

Summary
Peroxisome proliferator-activated receptors (PPARs) are pharmacological targets for the treatment of metabolic disorders. Previously, we demonstrated the antidiabetic effects of SR1664, a PPARγ modulator lacking classical transcriptional agonism, despite its poor pharmacokinetic properties. Here we report identification of the antagonist SR11023 as a potent insulin sensitizer with significant plasma exposure following oral administration in mice. To determine the structural mechanism of ligand-dependent antagonism of PPARγ, we employed an integrated approach combining solution-phase biophysical techniques to monitor activation helix (helix 12) conformational dynamics. While informative on receptor dynamics, HDX-MS and NMR data provide limited information regarding the specific orientations of structural elements. In contrast, label free quantitative crosslinking mass spectrometry (XL-MS) revealed that binding of SR11023 to PPARγ enhances interaction with co-repressor motifs by pushing H12 away from the agonist active conformation towards the H2-H3 loop region (i.e., the omega loop), revealing the molecular mechanism for active antagonism of PPARγ.
INTRODUCTION
Peroxisome proliferator-activated receptor gamma (PPARγ), a member of the nuclear receptor superfamily of transcriptional factors, has been implicated in type 2 diabetes, a disease characterized by a state of insulin resistance and glucose intolerance. PPARγ is the pharmacological target of the thiazolidinedione (TZD) class of antidiabetic drugs, including rosiglitazone and pioglitazone(Mayans, 2015; Wright et al., 2014). However, treatment of type 2 diabetes mellitus (T2DM) has moved away from administration of TZDs as safety concerns over their use has grown. While weight gain is associated with use of TZDs, the major safety concerns include edema, plasma volume expansion that is linked to cardiomegaly and heart failure, increased risk of bone fractures, and a slight increased risk for bladder cancer (specific to pioglitazone) (Kung and Henry, 2012; Rubenstrunk et al., 2007; Shi et al., 2011). As an attempt to dissociate efficacy from side effects, selective PPARγ partial agonist termed SPPARγMs were developed that exhibit significantly reduced receptor target gene expression without loss of anti-diabetic efficacy in rodent models of T2DM (Liu et al., 2005; Minoura et al., 2004). For example, MRL24, a potent PPARγ partial agonist, was shown to be anti-diabetic in db/db mice with no detectable cardiac hypertrophy, minimal increases in plasma volume, and no increase in extracellular fluid volume(Acton et al., 2009; Acton et al., 2005). Subsequent studies were conducted to examine this mechanism of action for both agonists and partial agonists. These studies demonstrated that the insulin sensitization afforded by partial agonist and TZD agonist treatment tightly correlate with the ability of these drugs to block the obesity-induced phosphorylation of PPARγ at S273 (pS273)(Choi et al., 2010). This work was followed by demonstration that the PPARγ antagonist SR1664, a compound that potently blocks pS273 but does not increase expression of pro-adipogenic PPARG target genes, was an efficacious insulin sensitizer in rodent models of diabetes(Choi et al., 2011). Unfortunately, SR1664 suffers from poor pharmacokinetic properties thus limiting its utility in chronic studies. To improve the pharmaceutical properties of SR1664, extensive SAR studies were carried out (Asteian et al., 2015). Here, we report that the optimized SR1664 analog SR11023 demonstrates significantly improved pharmacokinetic properties such that the compound can be administered orally at relatively low doses resulting in substantial drug levels in blood and white adipose tissue (WAT), the metabolic tissue where PPARγ activity is critical for systemic insulin sensitivity(Kintscher and Law, 2005; Sugii et al., 2009).
To assist the SAR studies, we sought to understand the structural basis for antagonism of PPARγ by probing the receptor global and local dynamics in the context of ligand binding, co-activator and co-repressor NR box motif recruitment. The C-terminal most helix of most NRs, H12 or activation helix (AH or AF2), is often described as the molecular switch governing the transitions between active and inactive conformations(Heldring et al., 2007). Binding of agonist ligands to the NR ligand binding pocket shifts H12 towards an active conformation to facilitate recruitment of transcriptional coactivators (e.g., p160 family members such as SRC1) that tether histone acylation activity relaxing chromatin allowing RNA polymerase II binding to the target gene. In contrast, repressive ligands (antagonists and inverse agonists) shift H12 towards an inactive conformation facilitating receptor interaction with co-repressor proteins such as N-CoR (nuclear receptor corepressor) or SMRT (silencing mediator for retinoid and thyroid hormone receptors). These corepressor proteins tether histone deacetylases to the transcriptional complex to promote condensation of chromatin to shut down transcription(Horlein et al., 1995; Hu and Lazar, 1999; Nagy et al., 1999). The receptor interaction domain (RID) of these coregulatory proteins contain highly conserved hydrophobic helical motifs called NR boxes that engage the region of H12 referred to as AF2 (activation function 2). The RID of coactivator proteins contain 5’ - LXXLL – 3’ motifs whereas corepressor proteins contain 5’ – LXXI/HIXXXL/I – 3’ motifs called CoRNR boxes(Xu et al., 2002b). While high resolution atomic structures exist for most ligand binding domains (LBD) of NRs, the orientation of H12 upon binding to repressive ligands and NR or CoRNR box peptides most often is not resolved by co-crystallography. To address this shortcoming, we employed an integrated approach using solution based techniques — chemical crosslinking mass spectrometry (XL-MS), hydrogen/deuterium exchange mass spectrometry (HDX-MS) and nuclear magnetic resonance (NMR) spectroscopy — to aid structural characterization of ligand binding-mediated coactivator/corepressor receptor interactions and H12 dynamics. XL-MS has been used to gain insights into structural rearrangement between protein subunits(Wu et al., 2013), protein conformational changes(Schmidt and Robinson, 2014), and the assembly of large protein complexes(Chen et al., 2010; Knutson et al., 2014). We demonstrate that SR11023 is a potent antagonist of PPARγ with strikingly improved pharmacokinetics over SR1664, blocks pS273 and lacks classic agonist properties. Unlike full agonist TZDs, binding of SR11023 to PPARγ only results in stabilization of H3. This is in accordance with a molecular docking model that unlike full and partial agonists, SR11023 adopts a distinct conformation residing beside H3 and facing away from AF2 surface. Solution based biophysical analyses further reveal that H12 does in fact act as a molecular switch governing the ligand-dependent activation of PPARγ. Instead of being trapped in one conformation as observed in numerous co-crystallographic studies, H12 possesses structural dynamics in solution and adopts unique ensembles of conformers depending on the receptor/ligand/NR-box peptide complex. Binding of SR11023 enhances the receptor interaction with co-repressor motifs by driving H12 away from agonist position towards the H2-H3 loop region (omega loop) of the protein. These results suggest a unique yet to be observed molecular mechanism for SR11023-mediated antagonism of PPARγ.
RESULTS
Pharmacological antagonism of PPARγ by SR11023:
The poor pharmacokinetic (PK) properties of SR1664 restricted its route of drug administration to intraperitoneal injection. Efforts to improve the PK properties and to develop SAR around the core biaryl indole scaffold of SR1664 led to the synthesis of a wide range of analogs. Compounds were screened in cell-based assays to identify compounds that exhibit minimal PPARγ transactivation activity (<10% at 1μM in Gal4-LBD assays and ~0% in full length PPRE assays) while retaining good binding affinity to PPARγ (IC50 < 250 nM in a competitive displacement assay). Compounds that met these criteria were evaluated in rodent PK studies to identify analogs that exhibited excellent plasma exposure following oral administration. Previously it has been demonstrated that the substitution of t-butyl for nitro at the para position of SR1664 led to identification of a series of inverse agonists (e.g., SR2595 and SR10171) that suppress receptor basal transcriptional activity; and although SR2595 and SR10171 exhibit beneficial effects on osteogenesis in multipotent mesenchymal stem cells (MSCs) and bone metabolism in mouse models, these compounds repress PPARG target gene expression suggesting different mechanism from the antagonist SR1664(Marciano et al., 2015; Stechschulte et al., 2016). Further optimization of the SR1664 scaffold, including substitution of cyclopropane for trifluoromethyl at the meta position of the left-hand benzyl group, resulted in identification of the antagonist SR11023 which exhibited good binding affinity (IC50 = 108nM) yet poor transactivation activity in the GAL4-PPARG assay (2.7μM, 10% Max.Stim), similar to that observed for SR1664 (4.5μM, 10% Max.Stim) (Table 1). Note that poor transactivation is not due to lack of cell penetration as both SR1664 and SR11023 significantly right-shift the dose response curve for rosiglitazone in the GAL4-PPARG assay (data not shown). In addition, in both human PPARα (hPPARα) and hPPARδ-Gal4 transactivation assays, SR11023 and rosiglitazone exhibited no transcriptional agonism (Figure S1A).
Table 1.
Compound structures and characteristics
| Compound | Chemical structures | IC50a (nM) PPARγ | EC50a (nM), Max.Stim. PPARγ Gal-4 | EC50a (nM) PPRE | EC50a (μM) PPARα GAL-4 | % inhibition p273- PPARγ (2μM, 20μM) |
|---|---|---|---|---|---|---|
| Rosiglitazone | 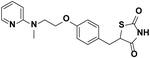 |
18 | 7.4, 100% | 10 | No activity | 85, 100 (Choi et al., 2011) |
| MRL24 | 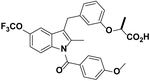 |
1 | 2, 21% (Acton et al., 2005) | NT | NT | More potent than Rosi to inhibit p273 (Choi et al., 2010) |
| SR1664 | 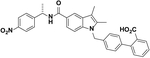 |
80 | 4501, 10% (Asteian et al., 2015) | No activity | 8.6 | 90, 100 (Choi et al., 2011) |
| SR11023 | 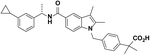 |
108 | 2761, 10% | No activity | No activity | 25, 77 |
% transactivation at 10 μM; NT: not tested
As previously shown, the insulin sensitization efficacy of antagonist ligands such as SR1664, correlates with their ability to reduce phosphorylation of PPARγ at S273 (pS273) (Choi et al., 2011). In an in vitro kinase assay, dose-dependent reduction in pS273 was observed for the SR11023 treated receptor (25% at 2μM and 75% at 20μM) as compared to DMSO only controls. This result suggested that SR11023 can reduce pS273 and should demonstrate antidiabetic properties in rodents (Table 1 and Figure S1B). Unlike rosiglitazone and similar to SR1664, SR11023 possesses minimal adipogenic activity in differentiated 3T3-L1 cells as demonstrated by the absence of lipid formation and lack of upregulation of the PPARγ-driven pro-adipogenic genes, e.g. aP2 and cd36 (Figure 1A and Figure 1B). To obtain an unbiased assessment of the compounds’ impact on gene expression in adipocytes, we performed RNA-seq of mRNA isolated from 3T3-L1 cells incubated with rosiglitazone, SR11023, or DMSO-only for 6 days. Rosiglitazone exhibited robust activation of a large gene set containing many known PPARγ target genes whereas SR11023 displayed very modest regulation of only a small subset of PPARγ controlled genes (Figure 1C), reminiscent of that observed for the antagonist SR1664 (Choi et al., 2011). However, unlike SR1664, SR11023 showed significant plasma exposure in C57BL/6 mice following oral administration; two hours after dosing either both compound at 40mg.kg−1, the plasma concentration of SR11023 reached 70μM whereas SR1664 plasma exposure was less than 1μM (Figure S1C). Thus, SR11023 is a potent antagonist of PPARγ with strikingly improved pharmacokinetics over SR1664, blocks pS273 and lacks classic agonist properties.
Figure 1. SR11023 functions as a non-agonist of PPARγ.

(A) Oil red staining of differentiated 3T3-L1 adipocytes in the presence of Rosiglitazone or SR11023. (B) Expression of adipocyte genes in differentiated 3T3-L1 cells. Results are expressed as mean +/− SEM. (C) Linear plot of RNA-seq data of differential gene expression in 3T3-L1 cells induced by 6-day ligand treatments. (D) Superimposition of SR11023, MRL24 and Rosiglitazone within PPARγ LBD. Top scoring in silico docking binding pose for SR11023 using chain A of PDB: 4R06 (atomic coordinates specific for PPARγ crystal structure from the Protein Structure Database). (E) Differential consolidation HDX data are mapped onto the PPARγ LBD structure, as shown by representation of altered conformational dynamics of receptor upon binding to Rosi, MRL24 and SR11023. Percentages of deuterium differences are color-coded according to the smooth color gradient key in the bottom. (F) TR-FRET analysis of His-PPARγ interaction with FITC-labeled peptides representative of NR binding motifs from either transcriptional co-activators (p300 and SRC1–2) or co-repressors (NCOR1–3 and SMRT-2) in the presence of increasing concentrations of Rosi, MRL24 and SR11023. GW9662, a potent antagonist of PPARγ, is used as a control ligand. See also Figure S1 and Figure S2A and S2B.
SR11023 has a distinct binding mode within PPARγ and enhances interaction with co-repressor peptides:
Next, we applied molecular docking, HDX-MS, and NMR to investigate SR11023 interactions with receptor. Using in silico docking, superimposition of SR11023 with the binding poses for rosiglitazone (PDB:1PRG) and the partial agonist MRL24 (PDB:2Q5P) within the ligand binding pocket (LBP) of PPARγ suggests that SR11023 adopts a relatively linear orientation facing away from the AF2 surface residing along H3. In contrast, both rosiglitazone and MRL24 display curved shaped conformations with the thiazolidinedione of rosiglitazone and the indole of MRL24 oriented towards AF2 surface. These distinct orientations within the LBP are consistent with results from HDX-MS analyses that reveal the exchange kinetics of the H11-H12 region of PPARγ was insensitive to SR11023 binding whereas rosiglitazone, and MRL24 to a lesser extent, stabilize the C-terminal end of H11 and H11-H12 loop region as demonstrated by reduced solvent exchange (Figure 1E and Figure S2A). Only binding of rosiglitazone to PPARγ results in full stabilization of H12 (Figure 1E and Figure S2A). Among these ligands, NMR analysis reveals that a NMR peak corresponding to Y473, which resides on H12, is only present for rosiglitazone, indicating H12 is dynamic on the μs-ms time scale in the apo form or when bound to MRL24 or SR11023 (Figure S2B). Binding of all three ligands to PPARγ afforded strong protection to solvent exchange in H3, residues 279–286 and 287–298, further supporting that SR11023 possesses high binding affinity with receptor comparable to that of rosiglitazone and MRL24 (Figure 1E and Figure S2A). In a TR-FRET peptide interaction assay, both rosiglitazone and MRL24 enhanced the association of PPARγ with NR box peptides derived from the coactivators p300 and SRC1 (SRC1–2), whereas in the presence of SR11023, there was no enhanced interaction. In contrast, in the presence of SR11023, receptor interaction with peptides derived from the corepressors NCOR1 (NCOR1–3) and SMRT (SMRT-2) was increased whereas binding of either rosiglitazone or MRL24 facilitated dissociation of these corepressor peptides (referred to as CoRNR box peptides) from PPARγ (Figure 1F).
XL-MS reveals mobility of PPARγ H12 influenced by SR11023 and corepressor peptide:
The structural switch of H12 in the LBD of NRs between an inactive towards an active conformation is considered a classical model of AF2 ligand-dependent activation(Heldring et al., 2007; le Maire et al., 2010). However, the orientation of H12 of PPARγ in non-active conformations is hard to resolve. Co-crystallography of apo and ligand bound LBD all reveal H12 in an active conformation (Bruning et al., 2007). Previous studies using HDX-MS and NMR have shown that the dynamics of H12 are differentially altered by ligands. However, these techniques, which examine receptor hydrogen bond networks and changes in chemical environment via chemical shift perturbations, do not provide insight into the structural orientation of H12. To address this, we applied label free quantitative crosslinking mass spectrometry to examine the solution state mobility of PPARγ H12 in the presence and absence of ligands, co-activator and or co-repressor peptides. XL-MS studies were performed with the BS3 crosslinker where two proximal lysine residues within the distance of ~24 Å can be crosslinked (6 Å side chain of each lysine and the 11.4 Å spacer of BS3). In several published studies, a distance range of 25–30 Å is considered as a reasonable crosslink limit, measured between alpha-carbon atoms of lysine (a tolerance about 3 Å is added to the theoretical maximum cross-linking distance of BS3) (Fischer et al., 2013; Merkley et al., 2014). A summary of identified XL peptides is shown in Table S1. XL-MS studies of apo PPARγ demonstrated that lysine474 (K474) which is part of a H12 peptide, K(474)DLY, was capable of forming crosslinks between several lysines including LQVIK(457)K (part of H11), AK(301)SIPGF (C-terminal region of H3), QEQSK(265)EVAIR and FK(275)HITPL (part of the H2-H3 loop). The MS/MS spectra of these XL peptides are shown in Figure S3. The ion intensities and peak areas of these crosslinked peptides in twenty different experimental conditions (3 replicates each condition) were manually calculated and compared. The results from this analysis are displayed in Figure 2. The highest peak intensity (derived from selected ion chromatograms (SICs)) across twenty XL samples (including 3 replicates) in each peptide panel - SVEAVQEITEY +2, K224-K232 XL peptide, K474-K457 XL peptide, K474-K257 XL peptide, K474-K265 XL peptide and K474-K301 XL peptide - were normalized as 100% bar plot. For all other peptides, their rest peak abundances within an indicated peptide panel were scaled accordingly. The intensity of the peptide that contains K224-K232 conjugation between H1-H2 was used as an internal control since this region of the receptor is not involved in ligand or NR box peptide binding. As expected, similar signal intensity was observed for this peptide in all conditions. An additional internal control was peptide SVEAVQEITEY (634.31 m/z, +2) which lacks a lysine residue and thus cannot be conjugated with BS3, afforded similar peak intensity across all samples (Figure 2A and B). These crosslinking results displayed in Figure 2 suggest that H12 of apo receptor is a dynamic ensemble of distinct conformers. This observation is in contrast to the locked conformation of H12 observed in the crystal structure of apo PPARγ (Bruning et al., 2007).
Figure 2. Influence of ligand and NR box peptide on H12 mobility revealed by BS3 crosslinking MS.

Different H12 orientations in PPARγ LBD are superimposed in the PPARγ LBD, as shown by an ensemble of structural models of different orientations of H12 (K474) conjugated to K457 (blue H12, Figure 2C), K275 (red H12, Figure 2D), K265 (orange H12, Figure 2E) and K301 (green H12, Figure 2F). Yellow, H3 residue region SVEAVQEITEY; Cyan: BS3 conjugated lysine residues shown in stick. (A) relative peak area of non-XL peptide SVEAVQEITEY. (B) relative peak area of K224-K232 XL peptide. Results are expressed as mean +/− SEM. (C-F) relative peak area of K474-K457, K474-K275, K474-K265 and K474-K301 XL peptide, respectively. All results are expressed as mean +/− SEM. Statistical analysis was performed by two-way ANOVA between indicated pairwise experiment (*** = p<0.001; ** = p<0.01; * = p<0.05). DynaXL program is used to generate input structure calculation scripts that is further used by the xplor-nih program for structure modeling of XL-MS data. The data is consistent with models for both PPARγ alone and the PPARγRXRα heterodimer complex. See also Figure S3 and Figure S4.
The conformational dynamics of the activation helix of PPARγ (H12) is influenced upon receptor binding to pharmaceutically distinct ligands and or sequence specific NR or CoRNR box peptides. The higher the MS signal intensity of a particular crosslinked peptide pair that includes conjugation between H12 and one distal lysine within PPARγ, the more the population of H12 conformers are orientated towards that lysine residue. On the other hand, the lower the MS signal intensity, that conformer is less populated. Label free quantitative XL-MS studies of PPARγ LBD in the presence of either rosiglitazone, MRL24, or SR11023 resulted in changes in the peak intensities of various crosslinked peptides as compared to those observed for apo receptor. As shown in Figure 2C, addition of the co-activator SRC1–2 NR box peptide to either apo receptor or ligand bound receptor selectively increased the abundance of the intensity of ions corresponding to the crosslink between K457-K474, suggesting that co-activator peptide binding drives increased populations of H12 towards H11 K457 residue. In the reported X-ray crystal structure, coactivator peptide binding reinforces H12 docking against H11 (Nolte et al., 1998). Although crosslinks between K474-K457 and K474-K301 were observed in the presence of all ligands, agonist and SRC1–2 binding afforded a statistically significant reduction in the intensity (population) of K474-K265 and K474-K275 crosslinks, suggesting that H12 in the agonist complex cannot effectively sample the longer-distance conformers; K474 to K275 at ~26 Å and K474 to K265 at ~35 Å (determined from PPARγ X-ray structure PDB: 2PRG) (Figure 2D and E). In contrast, SR11023 binding to receptor restored these longer distance crosslinks as represented by an increased intensity of K474-K275 and K474-K265 XL peptides (Figure 2D and E). Similar intensities of ions corresponding to crosslinks for K474-K265 and K474-K275 were observed in when comparing PPARγ alone (homodimer) or PPARγ/RXRα heterodimer, suggesting that these crosslinks are intra-molecular and not inter-molecular (Figure 2 and Figure S4). These data demonstrate that the mobility of H12 in the apo receptor is reduced upon ligand binding and that H12 adopts different orientations relative to AF2 when comparing agonist to antagonist bound receptor (Figure 3). The largest change in peak intensities for crosslinks was mediated by antagonist bound receptor in the presence of the co-repressor peptide NCOR1–3 (Figure 2F). In this experiment, the crosslink between K474-K301 was abrogated while the intensity of crosslinks between K474-K265/K275 increased with statistical significance, suggesting a stable re-orientation of H12 in proximity of H2-H3 loop region (Figure 2 D, E, F and Figure 3).
Figure 3. PPARγ H12 mobility based on XL-MS observation.

In the apo or inverse agonist bound state, H12 adopts an ensemble of multiple populations swing around H3. Binding of inverse agonist to PPARγ further stabilize the interaction with CoRNR, which displaces H12 from folding back to agonist position. In contrast, agonist binding directly stabilize H12 into an agonist position, which forms AF2 surface with H3 and H5 for CoA binding.
NMR and HDX-MS data provide complimentary information to the XL-MS data:
Further evidence for corepressor binding affecting the K474-K301structural perturbation was obtained by protein NMR, which revealed that addition of NCOR1–3 or SMRT2 peptide caused NMR chemical shift perturbations for residues within the AF-2 surface, including the site of crosslinking (K301) and other nearby residues (e.g., I303 and V307) (Figure 4A). Combined with the crosslinking data, these results indicating that binding of the co-repressor peptides, which have nearly identical affinity for PPARγ (Figure 4B), to this region can block BS3 crosslinks at K301. Although NCOR1–3 and SMRT-2 share a conserved CoRNR box motif 5’ – LXXI/HIXXXL/I – 3’, SMRT-2 was not as efficient as NCOR1–3 at altering H12 orientation (Figure 2). Additionally, HDX-MS analysis revealed that NCOR1–3, when bound to apo PPARγ, destabilized H12 to a greater extent than SMRT-2 (Figure S2A). It is likely that the NCOR1–3 peptide docks into the AF2 hydrophobic cleft in a slightly different orientation than the SMRT-2 peptide, which is supported by the NMR results showing differences in the peptide-bound peaks for residues in this region, resulting in enhanced ability to disrupt the charge-clamp within the AF2 surface. In general, the observations described above are consistent with the reported crystal structures of co-repressor and antagonist bound RAR LBD and PPARα LBD wherein NCOR or SMRT peptide binding occupies the H3-H5 binding groove within the AF2 surface and displaces AF2 helix from folding back to the agonist position(le Maire et al., 2010; Xu et al., 2002a). Taken together, differences in compound structure and the specific NR-box sequence combine to impact the mobility of AF2, which in turn impacts PPARγ transcriptional activity. Binding of SR11023 to PPARγ would blunt activation of its target genes by maintaining co-repressor interaction and keeping the receptor in an inactive conformation.
Figure 4. NMR chemical shift induced by co-repressor peptide.

(A) Differential 2D [1H,15N]-TROSY-HSQC NMR data comparing apo-PPARγ without or with 2 equivalents of NCoR1–3 or SMRT2 corepressor peptide. (B) Fluorescence polarization assay data showing the affinity of PPARγ for NCoR1–3 (240 nM) and SMRT2 (170 nM) are similar. Results are expressed as mean +/− SEM.
DISCUSSION
The TZD class of drugs function as full agonists of PPARγ and have been used to treat T2DM. However, their diminished clinical usage due to undesirable side effects has given rise to the necessity to develop the next generation of class of insulin sensitizers that afford dissociation of classical agonism(Rizos et al., 2009). The antidiabetic actions of rosiglitazone and MRL24 have been shown to tightly correlate with their abilities to block PPARγ pS273, were the degree of phosphorylation at this site is associated with obesity and impaired glucose tolerance (Banks et al., 2015; Choi et al., 2010). The discovery of SR1664 proved a therapeutic concept that it is possible to develop a PPARγ ligand blocking pS273 that is devoid of classical agonism, yet retains TZD-like antidiabetic activities in animal models (Choi et al., 2011). In this study, we profile a PPARγ modulator SR11023 that possesses therapeutic potential for treatment of T2DM. We show that SR11023 exhibits the ability to block pS273 yet has significantly improved pharmacokinetic properties compared to that of SR1664. Yet unlike TZDs, SR11023 is absent of pro-adipogenic behavior and lacks the classical agonism to upregulate PPARγ target gene expressions in 3T3-L1 cells.
We investigated the structural determinants of SR11023 mediated PPARγ inactivation and examined the role of H12 as a molecular switch governing ligand-dependent activation of PPARγ. Unlike TZDs, binding of SR11023 to PPARγ only results in stabilization of H3. This is in accordance with molecular docking model that unlike full and partial agonists, SR11023 adopts distinct conformation residing beside H3 and facing away from AF2 surface. While HDX-MS and NMR data provide molecular hydrogen bonding activities and chemical perturbations, both of them yield limited information regarding orientation of concrete structural elements. XL-MS provides an orthogonal approach to detect the mobility of H12 with respect to residue-residue distance, residue solvent exposure, and local conformational rearrangement. Label free quantitative XL-MS was used to profile four distinct H12 helix crosslinking reactions in 20 different protein states. One advantage of label free quantitation is that there are no limits to the number of samples that can be compared. In one study, the robustness and reproducibility of this quantitation method has been examined (Muller et al., 2018). Instead of being trapped in one conformation as observed in numerous co-crystallographic studies, H12 possesses structural dynamics in aqueous environment and its conformational landscape can vary in a ligand and coregulator dependent manner. For example, rosiglitazone binding to receptor blocks H12 from conjugation to H2-H3 loop region, yet these long-range crosslinks are restored when the receptor is bound to SR11023. GW9662, a potent covalent antagonist, drives H12 populations to K265 region nearly two times more than that observed with the reversible antagonist SR11023, further indicating the greater potency of GW9662 to drive the largest degree of H12 mobility. In addition, sequence specific NR-box peptides play a potential role in regulation of H12 conformational mobility. Co-activator peptide binding reinforces AF2 helix docking against H11 whereas co-repressor peptide binding displaces AF2 helix from folding back to the agonist position. HDX-MS shows that NCOR1–3 exhibits enhanced ability to allosterically destabilize C-terminal H11 and H12 region in apo receptor compared to that observed with SMRT-2 peptide. Correspondingly, the presence of NCOR1–3 drives higher conformational population of H12 proximal to H2-H3 loop region, suggesting the differential regulatory roles of NCOR1–3 and SMRT-2. Combined, these data suggest that binding of SR11023 enhances the receptor interaction with co-repressor motifs by driving resulting H12 away from the agonist position towards H2-H3 loop region to avoid clashing. This observation suggests a molecular mechanism for SR11023-mediated antagonism of PPARγ.
STAR METHODS
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| LanthaScreen Elite Tb-anti-His Antibody | Thermo Fisher Scientific | Cat:PV5863 |
| LanthaScreen Tb-anti-GST-antibody | Thermo Fisher Scientific | Cat:PV3550 |
| Bacterial and Virus Strains | ||
| BL21(DE3) E. coli cells | New England Biolabs | Cat:C2527 |
| Biological Samples | ||
| HEK293T | ATCC | Cat:CRL-3216 |
| 3T3-L1 fibroblasts | ATCC | Cat:CL-173 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 9-cis Retinoic Acid | Cayman Chemical | Cat:14587 |
| Ammonium Chloride, 99%15N | Cambridge Isotope Labs | Cat:NLM-467 |
| Deuterium Oxide, 99.9% D | Sigma-Aldrich | Cat:151882 |
| Dimethyl Sulfoxide-d6, 99.9% D | Cambridge Isotope Labs | Cat:DLM-10 |
| Fluorescein Isothiocyanate Isomer I | Sigma-Aldrich | Cat:4274 |
| HiLoad 16/600 Superdex 75 pg column | GE Healthcare Life Sciences | Cat:28989333 |
| HiLoad 16/600 Superdex 200 pg column | GE Healthcare Life Sciences | Cat:28989335 |
| HisTrap HP Ni NTA column | GE Healthcare Life Sciences | Cat:17–5248-02 |
| Hypersil GOLD C8 LC column | Thermo Fisher Scientific | Cat:25205 |
| Hypersil GOLD C18 LC column | Thermo Fisher Scientific | Cat:25012 |
| Oil Red dye | Sigma-Aldrich | Cat:O0625 |
| Rosiglitazone | Sigma-Aldrich | Cat:R2408 |
| SR11023 | Made by T.M.K. | Patent: WO2012170561 – US2012/0303769 |
| GST-PPARγ-LBD | Thermo Fisher Scientific | Cat:PV4545 |
| GST-PPARα-LBD | Thermo Fisher Scientific | Cat:PV4691 |
| Cdk5/p35 kinase | Milipore | Cat: 14–477 |
| Fluormone Pan-PPAR Green | Thermo Fisher Scientific | Cat:PV4896 |
| 3-iso-butyl-1-methylxanthine | Sigma-Aldrich | Cat:I7018 |
| Dexamethasone | Sigma-Aldrich | Cat:D4902 |
| Insulin | Sigma-Aldrich | Cat:I9278 |
| X-treme Gene 9 transfection reagent | Sigma-Aldrich | Cat: 6365779001 |
| SYBR green fluorescent dye | Bio-Rad | Cat: 1725271 |
| TR-FRET Nuclear receptor buffer F | Thermo Fisher Scientific | Cat: PV4547 |
| Pepsin column | (Busby et al., 2007) | http://www.sciencedirect.com/science/article/pii/S1387380606004040 |
| C8 trap column, 1 mm x 10 mm | Thermo Scientific | Cat:25205–011001 |
| Betasil C8 column, 5 μ 10×1 mm | Thermo Scientific | Cat:25203–051030 |
| DTT | Sigma | Cat:10708984001 |
| Iodoacetamide | Sigma | Cat:I1149 |
| Trypsin | Promega | Cat:V5111 |
| FITC-NH-ASKHKQLSELLRSGSS | LifeTein, LLC | N/A |
| FITC-NH-TNMGLEAIIRKALMGKYDQWEE | LifeTein, LLC | N/A |
| FITC-NH-LTERHKILHRLLQEGSPSD | LifeTein, LLC | N/A |
| FITC-NH-ASNLGLEDIIRKALMGSFD | LifeTein, LLC | N/A |
| NH-ASKHKQLSELLRSGSS | LifeTein, LLC | N/A |
| NH-TNMGLEAIIRKALMGKYDQWEE | LifeTein, LLC | N/A |
| NH-LTERHKILHRLLQEGSPSD | LifeTein, LLC | N/A |
| NH-ASNLGLEDIIRKALMGSFD | LifeTein, LLC | N/A |
| Spectra Multicolor Broad Range Protein Ladder | Thermo Fisher Scientific | Cat:26634 |
| BS3 crosslinker | Thermo Fisher Scientific | Cat:21580 |
| Chymotrypsin | Promega | Cat:V1061 |
| Human PPARγ LBD, residues 231–505; isoform 2 | (Hughes et al., 2012) | N/A |
| Human RXRα LBD, residues 223–462 | (Kojetin et al., 2015) | N/A |
| PPARγ LBD crystal structure | (Nolte et al., 1998) | PDB code: 2PRG |
| Critical Commercial Assays | ||
| Qiazol reagent | Qiagen | Cat:79306 |
| RNeasy Mini Kit | Qiagen | Cat:74104 |
| LanthaScreen TR-FRET PPARγ competitive binding assay kit | Thermo Fisher Scientific | Cat:PV4893 |
| High Capacity Reverse Transcription Kit | Applied Biosystems | Cat:4368814 |
| HTScan CDK/Cyc kinase assay kit | Cell Signaling Technology | Cat:7519 |
| RNeasy Mini Kit | Qiagen | Cat:74106 |
| TruSeq stranded mRNA sample prep protocol | Illumina | Cat:RS-122–2101 |
| Experimental Models: Cell Lines | ||
| HEK293T | ATCC | Cat:CRL-3216 |
| 3T3-L1 fibroblasts | ATCC | Cat:CL-173 |
| Software and Algorithms | ||
| HDX Workbench | (Pascal et al., 2012) | http://hdxworkbench.com |
| Mascot | Matrix Science | http://www.matrixscience.com/ |
| Plink2 | Pfind Studio (Yang et al., 2012) | http://pfind.ict.ac.cn/software/pLink1/index.html |
| Deposited mass spectrometry raw data | ||
| XL-MS data | PRIDE (https://www.ebi.ac.uk/pride/archive/) | Accession number: PXD010222 |
| HDX-MS data | Figshare (https://figshare.com) | https://figshare.com/s/d9fe41df3e945828e548 |
| Other | ||
| Q Exactive Mass spectrometer | Thermo Fisher Scientific | Cat:IQLAAEGAAPFALGMAZR |
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact PRG (pgriffin@scripps.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
The species/strain of experimental models of HEK293T cells are female Homo sapiens embryonic kidney 293 cells contains the SV40 T-antigen. HEK293T cells were cultured with Dublecco’s Modified Eagle’s Medium (DMEM) contained 10% Fetal Bovine Serum (heat inactivated), 2mM L-glutamine, 1% Penicillin/Streptomycin in 37oC, 5% CO2 incubator. The authentication of cell lines is ATCC (Cat: CRL-3216). The species/strain of experimental models of 3T3-L1 fibroblasts cells are male Mus musculus embryo fibroblasts. 3T3-L1 cells were cultured with Dublecco’s Modified Eagle’s Medium (DMEM) contained 10% Fetal Bovine Serum (heat inactivated), 2mM L-glutamine, 1% Penicillin/Streptomycin in 37oC, 5% CO2 incubator. The authentication of cell lines is ATCC (Cat: CL-173).
METHOD DETAILS
Synthesis of SR11023 method:
(S)-2-(4-((5-((1-(3-cyclopropylphenyl)ethyl)carbamoyl)-2,3-dimethyl-1H-indol-1-yl)methyl)phenyl)-2-methylpropanoic acid . Commercially available 2,3-dimethyl-1H-indole-5-carboxylic acid was N-alkylated with commercially available methyl 2-(4-(bromomethyl)phenyl)-2-methylpropanoate using NaH in DMF. The resulting acid was subsequently coupled with (S)-1-(3-cyclopropylphenyl) ethanamine using 2-(3H-[1,2,3]triazolo[4,5-b]pyridin-3-yl)-1,1,3,3-tetramethylisouronium hexafluorophosphate(V) (HATU) and diisopropylethylamine in CH2Cl2 to provide the amide. The methyl ester was finally hydrolysed using aqueous NaOH in ethanol to give the crude acid which was purified by flash chromatography (ethyl acetate/hexanes 10–100%) to give the title compound as a colorless solid. Electrospray ionization coupled with mass spectrometry (ESI-MS; m/z): 509.27 [M+H]+; 1H NMR (400 MHz, dimethylsulphoxide (DMSO)-d6): δ (p.p.m.) δ 12.34 (broad S, 1H), 8.63 (d, 1H, J=8.0 Hz), 8.12 (d, 1H, J=1.2 Hz), 7.65 (dd, 1H, J=8.4 Hz, 1.6 Hz), 7.44 (d, 1H, J=8.8 Hz), 7.28 (d, 1H, J=8.8 Hz), 7.24–7.18 (m, 4H), 6.97–6.92 (m, 3H), 5.43 (s, 2H), 5.20 (q, 1H, J=7.2 Hz), 2.54 (q, 2H, J=1.6 Hz), 2.33 (s, 2H), 2.30 (s, 2H), 1.95–1.92 (m, 1H), 1.51 (d, 3H, J=7.6 Hz), 1.44 (s, 6H), 0.97–0.95 (m, 2H), 0.69–0.67 (m, 2H); 13C NMR (100 MHz, d-DMSO) 177.82, 166.51, 145.26, 144.01, 143.40, 137.49, 136.44, 133.92, 128.06, 127.51, 125.96, 125.86 (2C), 125.17 (2C), 123.45, 123.14, 123.07, 120.24, 117.59, 108.64, 107.12 , 48.24, 45.51, 45.49, 26.30, 22.42, 15.09 (2C), 10.00, 9.27 (2C), 8.72.
Differentiation of 3T3-L1 cells
was induced with a cocktail consisting of 0.5μM 3-iso-butyl-1-methylxanthine (Cat: I7018), 1μM dexamethasone (Cat: D4902) and 5μg/ml insulin (Cat: I9278). Cells were treated with 1μM Rosiglitazone or 1μM SR11023 from the initial day of differentiation. After incubation for six days, cells were harvested for RNA extraction or stained with Oil Red dye (Cat: O0625) to visualize lipid droplets
Cell-based transactivation assay.
HEK293T cells were co-transfected in batch by adding 4.5 μg Gal4-PPARγ-LBD, 4.5ug GAL4-PPARα-LBD, or 4.5ug GAL4-mPPARα-LBD, and 4.5ugUAS-luciferase reporter. Transfections were conducted using the X-treme Gene 9 transfection reagent (Cat: 6365779001), using manufacturer’s protocol. After 18-h incubation at 37 °C, transfected cells were plated in triplicates in white 384-well plates (Perkin Elmer) at a density of 10,000 cells per well. After re-plating, cells were treated with either DMSO only or the indicated compounds in increasing doses from 2 pM–10 μM. After 18-h incubation, treated cells were developed with Brite Lite Plus (Perkin Elmer) and read in 384-well Luminescence Perkin Elmer EnVision Multilabel plate reader. Graphs were plotted as fold change of treated cells over DMSO-treated control cells.
PPARγ Binding Assays.
LanthaScreen TR-FRET PPARγ competitive binding assay (Cat:PV4893) was performed according to the manufacturer’s protocol. A mixture of 5 nM PPARG ligand binding domain (GST-PPARγ-LBD, Cat:PV4545) or PPARα ligand binding domain (GST-PPARα-LBD, Cat:PV4691), 5 μM Tb-GST-antibody (Cat:PV3550), 5 nM Fluormone Pan-PPAR Green (Cat:PV4896), and serial dilutions of compound beginning at 10 μM downwards was added to wells of black 384-well low-volume plates (Greiner) to a total volume of 18 μl. All dilutions were made in TR-FRET PPAR assay buffer. DMSO at 2% final concentration was used as a no-ligand control. Experiments were performed in triplicate and incubated for 2h in the dark before analysis in Perkin Elmer ViewLux ultra HTS microplate reader. The FRET signal was measured by excitation at 340 nm and emission at 520 nm for fluorescein and 490 nm for terbium. The fold change over DMSO was calculated by 520 nm/490 nm ratio. Graphs were plotted in GraphPad Prism as fold change of FRET signal for each compound over DMSO-only control. IC50s were determined from GraphPad Prism software.
Quantitative PCR.
Total RNA was extracted from tissues using Qiazol reagent (Cat:79306) and RNeasy Mini Kit (Cat:74104). Subsequently, cDNA was generated using High Capacity Reverse Transcription Kit (Applied Biosystems, Cat: 4368814). Quantitative PCR reactions were performed with SYBR green fluorescent dye (Cat: 1725271) using a 7900HT Fast Real-Time qPCR. Relative mRNA expression was determined by the ΔΔ-Ct method normalized to Tbp levels. The sequences of primers used in this study are found in Table S1.
In vitro kinase assay.
Active Cdk5/p35 kinase (Cat:14–477) were purchased from Millipore. In vitro CDK kinase assay was performed using the HTScan CDK/Cyc kinase assay kit (Cat:7519) according to the manufacturer’s instructions (Cell Signaling Technology). Briefly, 1 μg of immuno-purified WT PPARγ were incubated with active CDK kinase in kinase assay buffer (25 mM Tris-HCl pH 7.5, 5 mM β-glycerophosphate, 2 mM dithiothreitol (DTT), 0.1 mM Na3VO4, 10 mM MgCl2) containing 20 μM ATP for 15 min at 30 °C. SR11023 was pre-incubated with substrates for 30 min before performing assay. Phosphorylation of substrates after SDS–PAGE was analysed with anti-CDK substrate antibody to detect phospho-Ser in a K/R-S-P-K/R motif, which is the consensus motif for Cdk substrate proteins (Cell Signaling Technology).
Molecular docking.
The ICM Molsoft suite(Abagyan and Totrov, 1994) was used to dock compound SR11023 into the PPARγ LBD structure. PDB 3FUR with ligands and water removed was used as the starting model for docking. The PPARγ LBD structure was prepared for docking by protonation, deletion of water molecules, and energy minimization by means of the ICM force field and distance dependent dielectric potential with an RMS gradient of 0.1. PocketFinder within ICM was used to define the ligand binding pocket and was consistent with previously published X-ray structures. Default settings within the ICM docking module were used with a rectangular box centered at the LBD with a grid spacing of 0.5 Å. The top ranked docking for SR11023 was chosen for interpretation, as the conformations were very consistent among all the top scored dockings.
BS3 mediated crosslinking mass spectrometry.
PPARγ LBD (10 μM) (Hughes et al., 2012a) or PPARγ/RXRα heterodimer (Kojetin et al., 2015) complex with or without PPARγ ligand (10-fold excess)/SRC1–2/NCOR1–3/SMRT-2 (all peptides were 5-fold excess to protein) were incubated with BS3 (50-fold molar excess) (Cat:21580) for 1hr. Reaction was quenched with 50 mM Tris buffer and protein samples were overnight digested with trypsin (trypsin: protein ratio = 1:20, w/w) (Cat:V5111) and sequentially overnight digested with chymotrypsin (chymotrypsin: protein ratio = 1:20, w/w) (Cat:V1061) prior to analysis by LC-MS/MS. We specifically used chymotrypsin to introduce a specific cleavage between Tyrosine473 and Lysine474 for identification of a shorter peptide K474DLY, which resides on the C-terminus of AF2 α-helix. The lysine residue has a flexible 6 Å side chain while BS3 has an 8-atom spacer arm (11.4 Å). As a result, K474 could be cross-linked by BS3 to proximal lysine residues within the PPARγ LBD (within the distance of 24 Å). Plink2 software was used to analyze and identify the XL-peptides (Yang et al., 2012). The parameters for pLink2 search were as follows: three missed cleavage sites for trypsin/chymotrypsin per chain; peptide length 4 – 100 amino acid; pLink2 search results were filtered by requiring precursor tolerance (±10 p.p.m.) and fragment tolerance (±15 p.p.m.) and FDR below 5% was required for all identified XL-MS peaks. The ion intensities and peak areas of the crosslinked peptides from different experimental conditions were manually calculated and compared. The highest peak intensity across all indicated experimental conditions within each bar plot panel was normalized as 100% and other peak abundances were scaled accordingly. Experiments were triplicated. Statistical summary was from a two-way ANOVA between indicated pairwise experiment (*** = p<0.001; ** = p<0.01; * = p<0.05).
Hydrogen-deuterium exchange (HDX) detected by mass spectrometry.
Solution-phase amide HDX experiments were carried out with a fully automated system (CTC HTS PAL, LEAP Technologies, Carrboro, NC; housed inside a 4°C cabinet) as described previously with slight modifications (Chalmers et al., 2006).
Peptide Identification:
Peptides were identified using tandem MS (MS/MS) experiments performed with either a LTQ Orbitrap XL with ETD or a Q Exactive (Thermo Fisher Scientific, San Jose, CA) over a 70-min gradient. Product ion spectra were acquired in a data-dependent mode and the five most abundant ions were selected for the product ion analysis per scan event. The MS/MS *.raw data files were converted to *.mgf files and then submitted to MASCOT (version 2.3 Matrix Science, London, UK) for peptide identification. The maximum number of missed cleavages was set at 4 with the mass tolerance for precursor ions +/− 0.6 Da and for fragment ions +/− 8ppm. Oxidation to methionine was selected for variable modification. Pepsin was used for digestion and no specific enzyme was selected in the MASCOT during the search. Peptides included in the peptide set used for HDX detection had a MASCOT score of 20 or greater. The MS/MS MASCOT search was also performed against a decoy (reverse) sequence and false positives were ruled out if they did not pass a 1% false discovery rate. The MS/MS spectra of all the peptide ions from the MASCOT search were further manually inspected and only the unique charged ions with the highest MASCOT score were included in HDX peptide set.
HDX-MS analysis:
10 μM of the apo protein was mixed with 1:10 molar excess of ligand and incubated for 2 hours at 4°C for complex formation before subjecting them to HDX analysis. For the differential HDX experiments, 5 μl of either the apo or the liganded protein complex were mixed with 20 μl of D2O-containing HDX buffer (50 mM Tris, pH 8.0, 150 mM NaCl, and 2 mM DTT) and incubated at 4°C for 0s, 10s, 30s, 60s, 300s, 900s or 3,600s. Following on-exchange, unwanted forward- or back-exchange was minimized and the protein was denatured by the addition of 25 μl of a quench solution (1% v/v TFA in 3 M urea and 50 mM TCEP). Samples were then immediately passed through an immobilized pepsin column (prepared in house) (Busby et al., 2007) at 50 μl min-1 (0.1% v/v TFA, 15°C) and the resulting peptides were trapped and desalted on a 1.0 mm × 10 mm C8 trap column (Hypersil Gold, Thermo Fisher, Grand Island, NY). The bound peptides were then gradient-eluted (5–50% CH3CN v/v and 0.3% v/v formic acid) across a 1.0 mm × 50 mm C18 separation column (Hypersil Gold, Thermo Fisher, Grand Island, NY) for 6 min. Sample handling and peptide separation were conducted at 4°C. The eluted peptides were then subjected to electrospray ionization directly coupled to a high resolution Orbitrap mass spectrometer (LTQ Orbitrap XL with ETD, Q Exactive, or Exactive, Thermo Fisher Scientific, San Jose, CA). Each HDX experiment was carried out in triplicate with a single preparation of each protein-ligand complex. The intensity weighted mean m/z centroid value of each peptide envelope was calculated and subsequently converted into a percentage of deuterium incorporation. This is accomplished by determining the observed averages of the undeuterated and fully deuterated spectra using the conventional formula described elsewhere(Zhang and Smith, 1993). In the absence of a fully deuterated control, 100% deuterium incorporation was calculated theoretically, and corrections for back-exchange were made on the basis of an estimated 70% deuterium recovery and accounting for 79.9% final deuterium concentration in the sample (1:5 dilution in D2O HDX buffer). Statistical significance for the differential HDX data is determined by an unpaired t-test for each time point, a procedure that is integrated into the HDX Workbench software(Pascal et al., 2012).
Data Rendering:
The HDX data from all overlapping peptides were consolidated to individual amino acid values using a residue averaging approach. Briefly, for each residue, the deuterium incorporation values and peptide lengths from all overlapping peptides were assembled. A weighting function was applied in which shorter peptides were weighted more heavily and longer peptides were weighted less. Each of the weighted deuterium incorporation values were then averaged incorporating this weighting function to produce a single value for each amino acid. The initial two residues of each peptide, as well as prolines, were omitted from the calculations. This approach is similar to that previously described (Keppel and Weis, 2015).
NR box peptide recruitment assay.
A TR-FRET-based interaction assay was used. Tb-anti-His antibody (7.5 nM; Cat:PV5863) and a gradient of ligand concentrations were incubated in complete TR-FRET Nuclear receptor buffer F (Cat: PV4547) containing 7.5 nM purified His-PPARγ2 for 1 hr at room temperature. 450nM FITC-labeled peptides p300 (sequence: ASKHKQLSELLRSGSS), SMRT-2 (TNMGLEAIIRKALMGKYDQWEE), SRC1–2 (LTERHKILHRLLQEGSPSD), and NCOR1–3 (ASNLGLEDIIRKALMGSFD) were added and incubated for additional two hrs at room temperature (in dark). The FRET signal was measured by excitation at 340 nm and emission at 520 nm for fluorescein and 490 nm for terbium in Perkin Elmer ViewLux ultra HTS microplate reader. The fold change over DMSO was calculated by 520 nm/490 nm ratio. Graphs were plotted in GraphPad Prism as fold change of FRET signal for each compound over DMSO-only control.
mRNA-seq.
L1 cells RNA extraction protocol for RNA seq was derived from RNeasy Mini Kit (Cat:74106). Total RNA was quantified using the Qubit 2.0 Fluorometer (Invitrogen, Carlsbad, CA) and run on the Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA) for quality assessment. If the RNA profile is good quality with RNA Integrity Number (RIN) > 8.0, the samples are continued for processing. A RNase-free working environment is maintained and RNase-free tips, Eppendorf tubes and plates are utilized for the subsequent steps. Messenger RNA is selectively isolated from total RNA (typically 100–300ng input) using poly-T oligos attached to magnetic beads according to the TruSeq stranded mRNA sample prep protocol (Cat:RS-122–2101, Illumina, San Diego, CA). The enriched mRNA samples are chemically fragmented in a buffer containing divalent cations and heating at 94°C for 8 minutes. The fragmented RNA is random hexamer primed and reverse transcribed to generate the first strand cDNA. The second strand is synthesized after removing the RNA template and incorporating dUTP in place of dTTP. The incorporation of dUTP quenches the second strand during the PCR amplification step later and therefore the strand information is preserved. The ds cDNA is then end repaired and adenylated at their 3’ ends. A corresponding ‘T’ nucleotide on the adaptors is utilized for ligating the adaptor sequences to the ds cDNA. The adaptor ligated DNA is purified using magnetic Ampure XP beads and PCR amplified using 13 cycles to generate the final libraries. The final libraries are size selected and purified using 1.0 x Ampure XP beads to remove any primer dimers. The final library size is typically 200–600bp with insert sizes ranging from 80–450bp. The final libraries are validated by the bioanalyzer DNA chips, normalized to 1nM, pooled equally and loaded onto the NextSeq 500 flow cell at 1.8pM final concentration and sequenced using 2 × 75bp paired-end chemistry. On average, we generate 20–22 million reads pass filter (base quality score >Q30 suggesting less than 1 error in 1000bp).
NMR spectroscopy.
Two-dimensional (2D) [1H,15N]-transverse relaxation optimized spectroscopy (TROSY)-heteronuclear single quantum correlation (HSQC) data were collected at 298K using a Bruker 700 Mhz NMR instrument equipped with a QCI cryoprobe. Samples contained approximately 200 μM protein in a NMR buffer containing 50 mM potassium phosphate (pH 7.4), 20 mM potassium chloride, 1 mM TCEP, and 10% D2O; with or without 2 equivalents NCOR1–3 or SMRT2 peptide, or with or without 2 equivalents of rosiglitazone, MRL24, or SR11023. Data were processed and analyzed using Topspin 3.0 (Bruker) and NMRViewJ (OneMoon Scientific, Inc.) (Johnson, 2004). NMR analysis was limited to well resolved peaks for residues with chemical shift values similar to PPARγ bound to rosiglitazone (BMRB entry 17975) (Hughes et al., 2012b) using the minimum chemical shift procedure (Williamson, 2013).
Fluorescence polarization coregulatory binding assay.
His-PPARγ LBD was prepared by serial dilution in assay buffer (50 mM potassium phosphate, 20 mM potassium chloride, 1 mM TCEP, 0.01% Tween 20, pH 7.4) and plated in black 384-well plates (Greiner) with peptides (100 nM final concentration) derived from the SMRT2 corepressor (TRAP220–2; residues 638–656; TNMGLEAIIRKALMGKYDQWEE) or the NCoR corepressor (NCoR-D3; residues 2256–2278; TITAANFIDVIITRQIASDK) containing a N-terminal FITC label with a six-carbon linker (Ahx). The plate was incubated at 4 °C for 2 hr and FP measured on a Spectramax M5e multimode plate reader at 485 nm excitation and 538 nm emission wavelengths. Data were analyzed using GraphPad Prism (FP signal vs. ligand concentration) and fit to one-site binding equation
QUANTIFICATION AND STATISTICAL ANALYSIS
HDX-MS analysis
Data Statistics:
Deuterium uptake for each peptide is calculated as the average of % D for all on-exchange time points and the difference in average %D values between the apo and ligand bound samples is presented as a heat map with a color code given at the bottom of the Figure (warm colors for deprotection and cool colors for protection). Peptides are colored by the software automatically to display significant differences, determined either by a >5% difference (less or more protection) in average deuterium uptake between the two states, or by using the results of unpaired t-tests at each time point (p-value < 0.05 for any two time points or a p-value < 0.01 for any single time point). Peptides with non-significant changes between the two states are colored grey. The exchange at the first two residues for any given peptide is not colored. Each peptide bar in the heat map view displays the average Δ %D values, associated standard deviation, and the charge state. Additionally, overlapping peptides with a similar protection trend covering the same region are used to rule out data ambiguity.
Fluorescence polarization coregulatory binding assay/ NR box peptide recruitment assay/PPARγ Binding Assays.
Results are expressed as mean +/− SEM. The significance of differences between groups was evaluated by one-way analysis of variance (ANOVA) followed by a Dunnett’s test for multiple comparisons, or Student’s t-test with or without Bonferroni’s correction. Analyses were done with GraphPad Prism software (GraphPad).
Data and Software Availability
The mass spec data have been have been deposited to the ProteomeXchange Consortium via the PRIDE (Vizcaino et al., 2016) partner repository with the accession number: PXD010222. HDX-MS data have been deposited to Figshare (https://figshare.com/s/d9fe41df3e945828e548). HDX data was processed by HDX Workbench (Pascal et al., 2012). XL-MS data was processed by plink2 (Yang et al., 2012).
Supplementary Material
AKNOWLEDGEMENTS
This work was supported from the National Institutes of Health; NIH grants DK DK105825 (PI: P.R.G.), DK101871 (PI: D.J.K.), The Landenberger Foundation (J.Z.) and the Klorfine Family Fellowship (C.A.C.).
Footnotes
DECLARATION OF INTERESTS:T.M.K. and P.R.G. have WO2012170561 – US2012/0309769 patent for SR11023 reported in this manuscript.
REFERENCES
- Abagyan R, and Totrov M (1994). Biased probability Monte Carlo conformational searches and electrostatic calculations for peptides and proteins. J Mol Biol 235, 983–1002. [DOI] [PubMed] [Google Scholar]
- Acton JJ 3rd, Akiyama TE, Chang CH, Colwell L, Debenham S, Doebber T, Einstein M, Liu K, McCann ME, Moller DE, et al. (2009). Discovery of (2R)-2-(3-{3-[(4-Methoxyphenyl)carbonyl]-2-methyl-6-(trifluoromethoxy)-1H-indol-1 -yl}phenoxy)butanoic acid (MK-0533): a novel selective peroxisome proliferator-activated receptor gamma modulator for the treatment of type 2 diabetes mellitus with a reduced potential to increase plasma and extracellular fluid volume. Journal of medicinal chemistry 52, 3846–3854. [DOI] [PubMed] [Google Scholar]
- Acton JJ 3rd, Black RM, Jones AB, Moller DE, Colwell L, Doebber TW, Macnaul KL, Berger J, and Wood HB (2005). Benzoyl 2-methyl indoles as selective PPARgamma modulators. Bioorg Med Chem Lett 15, 357–362. [DOI] [PubMed] [Google Scholar]
- Asteian A, Blayo AL, He Y, Koenig M, Shin Y, Kuruvilla DS, Corzo CA, Cameron MD, Lin L, Ruiz C, et al. (2015). Design, Synthesis, and Biological Evaluation of Indole Biphenylcarboxylic Acids as PPARgamma Antagonists. ACS Med Chem Lett 6, 998–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banks AS, McAllister FE, Camporez JP, Zushin PJ, Jurczak MJ, Laznik-Bogoslavski D, Shulman GI, Gygi SP, and Spiegelman BM (2015). An ERK/Cdk5 axis controls the diabetogenic actions of PPARgamma. Nature 517, 391–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruning JB, Chalmers MJ, Prasad S, Busby SA, Kamenecka TM, He Y, Nettles KW, and Griffin PR (2007). Partial agonists activate PPARgamma using a helix 12 independent mechanism. Structure 15, 1258–1271. [DOI] [PubMed] [Google Scholar]
- Busby SA, Chalmers MJ, and Griffin PR (2007). Improving digestion efficiency under H/D exchange conditions with activated pepsinogen coupled columns. Int J Mass Spectrom 259, 130–139. [Google Scholar]
- Chalmers MJ, Busby SA, Pascal BD, He Y, Hendrickson CL, Marshall AG, and Griffin PR (2006). Probing protein ligand interactions by automated hydrogen/deuterium exchange mass spectrometry. Analytical chemistry 78, 1005–1014. [DOI] [PubMed] [Google Scholar]
- Chen ZA, Jawhari A, Fischer L, Buchen C, Tahir S, Kamenski T, Rasmussen M, Lariviere L, Bukowski-Wills JC, Nilges M, et al. (2010). Architecture of the RNA polymerase II-TFIIF complex revealed by cross-linking and mass spectrometry. The EMBO journal 29, 717–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JH, Banks AS, Estall JL, Kajimura S, Bostrom P, Laznik D, Ruas JL, Chalmers MJ, Kamenecka TM, Bluher M, et al. (2010). Anti-diabetic drugs inhibit obesity-linked phosphorylation of PPARgamma by Cdk5. Nature 466, 451–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JH, Banks AS, Kamenecka TM, Busby SA, Chalmers MJ, Kumar N, Kuruvilla DS, Shin Y, He Y, Bruning JB, et al. (2011). Antidiabetic actions of a non-agonist PPARgamma ligand blocking Cdk5-mediated phosphorylation. Nature 477, 477–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer L, Chen ZA, and Rappsilber J (2013). Quantitative cross-linking/mass spectrometry using isotope-labelled cross-linkers. J Proteomics 88, 120–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heldring N, Pike A, Andersson S, Matthews J, Cheng G, Hartman J, Tujague M, Strom A, Treuter E, Warner M, et al. (2007). Estrogen receptors: How do they signal and what are their targets. Physiological reviews 87, 905–931. [DOI] [PubMed] [Google Scholar]
- Horlein AJ, Naar AM, Heinzel T, Torchia J, Gloss B, Kurokawa R, Ryan A, Kamei Y, Soderstrom M, Glass CK, et al. (1995). Ligand-independent repression by the thyroid hormone receptor mediated by a nuclear receptor co-repressor. Nature 377, 397–404. [DOI] [PubMed] [Google Scholar]
- Hu X, and Lazar MA (1999). The CoRNR motif controls the recruitment of corepressors by nuclear hormone receptors. Nature 402, 93–96. [DOI] [PubMed] [Google Scholar]
- Hughes TS, Chalmers MJ, Novick S, Kuruvilla DS, Chang MR, Kamenecka TM, Rance M, Johnson BA, Burris TP, Griffin PR, et al. (2012a). Ligand and receptor dynamics contribute to the mechanism of graded PPARgamma agonism. Structure 20, 139–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes TS, Chalmers MJ, Novick S, Kuruvilla DS, Chang MR, Kamenecka TM, Rance M, Johnson BA, Burris TP, Griffin PR, et al. (2012b). Ligand and receptor dynamics contribute to the mechanism of graded PPARγ agonism. Structure 20, 139–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson BA (2004). Using NMRView to visualize and analyze the NMR spectra of macromolecules. Methods Mol Biol 278, 313–352. [DOI] [PubMed] [Google Scholar]
- Keppel TR, and Weis DD (2015). Mapping residual structure in intrinsically disordered proteins at residue resolution using millisecond hydrogen/deuterium exchange and residue averaging. Journal of the American Society for Mass Spectrometry 26, 547–554. [DOI] [PubMed] [Google Scholar]
- Kintscher U, and Law RE (2005). PPARgamma-mediated insulin sensitization: the importance of fat versus muscle. Am J Physiol Endocrinol Metab 288, E287–291. [DOI] [PubMed] [Google Scholar]
- Knutson BA, Luo J, Ranish J, and Hahn S (2014). Architecture of the Saccharomyces cerevisiae RNA polymerase I Core Factor complex. Nature structural & molecular biology 21, 810–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojetin DJ, Matta-Camacho E, Hughes TS, Srinivasan S, Nwachukwu JC, Cavett V, Nowak J, Chalmers MJ, Marciano DP, Kamenecka TM, et al. (2015). Structural mechanism for signal transduction in RXR nuclear receptor heterodimers. Nature communications 6, 8013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kung J, and Henry RR (2012). Thiazolidinedione safety. Expert opinion on drug safety 11, 565–579. [DOI] [PubMed] [Google Scholar]
- le Maire A, Teyssier C, Erb C, Grimaldi M, Alvarez S, de Lera AR, Balaguer P, Gronemeyer H, Royer CA, Germain P, et al. (2010). A unique secondary-structure switch controls constitutive gene repression by retinoic acid receptor. Nature structural & molecular biology 17, 801–807. [DOI] [PubMed] [Google Scholar]
- Liu K, Black RM, Acton JJ 3rd, Mosley R, Debenham S, Abola R, Yang M, Tschirret-Guth R, Colwell L, Liu C, et al. (2005). Selective PPARgamma modulators with improved pharmacological profiles. Bioorganic & medicinal chemistry letters 15, 2437–2440. [DOI] [PubMed] [Google Scholar]
- Marciano DP, Kuruvilla DS, Boregowda SV, Asteian A, Hughes TS, Garcia-Ordonez R, Corzo CA, Khan TM, Novick SJ, Park H, et al. (2015). Pharmacological repression of PPAR gamma promotes osteogenesis. Nature communications 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayans L (2015). Metabolic Syndrome: Insulin Resistance and Prediabetes. FP Essent 435, 11–16. [PubMed] [Google Scholar]
- Merkley ED, Rysavy S, Kahraman A, Hafen RP, Daggett V, and Adkins JN (2014). Distance restraints from crosslinking mass spectrometry: mining a molecular dynamics simulation database to evaluate lysine-lysine distances. Protein Sci 23, 747–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minoura H, Takeshita S, Ita M, Hirosumi J, Mabuchi M, Kawamura I, Nakajima S, Nakayama O, Kayakiri H, Oku T, et al. (2004). Pharmacological characteristics of a novel nonthiazolidinedione insulin sensitizer, FK614. European journal of pharmacology 494, 273–281. [DOI] [PubMed] [Google Scholar]
- Muller F, Fischer L, Chen ZA, Auchynnikava T, and Rappsilber J (2018). On the Reproducibility of Label-Free Quantitative Cross-Linking/Mass Spectrometry. J Am Soc Mass Spectrom 29, 405–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy L, Kao HY, Love JD, Li C, Banayo E, Gooch JT, Krishna V, Chatterjee K, Evans RM, and Schwabe JW (1999). Mechanism of corepressor binding and release from nuclear hormone receptors. Genes & development 13, 3209–3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolte RT, Wisely GB, Westin S, Cobb JE, Lambert MH, Kurokawa R, Rosenfeld MG, Willson TM, Glass CK, and Milburn MV (1998). Ligand binding and co-activator assembly of the peroxisome proliferator-activated receptor-gamma. Nature 395, 137–143. [DOI] [PubMed] [Google Scholar]
- Pascal BD, Willis S, Lauer JL, Landgraf RR, West GM, Marciano D, Novick S, Goswami D, Chalmers MJ, and Griffin PR (2012). HDX workbench: software for the analysis of H/D exchange MS data. J Am Soc Mass Spectrom 23, 1512–1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizos CV, Elisaf MS, Mikhailidis DP, and Liberopoulos EN (2009). How safe is the use of thiazolidinediones in clinical practice? Expert Opin Drug Saf 8, 15–32. [DOI] [PubMed] [Google Scholar]
- Rubenstrunk A, Hanf R, Hum DW, Fruchart JC, and Staels B (2007). Safety issues and prospects for future generations of PPAR modulators. Biochimica et biophysica acta 1771, 1065–1081. [DOI] [PubMed] [Google Scholar]
- Schmidt C, and Robinson CV (2014). A comparative cross-linking strategy to probe conformational changes in protein complexes. Nat Protoc 9, 2224–2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi L, Zhao Y, Szymanski K, Yau L, and Fonseca V (2011). Impact of thiazolidinedione safety warnings on medication use patterns and glycemic control among veterans with diabetes mellitus. Journal of diabetes and its complications 25, 143–150. [DOI] [PubMed] [Google Scholar]
- Stechschulte LA, Czernik PJ, Rotter ZC, Tausif FN, Corzo CA, Marciano DP, Asteian A, Zheng J, Bruning JB, Kamenecka TM, et al. (2016). PPARG Post-translational Modifications Regulate Bone Formation and Bone Resorption. EBioMedicine 10, 174–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugii S, Olson P, Sears DD, Saberi M, Atkins AR, Barish GD, Hong SH, Castro GL, Yin YQ, Nelson MC, et al. (2009). PPARgamma activation in adipocytes is sufficient for systemic insulin sensitization. Proc Natl Acad Sci U S A 106, 22504–22509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vizcaino JA, Csordas A, Del-Toro N, Dianes JA, Griss J, Lavidas I, Mayer G, Perez-Riverol Y, Reisinger F, Ternent T, et al. (2016). 2016 update of the PRIDE database and its related tools. Nucleic Acids Res 44, 11033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson MP (2013). Using chemical shift perturbation to characterise ligand binding. Prog Nucl Magn Reson Spectrosc 73, 1–16. [DOI] [PubMed] [Google Scholar]
- Wright MB, Bortolini M, Tadayyon M, and Bopst M (2014). Minireview: Challenges and opportunities in development of PPAR agonists. Molecular endocrinology 28, 1756–1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu B, Peisley A, Richards C, Yao H, Zeng X, Lin C, Chu F, Walz T, and Hur S (2013). Structural basis for dsRNA recognition, filament formation, and antiviral signal activation by MDA5. Cell 152, 276–289. [DOI] [PubMed] [Google Scholar]
- Xu HE, Stanley TB, Montana VG, Lambert MH, Shearer BG, Cobb JE, McKee DD, Galardi CM, Plunket KD, Nolte RT, et al. (2002a). Structural basis for antagonist-mediated recruitment of nuclear co-repressors by PPAR alpha. Nature 415, 813–817. [DOI] [PubMed] [Google Scholar]
- Xu HE, Stanley TB, Montana VG, Lambert MH, Shearer BG, Cobb JE, McKee DD, Galardi CM, Plunket KD, Nolte RT, et al. (2002b). Structural basis for antagonist-mediated recruitment of nuclear co-repressors by PPARalpha. Nature 415, 813–817. [DOI] [PubMed] [Google Scholar]
- Yang B, Wu YJ, Zhu M, Fan SB, Lin J, Zhang K, Li S, Chi H, Li YX, Chen HF, et al. (2012). Identification of cross-linked peptides from complex samples. Nat Methods 9, 904–906. [DOI] [PubMed] [Google Scholar]
- Zhang Z, and Smith DL (1993). Determination of amide hydrogen exchange by mass spectrometry: a new tool for protein structure elucidation. Protein science : a publication of the Protein Society 2, 522–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.