. It is unclear if these components are from polysaccharide contaminants or exist as free monosaccharide impurities.
Relative abundances for the glycosidic linkages identified within galactan were obtained by the analysis of four experimental replicates. A representative MRM chromatogram showing the major linkage abundances present within galactan is shown in Figure S-4. Table S-1 lists a total of 18 linkage residues identified with relative abundances > 0.1 %. Analysis of this standard reveals the presence of 4-galactose/4-mannose (50.4 ± 1.9 %) and T-galactose (25.1 ± 2.6 %) in high abundance. Despite the lack of resolution between 4-galactose and 4-mannose, it is likely that this abundance corresponds solely to 4-galactose based on the reported monosaccharide composition. The surprisingly high relative abundance of T-galactose may be due to the presence of free galactose monosaccharide present within the standard. Two co-eluting bisecting hexose linkage residues, 2,X-hexose and X,X-hexose (9.3 ± 2.1 %), and X,X-hexose (2.7 ± 1.0 %) were found, indicating the presence of an additional heterogeneous polysaccharide. Analysis of the standard also revealed non-hexose linkage residues such as 5-F-arabinose (0.4 ± 0.1 %), T-F-arabinose (5.3 ± 0.7 %), and X-deoxyhexose (2.3 ± 0.5 %). While linkages for rhamnose are not included in the linkage library, it is presumed that the identified X-deoxyhexose residue is a linear linkage of rhamnose. These results confirm the expected linkage composition of the monosaccharide residues present within galactan. Standard deviations were calculated to be < 2.6 %, demonstrating the method’s high reproducibility for the analysis of a purified polysaccharide.
We further evaluate the method’s capabilities for the characterization of polysaccharides within a complex matrix, such as whole carrots. Results by both UHPLC/MRM-MS and the standard GC-MS method were obtained by performing three experimental replicates using the same starting material. A total of 24 linkage residues were elucidated from the whole carrot samples using the LC-MS method. A representative MRM chromatogram for whole carrots is displayed in Figure 2B. Table 2 shows the detailed comparison of the relative abundances for identified glycosidic linkage residues within whole carrots by UHPLC/MRM-MS and the standard GC-MS method. Standard deviations were found to be ≤ 1.1 % and are comparable to the < 1.6 % standard deviation values obtained by the GC-MS analysis workflow. This illustrates reproducibility of the presented method for the analysis of insoluble starting material such as whole foods consisting of a mixture of carbohydrates. The analysis revealed high amounts of T-glucose (37.3 ± 0.6 %) and 4-glucose (19.9 ± 1.1 %) which is likely representative of the starch and cellulose components present within carrots63. The relatively high abundance of T-glucose could be a result of free glucose monomers or the presence of disaccharides, such as sucrose. Furthermore, linkages corresponding to T-F-arabinose (11.3 ± 0.7 %), 4-galactose/6-mannose (9.8 ± 0.1 %), 4-P-xylose (0.7 ± 0.0 %), and 5-F-arabinose (2.4 ± 0.0 %) were detected. These constituents are likely derived from other polysaccharides present within carrots.
Table 2.
Linkage analysis comparison of the same whole carrot (N=3) starting material by both UHPLC/MRM-MS and GC-MS.
| Linkage Residue | RT (mins) | Relative Abundance (%) | GC-MS Linkage Residue | GC-MS Relative Abundance (%) |
|---|---|---|---|---|
| T-Glucose | 12.36 | 37.3 ± 0.6 | T-Glucose | 58.9 ± 1.6 |
| 4-Glucose | 7.87 | 19.9 ± 1.1 | 4-Glucose | 25.9 ± 1.5 |
| T-F-Arabinose | 10.04 | 11.3 ± 0.7 | **T-F-Arabinose | ND |
| 4-Galactose/6-Mannose | 7.45 | 9.8 ± 0.1 | 4-Galactose | 6.9 ± 0.5 |
| T-Mannose | 12.92 | 4.6 ± 0.5 | T-Mannose | 0.6 ± 0.1 |
| 4-Mannose | 10.76 | 0.3 ± 0.0 | 4-Mannose | 1.8 ± 0.0 |
| T-Galactose | 11.78 | 3.3 ± 0.2 | T-Galactose | 1.4 ± 0.1 |
| 4-P-Xylose | 5.98 | 0.7 ± 0.0 | **4-P-Xylose | ND |
| 5-F-Arabinose | 6.12 | 2.4 ± 0.0 | **5-F-Arabinose | ND |
| 3-Glucose | 8.52 | 1.0 ± 0.2 | 3-Glucose | 0.5 ± 0.1 |
| T-P-Xylose | 8.29 | 0.9 ± 0.0 | **T-P-Xylose | ND |
| 3,4-P-Xylose | 5.22 | 0.8 ± 0.0 | **3,4-P-Xylose | ND |
| 2-Glucose | 9.51 | 0.8 ± 0.1 | 2-Glucose | 1.1 ± 0.3 |
| X-Deoxyhexose | 8.93 | 0.7 ± 0.2 | **X-Deoxyhexose | ND |
| 6-Galactose/6-Glucose | 6.68 | 0.6 ± 0.0 | **6-Glucose | ND |
| T-Fucose | 11.44 | 0.5 ± 0.0 | **T-Fucose | ND |
| T-Deoxyhexose | 11.98 | 0.5 ± 0.0 | **T-Deoxyhexose | ND |
| X,X-Hexose | 6.44 | 0.4 ± 0.1 | 4,6-Glucose | 0.6 ± 0.1 |
| 3-Mannose | 10.25 | 3.7 ± 0.1 | **3-Mannose | ND |
| T-Hexose | 13.67 | 0.2 ± 0.0 | **T-Hexose | ND |
| X,X-Hexose | 7.15 | 0.1 ± 0.0 | 3,4-Glucose | 0.7 ± 0.1 |
| X-Pentose | 7.15 | 0.1 ± 0.0 | **X-Pentose | ND |
| *2,4-Galactose | - | ND | 2,4-Galactose | 0.6 ± 0.1 |
| *2-Rhamnose | - | ND | 2-Rhamnose | 0.3 ± 0.0 |
| *2,4-Rhamnose | - | ND | 2,4-Rhamnose | 0.8 ± 0.0 |
Not detected by UHPLC/MRM-MS
Not detected by GC-MS
Analysis of whole carrots by GC-MS at a separate site resulted in identification of only 13 glycosidic linkage residues. A combination of differences in sample preparation and instrumental sensitivity between the two platforms may contribute to the discrepency in identified linkage residues. Results from the GC-MS workflow show identification of linkages mainly from hexose residues and < 0.8 % for deoxyhexose residues. The lack of arabinose and xylose residues detected by the GC-MS workflow is noteworthy given the relatively high abundances observed when using the UHPLC/MRM-MS workflow. Overall, the difference in the number of identified linkage residues between the two platforms exemplify the increased sensitivity advantage when performing the analysis using the presented workflow.
CONCLUSIONS
In this research, we developed a MRM method that is capable of rapidly profiling 22 unique glycosidic linkages present in oligosaccharides and polysaccharides. While the number of linkages was limited by the availability of standards, we are currently synthesizing new linkage standards that will significantly increase the total number in the near future. Although the use of acid hydrolysis after permethylation results in a loss of alpha/beta stereochemical information, the combination of these approaches followed by derivatization with PMP results in effective chromatographic separation of isomers during the analysis. Method validation was performed with the GC-MS method, which is still the gold standard for this type of analysis. Additionally, the high reproducibility of the analysis in experimental replicates of a polysaccharide suggests that the developed sample preparation method is sufficiently robust to profile branching residues together with the linear and terminal components within polysaccharides. This method further compliments our recent method on monosaccharide analysis that is also based on LC-MS analysis. The monosaccharide analysis method improved the previous existing GC-MS analysis method by being over 1000 more sensitive and five times faster in analysis time15. The presented UHPLC/MRM-MS method provides similar enhancements and is a powerful new tool for carbohydrate glycosidic linkage analysis. It will, we hope, renew interests in the development of other protecting groups and reducing-end labels for more specific applications.
Supplementary Material
ACKNOWLEDGMENT
Funding Sources
Funding provided by the Institute of General Medicine, of the National Institutes of Health is gratefully acknowledged (RO1GM049077). This work was supported by the Chemical Sciences, Geosciences and Biosciences Division, Office of Basic Energy Sciences, U.S. Department of Energy grant (DE-FG02-93ER20097) to Parastoo Azadi at the Complex Carbohydrate Research Center.
Footnotes
Supporting Information.
Supporting figures include the effect of permethylation reaction time for maltohexaose (Figure S-1), effect of hydrolysis time for amylopectin (Figure S-2), MRM chromatograms for 4-O-(b-D-galactopyranosyl)-D-galactopyranose and 2’-fucosyllactose (Figure S-3), an MRM chromatogram for galactan from Lupin (Figure S-4). A scheme for the proposed fragment ion structures of monosaccharide linkage residues with or without linkage at the 2-position is shown in Scheme S-1. A detailed comparison of relative abundances calculated from four experimental replicates of galactan are listed in Table S-1.
The authors declare no competing financial interest.
REFERENCES
- 1.Caffall KH; Mohnen D, The structure, function, and biosynthesis of plant cell wall pectic polysaccharides. Carbohydr Res 2009, 344 (14), 1879–900. [DOI] [PubMed] [Google Scholar]
- 2.Jequier E, Carbohydrates as a source of energy. Am J Clin Nutr 1994, 59 (3 Suppl), 682S–685S. [DOI] [PubMed] [Google Scholar]
- 3.Hart GW; Copeland RJ, Glycomics hits the big time. Cell 2010, 143 (5), 672–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Latge JP, The cell wall: a carbohydrate armour for the fungal cell. Mol Microbiol 2007, 66 (2), 279–90. [DOI] [PubMed] [Google Scholar]
- 5.Tran Thanh Van K; Toubart P; Cousson A; Darvill AG; Gollin DJ; Chelf P; Albersheim P, Manipulation of the morphogenetic pathways of tobacco explants by oligosaccharins. Nature 1985, 314 (6012), 615–617. [Google Scholar]
- 6.Barile D; Rastall RA, Human milk and related oligosaccharides as prebiotics. Curr Opin Biotechnol 2013, 24 (2), 214–9. [DOI] [PubMed] [Google Scholar]
- 7.Musilova S; Rada V; Vlkova E; Bunesova V, Beneficial effects of human milk oligosaccharides on gut microbiota. Benef Microbes 2014, 5 (3), 273–83. [DOI] [PubMed] [Google Scholar]
- 8.Smilowitz JT; Lebrilla CB; Mills DA; German JB; Freeman SL, Breast milk oligosaccharides: structure-function relationships in the neonate. Annu Rev Nutr 2014, 34, 143–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dwek RA, Glycobiology: Toward Understanding the Function of Sugars. Chemical Reviews 1996, 96 (2), 683–720. [DOI] [PubMed] [Google Scholar]
- 10.Kailemia MJ; Park D; Lebrilla CB, Glycans and glycoproteins as specific biomarkers for cancer. Anal Bioanal Chem 2017, 409 (2), 395–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moremen KW; Tiemeyer M; Nairn AV, Vertebrate protein glycosylation: diversity, synthesis and function. Nat Rev Mol Cell Biol 2012, 13 (7), 448–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Torello LA; Yates AJ; Thompson DK, Critical study of the alditol acetate method for quantitating small quantities of hexoses and hexosamines in gangliosides. Journal of Chromatography A 1980, 202 (2), 195–209. [DOI] [PubMed] [Google Scholar]
- 13.Richardson S; Gorton L, Characterisation of the substituent distribution in starch and cellulose derivatives. Analytica Chimica Acta 2003, 497 (1–2), 27–65. [Google Scholar]
- 14.Kailemia MJ; Ruhaak LR; Lebrilla CB; Amster IJ, Oligosaccharide analysis by mass spectrometry: a review of recent developments. Anal Chem 2014, 86 (1), 196–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xu G; Amicucci MJ; Cheng Z; Galermo AG; Lebrilla CB, Revisiting monosaccharide analysis - quantitation of a comprehensive set of monosaccharides using dynamic multiple reaction monitoring. Analyst 2017, 143 (1), 200–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.He Y; Zhang M; Shan M; Zeng P; Li X; Hao C; Dou H; Yang D; Feng N; Zhang L, Optimizing microwave-assisted hydrolysis conditions for monosaccharide composition analyses of different polysaccharides. Int J Biol Macromol 2018, 118 (Pt A), 327–332. [DOI] [PubMed] [Google Scholar]
- 17.Koh DW; Park JW; Lim JH; Yea MJ; Bang DY, A rapid method for simultaneous quantification of 13 sugars and sugar alcohols in food products by UPLC-ELSD. Food Chem 2018, 240, 694–700. [DOI] [PubMed] [Google Scholar]
- 18.Robinson RC; Poulsen NA; Barile D, Multiplexed bovine milk oligosaccharide analysis with aminoxy tandem mass tags. PLoS One 2018, 13 (4), e0196513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pereira GA; Arruda HS; Molina G; Pastore GM, Extraction optimization and profile analysis of oligosaccharides in banana pulp and peel. Journal of Food Processing and Preservation 2018, 42 (1). [Google Scholar]
- 20.Totten SM; Wu LD; Parker EA; Davis JC; Hua S; Stroble C; Ruhaak LR; Smilowitz JT; German JB; Lebrilla CB, Rapid-throughput glycomics applied to human milk oligosaccharide profiling for large human studies. Anal Bioanal Chem 2014, 406 (30), 7925–35. [DOI] [PubMed] [Google Scholar]
- 21.Davis JC; Totten SM; Huang JO; Nagshbandi S; Kirmiz N; Garrido DA; Lewis ZT; Wu LD; Smilowitz JT; German JB; Mills DA; Lebrilla CB, Identification of Oligosaccharides in Feces of Breast-fed Infants and Their Correlation with the Gut Microbial Community. Mol Cell Proteomics 2016, 15 (9), 2987–3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang X; Kim SM; Ruzanski R; Chen Y; Moses S; Ling WL; Li X; Wang SC; Li H; Ambrogelly A; Richardson D; Shameem M, Ultrafast and high-throughput N-glycan analysis for monoclonal antibodies. MAbs 2016, 8 (4), 706–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim YG; Jeong HJ; Jang KS; Yang YH; Song YS; Chung J; Kim BG, Rapid and high-throughput analysis of N-glycans from ovarian cancer serum using a 96-well plate platform. Anal Biochem 2009, 391 (2), 151–3. [DOI] [PubMed] [Google Scholar]
- 24.Kronewitter SR; de Leoz ML; Peacock KS; McBride KR; An HJ; Miyamoto S; Leiserowitz GS; Lebrilla CB, Human serum processing and analysis methods for rapid and reproducible N-glycan mass profiling. J Proteome Res 2010, 9 (10), 4952–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aldredge D; An HJ; Tang N; Waddell K; Lebrilla CB, Annotation of a serum N-glycan library for rapid identification of structures. J Proteome Res 2012, 11 (3), 1958–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sawardeker JS; Sloneker JH; Jeanes A, Quantitative Determination of Monosaccharides as Their Alditol Acetates by Gas Liquid Chromatography. Analytical Chemistry 1965, 37 (12), 1602–1604. [Google Scholar]
- 27.Björndal H; Hellerqvist CG; Lindberg B; Svensson S, Gas-Liquid Chromatography and Mass Spectrometry in Methylation Analysis of Polysaccharides. Angewandte Chemie International Edition in English 1970, 9 (8), 610–619. [Google Scholar]
- 28.Hakomori S, A Rapid Permethylation of Glycolipid, and Polysaccharide Catalyzed by Methylsulfinyl Carbanion in Dimethyl Sulfoxide. J Biochem 1964, 55, 205–8. [PubMed] [Google Scholar]
- 29.Sims IM; Carnachan SM; Bell TJ; Hinkley SFR, Methylation analysis of polysaccharides: Technical advice. Carbohydr Polym 2018, 188, 1–7. [DOI] [PubMed] [Google Scholar]
- 30.Biermann CJ; McGinnis GD, Analysis of carbohydrates by GLC and MS. CRC Press: Boca Raton, Fla., 1989. [Google Scholar]
- 31.Yokota H; Mori K; Yamaguchi H; Kaniwa H; Saisho N, Monosaccharide composition analysis of pamiteplase by anion exchange chromatography with pulsed amperometric detection. Journal of Pharmaceutical and Biomedical Analysis 1999, 21 (4), 767–774. [DOI] [PubMed] [Google Scholar]
- 32.Yu Ip CC; Manam V; Hepler R; Hennessey JP, Carbohydrate composition analysis of bacterial polysaccharides: Optimized acid hydrolysis conditions for HPAEC-PAD analysis. Analytical Biochemistry 1992, 201 (2), 343–349. [DOI] [PubMed] [Google Scholar]
- 33.Zhang Z; Khan NM; Nunez KM; Chess EK; Szabo CM, Complete monosaccharide analysis by high-performance anion-exchange chromatography with pulsed amperometric detection. Anal Chem 2012, 84 (9), 4104–10. [DOI] [PubMed] [Google Scholar]
- 34.Mao L; Shao S; Sun S; Wang Y; Xu P; Cai L, Purification, Physicochemical Characterization, and Bioactivities of Polysaccharides from Puerh Tea. Journal of Food and Nutrition Research 2014, 2 (12), 1007–1014. [Google Scholar]
- 35.Li H; Long C; Zhou J; Liu J; Wu X; Long M, Rapid analysis of mono-saccharides and oligo-saccharides in hydrolysates of lignocellulosic biomass by HPLC. Biotechnol Lett 2013, 35 (9), 1405–9. [DOI] [PubMed] [Google Scholar]
- 36.Grill E; Huber C; Oefner P; Vorndran A; Bonn G, Capillary zone electrophoresis ofp-aminobenzoic acid derivatives of aldoses, ketoses and uronic acids. Electrophoresis 1993, 14 (1), 1004–1010. [DOI] [PubMed] [Google Scholar]
- 37.Wu X; Jiang W; Lu J; Yu Y; Wu B, Analysis of the monosaccharide composition of water-soluble polysaccharides from Sargassum fusiforme by high performance liquid chromatography/electrospray ionisation mass spectrometry. Food Chem 2014, 145, 976–83. [DOI] [PubMed] [Google Scholar]
- 38.Zhang P; Wang Z; Xie M; Nie W; Huang L, Detection of carbohydrates using a pre-column derivatization reagent 1-(4-isopropyl) phenyl-3-methyl-5-pyrazolone by high-performance liquid chromatography coupled with electrospray ionization mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci 2010, 878 (15–16), 1135–44. [DOI] [PubMed] [Google Scholar]
- 39.Huang J; Kailemia MJ; Goonatilleke E; Parker EA; Hong Q; Sabia R; Smilowitz JT; German JB; Lebrilla CB, Quantitation of human milk proteins and their glycoforms using multiple reaction monitoring (MRM). Anal Bioanal Chem 2017, 409 (2), 589–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Song E; Pyreddy S; Mechref Y, Quantification of glycopeptides by multiple reaction monitoring liquid chromatography/tandem mass spectrometry. Rapid Commun Mass Spectrom 2012, 26 (17), 1941–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ross AR; Ambrose SJ; Cutler AJ; Feurtado JA; Kermode AR; Nelson K; Zhou R; Abrams SR, Determination of endogenous and supplied deuterated abscisic acid in plant tissues by high-performance liquid chromatography-electrospray ionization tandem mass spectrometry with multiple reaction monitoring. Anal Biochem 2004, 329 (2), 324–33. [DOI] [PubMed] [Google Scholar]
- 42.Bennett BD; Kimball EH; Gao M; Osterhout R; Van Dien SJ; Rabinowitz JD, Absolute metabolite concentrations and implied enzyme active site occupancy in Escherichia coli. Nat Chem Biol 2009, 5 (8), 593–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hong Q; Ruhaak LR; Totten SM; Smilowitz JT; German JB; Lebrilla CB, Label-free absolute quantitation of oligosaccharides using multiple reaction monitoring. Anal Chem 2014, 86 (5), 2640–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xu G; Davis JC; Goonatilleke E; Smilowitz JT; German JB; Lebrilla CB, Absolute Quantitation of Human Milk Oligosaccharides Reveals Phenotypic Variations during Lactation. J Nutr 2017, 147 (1), 117–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hammad LA; Saleh MM; Novotny MV; Mechref Y, Multiple-reaction monitoring liquid chromatography mass spectrometry for monosaccharide compositional analysis of glycoproteins. J Am Soc Mass Spectrom 2009, 20 (6), 1224–34. [DOI] [PubMed] [Google Scholar]
- 46.Hammad LA; Derryberry DZ; Jmeian YR; Mechref Y, Quantification of monosaccharides through multiple-reaction monitoring liquid chromatography/mass spectrometry using an aminopropyl column. Rapid Commun Mass Spectrom 2010, 24 (11), 1565–74. [DOI] [PubMed] [Google Scholar]
- 47.Han J; Lin K; Sequria C; Yang J; Borchers CH, Quantitation of low molecular weight sugars by chemical derivatization-liquid chromatography/multiple reaction monitoring/mass spectrometry. Electrophoresis 2016, 37 (13), 1851–60. [DOI] [PubMed] [Google Scholar]
- 48.Doares SH; Albersheim P; Darvill AG, An Improved Method for the Preparation of Standards for Glycosyl-Linkage Analysis of Complex Carbohydrates. Carbohyd Res 1991, 210, 311–317. [Google Scholar]
- 49.Sweeley CC; Bentley R; Makita M; Wells WW, Gas-Liquid Chromatography of Trimethylsilyl Derivatives of Sugars and Related Substances. Journal of the American Chemical Society 1963, 85 (16), 2497–2507. [Google Scholar]
- 50.Ruiz-Matute AI; Hernandez-Hernandez O; Rodriguez-Sanchez S; Sanz ML; Martinez-Castro I, Derivatization of carbohydrates for GC and GC-MS analyses. J Chromatogr B Analyt Technol Biomed Life Sci 2011, 879 (17–18), 1226–40. [DOI] [PubMed] [Google Scholar]
- 51.Sassaki GL; Iacomini M; Gorin PAJ, Methylation-GC-MS analysis of arabinofuranose- and galactofuranose-containing structures: rapid synthesis of partially O-methylated alditol acetate standards. Anais da Academia Brasileira de Ciências 2005, 77 (2), 223–234. [Google Scholar]
- 52.Stellner K; Saito H; Hakomori S-I, Determination of aminosugar linkages in glycolipids by methylation. Archives of Biochemistry and Biophysics 1973, 155 (2), 464–472. [DOI] [PubMed] [Google Scholar]
- 53.Wang W; Chen F; Wang Y; Wang L; Fu H; Zheng F; Beecher L, Optimization of reactions between reducing sugars and 1-phenyl-3-methyl-5-pyrazolone (PMP) by response surface methodology. Food Chem 2018, 254, 158–164. [DOI] [PubMed] [Google Scholar]
- 54.Lv Y; Yang X; Zhao Y; Ruan Y; Yang Y; Wang Z, Separation and quantification of component monosaccharides of the tea polysaccharides from Gynostemma pentaphyllum by HPLC with indirect UV detection. Food Chemistry 2009, 112 (3), 742–746. [Google Scholar]
- 55.Li J; Sun J; Wang Z; Huang L, Simultaneous Determination of Aldoses and Uronic Acids of Citrus Pectin by LC with Precolumn Derivatization and UV Detection. Chromatographia 2010, 72 (9–10), 849–855. [Google Scholar]
- 56.Honda S; Akao E; Suzuki S; Okuda M; Kakehi K; Nakamura J, High-performance liquid chromatography of reducing carbohydrates as strongly ultraviolet-absorbing and electrochemically sensitive 1-phenyl-3-methyl5-pyrazolone derivatives. Analytical Biochemistry 1989, 180 (2), 351–357. [DOI] [PubMed] [Google Scholar]
- 57.Stepan H; Staudacher E, Optimization of monosaccharide determination using anthranilic acid and 1-phenyl-3-methyl-5-pyrazolone for gastropod analysis. Anal Biochem 2011, 418 (1), 24–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Saba JA; Shen X; Jamieson JC; Perreault H, Investigation of different combinations of derivatization, separation methods and electrospray ionization mass spectrometry for standard oligosaccharides and glycans from ovalbumin. J Mass Spectrom 2001, 36 (5), 563–74. [DOI] [PubMed] [Google Scholar]
- 59.Strydom DJ, Chromatographic separation of 1-phenyl-3-methyl-5-pyrazolone-derivatized neutral, acidic and basic aldoses. Journal of Chromatography A 1994, 678 (1), 17–23. [Google Scholar]
- 60.Cancilla MT; Penn SG; Lebrilla CB, Alkaline Degradation of Oligosaccharides Coupled with Matrix-Assisted Laser Desorption/Ionization Fourier Transform Mass Spectrometry: A Method for Sequencing Oligosaccharides. Analytical Chemistry 1998, 70 (4), 663–672. [DOI] [PubMed] [Google Scholar]
- 61.Timell TE, The Acid Hydrolysis of Glycosides: I. General Conditions and the Effect of the Nature of the Aglycone. Canadian Journal of Chemistry 1964, 42 (6), 1456–1472. [Google Scholar]
- 62.Wang QC; Zhao X; Pu JH; Luan XH, Influences of acidic reaction and hydrolytic conditions on monosaccharide composition analysis of acidic, neutral and basic polysaccharides. Carbohydr Polym 2016, 143, 296–300. [DOI] [PubMed] [Google Scholar]
- 63.Theander O; Åman P, Studies on dietary fibre. A method for the analysis and chemical characterisation of total dietary fibre. Journal of the Science of Food and Agriculture 1982, 33 (4), 340–344. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.