

Abstract
ETHE1 is a member of a growing subclass of nonheme Fe enzymes that catalyzes transformations of sulfur containing substrates without a cofactor. ETHE1 dioxygenates glutathione persulfide (GSSH) to glutathione (GSH) and sulfite in a reaction that is similar to that of cysteine dioxygenase (CDO), but with monodentate (vs. biden-tate) substrate coordination and a 2-His/1-Asp (vs. 3-His) ligand set. In this study, we demonstrate that GSS− binds directly to the iron active site causing coordination unsaturation to prime the site for O2 activation. Nitrosyl complexes without and with GSSH were generated and spectroscopically characterized as unreactive analogs for the invoked ferric superoxide intermediate. New spectral features from persulfide binding to the FeIII include the appearance of a low energy FeIII ligand field transition, an energy shift of a NO− to FeIII CT transition, and two new GSS− to FeIII CT transitions. Time-dependent density functional theory calculations were used to simulate the experimental spectra to determine the persulfide orientation. Correlation of these spectral features with those of monodentate cysteine binding in isopenicillin N synthase (IPNS) shows that the persulfide is a poorer donor, but still results in an equivalent frontier molecular orbital for reactivity. The ETHE1 persulfide dioxygenation reaction coordinate was calculated, and while the initial steps are similar to the reaction coordinate of CDO, an additional hydrolysis step is required in ETHE1 to break the S-S bond. Unlike ETHE1 and CDO, which both oxygenate sulfur, IPNS oxidizes sulfur through an initial H-atom abstraction. Thus, factors that determine oxygenase vs. oxidase reactivity were evaluated. In general, sulfur oxygenation is thermodynamically favored and has a lower barrier for reactivity. However, in IPNS, second sphere residues in the active site pocket constrain the substrate raising the barrier for sulfur oxygenation relative to oxidation via H-atom abstraction.
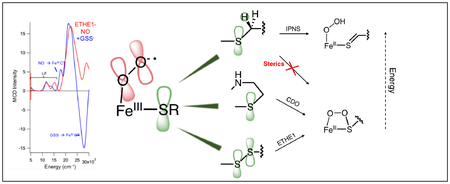
Graphical TOC Entry
Introduction
Mononuclear nonheme iron (NHFe) enzymes catalyze biologically important chemical transformations including those related to antibiotic and neurotransmitter biosynthesis, hypoxia regulation, bioremediation and DNA repair.1–6 One subclass of these enzymes activates O2 utilizing non redox active substrates,7 and a growing number of these act on sulfur containing substrates including isopenicillin N-synthase (IPNS), cysteine dioxygenase (CDO), persulfide dioxygenase (ETHE1) and sulfoxide synthase (EgtB, OvoA). These enzymes perform different reactions, including sulfur oxidation (IPNS) and sulfur oxygenation (CDO, ETHE1).8–12
IPNS, which is involved in penicillin biosynthesis, catalyzes an oxidative double ring closure of its tripeptide substrate (δ-(L-α-aminoadipoyl)-L-cysteinyl-D-valine, ACV) to form isopenicillin N and reducing O2 to water in the process (Scheme 1A).8,13–16 The FeII is co-ordinated by the common 2-His/1-Asp facial triad, and ACV binds monodentate via its cysteine thiolate, displacing two of the three water ligands in a five coordinate site.17 Charge donation from the thiolate to the Fe lowers its 1 electron (e−) reduction potential to favor O2 binding as superoxide,18 and recently, this ferric superoxide species was detected experimentally.19 Calculated reaction coordinates indicate that this intermediate abstracts a hydrogen atom from the substrate β-cysteinyl carbon to form a ferrous hydroperoxo species and a C-S double bond.20,21 IPNS exhibits sulfur oxygenase activity with a non-native substrate, but after the first ring closure, which mirrors the native substrate reactivity.22
Scheme 1:

(A) Conversion of ACV to Isopenicillin N by IPNS (B) Conversion of Cysteine to Cysteine Sulfinic Acid by CDO (C) Conversion of Glutathione Persulfide to Glutathione and Sulfite by ETHE1
CDO dioxygenates cysteine to form cysteine sulfinic acid (CSA) in the first step of cysteine catabolism (Scheme 1B).9,23 The substrate coordinates the ferrous site in a bidentate mode via the cysteine thiolate and amine groups, and along with the 3-His triad present in this enzyme, this leaves one coordination position available on the Fe for O2 binding.24 Reaction coordinate calculations of the CDO mechanism invoke binding of O2 to generate a ferric superoxo intermediate that performs an electrophilic attack of the distal oxygen on the sulfur atom, forming a peroxo-bridged structure. Heterolytic O-O bond cleavage follows to form a ferryl oxo and sulfoxide.25–28 Recently, a short-lived intermediate was detected in the reaction of CDO/Cys with O2 with absorption maxima at 500 and 640 nm. The intermediate could not be further characterized, but time-dependent density functional theory (TD-DFT) calculations predict it to be the peroxo-bridged intermediate that precedes the ferryl oxo/sulfoxide.29
Much less is understood about the mechanism of the persulfide dioxygenase ETHE1, which is important in mitochondrial sulfide oxidation. Mutations in the gene encoding ETHE1 result in ethylmalonic encephalopathy - an autosomal recessive disorder that causes severe gastrointestinal and neurological symptoms resulting in premature death.30–32 ETHE1 catalyzes the dioxygenation of glutathione persulfide (GSSH) to give glutathione (GSH) and sulfite (Scheme 1C). Crystallography on the resting form of human ETHE1 shows that the ferrous center is coordinated by the common 2-His/1-Asp facial triad motif and three water molecules in a six coordinate (6C) site.33 Substrate has not been co-crystallized with ETHE1 due to instability of the persulfide, and both bidentate34 and monodentate33 binding modes have been suggested in the literature. A product (GSH) bound form of a bacterial persulfide dioxygenase has been crystallized showing monodentate coordination via the thiolate to a ferric center, but it is unclear if this structure is catalytically relevant.35 It has been suggested that the bidentate Cys coordination and the 3-His triad in CDO are critical to its ability to dioxygenate sulfur.26 Since ETHE1 catalyzes a sulfur dioxygenation reaction like that of CDO, but utilizing a 2-His/1-(Asp/Glu) facial triad and with monodentate substrate coordination like IPNS (vide infra), it provides the opportunity to evaluate the factors that lead to sulfur oxidation vs. oxygenation.
Interest in studying the oxygen adduct in the first step of NHFe enzymatic reactions has prompted use of nitric oxide as an unreactive O2 surrogate.1,13,36 NO binds at the FeII center to create a stable, chromophoric and EPR (electron paramagnetic resonance) active species (generally S = 3/2) that can be studied with multiple spectroscopic techniques. Extensive analysis of high spin (HS) (S = 3/2) {FeNO}7 model complexes has resulted in its electronic structure description as a HS FeIII (S = 5/2) antiferromagnetically (AF) coupled to NO− (S = 1).37,38 The {FeNO}7 complexes of IPNS and CDO have been formed and characterized spectroscopically and computationally. IPNS-ACV-NO is a HS (S = 3/2) species with an electronic structure equivalent to that determined for the model complexes.13,17 Relative to model complexes, IPNS-ACV-NO exhibits new charge transfer (CT) bands due to the thiolate coordination that have provided insight into the Cys-FeIII bond and its activation of a key frontier molecular orbital (FMO) for reactivity. In contrast to nitrosyl complexes of other enzymes, CDO-Cys-NO has been found to be a low spin (LS) S = 1/2 complex with an electronic structure description of LS FeII (S = 0) with NO• (S = 1/2), which has been invoked in reaction coordinate calculations.26,39
In the present study, we used VTVH MCD (variable temperature, variable field magnetic circular dichroism) to determine whether GSSH binding opened a coordination position on the FeII for O2 binding as has been observed in other NHFe enzymes.7 We formed the {FeNO}7 complexes without and with substrate and determined the new spectral features from the bound persulfide. Using DFT we were able to define the orientation of the persulfide bond relative to the other Fe ligands, and we described the persulfide-FeIII bond for comparison to the thiolate-FeIII bond defined for IPNS.18 We computationally extended this to the {FeO2}8 complex and identified the key FMO that initiates the ETHE1 sulfur dioxygenation reaction. The calculated reaction coordinate showed similarities to those defined for CDO, which utilizes a 3-His triad and bidentate bound substrate.25–28 However, the ETHE1 active site structure defined here is similar to that of IPNS, which catalyzes a sulfur oxidation on a monodentate bound substrate, utilizing a 2-His/1-Asp facial triad. This led us to perform a series of calculations that identified how these sulfur transforming enzymes select for sulfur oxidation vs. oxygenation.
Results and Analysis
Ferrous ETHE1 Spectroscopy
CD and MCD.
Near-IR CD (circular dichroism) and MCD (magnetic CD) spectroscopies were used to study FeII-ETHE1 and the effect of substrate binding. All MCD samples were prepared with deuterated sucrose as a glassing agent, because deuterated glycerol was found to perturb the FeII-ETHE1 CD spectrum (Figure S1). The room temperature (RT) CD and +7 T, 5 K MCD spectra of FeII-ETHE1 are shown in Figure 1. From MCD studies of ferrous model complexes in different coordination environments, FeII in a distorted octahedral site has two ligand field (LF) transitions to the 5E set in the near-IR region centered around ~10,000 cm−1 and split by ~2,000 cm−1. In a square pyramidal five coordinate (5C) site, these transitions further split to show one at ~5,000 and one at ~10,000 cm−1. Distortion to a trigonal bipyramidal site shifts these transitions to <5,000 and <10,000 cm−1, and for distorted tetrahedral sites, only low energy transitions are observed (~5,000 – 7,000 cm−1).1 There are two transitions in the CD spectrum of FeII-ETHE1 at 8,200 and 11,000 cm−1 (Figure 1, top). Upon cooling to 5 K, these two transitions sharpen and shift to 8,800 and 11,000 cm−1 in MCD (Figure 1, bottom). The energies of these transitions give a 5E splitting of 2,200 cm−1 that is in the range of 6C FeII sites in NHFe enzymes.
Figure 1:

Room temperature CD (top) and +7 T, 5 K MCD (bottom) spectra of FeII-ETHE1 with Gaussian fits.
FeII-ETHE1 was titrated with GSSH until the spectrum stopped changing, indicating saturation of the active site with substrate. The resulting RT CD spectrum (Figure 2A, green) is dramatically different from that of resting enzyme (Figure 1, top) with a broad, negative feature centered at ~10,000 cm−1. To ensure that the observed spectral change resulted from GSS− binding to the active site, controls were performed using the contaminants present from substrate preparation, GSH and HS−. The CD and MCD spectra of FeII-ETHE1 in the presence of GSH and HS− (Figures S2 and S3, respectively) are identical to those of FeII-ETHE1. This is seen clearly in the overlay of the MCD spectra in Figure S4. Thus, the changes observed upon substrate addition are due only to GSS− binding. The broad 10,000 cm−1 feature in RT CD is composed of two transitions that sharpen and shift up in energy upon cooling in the low temperature (LT) CD spectrum (Figure 2A, black). Application of a magnetic field gives the MCD spectrum in Figure 2A (blue). Because the selection rules governing CD and MCD are different, the sign and intensity of transitions vary between the methods. However, the transitions observed in both CD and MCD must have the same energy and bandwidth if they are collected at the same temperature (as in Figure 2A, black and blue spectra). There are at least three transitions in the MCD spectrum labeled bands 1, 2 and 4, and simultaneous fitting with the LT CD spectrum requires that a fourth transition be included (Band 3, at 10,450 cm−1), although it is not intense in MCD. Note that band 4 is not from unconverted resting enzyme (see Figure S5). Thus, the (FeII/GSS−)-ETHE1 MCD spectrum exhibits four transitions. Because a single FeII site can only have two LF transitions in this energy region, there must be two substrate bound FeII sites contributing to the blue spectrum in Figure 2A. The possibility that bands 3 and 4 are from a 6C site can be excluded, because their splitting (850 cm−1) is smaller than the minimum splitting of 1,200 cm−1 that results from a rigorously octahedral FeII site having an excited state Jahn-Teller effect.40 This leaves two possible combinations for the LF splitting of the d orbitals. Either bands 1 and 3, and bands 2 and 4 can be paired for two distinct 5C sites or bands 1 and 4, and bands 2 and 3 pair for one 5C and one 6C site, respectively.
Figure 2:

(A) Room temperature CD (green), 5K CD (black) and +5T, 5K MCD (blue) spectra of (FeII/GSS−)-ETHE1 with Gaussian fits. Intensity at 6,000 cm−1 in black spectrum is due to a baseline issue from glass strain. Arrows in blue spectrum indicate where saturation magnetization data were collected (B) at 6,450 cm−1 and (C) at 8,900 cm−1.
VTVH MCD.
To assess whether the two sites contributing to the (FeII/GSS−)-ETHE1 MCD spectrum are 5C/5C or 6C/5C, VTVH MCD was used to define their ground state splittings. Saturation magnetization data could be collected on bands 1 and 2 at energies indicated by the arrows in Figure 2A, blue spectrum. Their saturation magnetization curves do not overlay within error showing that they are associated with different FeII sites. VTVH MCD data were not collected on band 3 due to its very low intensity in MCD. Data were collected on band 4, however due to its low intensity, only the low temperature (2K) isotherm had good enough error bars to fit (Figure S6). The VTVH MCD data were fit to a non-Kramers doublet model, which in turn gives the LF splitting of the FeII dπ orbitals, where Δ defines the axial splitting (Δ = dxz,yz - dxy) and V the rhombic splitting (V = dxz - dyz).1 The data collected at 8,900 cm−1 on band 2 (Figure 2C) are best fit to an axial site with Δ = +700 cm−1 and |V/2Δ| = 0.16. These dπ orbital splittings are inset in Figure 2C and are consistent with a distorted 6C site. The other transition from the 6C site must be band 3 (at ~10,500 cm−1), based on the 5E splitting predicted by LF theory. VTVH MCD data collected at 6,450 cm−1 on band 1 (Figure 2B) are fit with Δ = +1,000 cm−1 and | V/2 | = 0.22. The larger tetragonal splitting is consistent with a 5C site, and therefore, the other LF transition is assigned as band 4 (11,300 cm−1). The fit to the data collected on band 4 could not distinguish between the different ground states associated with bands 1 and 2 (Figure S6). Importantly, the four transitions observed in the CD/MCD spectra of (FeII/GSS−)-ETHE1 represent a mixture of two substrate bound FeII sites, one 6C and one 5C. The presence of a 5C site indicates that substrate binding opens a coordination position on the FeII for O2 activation. A summary of the ground (Δ and |V |) and excited (Δ5E) state splittings for these two sites is presented in Table 1.
Table 1:
Ground and excited state splitting parameters for the two sites in the (FeII/GSS−)- ETHE1 MCD spectrum.
| Parameter | 6C Component | 5C Component |
|---|---|---|
| Δ5E (cm−1) | 2,000 | 4,300 |
| Δ(cm−1) | + 700 | + 1,000 |
| |V/2Δ| | 0.16 | 0.22 |
| |V| (cm−1) | 220 | 440 |
{FeNO}7 Spectroscopy
Ground State Spectroscopy.
The {FeNO}7 complexes of ETHE1 were generated without and with substrate, and their EPR spectra are shown in Figure 3A. The ETHE1-NO complex exhibits effective g’ values of gx’ = 4.10, gy’ = 3.95 and gz’ = 2.00 and an |E/D| of 0.013, indicating that it is a HS S = 3/2, nearly axial species (blue spectrum), consistent with NO adducts of NHFe model complexes38 and enzymes in the absence of substrate with both facial and 3-His triads.18,39 The ETHE1-GSS-NO EPR signal indicates that the substrate bound complex also has a HS S = 3/2 ground state with gx’ = 4.10, gy’ = 3.97 and gz’ =2.01 and an |E/D| of 0.014 (Figure 3, orange spectrum). These EPR spectra are described by the spin Hamiltonian in Equation 1, where g0 = 2.0023 and D and E are the axial and rhombic zero-field splitting (ZFS) parameters, respectively.
Figure 3:

(A) EPR spectra and corresponding fits for ETHE1-NO (blue) and ETHE1-GSS NO (orange). Inverse temperature dependence of the integrated area of the EPR signal and best fit to the data for (B) ETHE1-NO and (C) ETHE1-GSS-NO.
| (1) |
The spin Hamiltonian splits the Ms = ± 1/2, ± 3/2 doublets by the amount 2D. Temperature dependent EPR spectra were collected for both {FeNO}7 complexes and show decreasing EPR intensity with increasing temperature under non-saturating conditions. The inverse relationship of signal intensity to temperature indicates that D is positive with the Ms = ± 1/2 doublet lowest in energy. To determine D, the integrated area of the EPR signal was fit to the Curie law including its Boltzmann population (Equation 2), where C is the Curie constant, T is the temperature in Kelvin, and k is the Boltzmann constant. This gives a D of +8.6 ± 1.6 cm−1 for ETHE1-NO (Figure 3B) and +6.6 ± 1.6 cm−1 for ETHE1-GSS-NO (Figure 3C).
| (2) |
A decrease in the axial ZFS parameter (D) with substrate binding has been observed in other NHFe systems and has been attributed to strong equatorial donation of the substrate into the FeIII dπ orbitals.18,41 These data show that even with substrate bound, the {FeNO}7 complex is HS (in contrast to CDO). These experimental spin Hamiltonian parameters are used below to fit the VTVH MCD for the assignment of electronic transitions.
Excited State Spectroscopy.
Room temperature absorbance (Abs) spectra were collected for ETHE1-NO and ETHE1-GSS-NO to identify the new and perturbed spectral features that result from persulfide binding (Figure 4). The ETHE1-NO spectrum (Figure 4, blue) resembles that of FeEDTA-NO, which showed two spin forbidden FeIII LF transitions at ~15,000 cm−1 (ϵ ~100–200 M−1 cm−1) and three NO− to FeIII ligand-to-metal CT (LMCT) transitions at 20,000–25,000 cm−1 (ϵ ~500–1,000 M−1 cm−1).38 The FeIII d-d LF transitions are an order of magnitude more intense than in other mononuclear ferric complexes, because they gain intensity dominantly through spin-orbit coupling (SOC) to the low energy NO-to FeIII CT transitions.38 Starting from low energy, the LMCT transitions are assigned as NO− 2π* in-plane (ip) → Fe dxy, NO− 2π* ip → Fe dyz and NO− 2π* out-of-plane (oop) →Fe dxz charge transfer transitions, where the ip and oop labels refer to the plane of the Fe-N-O bond (The Fe-NO is bent by ~150° in the yz plane). Compared to the Abs spectrum of ETHE1-NO, the ETHE1-GSS-NO spectrum (Figure 4, orange) has a new feature at ~17,500 cm−1, and shows an overall increase in intensity at higher energies (20,000 – 30,000 cm−1). These spectral differences were analyzed using MCD spectroscopy.
Figure 4:

Room temperature UV-Vis absorbance spectra of ETHE1-NO with the FeIIETHE1 spectrum subtracted (blue) and ETHE1-GSS-NO with the (FeII/GSS−)-ETHE1 spectrum subtracted (orange). Gaussian resolution of bands shown.
The +7 T, 5 K MCD spectrum of ETHE1-NO is shown in Figure 5A. Based on their energies and low intensities, bands 1 and 2 are assigned as the spin forbidden FeIII LF transitions. In addition, bands 3–5 have similar energies and intensities as the NO− to FeIII CT transitions in FeEDTA-NO. Using the ZFS parameters defined by EPR, saturation magnetization data can be fit to determine the polarizations of the transitions observed in MCD relative to the coordinate system defined by the ZFS (i.e. the z-axis along the Fe-N bond).42 Fits to VTVH MCD data collected at ~22, 000 cm−1 on band 4 in ETHE1-NO (arrow in Figure 5A; Figure S7) indicate that this transition is 72% z-polarized. Since LMCT transitions are dominantly polarized along the direction of the metal-ligand bond and the strong Fe-NO bond defines the ZFS z-axis of the FeIII site, band 4 is confirmed as one of the NO− to FeIII CT transitions. Based on their energies and intensities, bands 3 and 5 are assigned as the other two NO−to FeIII CT transitions. There are two additional transitions in the ETHE1-NO spectrum (bands 6 and 7) that are not seen in the FeEDTA-NO spectrum, which was not recorded beyond 25,000 cm−1. These are assigned using TD-DFT calculations in the next section.
Figure 5:

The +7 T, 5 K MCD spectra of (A) ETHE1-NO and (B) ETHE1-GSS-NO with Gaussian fits to the spectra shown and transitions numbered consistent with the numbering in Figure 4. Arrows indicate energies at which VTVH MCD data were collected. VTVH MCD isotherms for ETHE1-GSS-NO and best fits to the data collected at (C) 17,600 cm−1 and (D) 25,800 cm−1.
The +7 T, 5 K MCD spectrum of ETHE1-GSS-NO is shown in Figure 5B. The most notable differences relative to the MCD spectrum of ETHE1-NO are the high energy tail of a low energy transition (band 1 at 5,000 cm−1), a transition at ~18, 000 cm−1 (band 5) and a large, negative feature centered at ~27, 000 cm−1. The RT Abs, RT CD, LT CD and MCD spectra of ETHE1-GSS-NO were simultaneously fit with Gaussians allowing for bands to shift and sharpen at LT (Figure S8), and their spectral assignment from low to high energy follows. VTVH MCD with good signal-to-noise could not be collected on band 1 due to its low intensity. A similar low energy transition was observed in the substrate bound {FeNO}7 S = 3/2 complex of another NHFe enzyme, (4-hydroxyphenyl)pyruvate dioxygenase (HPPD), and was determined to be a spin forbidden FeIII d-d transition that increased in energy relative to HPPD-NO due to strong donation of the α-ketoacid substrate in the equatorial plane.41 In ETHE1, the substrate also binds in the x,y-plane and is expected to have a similar effect. Bands 2 and 3 in the ETHE1-GSS-NO MCD spectrum correlate in energy and intensity with the two LF transitions that are typically observed in {FeNO}7 complexes, and therefore, bands 1–3 are assigned as FeIII LF transitions.
Although band 4 has very little intensity in MCD, it has intensity in CD and is required by the simultaneous fit of the spectra (see Figure S8). In MCD, this negative band is most evident at low temperature (2.2 K) and high field (7T, Figure S9), and it is a new band from persulfide binding. VTVH MCD cannot be collected on this band due to its low intensity; however, a similar pattern of negative/positive transitions (band 4/band 5) was observed in in the MCD spectrum of IPNS-ACV-NO and assigned as a pseudo-A term, which has specific requirements for MCD intensity. Analysis of band 4 is developed in the next section.
Band 5 in ETHE1-GSS-NO is centered at ~17, 800 cm−1 and does not have a corresponding transition in the ETHE1-NO Abs or MCD spectra. VTVH MCD fits this transition to 86% z-polarization, and it is therefore assigned as one of the three NO− to FeIII CT transitions (Figure 5C). This corresponds to a shift to lower energy of this transition by ~2, 000 cm−1 upon substrate binding. A ~5, 000 cm−1 shift to lower energy of a NO− to FeIII CT transition was observed in IPNS-ACV-NO and was due to a configuration interaction between a substrate thiolate orbital and a NO− 2π* orbital.18 Band 6 is also 86% z-polarized from VTVH MCD (Figure S10), resulting in the assignment of bands 5–7 as the three NO− to FeIII CT transitions. Band 8 in the ETHE1-GSS-NO MCD spectrum appears at a similar energy and with a similar intensity to band 6 in ETHE1-NO and is assigned using TD-DFT.
Bands 9 and 10 in Figure 5B are part of a large negative feature that arises from substrate binding. The Abs spectrum of ETHE1-GSS-NO shows broad increased intensity relative to ETHE1-NO in this energy region (Figure 4, 25,000 – 30,000 cm−1). VTVH MCD data were collected at multiple energies on either side of this feature. Data collected at 25,800 cm−1 were fit to 39% x,y-polarization (Figure 5D), and moving to higher or lower energies resulted in a greater percentage of z-polarization. Overlap with nearby, z-polarized transitions precludes an accurate determination of this band’s polarization, but due to its significant x,y-character, lack of a counterpart in the ETHE1-NO MCD spectrum and the presence of new Abs intensity in this region associated with persulfide binding (Figure 4, orange), band 9 is assigned as a LMCT transition from the persulfide substrate to the HS FeIII.
In summary, persulfide binding to ETHE1-NO results in the appearance of at least four new features (bands 1, 4, 5 and 9). Band 1 is a spin forbidden FeIII LF transition that increased in energy relative to its ETHE1-NO counterpart. Band 5 is an NO− to FeIII CT transition that has been lowered in energy relative to the transition in ETHE1-NO, and bands 4 and 9 are persulfide to FeIII CT transitions. These new features are used to define the persulfide-FeIII binding interaction in the next section.
TD-DFT Analysis of ETHE1-GSS-NO: Defining the Electronic Structure and Orientation of the Persulfide-FeIII Bond
To understand the binding interaction of the persulfide substrate with the FeIII site, first the molecular orbitals (MOs) of the unbound persulfide anion are defined and compared to those of the thiolate anion. A truncated glutathione persulfide and the corresponding thiolate were optimized and their FMOs are given in Figure 6. For thiolate, the highest occupied MOs (HOMOs) are a pair of p orbitals that are nearly degenerate (split by 0.09 eV) and perpendicular to the S-C bond. In contrast, the HOMOs in persulfide are two SS-π* antibonding orbitals that are split by 0.8 eV. The lower energy of these two orbitals has electron density localized primarily on the terminal sulfur and oriented in the S-S-C plane (π*ip). The higher energy MO has electron density delocalized over both sulfur atoms and oriented perpendicular to the S-S-C plane (π*oop). The increased antibonding character in the π*oop orbital raises its energy relative to the π*ip orbital. The persulfide lowest unoccupied MO (LUMO) is a low lying SS− σ*orbital that is 3.4 eV above the HOMO. In contrast, the unoccupied C-S σ*orbital of the thiolate is significantly higher in energy (5.6 eV).
Figure 6:

Frontier molecular orbitals in glutathione thiolate (left) and persulfide (right). SP and SD refer to the anionic persulfide sulfur and the persulfide sulfur that bridges this sulfur and the nearest carbon atom, respectively.
The binding mode of the GSS− substrate to the iron center is not known, and both biden-tate (via persulfide and deprotonated amide to form a six membered ring)34 and monodentate (via persulfide)33 binding modes have been suggested in the literature. The GSS− substrate was docked in the active site pocket of the FeII-ETHE1 crystal structure,33,43 and due to the size of the tripeptide substrate and constraints of the active site pocket, only monodentate binding modes resulted. These structures included an electrostatic interaction between the GSS− glycinyl carboxylate and Arg214 and a hydrogen bond (H-bond) between the GSS− cysteinyl amide and Tyr197 in good agreement with a product bound crystal structure35 and another substrate docking model.33 Monodentate persulfide binding is also consistent with persulfide Cys and 3-mercaptopropionic acid persulfide bound crystals of CDO.44 The local environment around the Fe accommodated multiple, energetically reasonable orientations of the persulfide moiety relative to the Fe and its other ligands. However, the EPR spectrum of ETHE1-GSS-NO (Figure 3A, orange) showed that a single substrate orientation is present in solution. To determine the persulfide orientation relative to the Fe-NO bond, two limiting N(NO)-Fe-S-S dihedral angles (90° and 180°) were considered for comparison to the spectroscopic data (Figure 7).
Figure 7:

DFT optimized structures of two limiting orientations of the persulfide moiety relative to the Fe-NO bond. The second sphere residues are not shown for clarity.
Starting with the docked substrate structure, a truncated model of the active site was generated for DFT calculations that included the three first sphere facial triad residues (His79, His135 and Asp154) and two second sphere H-bonding residues (Asp83 and Tyr197). Based on the docking analysis, the substrate replaced the water trans to His79. There are two other exchangeable sites that NO can occupy. Because of a strong H-bond between Asp83 and the water trans to His135, the NO replaced the water trans to Asp154 in the more labile site. The water molecule trans to His135 was retained for a 6C site, because the ligand field transition energies of ETHE1-GSS-NO (Figure 5B) are similar to those of other 6C {FeNO}7 model38 and enzyme17,18 complexes in the literature. The docked substrate was replaced with a water molecule in the ETHE1-NO structure, as this site is also expected to be 6C due to its ligand field transition energies and the low conversion to form the nitrosyl complex in the absence of substrate. Structures of ETHE1-NO and the two limiting orientations of ETHE1-GSS-NO were geometry optimized using a functional and basis set that was experimentally validated for {FeNO}7 complexes (the BP86 functional with 10% Hartree-Fock exchange with the Pople triple-ζ basis set (6–311G*) on Fe, NO, SS and all other heteroatoms bound to the Fe and a double- ζ basis set (6–31G*) on the remaining atoms).45 The amino acid residues were truncated and terminated by a hydrogen on the α-carbon, and the α -carbons were constrained to mimic the protein backbone.
The ETHE1-NO optimized structure has an S = 3/2 ground state with an electronic structure of five α Fe d electrons AF coupled to two β NO− electrons (Figure S11). The 90° and 180° substrate bound structures both reproduce the experimental S = 3/2 ground state with the same electronic structure description as ETHE1-NO (Figures S12 and S13, respectively). In both, the lower energy SS− π*ip orbital has a pseudo-σ interaction with the Fe, and the higher energy SS− π*oop orbital has a ˇ bonding interaction that differs between the two orientations. With a N(NO)-Fe-S-S dihedral of 90°, the SS− π*oop orbital is parallel to the Fe-NO bond and overlaps the Fe dxz and dyz orbitals (Figure 8A). This can be quantified by the amount of occupied SS− character mixed into these unoccupied Fe dπ orbitals with 1% in Fe dxz and 3% in Fe dyz. With a N(NO)-Fe-S-S dihedral of 180°, the SS−π*oop orbital is oriented in the x,y-plane and has good overlap with Fe dxy, which has 11% persulfide character (Figure 8B).
Figure 8:

Occupied SS π*oop orbital showing overlap with Fe d orbitals in the (A) 90° and (B) 180° orientations. The second sphere residues are not shown for clarity. TD-DFT calculated NO− and SS− to Fe d CT transitions for the (C) 90° and (D) 180° orientations.
TD-DFT calculations were used to identify the CT transitions arising from these limiting substrate orientations for comparison to the experimental spectrum in Figure 4. The TDDFT predicted Abs spectrum of ETHE1-NO was used as a reference (Figure S14) with its calculated NO− π*ip to Fe dxy and NO− π*ip to Fe dyz transitions at ~14, 000 cm−1 and the more intense NO− π*oop to Fe dxz transition at ~18, 400 cm−1. Although these energies are lower than those observed experimentally, the relative intensities and ordering of these transitions are consistent with the experimental spectrum. The TD-DFT predicted Abs spectra of the two limiting orientations of bound persulfide show significant differences. At low energy in the 90° orientation (Figure 8C), there is a SS− π*oop(π) to Fe dyz CT transition and a much weaker SS− π*oop(π) to Fe dxz transition. Increasing in energy, these are followed by the NO− to FeIII CT transitions, and at higher energy, there are two intense persulfide to Fe CT transitions: SS− π*oop(π) to Fe dσ and SS− π*ip(σ) to Fe dπ. Note that the low energy persulfide LMCT at 9,500 cm−1 is less intense than the most intense NO− to FeIII CT transition (18,000 cm−1). At low energy in the 180° orientation (Figure 8D), there is an intense SS− π*oop(π) to Fe dxy CT transition (11,800 cm−1) and a weaker SS− π*oop(π) to Fe dyz transition. Following the NO− to FeIII CT transitions, there are two higher energy persulfide to Fe CT transitions: a weak SS− π*oop(π) to Fe dσ (23,400 cm−1) and an intense SS− π*ip(σ) to Fe dπ (24,000 cm−1). For the 180° structure, the low energy persulfide LMCT is more intense than the most intense NO− to FeIII CT transition (17,200 cm−1) and dominates the spectrum.
These calculated TD-DFT spectra were compared to experiment to define the persulfide orientation in the active site. The experimental Abs/MCD spectra of ETHE1-GSS-NO (Figure 4 and 5B) show a weak persulfide LMCT at low energy, followed by the three NOLMCT transitions, one of which is shifted down by ~2, 000 cm−1. There is also an intense persulfide LMCT transition at higher energy (26,300 cm−1). Both persulfide orientations reproduce the intense SS− to FeIII CT transition at high energy. However, the 180° orientation predicts an intense low energy persulfide LMCT transition that dominates the spectrum, which is not observed experimentally. The weak low energy SS− to FeIII CT transition predicted in the 90° orientation is consistent with experiment and reflects the low mixing of SS− π*oop into Fe dxz,yz due to their strong dπ mixing with the NO− (Figure 8A). Thus, the solution orientation of the persulfide moiety relative to the Fe-NO bond must be closer to the 90° orientation, which was thus used to model the ETHE1-GSS-NO active site.
With the substrate orientation defined and an electronic structure description developed, the ETHE1-GSS-NO MCD spectrum (Figure 5B) can be more rigorously assigned. Band 4 is a new feature from substrate binding and is the negative component of a pseudo-A term; the positive component is a NO− to FeIII CT transition (band 5). A pseudo-A term requires that two CT transitions with perpendicular polarization spin-orbit couple in a third mutually perpendicular direction.18,42 Since SOC is mostly a single center, 1 e− operator, they must share a common donor or acceptor orbital. For CT transitions from different ligands, they must excite into the same acceptor orbital, and the angular momentum operator of SOC in the third mutually perpendicular direction must rotate the donor orbitals into each other on the Fe center. The NO− (z-polarized) and SS− (y-polarized) to Fe CT transitions both excite an electron into the Fe dyz orbital. In the experimentally validated persulfide bound structure, the Fe-N-O bend bisects the Fe-SS and Fe-NHis bonds. As a result, the SS− π*oop(π) orbital overlaps both the Fe dxz and dyz orbitals (Figure 8A). The NO− π*ip orbital overlaps both the Fe dxy and dyz orbitals. The Lx operator rotates dxy (mixed into NO− π*ip) into dxz (mixed into SS− π*oop(π)). This SOC mechanism leads to the derivative shaped pseudo-A term in the MCD spectrum, and band 4 is assigned as a SS− to Fe dyz CT transition. The higher energy bands are assigned in the SI.
Persulfide donation to the FeIII in ETHE1-GSS-NO can be compared to our previous studies of thiolate donation in IPNS-ACV-NO, where the thiolate of the ACV substrate had pseudo-σ and π bonding interactions with the FeIII center.18 In the DFT optimized structure of IPNS-ACV-NO, the Fe dxy unoccupied β orbital had substantial (7.5%) S character that was responsible for the intense CT transition observed at ~20,000 cm−1 in the experimental Abs spectrum. In addition, the NO− π*ip to Fe dxy CT transition was shifted down in energy by ~5, 000 cm−1 relative to this transition in {FeNO}7 complexes of enzymes with facial triad ligation. This shift in energy was due to a configuration interaction between the thiolate pseudo-σ and NO− π*ip orbitals mediated by the Fe dz2 orbital and was reflected in the significant amount of S pseudo-σ character mixed into the NO− π*ip unoccupied α orbital(10.5%). In ETHE1-GSS-NO, the NO− π*ip to Fe dxy CT transition also shifts down in energy, but by less (~2, 000 cm−1), and this is reflected in the smaller amount of SS− π*ip(σ) character in the unoccupied α NO− π*ip orbital (4.9%). In the experimental ETHE1-GSS-NO spectra, the SS− π*oop(π) to Fe dyz CT transition is weak due to the low persulfide mixing into the unoccupied β Fe dyz orbital (3%). In the free ligands, the thiolate p orbitals are localized on the S atom that binds to Fe, while the persulfide π*oop orbital is delocalized over both S atoms (Figure 6), with only the distal sulfur binding to the Fe. In addition, the DFT calculated Fe-S bond is long in ETHE1-GSS-NO (2.55 Å) relative to IPNS-ACV-NO (2.33 Å, vide infra). The delocalization of electron density over both sulfur atoms and the long Fe-SS bond result in poor overlap between SS− π*oop and Fe dyz, responsible for the relatively weak CT band observed in the ETHE1-GSS-NO spectra. Thus, persulfide is a weaker donor than thiolate. However, persulfide is still capable of activating reaction with O2, and this reaction coordinate is evaluated below.
ETHE1 Sulfur Dioxygenation Reaction Coordinate
The experimentally validated ETHE1-GSS-NO structure was used as a starting point for the native sulfur dioxygenation reaction coordinate. First, NO was replaced with O2 and optimized on the S = 1, 2 and 3 surfaces, with the S = 2 spin state lowest and the S = 1 spin state 1.3 kcal/mol higher in Gibbs free energy. The MO diagram for the S = 2 ground state is shown in Figure S15 and is best described as HS FeIII (S = 5/2) AF coupled to O2•− (S = 1/2). The O2 is oriented above the bound sulfur atom, and its FMO is the O2•− π* ip orbital. Importantly, this is the same FMO calculated for IPNS-ACV-O2, where thiolate donation was shown to lower the energy of the O2•− π* oop relative to the π* ip orbital to direct reactivity.18 In ETHE1-GSS-O2, persulfide donation is still enough to result in the O2•− π* ip FMO, which has good overlap with the SS− π* oop (π) orbital.
The ETHE1 sulfur oxygenation reaction coordinate was calculated on the S = 1 and S = 2 surfaces and is displayed in Figure 9. It begins with an electrophilic attack of the ferric superoxo (I1) on the coordinated sulfur atom that involves the π* ip FMO of the O2•− interacting with the π* oop of the coordinated persulfide (Figure 10, top row). Note that if the persulfide was in the 180° orientation, there would not be good overlap between the oxidizable SS− π*oop orbital and the O2•− π* ip FMO (Figure 8B). The barrier for this electrophilic attack is 11 kcal/mol higher on the triplet than on the quintet surface, paralleling the CDO reaction coordinate.25 This FeII peroxo-bridged transition state (TS1,2, S = 2) is characterized by a long 2.8 Å Fe-SS bond and resembles the second intermediate in the CDO reaction coordinate.25,27 For ETHE1, O-O cleavage is barrierless to form the second intermediate (I2), which is best described as an FeIV=O with a coordinated persulfenate. For I2, the triplet state is lowest (intermediate spin FeIV=O (S = 1) and persulfenate (S =0)), and the quintet state (HS FeIV=O (S = 2) and persulfenate (S = 0)) is 10.6 kcal/mol higher in energy, also consistent with CDO calculations.25,27
Figure 9:

Calculated sulfur dioxygenation reaction coordinate of ETHE1.
Figure 10:

FMOs involved in electrophilic attack of the ferric superoxo on the coordinated persulfide sulfur (top) and those involved in the second oxygen atom transfer step (bottom).
To transfer the second oxygen, the oxo ligand of the FeIV=O unit requires overlap with the remaining oxidizable SS-O π* orbital. This sulfur orbital is involved in a bonding interaction with the Fe, and therefore a geometric rearrangement of the substrate is needed for oxo transfer. In previous studies of CDO, a rearrangement via rotation of the S-O moiety to coordinate Fe through the oxygen atom of the sulfenate produces a species with the S-O π* primed for the second oxygen atom transfer (OAT).25 Alternatively, the equivalent O-coordinated species (I3) can be achieved through an intermediate η2-SO side-on coordination step (box in Figure 9). The oxygen coordinated FeIV=O structure I3 is 2.1 kcal/mol lower on the S = 2 than the S = 1 surface, so this rearrangement step also involves a spin crossover to the S = 2 surface, which remains the lowest energy spin state for the remainder of the reaction.
In the next OAT step two electrons are transferred in a stepwise fashion from the S to break the Fe=O bond and form the second S-O bond. This sequential transfer of an α electron into dz2 followed by a β electron into dxz,yz was previously observed for the OAT to sulfur calculated for the [FeIV=O(TBC)(CH3CN]2+ model complex.46 The direct σ-attack via the oxo pz orbital is not available for ETHE1, because the S atom is not oriented along the Fe-O unit and lacks overlap with dz2. Instead, the Fe-O bond elongates gaining HS FeIII-oxyl character, and an electron is transferred from the S=O unit via a π-channel. At the transition state (TS3,4) the Fe-O bond has significantly elongated (from 1.63 to 1.77 Å), leading to population of the dz2 orbital with an α electron from the oxo. This is followed by transfer of a β electron from the S into an Fe dπ orbital. The barrier for this step is 14.9 kcal/mol on the quintet surface and results in an intermediate (I4) that has the dioxygenated persulfide bound to the Fe via the second transferred oxygen atom and is downhill by ~20 kcal/mol relative to I3. The S-S bond has elongated from 2.09 Å in the persulfenate intermediate (I3) to 2.37 Å in I4. Further, the I4 HOMO has significant S-S σ* character indicating that this bond is primed for heterolytic cleavage. The final step of the ETHE1 reaction is hydrolysis to break this elongated S-S bond to form glutathione and sulfite (P), which is further downhill by 6 kcal/mol. Interestingly, ETHE1 was able to dioxygenate a substrate alternative where the persulfide S-S moiety was replaced by a C-S. However, the resulting C-SO2 bond could not be hydrolyzed, and this product remained bound to the Fe resulting in enzyme inhibition.47 Thus, the final hydrolysis step is critical for sustained catalytic activity in ETHE1.
H-Atom Abstraction and Sulfur Oxygenation Reactions in IPNS and CDO
Aside from the final hydrolysis step, the ETHE1 reaction coordinate parallels those calculated for CDO25–28 despite the differences in Fe binding triad (2-His/1-Asp vs. 3-His) and substrate coordination mode (monodentate vs. bidentate). This prompted us to evaluate the factors that select for H-atom abstraction (HAA) vs. electrophilic sulfur attack (SOX) in the first steps of the sulfur oxidation and oxygenation reactions, respectively. The first steps of the native (to parallel the literature) and non-native reactions in IPNS and CDO were calculated to understand this differential selectivity.
An IPNS active site model was generated by replacing NO with O2 in the IPNS-ACVNO crystal structure17 and geometry optimizing on the S = 1, 2 and 3 surfaces. The energy difference between the lower energy triplet and quintet states is within the 2 kcal/mol error in DFT (quintet higher by 0.8 kcal/mol). The triplet and quintet MO diagrams are shown in Figures S16 and S17, respectively. From kinetic isotope experiments, the native reaction of IPNS involves an HAA from the cysteinyl β-carbon of its ACV substrate,8 and from quantum chemical computations, the reaction proceeds on the quintet surface.20,48 In the present study, the HAA step was calculated on the triplet and quintet surfaces, giving the energetics shown in Figure 11A with details summarized in Table S1. The S = 2 O2 adduct (5I1) is a ferric superoxide (HS FeIII (S = 5/2) AF coupled to O2•− (S = 1/2)), with an O2•−π* ip FMO that is primed for HAA (Figure 12, left). The first transition state has an S = 2 and is uphill 13.4 kcal/mol (5TS1), consistent with the previously calculated barrier of 14.6 kcal/mol20 and the experimental barrier of 16.8 kcal/mol.49 The next intermediate (5I2) is an FeII-hydroperoxide with substrate thioaldehyde and is thermodynamically uphill by 13.3 kcal/mol. Lundberg et al. calculate a first product that is much more stable (−4.6 kcal/mol relative to their septet ground state).20 Parallel calculations performed with the B3LYP functional showed that the large difference in product stability is due to the difference in functional and an additional strong H-bond between the hydroperoxide and deprotonated carboxylate of the substrate valine that is not present here.
Figure 11:

Calculated first steps in the (A) IPNS and (B) CDO native reactions. Both reactions were calculated on the S = 1 (red lines) and S = 2 (green lines) surfaces. Energies are Gibbs free energies. Schematic structures of the intermediates and transition states along the native reactions are included below the coordinate.
Figure 12:

FMOs of IPNS-ACV-O2 with Val side chain and water deleted for clarity (left) and CDO-Cys-O2 with second sphere His deleted for clarity (right).
Starting with the CDO/Cys crystal structure,24 a CDO-Cys-NO active site model was optimized that reproduced the LS (S = 1/2) ground state.39 NO was replaced with O2, and the spin-unrestricted MO energy level diagrams for the ground state triplet and quintet (+6.4 kcal/mol above triplet) optimized structures are shown in Figures S18 and S19, respectively. CDO sulfur oxygenation reaction coordinates in the literature proceed on both the S = 1 and S = 2 surfaces and begin with end-on O2 binding followed by electrophilic attack of the distal oxygen on the coordinated Cys sulfur.25,27–29 Here, the first step of the native CDO reaction was calculated on both the triplet and quintet surfaces and is detailed in Figure 11B and Table S2. The FMO of the S = 2 FeIII-superoxide formed from O2 binding (5I1) is the O2•− π* ip orbital that is primed for electrophilic sulfur attack (Figure 12, right). The first transition state has an S = 2 with a barrier of 15.3 kcal/mol in free energy (5TS1), indicating that there has been a spin crossover from the S = 1 surface. This is in good agreement with the experimental barrier of 17.8 kcal/mol for the human form of the enzyme.24 The next intermediate (3I2) has O2 cleaved into an FeIV-oxo and a singly oxygenated sulfur that is still coordinated to the Fe with the S = 1 surface lowest in energy (intermediate spin FeIV=O (S = 1)). Previous studies have calculated the second intermediate to be an Fe-O-O-S bridged structure (S = 2), with a small barrier to convert to this FeIV-oxo/monooxygenated sulfur species (S = 1).25,27 Parallel calculations using the B3LYP functional were able to reproduce this Fe-O-O-S bridged intermediate and small barrier for O-O cleavage. Using BP86 with 10% Hartree-Fock, the dioxygen cleavage is barrierless and results in the FeIV- oxo/monooxygenated cysteine, which is downhill by ~20 kcal/mol.
Thus, both IPNS and CDO have similar barrier heights for the first steps in their native sulfur oxidation (via HAA) and sulfur oxygenation reactions, respectively. To understand why IPNS does not oxygenate its thiolate substrate and why CDO does not perform an H-atom abstraction on its thiolate substrate, these non-native reaction coordinates were investigated, and the results are summarized in Figures 13 (A for IPNS and B for CDO) and S20 and Tables S3 and S4. The first step of the non-native sulfur oxygenation reaction in IPNS results in a monooxygenated sulfur product like in CDO. This step was calculated on the triplet and quintet surfaces (Figure S20A and Table S3), with the barrier on the quintet surface lower by ~10 kcal/mol. A comparison of the HAA and SOX reactions in IPNS on the S = 2 surface is shown in Figure 13A (with the S oxygenation pictured at the bottom for comparison to the HAA pictured at the bottom of Figure 11A). The barrier for HAA is 4 kcal/mol lower than for SOX, reflecting the native reactivity of IPNS. In CDO, the first step of the non-native sulfur oxidation reaction via HAA produces an FeII-hydroperoxide and a C-S double bond like in IPNS. The barrier for this step is 4.8 kcal/mol lower on the triplet than on the quintet surface (Figure S20B and Table S4). However, the barriers for HAA on both spin states are higher in energy than that for the native SOX reaction. A comparison of the HAA and SOX reactions in CDO on the S = 2 surface is shown in Figure 13B (structures associated with HAA pictured at the bottom for comparison to the SOX structures shown at the bottom of Figure 11B), with the barrier for SOX 4.6 kcal/mol lower than for HAA, reflecting the native reactivity of CDO. While the barriers for H-atom abstraction in IPNS and CDO are almost identical, the barrier for sulfur oxygenation is 8.2 kcal/mol higher in IPNS than in CDO.
Figure 13:

Calculated first steps in the (A) IPNS and (B) CDO sulfur oxidation via HAA (blue) and sulfur oxygenation (orange) reactions. Energies are Gibbs free energies. Schematic structures of the intermediates and transition states along the non-native reactions are included below the coordinate.
Analysis of the IPNS sulfur oxygenation transition state shows significant steric interaction between the Fe-O-O-S bridged structure and the Val side chain of the truncated ACV substrate (Figure 14A, green). The Val β-carbon (in addition to the Val carboxylate carbon and the Cys amine nitrogen) was frozen to prevent significant rearrangements of this side chain that is already in van der Waals contact with the protein pocket (Figure S21, left). When only the Val β-carbon constraint is released, the barrier for HAA is effectively unchanged (decreases by < 1 kcal/mol), but the barrier for SOX decreases by ~5 kcal/mol, resulting in comparable barriers for SOX (12.3 kcal/mol) and HAA (13.4 kcal/mol). An overlay of the SOX transition states with (Figure 14A, green) and without (magenta) the Val β-carbon constrained shows significant rearrangement of the unconstrained Val side chain. This motion minimizes its steric interaction with the Fe-O-O-S bridged structure and allows the bridged structure to distort to a lower energy orientation, lowering the overall barrier for oxygenation. However, this reorientation of the substrate is limited in the protein pocket of IPNS due to steric interactions with the side chains of Pro283 and Leu223 (Figure S21, right).
Figure 14:

(A) First transition state structures of the IPNS S = 2 sulfur oxygenation reaction with (green) and without (magenta) the substrate Val β-carbon constrained. Other Fe ligands not shown for clarity. (B) Calculated first step in the non-native sulfur oxygenation reaction of IPNS (S = 2) with (green) and without (magenta) the substrate Val β-carbon frozen. Energies are Gibbs free energies. Differences in the energies of the first intermediate (I1) are due to different orientations of the superoxide that result from the intrinsic reaction coordinates.
The SOX reaction is thermodynamically favored over HAA as it forms a more stable first product in both IPNS and CDO. In CDO, the SOX reaction on its chelate substrate also has a lower barrier. In IPNS, where ACV is constrained to reflect van der Waals contacts with the active site pocket, HAA has a lower barrier than SOX. When these constraints are relaxed, the SOX reaction becomes energetically favored in IPNS. In ETHE1, the second sphere H-bonds to the substrate are not located near the persulfide, and the local environment around the Fe is not as constrained as in IPNS. This is evidenced by the multiple orientations of the persulfide moiety that are possible from substrate docking (Figure 7). As a result, the persulfide substrate forms an Fe-O-O-S ring structure with a barrier that is comparable to that calculated for the parallel native reaction in CDO. Further, the nearest GSS− methylene is not available for HAA due to its extended distance from the Fe. The sulfur oxygenation is then thermodynamically and kinetically favored for ETHE1.
Discussion
Near-IR CD, MCD and VTVH MCD spectroscopies have defined the FeII-ETHE1 active site and showed that persulfide binding results in a coordinatively unsaturated FeII site for O2 activation. This activation was evaluated by spectroscopic and computational analysis of the ETHE1 {FeNO}7 complexes. At least four new spectroscopic features are associated with persulfide binding to the {FeNO}7, including a low energy LF transition (band 1 in Figure 5B) that had increased in energy relative to the band in ETHE1-NO, a NO− to FeIII CT transition (band 5) that had decreased in energy relative to this CT transition in ETHE1-NO, and two new persulfide to FeIII CT transitions (bands 4 and 9). These define the orientation of the persulfide binding to the Fe (90° in Figure 7) and the nature of the persulfide-FeIII bond.
Compared to thiolate binding in IPNS,18 persulfide is a weaker donor ligand. This weaker interaction resulted in the shift to lower energy of a NO− to FeIII CT transition by ~2, 000 cm−1, relative to the ~5, 000 cm−1 shift observed upon thiolate binding in IPNS. Further, the ETHE1-GSS-NO spectra showed a weak persulfide to FeIII CT transition, in contrast to the intense thiolate to FeIII CT transition in the IPNS-ACV-NO spectrum. This weaker intensity arises from a longer Fe-S(persulfide) bond, delocalization of electron density over both S atoms of the persulfide HOMO donor orbital (Scheme 2), and excitation into an acceptor orbital (dyz) that has a strong interaction with the NO−. O2 binding energetics were strongly influenced by the strong thiolate donation in IPNS, showing that absent the ACV substrate, O2 binding was endergonic. Thiolate charge donation from the bound substrate lowered the 1 e− reduction potential of the Fe such that O2 binding became exergonic (ΔG = −7.0 kcal/mol).18 Equivalent calculations were performed to define the effect of the persulfide donation on O2 binding to ETHE1. O2 binding to the resting site is uphill (ΔG = +8.9 kcal/mol), and charge donation from the bound persulfide substrate (GSS−) makes O2 binding only slightly exergonic (ΔG = −1.5 kcal/mol). For comparison, binding of the corresponding thiolate (GS−) results in O2 binding becoming even more favorable (ΔG = −6.9 kcal/mol). This indicates that while persulfide is a poorer donor to the Fe than thiolate, its charge donation is sufficient to make the 1 e− reduction of O2 to bind as superoxide favorable.
Scheme 2:

Comparison of donor interactions between (A) thiolate in IPNS-ACV-NO and (B) persulfide in ETHE1-GSS-NO.
With a definition of the persulfide-FeIII bond, the sulfur dioxygenation reaction coordinate for ETHE1 was calculated. NO was replaced with O2 in the experimentally calibrated DFT structure and resulted in an S = 2 ground state deriving from a HS FeIII (S = 5/2) antiferromagnetically coupled to O2•− (S = 1/2). In ETHE1-GSS-O2 the FMO is the O2•− π*ip orbital that has good overlap with the SS− π*oop occupied orbital (Figure 10 and Scheme 3A). This is the same FMO observed for IPNS-ACV-O2 and CDO-Cys-O2, showing that despite weaker interaction with the Fe, the persulfide donor is able to generate the same FMO as the thiolate donor. The reaction in ETHE1 (Figure 9) is initiated by electrophilic attack of this O2•− π*ip FMO on the bound persulfide to form an Fe-O-O-S bridged transition state (TS1,2). From this transition state, O-O cleavage is barrierless and the resulting intermediate (I2) is a triplet FeIV=O with a coordinated persulfenate. The persulfenate then rearranges to oxygen coordination (I3) resulting in a spin crossover back to the S = 2 surface to enable the second oxygen atom transfer to the proximal sulfur. The FeIV=O performs an electrophilic attack on the remaining sulfur lone pair to form the dioxygenated persulfide bound through the second transferred oxygen atom (Scheme 3B). The orientation of the persulfide precludes direct σ-attack, but Fe-O bond elongation populates Fe dz2 (α e−) to gain FeIII-oxyl character and activate a π-channel for a β electron transfer. The next intermediate (I4) has an elongated persulfide bond with significant S-S σ* character that is primed for hydrolysis to form a bisulfite bound to Fe and unbound GSH in the active site pocket.
Scheme 3:

(A) Overlap between O2− π* ip FMO primed for electrophilic attack on the occupied SS− π*oop and (B) Elongation of the Fe-O bond in I3 activates a π-channel for electrophilic attack on the S for OAT.
Aside from the final persulfide bond cleavage step, the ETHE1 reaction coordinate is similar to those calculated for CDO,25,27 despite the different substrate coordination modes and protein derived ligand sets between the two FeII sites. Like ETHE1, IPNS has a monodentate bound substrate and facial triad ligation, but performs a sulfur oxidation reaction instead of sulfur oxygenation. For CDO, sulfur oxygenation has a lower barrier and is thermodynamically favored. For IPNS, although sulfur oxygenation is also thermodynamically favored, H-atom abstraction has a lower barrier, leading to its native reactivity. HAA has a similar barrier in both enzymes, but the barrier for sulfur oxygenation is ~8 kcal/mol higher in IPNS than in CDO due to steric interactions between the Fe-O-O-S transition state structure and the substrate valine that is in van der Waals contact with the protein pocket (Figures 14A and S21A). In summary, charge donation by substrate S− binding in this subclass of NHFe enzymes (ETHE1, CDO, IPNS) activates the FeII to form a ferric superoxide intermediate with an in-plane π* FMO activated for electrophilic attack on the substrate. Sulfur oxygenation is the energetically favored reaction, but involves a first transition state that can be controlled by the protein environment to direct selectivity for substrate oxidation vs. oxygenation.
Experimental
Sample Preparation
Human ETHE1 was expressed and purified according to previously published procedures.10 All samples were prepared under an inert atmosphere inside an N2 purged wet box. ETHE1 was made anaerobic by purging under a nitrogen atmosphere on a Schlenk line at 0 °C for approximately 1 h. Buffer, GSSH, sodium dithionite and 6-(2-Hydroxy-1-methyl-2-nitrosohydrazino)-N-methyl-1-hexanamine (NONOate) were made anaerobic by purging with nitrogen on a Schlenk line. ETHE1 was treated with 5–10 equivalents of sodium dithionite to reduce the iron in the sample, then exchanged into deuterated Tris bu˙er (50 mM, pD = 7.5) and concentrated to 2–3 mM using an Amicon Ultra centrifugal filter (10 kDa). GSSH was prepared under anaerobic conditions and kept in sealed vials that were used within two weeks of the initial preparation to limit disproportionation into GSH and HS−. The concentration of GSSH was determined by a cyanolysis assay with colorimetric detection of the resulting ferric thiocyanate complex.10 To generate the substrate bound samples, GSSH was added to a concentration of 5–10 times the protein concentration. Solid NONOate was dissolved in 100 mM NaOH and added to FeII-ETHE1 and (FeII/GSS−)-ETHE1 to a concentration of 2–4 times that of the protein. For preparation of MCD cells, the protein was exchanged into deuterated Tris bu˙er that was saturated with deuterated sucrose for a glassing agent. GSSH and NONOate were also prepared as sucrose saturated solutions for these samples. CD control experiments showed no effect of sucrose on the Fe site.
Spectroscopic Methods
Absorption spectra were measured with an Agilent HP8453 diode array spectrophotometer at room temperature. Near-IR (600 – 2000 nm) CD and MCD data were recorded with a Jasco J-730 spectropolarimeter with a liquid N2 cooled InSb detector (Teledyne Judson Technologies) and an Oxford Instruments SM-4000–7T superconducting magnet. UV-Vis (300 −900 nm) CD and MCD data were recorded with a Jasco J-810D spectropolarimeter with an extended S-20 photomultiplier tube and an Oxford Instruments SM-4000–7T superconducting magnet. MCD spectra were corrected for baseline and CD features by subtracting a 0 T scan at each temperature. The sample temperature was measured using a calibrated Cernox resistor (Lakeshore Cryogenics, calibrated 1.5 – 300 K) that was inserted into the MCD cell. VTVH MCD data were normalized to the maximum intensity over all isotherms for a given wavelength. X-band EPR spectra were taken on a Bruker EMX spectrometer with a Bruker ER 041XG/ER microwave bridge and 4116 DM cavity. Spectra were collected at temperatures between 4 and 50 K using an Oxford ITC503 temperature controller with an ESR 900 continuous flow cryostat.
Computational Methods
The ETHE1-GSSH enzyme substrate complex was generated using the FeII-ETHE1 crystal structure (PDB ID: 4CHL)33 and the glutathione persulfide structure from PubChem via the Maestro Schrödinger software package (using the default parameters unless otherwise noted). The ETHE1 crystal structure was prepared using the Protein Preparation tool, GSSH was prepared using the LigPrep tool, the grid for ligand docking was created using a Receptor Grid Generation tool (coordinates: x = −5.41 y = −55.02 z = −15.11; size: 20 Å), and ligand docking was performed using the Glide Docking tool at the extra precision level. Docking structures were analyzed based on the Glide scoring function.43 Favorable scores of similar magnitude were found for the various rotamers of the persulfide group and two limiting orientations were chosen.
The starting geometries of the two limiting substrate orientations in the ETHE1-GSS-NO complex were generated from these docked structures. The protein derived ligands that were included (Asp154, His79, His135, Asp83 and Tyr197) were truncated to their α carbons. The GSS− substrate was truncated to remove the glycine and cysteinyl carbonyl. The glutamate was removed except for its carbonyl, and truncated segments were replaced by H. The coordinates of the α-carbons of the protein derived ligands were frozen to mimic the constraints of the protein backbone. The N(NO)-Fe-S-S dihedral angles for the two limiting substrate orientations were frozen. The IPNS-ACV-NO structure was taken from the IPNS-ACV-NO crystal structure (PDB ID: 1BLZ).17 The protein derived ligands that were included (His214, His270 and Asp216) were truncated to their α carbons, replaced with hydrogens and the α carbons were frozen. In addition, the ACV substrate was truncated to remove the six carbon aminoadipoyl chain. In one set of calculations, the cysteine nitrogen, valine carbonyl carbon and valine β carbon of the substrate were frozen. In another, only the cysteine nitrogen and valine carbonyl carbon were frozen. The CDO-Cys-NO structure was modeled from the substrate bound crystal structure (PDB ID: 2IC1) with NO placed in the only free coordination position.24 The protein derived ligands that were included (His86, His88, His140 and His155) were truncated to their α carbons, replaced with hydrogens and the α carbons were frozen. There were no atoms frozen in the substrate as it is bound bidentate, but the cysteine carboxylic acid was protonated to mimic a salt bridge to an asparagine in the crystal.
DFT calculations were performed using the Gaussian 09 software package, revision D.01.50 The unrestricted BP86 functional51 with 10% Hartree-Fock exchange under tight convergence criteria was used for all calculations, as it had been previously calibrated for {FeNO}7 complexes. The basis set consisted of 6–311G* on the Fe, NO/O2, SS, and all heteroatoms bound to the Fe and 6–31G* on the remaining atoms. Solvation effects were included using the Polarized Continuum Model (PCM)52 with a dielectric constant ϵ = 4.0 to model the protein environment. Frequencies and thermodynamic parameters were calculated using the same split basis set (6–311G*/6–31G*), and modes associated with motion of the frozen α carbons were removed. Molecular orbital compositions were calculated using QMForge,53 and molecular orbital contours were generated using Avogadro54 and/or LUMO.55 TD-DFT calculations were were performed (with Gaussian 09 using BP86 with 10% HF exchange and the split 6–311G*/6–31G* basis set as described above) to compare to the experimental spectra.
Supplementary Material
Acknowledgement
This research was supported by the U.S. National Institute of Health grants GM 40392 (E.I.S.) and GM 11245 (R.B.).
Footnotes
Supporting Information Available
CD and MCD spectra for FeII-ETHE1 control experiments (Figures S1–S4); fit to (FeII/GSS−)-ETHE1 MCD spectrum including unconverted resting enzyme (Figure S5); saturation magnetization data collected on (FeII/GSS−)-ETHE1 band 4 with fits to ground states from bands 1 and 2 (Figure S6); saturation magnetization data collected on ETHE1-NO at 22,000 cm−1 with best fit (Figure S7); simultaneous fit for ETHE1-GSS-NO Abs, RT CD, LT CD and MCD data (Figure S8); MCD spectrum of ETHE1-GSS-NO showing negative band 4 at low temperature and high field (Figure S9); saturation magnetization data collected on ETHE1-GSS-NO at 19,800 cm−1 with best fit (Figure S10); MO diagrams for optimized ETHE1 {FeNO}7 models (Figures S11-S13); TD-DFT calculated transitions for ETHE1-NO (Figure S14); TD-DFT assignment of high energy transitions in ETHE1 {FeNO}7 spectra (Page S6); MO diagram for optimized ETHE1-GSS-O2 model (Figure S15); energies, geometries and spin densities for structures calculated in IPNS and CDO reactions (Tables S1–S4); MO diagrams for optimized {FeO2}8 models of IPNS and CDO (Figures S16-S19); energy level diagrams for IPNS and CDO non-native reactions (Figure S20); structures showing second sphere interactions in IPNS (Figure S21); Cartesian coordinates of all geometry optimized structures (Tables S5–S31). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).Solomon EI; Brunold TC; Davis MI; Kemsley JN; Lee S-K; Lehnert N; Neese F; Skulan AJ; Yang Y-S; Zhou J Geometric and electronic structure/function correlations in non-heme iron enzymes. Chemical reviews 2000, 100, 235–350. [DOI] [PubMed] [Google Scholar]
- (2).Baldwin J; Abraham E The biosynthesis of penicillins and cephalosporins. Natural product reports 1988, 5, 129–145. [DOI] [PubMed] [Google Scholar]
- (3).Fitzpatrick PF The aromatic amino acid hydroxylases. Advances in Enzymology and Related Areas of Molecular Biology, Volume 74 2000, 235–294. [DOI] [PubMed] [Google Scholar]
- (4).Hrywna Y; Tsoi TV; Maltseva OV; Quensen JF; Tiedje JM Construction and characterization of two recombinant bacteria that grow on ortho-and para-substituted chlorobiphenyls. Applied and environmental microbiology 1999, 65, 2163–2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Mahon PC; Hirota K; Semenza GL FIH-1: a novel protein that interacts with HIF-1 and VHL to mediate repression of HIF-1 transcriptional activity. Genes & development 2001, 15, 2675–2686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Kataoka H; Yamamoto Y; Sekiguchi M A new gene (alkB) of Escherichia coli that controls sensitivity to methyl methane sulfonate. Journal of bacteriology 1983, 153, 1301–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Solomon EI; Goudarzi S; Sutherlin KD O2 activation by non-heme iron enzymes. Biochemistry 2016, 55, 6363–6374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Baldwin JE; Adlington RM; Moroney SE; Field LD; Ting H-H Stepwise ring closure in penicillin biosynthesis. Intitial -lactam formation. Journal of the Chemical Society, Chemical Communications 1984, 984–986. [Google Scholar]
- (9).Joseph CA; Maroney MJ Cysteine dioxygenase: structure and mechanism. Chemical Communications 2007, 3338–3349. [DOI] [PubMed] [Google Scholar]
- (10).Kabil O; Banerjee R Characterization of patient mutations in human persulfide dioxygenase (ETHE1) involved in H2S catabolism. Journal of Biological Chemistry 2012, 287, 44561–44567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Seebeck FP In vitro reconstitution of mycobacterial ergothioneine biosynthesis. Journal of the American Chemical Society 2010, 132, 6632–6633. [DOI] [PubMed] [Google Scholar]
- (12).Song H; Her AS; Raso F; Zhen Z; Huo Y; Liu P Cysteine oxidation reactions catalyzed by a mononuclear non-heme iron enzyme (OvoA) in ovothiol biosynthesis. Organic letters 2014, 16, 2122–2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Chen V; Orville A; Harpel M; Frolik C; Surerus K; Münck E; Lipscomb J Spectroscopic studies of isopenicillin N synthase. A mononuclear nonheme Fe2+ oxidase with metal coordination sites for small molecules and substrate. Journal of Biological Chemistry 1989, 264, 21677–21681. [PubMed] [Google Scholar]
- (14).Orville AM; Chen VJ; Kriauciunas A; Harpel MR; Fox BG; Munck E; Lipscomb JD Thiolate ligation of the active site iron (II) of isopenicillin N synthase derives from substrate rather than endogenous cysteine: spectroscopic studies of site-specific Cys. fwdarw. Ser mutated enzymes. Biochemistry 1992, 31, 4602–4612. [DOI] [PubMed] [Google Scholar]
- (15).Scott RA; Wang S; Eidsness MK; Kriauciunas A; Frolik CA; Chen VJ X-ray absorption spectroscopic studies of the high-spin iron (II) active site of isopenicillin N synthase: evidence for iron-sulfur interaction in the enzyme-substrate complex. Biochemistry 1992, 31, 4596–4601. [DOI] [PubMed] [Google Scholar]
- (16).Roach PL; Clifton IJ; Hensgens CM; Shibata N; Long AJ; Strange RW; Hasnain SS; Schofield CJ; Baldwin JE; Hajdu J Anaerobic Crystallisation of an Isopenicillin N Synthase· Fe (II)· Substrate Complex Demonstrated by X-Ray Studies. The FEBS Journal 1996, 242, 736–740. [DOI] [PubMed] [Google Scholar]
- (17).Roach PL; Clifton IJ; Hensgens CM; Shibata N; Schofield CJ; Hajdu J; Baldwin JE Structure of isopenicillinN synthase complexed with substrate and the mechanism of penicillin formation. Nature 1997, 387, 827. [DOI] [PubMed] [Google Scholar]
- (18).Brown CD; Neidig ML; Neibergall MB; Lipscomb JD; Solomon EI VTVH-MCD and DFT studies of thiolate bonding to {FeNO} 7/{FeO2} 8 complexes of isopenicillin N synthase: substrate determination of oxidase versus oxygenase activity in nonheme Fe enzymes. Journal of the American Chemical Society 2007, 129, 7427–7438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Tamanaha E; Zhang B; Guo Y; Chang W.-c.; Barr EW; Xing G; St Clair J; Ye S; Neese F; Bollinger JM Jr; Krebs C Spectroscopic Evidence for the Two C–H-Cleaving Intermediates of Aspergillus nidulans Isopenicillin N Synthase. Journal of the American Chemical Society 2016, 138, 8862–8874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Lundberg M; Siegbahn PE; Morokuma K The mechanism for isopenicillin N synthase from density-functional modeling highlights the similarities with other enzymes in the 2-His-1-carboxylate family. Biochemistry 2008, 47, 1031–1042. [DOI] [PubMed] [Google Scholar]
- (21).Lundberg M; Kawatsu T; Vreven T; Frisch MJ; Morokuma K Transition States in a Protein Environment- ONIOM QM: MM Modeling of Isopenicillin N Synthesis. Journal of chemical theory and computation 2009, 5, 222–234. [DOI] [PubMed] [Google Scholar]
- (22).Burzla NI; Rutledge PJ; Clifton IJ; Hensgens CM; Pickford M; Adlington RM; Roach PL; Baldwin JE The reaction cycle of isopenicillin N synthase observed by X-ray diraction. Nature 1999, 401, 721. [DOI] [PubMed] [Google Scholar]
- (23).Stipanuk MH Sulfur amino acid metabolism: pathways for production and removal of homocysteine and cysteine. Annu. Rev. Nutr 2004, 24, 539–577. [DOI] [PubMed] [Google Scholar]
- (24).Ye S; Wu X; Wei L; Tang D; Sun P; Bartlam M; Rao Z An Insight into the Mechanism of Human Cysteine Dioxygenase KEY ROLES OF THE THIOETHER-BONDED TYROSINE-CYSTEINE COFACTOR. Journal of Biological Chemistry 2007, 282, 3391–3402. [DOI] [PubMed] [Google Scholar]
- (25).Kumar D; Thiel W; de Visser SP Theoretical study on the mechanism of the oxygen activation process in cysteine dioxygenase enzymes. Journal of the American Chemical Society 2011, 133, 3869–3882. [DOI] [PubMed] [Google Scholar]
- (26).de Visser SP; Straganz GD Why do cysteine dioxygenase enzymes contain a 3-His ligand motif rather than a 2His/1Asp motif like most nonheme dioxygenases? The Journal of Physical Chemistry A 2009, 113, 1835–1846. [DOI] [PubMed] [Google Scholar]
- (27).Blaesi EJ; Gardner JD; Fox BG; Brunold TC Spectroscopic and computational characterization of the NO adduct of substrate-bound Fe (II) cysteine dioxygenase: Insights into the mechanism of O2 activation. Biochemistry 2013, 52, 6040–6051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Blaesi EJ; Fox BG; Brunold TC Spectroscopic and computational investigation of iron (III) cysteine dioxygenase: implications for the nature of the putative superoxo-Fe (III) intermediate. Biochemistry 2014, 53, 5759–5770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Tchesnokov E; Faponle A; Davies C; Quesne M; Turner R; Fellner M; Souness R; Wilbanks S; de Visser S; Jameson G An iron–oxygen intermediate formed during the catalytic cycle of cysteine dioxygenase. Chemical Communications 2016, 52, 8814–8817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Burlina A; Zacchello F; Dionisi-Vici C; Bertini E; Sabetta G; Bennet M; Hale D; Schmidt-Sommerfeld E; Rinaldo P New clinical phenotype of branched-chain acyl-CoA oxidation defect. The Lancet 1991, 338, 1522–1523. [DOI] [PubMed] [Google Scholar]
- (31).Garcia-Silva M; Campos Y; Ribes A; Briones P; Cabello A; Borbujo JS; Arenas J; Garavaglia B Encephalopathy, petechiae, and acrocyanosis with ethylmalonic aciduria associated with muscle cytochrome c oxidase deficiency. The Journal of pediatrics 1994, 125, 843. [PubMed] [Google Scholar]
- (32).Tiranti V; Viscomi C; Hildebrandt T; Di Meo I; Mineri R; Tiveron C; Levitt MD; Prelle A; Fagiolari G; Rimoldi M; Zeviani M Loss of ETHE1, a mitochondrial dioxygenase, causes fatal sulfide toxicity in ethylmalonic encephalopathy. Nature medicine 2009, 15, 200. [DOI] [PubMed] [Google Scholar]
- (33).Pettinati I; Brem J; McDonough MA; Schofield CJ Crystal structure of human persulfide dioxygenase: structural basis of ethylmalonic encephalopathy. Human molecular genetics 2015, 24, 2458–2469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Lin B; Ma G; Liu Y Mechanism of the Glutathione Persulfide Oxidation Process Catalyzed by Ethylmalonic Encephalopathy Protein 1. ACS Catalysis 2016, 6, 7010–7020. [Google Scholar]
- (35).Sattler SA; Wang X; Lewis KM; DeHan PJ; Park C-M; Xin Y; Liu H; Xian M; Xun L; Kang C Characterizations of two bacterial persulfide dioxygenases of the metallo-β-lactamase superfamily. Journal of Biological Chemistry 2015, 290, 18914–18923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Arciero D; Lipscomb J; Huynh B; Kent T; Münck E EPR and Mössbauer studies of protocatechuate 4, 5-dioxygenase. Characterization of a new Fe2+ environment. Journal of Biological Chemistry 1983, 258, 14981–14991. [PubMed] [Google Scholar]
- (37).Zhang Y; Pavlosky MA; Brown CA; Westre TE; Hedman B; Hodgson KO; Solomon EI Spectroscopic and theoretical description of the electronic structure of the S= 3/2 nitrosyl complex of non-heme iron enzymes. Journal of the American Chemical Society 1992, 114, 9189–9191. [Google Scholar]
- (38).Brown CA; Pavlosky MA; Westre TE; Zhang Y; Hedman B; Hodgson KO; Solomon EI Spectroscopic and theoretical description of the electronic structure of S= 3/2 iron-nitrosyl complexes and their relation to O2 activation by non-heme iron enzyme active sites. Journal of the American Chemical Society 1995, 117, 715–732. [Google Scholar]
- (39).Pierce BS; Gardner JD; Bailey LJ; Brunold TC; Fox BG Characterization of the nitrosyl adduct of substrate-bound mouse cysteine dioxygenase by electron para-magnetic resonance: electronic structure of the active site and mechanistic implications. Biochemistry 2007, 46, 8569–8578. [DOI] [PubMed] [Google Scholar]
- (40).Solomon EI; Pavel EG; Loeb KE; Campochiaro C Magnetic circular dichroism spectroscopy as a probe of the geometric and electronic structure of non-heme ferrous enzymes. Coordination Chemistry Reviews 1995, 144, 369–460. [Google Scholar]
- (41).Diebold AR; Brown-Marshall CD; Neidig ML; Brownlee JM; Moran GR; Solomon EI Activation of -keto acid-dependent dioxygenases: application of an {FeNO} 7/{FeO2} 8 methodology for characterizing the initial steps of O2 activation. Journal of the American Chemical Society 2011, 133, 18148–18160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Neese F; Solomon EI MCD C-term signs, saturation behavior, and determination of band polarizations in randomly oriented systems with spin S? 1/2. Applications to S= 1/2 and S= 5/2. Inorganic chemistry 1999, 38, 1847–1865. [DOI] [PubMed] [Google Scholar]
- (43).Friesner RA; Banks JL; Murphy RB; Halgren TA; Klicic JJ; Mainz DT; Repasky MP; Knoll EH; Shelley M; Perry JK; Shaw DE; Perry F; Shenkin PS Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. Journal of medicinal chemistry 2004, 47, 1739–1749. [DOI] [PubMed] [Google Scholar]
- (44).Souness RJ; Klemann T; Tchesnokov EP; Wilbanks SM; Jameson GB; Jameson GN Mechanistic implications of persulfenate and persulfide binding in the active site of cysteine dioxygenase. Biochemistry 2013, 52, 7606–7617. [DOI] [PubMed] [Google Scholar]
- (45).Schenk G; Pau MY; Solomon EI Comparison between the geometric and electronic structures and reactivities of {FeNO} 7 and {FeO2} 8 complexes: A density functional theory study. Journal of the American Chemical Society 2004, 126, 505–515. [DOI] [PubMed] [Google Scholar]
- (46).Wilson SA; Chen J; Hong S; Lee Y-M; Clémancey M; Garcia-Serres R; Nomura T; Ogura T; Latour J-M; Hedman B; Hodgson KO; Nam W; Solomon EI [FeIV=O(TBC)(CH3CN)]2+: Comparative Reactivity of Iron (IV)-Oxo Species with Constrained Equatorial Cyclam Ligation. Journal of the American Chemical Society 2012, 134, 11791–11806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Kabil O; Motl N; Strack M; Seravalli J; Metzler-Nolte N; Banerjee R Mechanism-based inhibition of human persulfide dioxygenase by -glutamylhomocysteinyl-glycine. Journal of Biological Chemistry 2018, jbc–RA118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Brown-Marshall CD; Diebold AR; Solomon EI Reaction coordinate of isopenicillin N synthase: oxidase versus oxygenase activity. Biochemistry 2010, 49, 1176–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Kriauciunas A; Frolik C; Hassell T; Skatrud P; Johnson M; Holbrook N; Chen V The functional role of cysteines in isopenicillin N synthase. Correlation of cysteine reactivities toward sulfhydryl reagents with kinetic properties of cysteine mutants. Journal of Biological Chemistry 1991, 266, 11779–11788. [PubMed] [Google Scholar]
- (50).Frisch MJ; Trucks GW; Schlegel HB; Scuseria GE; Robb MA; Cheese-man JR; Scalmani G; Barone V; Mennucci B; Petersson GA; Nakatsuji H; Caricato M; Li X; Hratchian HP; Izmaylov AF; Bloino J; Zheng G; Sonnenberg JL; Hada M; Ehara M; Toyota K; Fukuda R; Hasegawa J; Ishida M; Nakajima T; Honda Y; Kitao O; Nakai H; Vreven T; Montgomery JA Jr.; Peralta JE; Ogliaro F; Bearpark M; Heyd JJ; Brothers E; Kudin KN; Staroverov VN; Kobayashi R; Normand J; Raghavachari K; Rendell A; Bu-rant JC; Iyengar SS; Tomasi J; Cossi M; Rega N; Millam JM; Klene M; Knox JE; Cross JB; Bakken V; Adamo C; Jaramillo J; Gomperts R; Strat-mann RE; Yazyev O; Austin AJ; Cammi R; Pomelli C; Ochterski JW; Martin RL; Morokuma K; Zakrzewski VG; Voth GA; Salvador P; Dannenberg JJ; Dapprich S; Daniels AD; Farkas Ö; Foresman JB; Ortiz JV; Cioslowski J; Fox DJ Gaussian 09 Revision E.01. Gaussian Inc. Wallingford CT: 2009. [Google Scholar]
- (51).Becke AD Density-functional exchange-energy approximation with correct asymptotic behavior. Physical review A 1988, 38, 3098. [DOI] [PubMed] [Google Scholar]
- (52).Cramer CJ; Truhlar DG Implicit solvation models: equilibria, structure, spectra, and dynamics. Chemical Reviews 1999, 99, 2161–2200. [DOI] [PubMed] [Google Scholar]
- (53).Tenderholt AL QMForge, Version 2.1. 2007.
- (54).Avogadro: an open-source molecular builder and visualization tool. Version 1.2.0.
- (55).Kieber-Emmons MT LUMO. 2012; Version 1.0.1.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.