

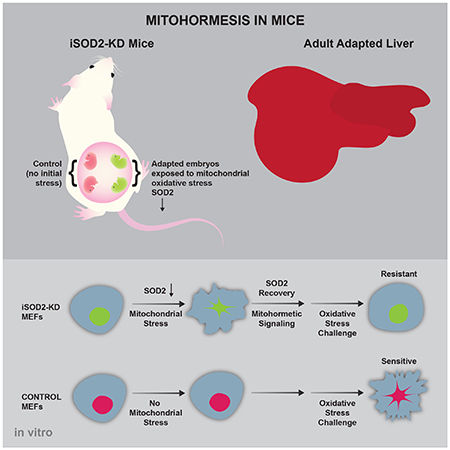
SUMMARY
Transient mitochondrial stress can promote beneficial physiological responses and longevity, termed “mitohormesis.” To interrogate mitohormetic pathways in mammals, we generated mice in which mitochondrial superoxide dismutase 2 (SOD2) can be knocked-down in an inducible and reversible manner (iSOD2-KD). Depleting SOD2 only during embryonic development did not cause post-natal lethality, allowing us to probe adaptive responses to mitochondrial oxidant stress in adult mice. Liver from adapted mice had increased mitochondrial biogenesis and antioxidant gene expression and fewer reactive oxygen species. Gene expression analysis implicated non-canonical activation of the Nrf2 antioxidant and PPARγ-PGC-1α mitochondrial signaling pathways in this response. Transient SOD2 knock-down in embryonic fibroblasts from iSOD2-KD mice also resulted in adaptive mitochondrial changes, enhanced antioxidant capacity, and resistance to a subsequent oxidant challenge. We propose that mitohormesis in response to mitochondrial oxidative stress in mice involves sustained activation of mitochondrial and antioxidant signaling pathways to establish a heightened basal antioxidant state.
Keywords: mitochondria, oxidative stress, signaling, superoxide dismutase, hormesis, reactive oxygen species, mtDNA, PPARγ, Nrf2
eTOC
A mouse model of inducible SOD2 gene knock-down reveals the existence of stable, adaptive responses in adult liver to mitochondrial oxidant stress during embryogenesis and in primary embryonic fibroblasts. These mitohormetic responses involve mitochondrial remodeling and priming of antioxidant pathways via basal activation of PPARγ, PGC-1α and Nrf2 signaling.
Summary
INTRODUCTION
Mitochondria are essential organelles at the crossroads of metabolism, redox homeostasis, and cell death (Nunnari and Suomalainen, 2012). While best known for their roles in ATP production and apoptosis, mitochondria have many additional functions including signaling, cell differentiation, and immunity (Chandel, 2015; Shadel and Horvath, 2015; West et al., 2011). Consequently, mitochondria contribute to disease and aging in a variety of ways. Mitochondria are a major source of reactive oxygen species (ROS), which are generated by the electron transport chain (ETC) and other mitochondrial enzymes (Brand, 2010). Mitochondrial superoxide is a ROS generated on both sides of the inner mitochondrial membrane and can be converted to hydrogen peroxide by superoxide dismutase 1 and 2 (SOD1 and SOD2) in the intermembrane space and matrix, respectively (Brand, 2010). The resultant mitochondrial hydrogen peroxide can be detoxified by mitochondrial enzymes (e.g. glutathione peroxidase). In addition to causing oxidative damage, mitochondrial hydrogen peroxide and superoxide have cellular signaling roles that continue to be elucidated (Shadel and Horvath, 2015).
Previous studies used conventional gene knock-out approaches to probe the role of SOD2 in mammals and to understand how increased mitochondrial oxidative stress impacts disease and aging. In mice, complete Sod2 knock-out results in lethality 4-14 days post birth due to dilated cardiomyopathy, fatty liver, and neurological dysfunction (Lebovitz et al., 1996; Li et al., 1995). These mice also exhibit signs of premature skin aging (Velarde et al., 2012). Heterozygous Sod2 knock-out (Sod2+/) mice have been studied to circumvent lethality and assess the role of moderately increased mitochondrial oxidative stress. These mice exhibit an accelerated decline in mitochondrial function, enhanced sensitivity to apoptosis and increased nuclear DNA damage (Kokoszka et al., 2001). Despite these phenotypes, Sod2+/ animals do not age prematurely (Van Remmen et al., 2003; Zhang et al., 2009), but they are significantly more prone to cancer, seizures, and other pathology (Jang and Van Remmen, 2009; Liang and Patel, 2004; Van Remmen et al., 2003). Others have used conditional Sod2 gene knock-out to analyze the role of mitochondrial oxidative stress in specific tissues (Biswal et al., 2016; Case and Domann, 2012; Sunagawa et al., 2014). One limitation to all of these studies is that SOD2 is reduced or absent from the beginning of embryogenesis or irreversibly lost once recombination has occurred. Thus, acute SOD2 deficiency for controlled timeframes in physiological settings (e.g. development) cannot be assessed. Likewise, probing adaptive signaling responses to transient mitochondrial oxidative stress is not possible, which is critical to study mitochondrial ROS signaling.
Various forms of mitochondrial stress are now appreciated to cause adaptive responses that can benefit organisms through “mitohormesis” (Ristow and Zarse, 2010; Yun and Finkel, 2014). For example, inhibition of respiration induces mitochondrial unfolded protein stress and can extend lifespan in invertebrate model organisms (Dillin et al., 2002; Feng et al., 2001; Merkwirth et al., 2016; Owusu-Ansah et al., 2013; Tian et al., 2016). Interestingly, mitochondrial stress during specific points in development can induce mitohormesis through both cell-autonomous and cell-non-autonomous signaling mechanisms (Durieux et al., 2011; Riera et al., 2016). Increased mitochondrial ROS caused by ETC mutations or knock-down of SOD2 also extends lifespan in C. elegans (Lee et al., 2010; Yang and Hekimi, 2010; Van Raamsdonk and Hekimi, 2009; Yang and Hekimi, 2010). In addition, we showed that extension of yeast chronological lifespan in strains with reduced mTORC1 signaling is associated with an increase in mitochondrial gene expression and respiration (Bonawitz et al., 2007). In this case, longevity is promoted by increased mitochondrial ROS that signal through the yeast homologs of ATM and Chk2 kinases to silence subtelomeric chromatin via increased histone methylation (Pan et al., 2011; Schroeder et al., 2013). Importantly, inducing mitochondrial ROS transiently is sufficient to extend lifespan via this pathway. Herein, we describe adaptive changes to transient mitochondrial oxidative stress in mammalian cells and liver using a mouse model in which the Sod2 gene can be knocked down in an inducible and reversible manner.
RESULTS
Mitochondrial oxidative stress induced by knock-down of the Sod2 gene during embryogenesis is reversible and non-lethal
Using the system developed by Premsrirut et al. (Premsrirut et al., 2011) (see Experimental Procedures), we generated an inducible and reversible mouse model of mitochondrial oxidative stress via shRNA-mediated knock-down of the Sod2 gene (iSOD2-KD mice). To facilitate knock-down in embryos, and mouse embryonic fibroblasts (MEFs) for in vitro studies, we chose a system where the reverse tetracycline-controlled transactivator (rtTA) is expressed from the Rosa26 promoter, which is robustly expressed in embryonic tissues. To account for off-target effects, we generated two iSOD2-KD mouse strains, each with an shRNA that targets a different sequence in the Sod2 gene (see Star Methods).
Due to the reported lethality of SOD2 knock-out mice (Lebovitz et al., 1996; Li et al., 1995), we initially limited SOD2 knock-down to a short (4-day) window during embryogenesis, which is the minimal time required to see complete knock-down of other genes using this system (Premsrirut et al., 2011). To avoid inhibition of embryo implantation and to allow cardiac tissues to establish (day 7-8e), we administered DOX to pregnant females in their food from days 8.5-12.5e (Figure 1A) (Li et al., 1995). Strong knock-down of SOD2 protein was observed in embryos from mothers treated with DOX from days 8.5-12.5e (Figure 1B). Negative controls were single-transgenic littermate embryos (harboring the rtTA transgene, but not the Sod2 shRNA transgene) and hence were treated identically with DOX. Furthermore, no changes in SOD2 levels were seen in embryos that were not treated with DOX (Figure S1A) and DOX had no effect on mitochondrial oxidative phosphorylation (OXPHOS) complex subunits, including mtDNA-encoded proteins (Figure S1B). These data indicate mitochondrial translation was not inhibited by DOX under these conditions. As expected, SOD2 knock-down in embryos resulted in significant oxidative stress compared to negative controls, as demonstrated by lower aconitase activity, a mitochondrial enzyme sensitive to superoxide (Figure 1C). We also observed an increase in MDA, a marker of lipid peroxidation (Figure 1D). Similar levels of SOD2 knock-down, aconitase inactivation, and lipid peroxidation were observed in embryos from strains expressing the second shRNA targeting Sod2.
Figure 1. Generation of transient mitochondrial oxidative stress via inducible knock-down of SOD2 during mouse embryogenesis.
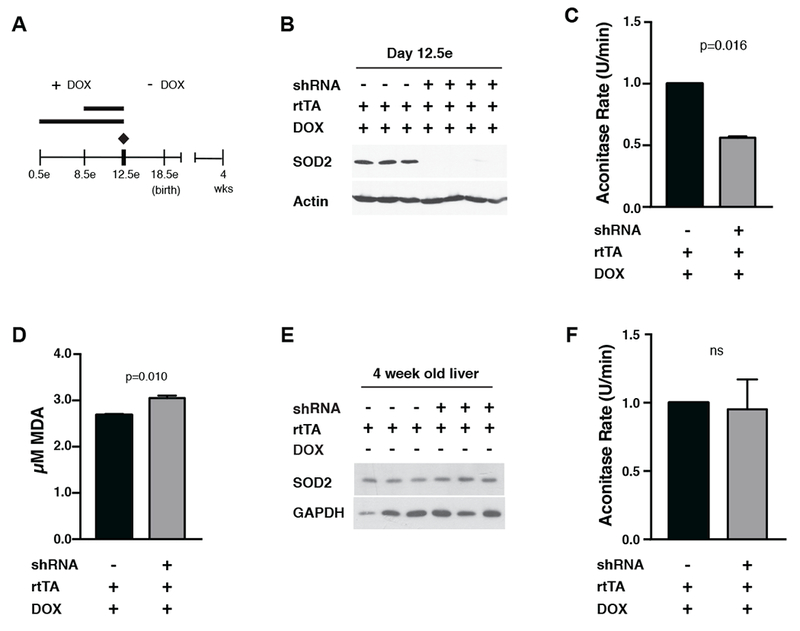
A) Timeline of SOD2 knock-down and recovery in iSOD2-KD mice. As indicated, doxycycline (DOX) was fed to pregnant mothers to achieve knock-down in embryos from day 8.5e to day 12.5e or from day 0.5e to day 12.5e. At day 12.5e, DOX was discontinued, and the pups were aged to 4 weeks. In all cases, negative controls were DOX-treated littermates only expressing the rtTA transgene.
B) Western blot of SOD2 in day 12.5 embryos relative to negative controls.
C) Mitochondrial aconitase activity (U/min) in day 12.5 embryos relative to negative controls. 100% equals aconitase activity of 5.720 nmol/min/ml.
D) TBARS assay for malondialdehyde (MDA) in day 12.5 embryos relative to negative controls.
E) Western blots for SOD2 in adapted liver relative to negative controls. GAPDH was probed as a control for loading.
F) Mitochondrial aconitase activity (U/min) of adapted liver relative to negative controls. Error bars represent +/−SEM. 100% equals aconitase activity of 2.969 nmol/min/ml.
All calculations for statistical significance were completed using a non-parametric, unpaired, two-tailed t-test. n=6.
Since pups that had SOD2 knock-down during days 8.5-12.5e were born in expected numbers and developed indistinguishably from negative-control littermates, we extended the SOD2 knock-down window to days 0.5-12.5e (Figure 1A). Again, pups were born in expected numbers and were indistinguishable from negative controls in appearance and weight gain (Figure S1C). To address reversibility of the system, pups born from these mothers were aged for four weeks in the absence of DOX (Figure 1A) and analyzed. SOD2 levels in these “adapted” animals were identical to negative controls in liver (Figure 1E), and liver mitochondria from these animals had normal aconitase activity (Figure 1F). These data demonstrate the versatility and reversibility of the iSOD2-KD mouse model system of mitochondrial oxidative stress.
Mitochondrial oxidative stress during embryogenesis promotes a sustained, adaptive mitochondrial and antioxidant responses in liver
Previous studies in yeast, worms, and flies have shown that prior exposure to acute mitochondrial stress can elicit adaptive cellular and tissue responses (see Introduction). Therefore, we hypothesized that tissues from mice that experienced mitochondrial oxidative stress during embryogenesis would also exhibit adaptive responses compared to negative-control littermates. To probe for adaptive responses, we performed gene-expression profiling on livers from 4-week-old adapted iSOD2 mice and negative controls. To control for off-target effects, livers were isolated from both iSOD2 mouse strains (that express different SOD2 shRNAs). We identified 367 genes whose expression changed significantly and similarly in liver of both iSOD2 mouse strains compared to negative controls (Figure 2A, Table S1). We then analyzed this gene set for common upstream cis-regulatory elements, which revealed an enrichment of sites that could bind PPARγ, PPARα, PCG-1α, or Nrf2 (Figure 2A and Figure S2A), suggesting alterations in mitochondrial biogenesis and antioxidant responses in the adapted livers. We confirmed these global profiling results by qRT-PCR of mRNAs for upregulated genes. This included the highest upregulated gene, ARK1C18, two additional Nrf2 targets, GSTT1 and GSTT3, two PPARα targets, mitochondrial UCP2 and PTGR1, and PGC-1α (Figure 2B). PGC-1α was also increased at the protein level (Figure 2C), and we observed increased steady-state amounts of OXPHOS subunits, TFAM and VDAC, consistent with activation of a PGC-1α-driven mitochondrial biogenesis program (Figure 2D). Also consistent with an adaptive antioxidant response, liver from adapted iSOD2 mice had increased levels of the Nrf2-regulated antioxidant protein NQO1 (Figure 2C) and other antioxidant genes upregulated that were not identified by gene expression profiling (Figure S2B). They also produced less hydrogen peroxide in vitro (Figure 2E). In addition, hepatocytes isolated from adapted mice exhibited decreased CellRox staining by flow cytometry (Figure 2F), consistent with lower total cellular ROS, and a trend towards decreased MitoSox staining (Figure S2C), suggesting lower mitochondrial superoxide production. They also had lower basal and spare mitochondrial respiratory capacity (Figure 2G) and increased mitochondrial content as judged by EM (Figure 2H). In summary, liver and isolated hepatocytes from iSOD2 mice exhibit stable, adaptive mitochondrial and antioxidant responses to embryonic exposure to mitochondrial oxidative stress.
Figure 2. Adaptive changes in liver of mice that experienced SOD2 knock-down only during embryogenesis.
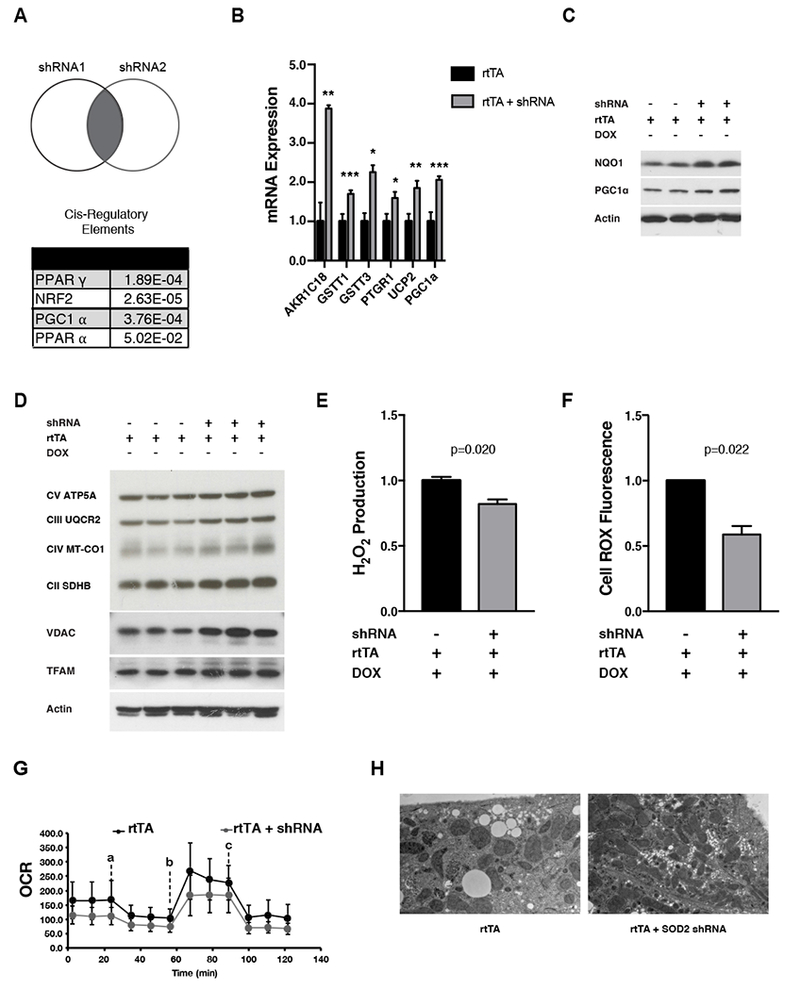
A) Venn Diagram depicting the differentially expressed genes from adapted livers (from two strains, each containing a different shRNA) after knock-down with each shRNA (bottom). Predicted transcription factors that drive the observed changes in gene expression based on analysis of cis-regulatory elements enriched in the promoters. PPARγ, Peroxisome Proliferator-activated receptor gamma; Nrf2, Nuclear factor, erythriod-2 like 2; PGC-1α, Peroxisome Proliferator-activated receptor gamma coactivator 1-alpha; PPARα, Peroxisome Proliferator-activated receptor alpha.
B) RNA expression levels (normalized to tubulin RNA) of top upregulated genes in adapted liver from iSOD2 mice. Error bars represent +/− SEM. ARK1C18, aldo-keto reductase family 1, member C18; GSTT1, glutathione s-transferase 1; GSTT3, glutathione s-transferase theta 3; PTGR1, prostaglandin reductase 1; UCP2, uncoupling protein 2; PGC-1 α, Peroxisome Proliferator-activated receptor gamma coactivator 1-alpha.
C) Western blots of NQO1 and PCG-1α in adapted liver relative to negative controls. Actin was probes as a loading control.
D) Western blot of OXPHOS complexes, VDAC, and TFAM in adapted liver relative to negative controls. Actin was probed as a loading control.
E) Hydrogen peroxide (Amplex Red Assay) present in adapted liver relative to negative controls.
F) Total ROS (CellROX) in adapted iSOD2 hepatocytes relative to negative controls.
G) Mitochondrial oxygen consumption rate (OCR; pmol O2/min) in adapted hepatocytes relative to negative controls. Error bars represent mean +/− SD of eight technical replicates. Inhibitors used in the Seahorse analysis were added as indicated VM oligomycin (a), 1 μM FCCP (b), and 0.5 μM antimycin A + 0.5 μM rotenone (c). n=3 rtTA controls, n=2 adapted hepatocytes
H) Electron microscopy of adapted hepatocytes relative to negative controls. Scale bar = 2 μM.
All calculations for statistical significance were completed using a non-parametric, unpaired, two-tailed t-test. n=2-12.
Transient mitochondrial oxidative stress produces beneficial adaptive mitochondrial and antioxidant responses in primary fibroblasts
Encouraged by our results in adapted liver, we next endeavored to develop an in vitro model of adaptive responses to mitochondrial oxidative stress. Primary MEFs isolated from both strains of iSOD2-KD mice (but that had not been exposed to DOX in vivo) were treated with DOX in vitro to knock-down SOD2. Significant knock-down at the protein level was observed by day 4 of DOX treatment (Figure 3A). These cells had more MitoSOX and CellRox staining (Figure 3B and S3A), lower aconitase activity (Figure 3C), and a trend toward lower total cellular ATP (Figure S3B), consistent with increased mitochondrial and oxidative stress. Control MEFs (not treated with DOX) had similar levels of SOD2 protein (Figure S3C) despite some “leaky” expression of the shRNA transgene (monitored by IRES-GFP expression from the shRNA transgene; Figure S3D). Also, consistent with previous reports (Flynn et al., 2011), SOD2 knockdown led to reduced mitochondrial spare respiratory capacity (Figure 3D), which may be linked to lower complex I and II abundance and lower amounts of mtDNA and TFAM (Figure 3E and 3F) that are needed to express mtDNA-encoded OXPHOS subunits. Finally, as expected (Ma, 2013; Van Raamsdonk and Hekimi, 2009), SOD2 knock-down cells resulted in upregulation of antioxidant genes. For example, we observed an increase in HMOX1, CGLM, NQO1, and GRX (Figure 3G) at the RNA level and mitochondrial peroxiredoxin 3 (PRDX3) at the protein level (Figure 3E). These data confirm that knock-down of SOD2 induces significant mitochondrial and cellular oxidative stress and a corresponding antioxidant response, setting the stage to interrogate adaptive responses to this form of mitochondrial stress in vitro.
Figure 3. Inducible SOD2 knock-down in mouse embryonic fibroblasts (MEFs) causes mitochondrial and cellular oxidative stress.
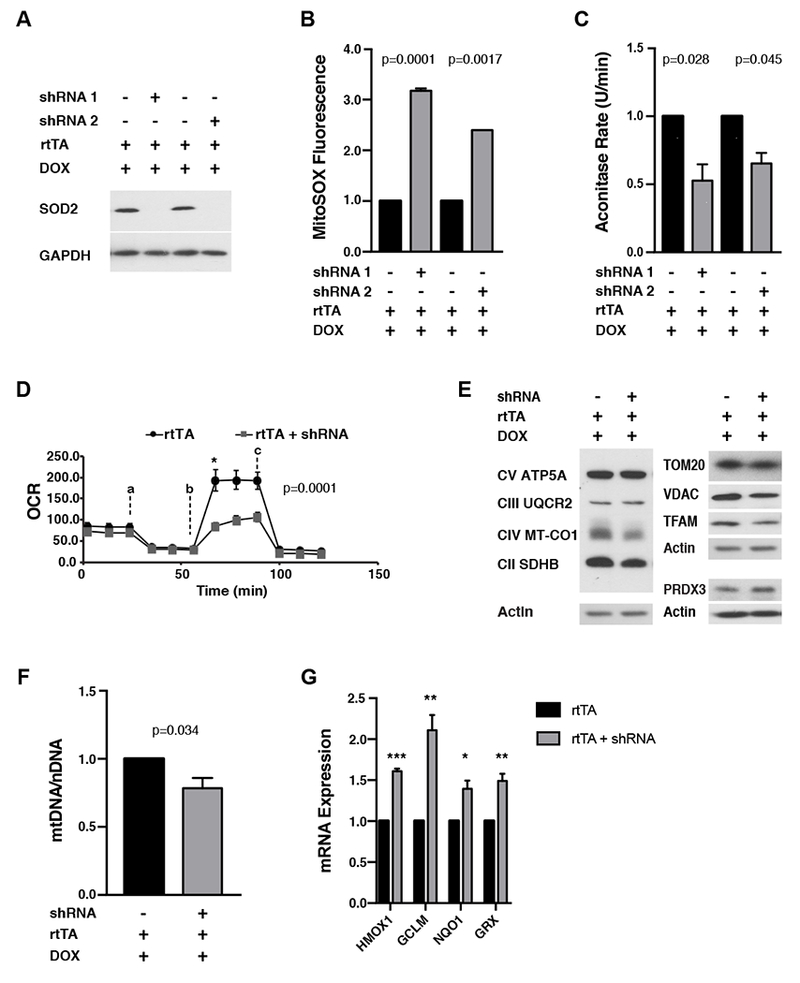
A) Western blot for SOD2 in SOD2 knock-down MEFs relative to negative controls. GAPDH was probed as a loading control.
B) MitoSox staining in SOD2 knock-down MEFs relative to negative controls.
C) Mitochondrial aconitase activity (U/min) in SOD2 knock-down MEFs relative to negative controls. 100% equals aconitase activity of 4.244 nmol/min/ml.
D) Mitochondrial oxygen consumption rate (OCR) in SOD2 knock-down MEFs compared to negative controls. Error bars represent +/− SD of six technical replicates. Inhibitors used in the Seahorse analysis were added as indicated 1μM oligomycin (a), 1 μM FCCP (b) and 0.5 μM antimycin A + 0.5 μM rotenone (c).◻E) Western blots of the indicated mitochondrial proteins in SOD2 knock-down MEFs relative to negative controls.
F) Relative mtDNA copy number in SOD2 knock-down MEFs relative to negative controls. Error bars represent +/− SEM.
G) Expression of antioxidant enzymes (normalized to tubulin RNA) in SOD2-KD MEFs relative to negative controls. HMOX1, Heme oxygenase 1; GCLM, glutamate-cysteine ligase modifier; NQO1, NAD(P)H quinone dehydrogenase 1; GRX, glutathione reductase.
All calculations for statistical significance were completed using a non-parametric, unpaired, two-tailed t-test. n=6-8.
We next induced SOD2 knock-down in MEFs for four days (as in Figure 3), but then removed DOX to allow SOD2 levels to recover (Figure 4A). SOD2 protein returned to normal 6-7 days after DOX removal (days 10-11, Figure 4A). Hence, we probed for adaptive responses to mitochondrial stress at day 12, after SOD2 had fully recovered for ~48 hours. Compared to identically DOX-treated negative controls, adapted cells had less MitoSox staining (Figure 4B) and hydrogen peroxide production (Figure 4C). There was no significant change in CellRox staining (Figure S4A), ATP levels (Figure S4B), mitochondrial OXPHOS complex subunit abundance (Figure 4D, top), TOM20 or VDAC (Figure 4D, bottom), or mitochondrial morphology (Figure S4C and S4D). However, the adapted cells displayed an increase in TFAM (Figure 4D, bottom) and mtDNA (Figure 4E), as well as a decrease in both basal mitochondrial oxygen consumption and spare respiratory capacity (Figure 4F). We also observed sustained upregulation of several antioxidant enzymes in the adapted cells at the mRNA and protein level (Figure 4G and 4H). These antioxidants overlapped to a significant degree with those upregulated in adapted liver, including known Nrf2 targets (Figure 2). However, while NQO1 protein was upregulated in both adapted liver and MEFs (Fig 2C and 4H), only its mRNA was upregulated in MEFs (Figure 4G). Additionally, HMOX1 mRNA and protein were upregulated in adapted MEFs, but not liver, (Figure 4G and 4H). Finally, to test if these adaptive changes were of physiological significance, we exposed the adapted MEFs to an oxidant challenge with menadione, a redox-cycling compound that increases mitochondrial superoxide production and oxidative stress. MEFs that experienced transient mitochondrial stress from SOD2 knock-down were more resistant to cell death induced by menadione compared to identically treated negative controls (Figure 4I).
Figure 4. Analysis of adaptive responses to transient SOD2 knock-down in MEFs from iSOD2-KD mice.
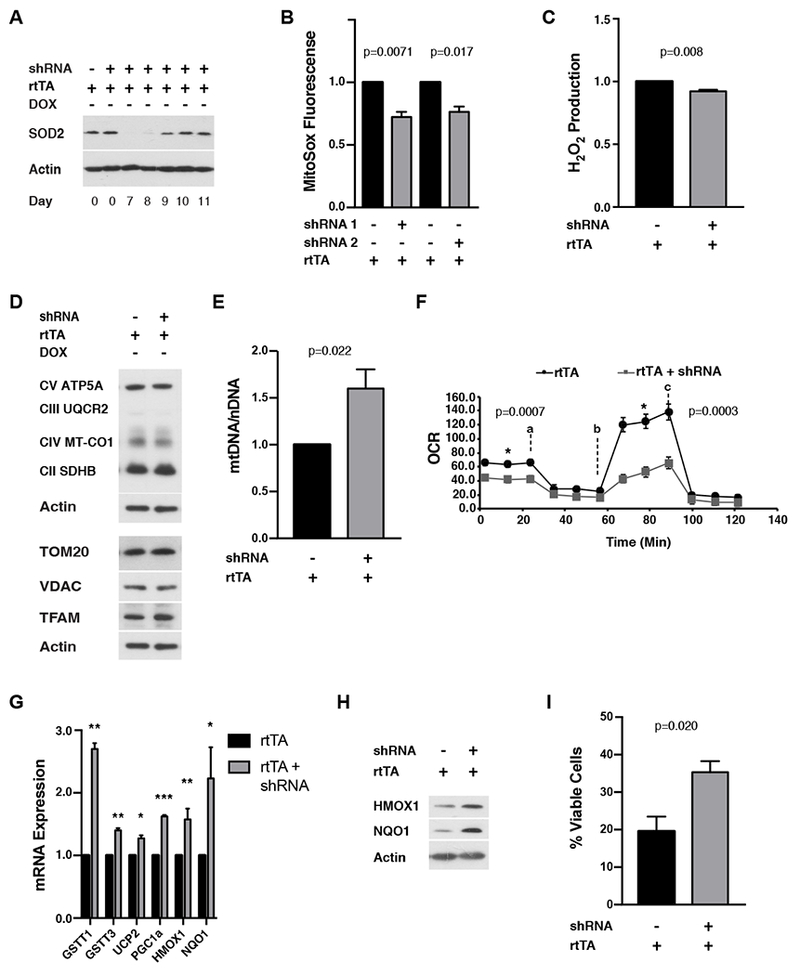
A) Western blot of SOD2 after DOX withdrawal to visualize the kinetics of SOD2 recovery. Actin was probed as a loading control.
B) MitoSox staining adapted MEFs (day 12) relative to negative controls.
C) Hydrogen peroxide (Amplex Red assay) of adapted MEFs (day 12) relative to negative controls.D) Western blots of indicated mitochondrial proteins in adapted MEFs (day 12). Actin was probed as a loading control.
E) Relative mtDNA copy number in adapted MEFs (day 12).
F) Mitochondrial oxygen consumption rate (OCR) in adapted MEFs (day 12) compared to negative controls. Error bars represent +/− SD of six technical replicates. Inhibitors used in the Seahorse analysis were added as indicated 1μM oligomycin (a), 1 μM FCCP (b), and 0.5 μM antimycin A + 0.5 μM rotenone (c).
G) RNA expression of antioxidant and mitochondrial enzymes (normalized to tubulin) in adapted MEFs (day 12). Error bars represent +/− SEM. GSTT1, glutathione s-transferase theta 1; GSTT3, glutathione s-transferase 3; UCP2, uncoupling protein 2; PGC1a, Peroxisome Proliferator-activated receptor gamma coactivator 1-alpha; HMOX1, Heme oxygenase 1; NQO1, NAD(P)H quinone dehydrogenase 1.
H) Western blot for the indicated antioxidant enzymes in adapted MEFs (day 12). Actin was probed as a loading control.
I) Percent viable cells after 24 hours treatment with 50 μM menadione in adapted MEFs relative to negative controls.
All calculations for statistical significance were completed using a non-parametric, unpaired, two-tailed t-test. n=4-9.
DISCUSSION
In this study, we describe development of the iSOD2-KD mouse model that allows knock-down of the mitochondrial antioxidant enzyme SOD2 to generate mitochondrial oxidative stress in an inducible and reversible manner. The utility of this model is demonstrated by our ability to strongly deplete SOD2 protein levels for controlled durations in embryos in vivo and MEFs in vitro. This capability allowed us to systematically analyze sustained, adaptive cellular and tissue responses to prior exposure to mitochondrial oxidative stress (i.e. mitohormesis).
Elimination of Sod2 expression by conventional gene knock-out studies results in lethality shortly after birth (Li et al. 1995; Lebovitz et al. 1996). Using the iSOD-KD mice, we demonstrate that strong depletion of SOD2 protein until day 12.5e in utero can be tolerated, even though significant oxidative stress was endured in embryos beginning after four days of SOD2 knock-down (Figure 1). Animals that experienced SOD2 knockdown during embryogenesis are born, gain weight normally and, to at least 4-weeks of age, are grossly indistinguishable from single-transgenic littermate controls (Figure S1C). These results demonstrate that embryos can withstand significant oxidative stress and still generate viable animals, and that lethality from loss of SOD2 is due to oxidative damage occurring after day e12.5 and likely during the post-natal period. Thus, the ability to analyze the requirement for SOD2 at different stages of development or for defined time windows in adults is one major advantage of the iSOD2-KD model system. Additionally, with iSOD2-KD mice, the amount and duration of mitochondrial oxidative stress during development or adulthood can be controlled by the investigator, allowing acute and adaptive responses to this form of mitochondrial stress to be analyzed for the first time. Finally, simple crosses can place the SOD2 shRNA under control of the rtTA driven by tissue-specific or other conditional promoters to further customize the timing, duration, and anatomical location of the mitochondrial oxidative stress.
The ability to reversibly knock-down SOD2 in mice allowed us to ask whether mitochondrial oxidative stress experienced only during embryogenesis can cause stable, adaptive changes in tissues of adult mice. This was addressed by analyzing liver from 4-week-old mice that experienced embryonic SOD2 knock-down. The global gene expression changes we observed in adapted liver varied considerably between strains that expressed the two different SOD2 shRNAs, likely due to the precise timing and extent of knock-down. Nonetheless, analysis of the common set of genes that changed predicted regulation by PPARγ, PPARα, PGC-1α and Nrf2 (Figure 2A and S2A). Consistent with this prediction, adapted liver had 1) known Nrf2-dependent antioxidant genes upregulated at the RNA and/or protein level (Figure 2B and 2C), 2) lower hydrogen peroxide production (Figure 2E), and 3) an increase in mitochondrial biogenesis indicated by several measures (mtDNA, TFAM, OXPHOS complexes; Figure 2D), consistent with a PPARγ/PGC-1α-driven gene program. Even hepatocytes isolated from liver of adapted mice exhibited lower mitochondrial respiration and cellular ROS, despite an upregulation of mitochondrial biogenesis as judged by EM (Figures 2F-2H). From these results, we conclude that mitochondrial oxidative stress during embryogenesis results in adaptive remodeling of liver to upregulate mitochondrial biogenesis, yet reduce respiration, so that fewer ROS are produced. In parallel, we speculate that upregulation of antioxidant gene expression has occurred to combat the potential for future oxidative stress. Importantly, we conclude that this is not a typical mitochondrial biogenesis and antioxidant response by PPARγ/PGC-1α and Nrf2, respectively, but rather non-canonical activation of a subset of the genes controlled by these factors to achieve heightened basal antioxidant state. For example, adapted livers only exhibited upregulation of a subset of Nrf2-regulated genes, which did not include a well-characterized target, HMOX1 (HO-1). In addition, adapted liver had increased steady-state amounts of the Nrf2 target NQO1, without a corresponding increase in its mRNA typical of a Nrf2-mediated transcriptional response. A similar case can be made that the PPARγ/PGC-1α mitochondria/antioxidant program is not fully engaged, since only a subset of targets of this pathway are upregulated. Furthermore, since PPARα target genes were also regulated in adapted livers (Figure 2A), we propose that crosstalk between PPARα and PPARγ, PGC-1α and Nrf2 is important for this adaptive response. Finally, the highest up-regulated gene in adapted liver is the PPARα target AKRC18, which is a NADPH-dependent aldo-keto reductase (Wang et al. 2010; Vergnes et al. 2003). Thus, it is tempting to speculate that its activities (e.g. as a 20α-hydroxysteroid dehydrogenase) might alter hormonal signaling via depletion of progesterone or modulation of corticosterones as part of the mitohormetic response to increased mitochondrial ROS.
Our results in primary MEFs in vitro also demonstrate that transient mitochondrial oxidative stress results in stable adaptive changes in mitochondria and cellular antioxidant capacity. After confirming that induced knock-down of SOD2 resulted in expected cellular phenotypes, including mitochondrial defects (e.g. altered OXPHOS subunit abundance, loss of spare respiratory capacity, and mtDNA depletion) and indicators of mitochondrial and cellular oxidative stress (Figure 3), we asked how cells adapted to this stress by allowing SOD2 levels recover (Figure 4A). The mitochondrial changes we observed in adapted MEFs were a decrease in basal oxygen consumption and spare respiratory capacity (Figure 4F) and a sustained increase in mtDNA and TFAM (Figure 4E and 4D). These changes appear to occur in the absence of any major changes in mitochondrial abundance or morphology (Figure 4D and S4C-D). An increase in mtDNA copy number largely uncoupled from mitochondrial mass has been reported previously in cells exposed to ionizing radiation (Eaton et al., 2007). This suggests increased mtDNA copy number may be a common adaptive mitochondrial stress response that ultimately protects cells from mtDNA damage. Adapted MEFs also exhibited a poised antioxidant state, characterized by increased antioxidant gene expression (Figures 4G, 4H and S4E) that mirrored that in adapted liver (Figure 2B and 2C). This included several glutathione S-transferase genes and the rate-limiting enzyme in glutathione biosynthesis (GCLM). NQO1 was also upregulated in both adapted liver and MEFs. This enzyme reduces quinones to prevent their ability to produce ROS, promotes CoQ and vitamin E antioxidant capacity, and protects mitochondria from oxidative stress, suggesting it is a key component of the adaptive response to acute mitochondrial oxidative stress (Kim et al., 2013; Ross and Siegel, 2017). Lastly, HMOX1 was upregulated at the RNA and protein level in adapted MEFs, but not at all in adapted liver. This suggests there is likely cell-type-specific adaptive programs that respond to transient mitochondrial oxidative stress.
At the organismal level, mitohormesis in response to mitochondrial respiration inhibition or oxidative stress has been documented in invertebrate model systems, primarily in the context of aging and longevity (Dillin et al., 2002; Lee et al., 2010b; Ristow and Zarse, 2010; Schulz et al., 2007; Yang and Hekimi, 2010b; Yee et al., 2014). Our own studies demonstrated that extension of yeast chronological lifespan by reduced mTORC1 signaling involves an increase in mitochondrial ROS that promotes adaptive epigenetic and transcriptional responses in the nucleus (Bonawitz et al., 2007; Pan et al., 2011; Schroeder et al., 2013). Similarly, the current study demonstrates that an increase in mitochondrial oxidative stress induced by SOD2 knock-down leads to a mitohormetic response in cultured mammalian cells that involves mitochondrial remodeling that increases mtDNA copy number, decrease basal respiration, and reduces mitochondrial ROS production. We propose that this reprogramming, coupled with sustained antioxidant gene expression, underlies our observation that SOD2 adapted cells are resistant to a subsequent, lethal oxidant challenge (Figure 4I). This, coupled with the similar responses in adapted liver from iSOD2 mice (Figure 2 and S2), is a clear demonstration of mitohormesis in mice. Determining if this mitohormetic response can enhance stress resistance or delay aging in mammals and which epigenetic changes underlie the stable changes in nuclear gene expression are particularly exciting avenues to pursue in future studies in the iSOD2-KD mouse model system.
Limitations of Study.
There are some caveats to consider when interpreting the results of this study. First, the results presented are from the initial analysis of a single tissue, liver, from mice that experienced mitochondrial oxidant stress during embryogenesis. Thus, it is likely that the responses observed are somewhat idiosyncratic to liver and may not be generalizable to other tissues. Likewise, the in vitro mitohormesis model in MEFs may not accurately reflect how different cell types will respond. We also note that, since SOD2 knock-down was done over a specific timeframe during embryogenesis and we only assessed SOD2 knock-down and oxidative stress in whole embryos, it is not clear yet which cells or tissues in the embryo were experiencing stress and generating the mitohormetic signal. Finally, the iSOD2 system uses doxycycline to induce the SOD2 shRNA, which is a drug that can inhibit mitochondrial translation under some conditions. We probed for this possibility and not see any evidence that this was the case. Furthermore, potential effects of the drug are controlled for in one sense because the negative controls employed were cells or embryos exposed to the same doxycycline concentration as the experimental groups, but had no shRNA to knock-down SOD2. Nonetheless, we cannot formally exclude the possibility of synergy between mild effects of doxycycline and stress from SOD2 knockdown per se on the phenotypes observed.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Gerald Shadel (gshadel@salk.edu). Transfer of materials may require execution of a materials transfer agreement.
EXPERIMENTAL MODEL DETAILS
iSOD2-KD mice and MEFs
The iSOD2-KD mice were generated as described (Dow et al., 2012; Premsrirut et al., 2011) from embryonic stem cells obtained from Mirimus Inc. that harbored two transgenes, 1) a reverse tetracycline-controlled transactivator 2 (rtTA2) protein expressed from the Rosa26 promoter (Rosa26-rtTA) on chromosome 6 and 2) and an shRNA targeting the SOD2 gene driven by a promoter with a tetracycline response element inserted at the Collal locus on chromosome 11. The shRNA construct also has an IRES-GFP to monitor expression of the shRNA by fluorescence. After screening multiple shRNAs for efficient SOD2 knock-down in cultured cells in collaboration with Mirimus, we found two that yielded significant (>90%) reduction of SOD2 expression at the mRNA and protein level (#582 and #835, see sequences below), but that targeted different regions of the SOD2 gene. Mirimus then generated the two ES cell lines in a mixed C57BL6/129SVJ background, and Yale Animal Genomic Services generated the corresponding transgenic mice using standard microinjection techniques. Founder animals have been extensively backcrossed to the C57BL6/J background, but the MEFs used in this study are from the third or fourth backcross.
All animals were housed in an AAALAC-accredited facility overseen by the Yale Animal Resource Center, which provides clinical veterinary services on a 24-hour basis. All animal work was performed according to protocols approved by the Yale University Institutional Animal Care and Use Committee (IACUC). Animals were housed at the Yale Animal Resource Center facility room kept at 72 +/− 2 °;F with a 12-hour light-dark cycle. Individually ventilated cages lined with shredded, corn-cob bedding (Harlan Teklad) were used and the housing density did not exceed five animals per cage. The animals had ad lib access to chow (Harlan Teklad Global 2018S) and water.
For embryonic SOD2 knock-down wild-type C57BL/6 females were bred with transgenic males that were homozygous for the Rosa26-rtTA2 transgene and heterozygous for the TRE-SOD2-shRNA. This breeding schematic ensures all pups are heterozygous for the Rosa26-rtTA2 transgene and a 50% of them are heterozygous for the TRE-SOD2-shRNA. The single transgenic Rosa26-rtTA2 pups were used as negative controls. To knock-down SOD2 in utero, pregnant dams were fed doxycycline-containing chow (Harlan Teklad, 625mg/kg) from either day 8.5e to 12.5e or 0.5e to 12.5e (see Figure 1A). The aforementioned breeding scheme ensured experimental and control embryos experienced the same exposure to DOX. Toe DNA was purified for genotyping using the Qiagen DNeasy Blood and Tissue Kit and the transgenes were amplified using the PCR primers below.
All experimental animals were male and four weeks of age at time of sacrifice.
Primers and shRNA sequences
Col1A1 Rev45 Reverse 5’-3’ CACCCTGAAAACTTTGCCCC
ROSA C 5’-3’ GGAGCGGGAGAAATGGATATG
ROSA B 5’-3’ GCGAAGAGTTTGTCCTCAACC
ROSA D 5’-3’ TCAGTAAGGGAGCTGCAGTGG
shRNA 835 Forward 5’-3’ AAGCCACAGATGTATCTTTCAGTA A
shRNA 582 Forward 5’-3’ AAGCCACAGATGTATTAAACTTCT C
Sod2_835 shRNA 1, 5’GAAGGCTCGAGAAGGTATATTGCTGTTGACAGTGAGCGATGGGAGAATGTTACTGAAAGATAGTGAAGCCACAGATGTATCTTTCAGTAACATTCTCCCAGTGCCTACTGCCTCGGACTTCAAGGGGCTAGAATTCGAGCA-3’
Sod2_582 shRNA 2, 5′GAAGGCTCGAGAAGGTATATTGCTGTTGACAGTGAGCGCTGGGTCTTTTGAGAAGTTTAATAGTGAAGCCACAGATGTATTAAACTTCTCAAAAGACCCAATGCCTACTGCCTCGGACTTCAAGGGGCTAGAATTCGAGCA-3’
Hepatocyte Isolation
Liver hepatocytes were isolated and cultured by the Yale Liver Center in William’s E Medium. Hepatocytes were plated at 100% confluence in Aligent Seahorse XFe96 microplates for Seahorse analysis and in 10-cm dishes for EM studies. All remaining hepatocytes were stained and analyzed by flow cytometry in suspension.
Cell Culture
Mouse embryonic fibroblasts (MEFs) were generated from day 13.5e embryos and grown in DMEM media (Gibco) with glutamine (584 mg/L), glucose (4.5 g/L), and 10% FBS (Life Technologies Lot Catalog Number 16000, Lot number 1566362). Cells were incubated at 37°C and 5% CO2. Cells were treated with a final concentration of 1μg/mL DOX to induce SOD2 knock-down. Cells were split at ~80% confluency and only cells that were <6 passages were analyzed. All cell lines were male.
METHOD DETAILS
Microarray Analysis
RNA was prepared from 25 mg of homogenized liver using the Qiagen RNeasy Kit Plus DNA exclusion columns. Three iSOD2 adapted liver samples from shRNA 1 and three samples from shRNA 2 were analyzed. Four total rtTA-only littermate controls were analyzed, including three from the shRNA 2 cohort and 1 from the shRNA 1 cohort. Approximately 1 mg of RNA was sent to the Yale Center for Genome Analysis for microarray analysis on Affymetrix GeneChip Mouse 2.0 ST array plates. Only samples which had a RIN value >8 were used analyzed. Data were normalized with Genome Expression Console. PCA analysis was conducted in Genome Expression Console, and all data were exported into Affymetrix Transcriptome Software to obtain a list of differentially expressed genes. Differentially expressed genes (fold change greater than 1.01) were analyzed in Ingenuity Pathway Analysis Software (Figure 6.1) and Enrichr. Samples were gated to only include data which was statically significant (p value < 0.05). Gene lists from both SOD2 targeting shRNAs were compared to identify common, differentially regulated genes. The complete list of common, differentially regulated genes and their corresponding fold-change values is in Table S1.
Flow Cytometry
Fifty thousand cells per well were plated in triplicate in a six well dish. The next day, cells were treated with MitoSOX (ThermoFisher Scientific, final concentration 5 μM), CellROX (ThermoFisher Scientific, final concentration 5 μM), for 20 minutes at 37°C. Cells were then trypsinized and resuspended in 2% FBS in PBS for analysis on a Stratedigm 13L cytometer. Samples were analyzed/compensated using FloJo software.
Mitochondrial Isolation and Aconitase Assay
Two ~80% confluent 15-cm dishes of MEFs or ~200 mg of liver were used to prepare mitochondria as described (Frezza et al., 2007). Mitochondrial pellets were lysed by three subsequent freeze-thaw cycles in 200μL of 50 mM Tris buffer (pH 7.5). Samples were normalized by protein concentration (0.5-0.75 μg/μl per sample) and assayed for aconitase activity using the Aconitase Assay Kit (Cayman Chemical). Manufacturer’s instructions were followed, except the isocitrate dehydrogenase was pelleted at maximum speed for 5 minutes before use to remove any insoluble precipitate.
Seahorse Analysis
Ten thousand cells per well were plated in the Aligent Seahorse XFe96 microplates and left to settle overnight before analysis with the FluxPax kits according to company standards. Six replicates were plated per sample. Mitochondrial Stress Tests were conducted following Seahorse guidelines (Agilent Technologies). For the assay base, media was supplemented with 10mM Glucose (Gibco), 2mM GlutaMAX (Gibco), and 1mM pyruvate (Gibco). Inhibitors were used at the following concentrations: 1uM oligomycin (Sigma), 1uM FCCP (Sigma), and 0.5uM antimycin A (Sigma) + 0.5uM rotenone (Sigma). Analyses were conducted using Wave software and XF Report Generators (Agilent Technologies).
ATP Production
MEFs were cultured until 80% confluency in 15 cm dishes. To determine ATP production in the cells, 1 million cells were pelleted and analyzed by the ATP Assay Kit (Sigma) as per manufacturer’s specifications. Samples were normalized to cellular protein level after cell lysing.
Analysis of mtDNA Copy Number and RNA Transcripts
To quantify the relative mtDNA copy number, one half of an ~80% confluent 15-cm dish of MEFs was pelleted, washed in 1X PBS, and lysed in 250 μL of 50mM NaOH. Samples were boiled for 10 minutes and neutralized with 25 μL of 1mM Tris buffer (pH 8.0).
Samples were centrifuged at 13,000 g for 1 minute, and the DNA concentration in the supernatant was measured using a nanodrop spectrophotometer. Samples were diluted to 10 ng/μL for mtDNA copy number analysis by PCR using mtDNA and nuclear DNA primers as described (West et al., 2015).
For RNA transcript analysis, half of an ~80% confluent 15-cm dish of MEFs was used to prepare RNA using the Qiagen RNeasey Mini Plus Kit. RNA (500-1000 ng) was then used to generate cDNA using the High-Capacity cDNA Reverse Transcription Kit (Thermo Fischer Scientific). The resulting cDNA was diluted 1:25 in water and used to perform qRT-PCR with SYBR green master mix (Applied Biosystems). Primers were obtained from the mouse Primer Depot and are described below.
AKR1C18 Forward 5′-3′ CAATATGGCAGAACCCAACAT
AKR1C18 Reverse 5′-3′ GCTTTGGCACCTATGCAACT
GSTT1 Forward 5’-3’ CTCACCAAGGAAAACAGGGA
GSTT1 Reverse 5’-3’ AGGCTCGTGCTCGTGTAGA
GSTT3 Forward 5’-3’ CCCCATCATTCACCATATCC
GSTT3 Reverse 5’-3’ GGATGGAGACCTCACCCTTT
PTGR Forward 5’-3’ AATCGTTCCTTTTGGGAAGG
PTGR Reverse 5’-3’ AAGCCCTGTTCCTCTCTGTG
HMOX1 Forward 5’-3’ CCTTCAAGGCCTCAGACAAA
HMOX1 Reverse 5’-3’ GAGCCTGAATCGAGCAGAAC
GCLM Forward 5’-3’ TTGG GAACT C CATT CATT CA
GCLM Reverse 5’-3’ CGGGAACCTGCTCAACTG
NQO1 Forward 5’-3’ GTCTTCTCTGAATGGGCCAG
NQO1 Reverse 5’-3’ CCAATCAGCGTTCGGTATTA
GRX Forward 5’-3’ ATCGTGCATGAATTCCGAGT
GRX Reverse 5’-3’ GGTGGTGGAGAGTCACAAGC
UCP2 Forward 5’-3’ CAGCGCCAGATGAGCTTTG
UCP2 Reverse 5’-3’ GGAAGCGGACCTTTACCACA
PGC1α Forward 5’-3’ TGAGGACCGCTAGCAAGTTT
PGC1α Reverse 5’-3’ TGAAGTGGTGTAGCGACCAA
GSTmu Forward 5’-3’ AACACAGGTCTTGGGAGGAA
GSTmu Reverse 5’-3’ CGTATGTTTGAGCCCAAGTG
GSTP1 Forward 5’-3’ GGGCCTTCACGTAGTCATTC
GSTP1 Reverse 5’-3’ ATGGGAAAAACCAGAGGGAG
GSTA1 Forward 5’-3’ CTCTTCAAACTCCACCCCTG
GSTA1 Reverse 5’-3’ TGGAGAAGAAGCCAGGACTC
Western Blotting
One half of an 80% confluent 15-cm dish of transgenic MEFs was lysed in 100 μL RIPA buffer plus protease and phosphatase inhibitors. Samples were incubated on ice for 10 minutes and centrifuged at 13,000×g for ten minutes at 4°3. Samples were normalized to 5 μg/μL and ran on a 12% tris-glycine gel. Approximately 20 μg of protein was loaded in to each well. Gels were transferred in transfer buffer with 20% methanol at 90V for 60 minutes at 4°C to a PVDF membrane activated with methanol. After transfer, membranes were washed once with water, rinsed in methanol for one minute, and dried at 37°3. Membranes were incubated with antibodies at 1:1000 dilution overnight at 4°C unless otherwise specified. Antibodies used are: SOD2 (Enzo sciences 1:5,000), OXPHOS Cocktail (Abcam), mt-COX3 (Santa Cruz), GAPDH (Life Technologies, 1:10,000), HSP60 (Cell Signaling), Actin (Sigma, 1:50,000), NQO1 (Abcam), PGC-1α (Novus Biologicals), VDAC (Abcam), TFAM (generous gift from the Wallace Lab, 1:2,000), PRX3 (R&D systems 1:300), HMOX1 (OWL ID 58476). Secondary antibodies were used at 1:2,000, and blots were developed with Milipore Crescendo Western HRP Substrate.
Cell Viability
Cells plated at twenty-five thousand cells/mL in a 6-well dish in triplicate were treated the next day with 50 μM menadione or vehicle control (ethanol) for 24 hours. Live cells were trypsinized and counted via hemocytometer (Reichard Bright Line) after washing the plate once with 1X PBS.
Lipid Peroxidation
Whole embryos were homogenized by dounce in 200 μL IBC buffer as described in mitochondrial purification section above. Homogenate (50 μL) was combined with 2X RIPA buffer and incubated on ice for 10 minutes. Samples were centrifuged at 13,000×g for 10 minutes at 4°C. Lipid peroxidation in the supernatant was measured with the TBARS Assay Kit (Cayman Chemical) as specified in the provided protocol.
Confocal Microscopy
Cells grown on coverslips (2,500 cells/mL) were fixed in a 37°C solution of 4% paraformaldehyde in PBS for 15 minutes at room temperature, followed by permeabilization with 0.1% Triton X-100 in PBS for 10 minutes at room temperature. Coverslips were blocked for one hour with PBS containing 5% FBS. Primary antibodies against HSP60 (Santa Cruz Biotechnology, 1:100 dilution) were diluted in blocking buffer, and coverslips were incubated at 4°C overni ght. Primary antibodies were removed by washing 4 times for 5 minutes each with PBS. Secondary antibodies (1:500, Alexa fluorophores 546 and 647, Invitrogen) were incubated in blocking buffer for 1 hour at room temperature and then removed by 4 × 5-minute washes in PBS. Coverslips were mounted onto slides using Prolong Antifade (Invitrogen). Images were acquired using a Leica SP5 microscope and a 100× oil objective. Fluorophores were imaged after 561 and 633 laser excitation. Images were acquired with a resolution of 512×512 pixels and 12-bit depth. Z-stacks were acquired and processed to generate average image intensity projections using ImageJ. Samples were blinded for confocal imaging. All samples were included in analysis.
Electron Microscopy
Hepatocytes were plated at confluence in 10-cm dishes by the Yale Liver Center and left to settle overnight. Adhered hepatocytes were fixed in 2.5% glutaraldehyde in 0.1M Sodium cacodylate pH 7.4 for half an hour at room temperature and then for 2 hours at 4°C on a rocking incubator. Fixed hepatocytes were then washed three times for five minutes each in sodium cacodylate buffer. Samples were delivered to the Yale Core Electron Microscopy Facilities, where they were fixed, embedded, and imaged.
Amplex Red Assay
Mitochondria from purified mouse liver were normalized to μg/μL of total protein (via Biorad Protein Assay Kit) in PBS. Amplex Red Assay (ThermoFisher Scientific) was conducted per manufacturer’s instructions to measure hydrogen peroxide production.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical Analysis
Statistical significance was calculated in Prism 7.0 using a non-parametric, unpaired, two-tailed t-test. No tests were used to determine if data met assumptions of the statistical approach.
DATA AVAILABILITY
The microarray data has been deposited under the GEO accession number GSE116676.
Supplementary Material
Table S1. Differentially Expressed Genes in Adapted Liver, Related to Figure 2
HIGHLIGHTS.
Inducible and reversible SOD2 knock-down mice (iSOD2-KD) have been developed
Embryonic mitochondrial oxidant stress results in adaptive changes in adult liver
Mitohormesis in adapted liver involves basal activation of PPARγ, PGC-1α and Nrf2
Mitohormesis in iSOD2 embryonic fibroblasts provides resistance to oxidative stress
Acknowledgements
This work was supported by NIH grant R21/R33 ES025636 to G.S.S.; he was also supported as the Joseph A. and Lucille K. Madri Professor of Experimental Pathology and currently holds the Audrey Geisel Chair in Biomedical Science. C.S.C. was supported by NIH T32 Chemical Biology Training Grant 5T32GM06754 3-12, M.A.H by NIH F32 AG05299, and B.E.C. by NIH F32 NS077723. Mouse hepatocytes were generated in the Yale Liver Center, which is supported by NIH Grant P30 DK34989. We wish to thank Dr. Prem Premsrirut at Mirimus, Inc. for help in designing the shRNA constructs and ES cells, and Yale Animal Genomic Services for help generating the iSOD2-KD mice.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests. The authors declare no competing interests.
REFERENCES
- Biswal MR, Ildefonso CJ, Mao H, Seo SJ, Wang Z, Li H, Le YZ, and Lewin AS (2016). Conditional Induction of Oxidative Stress in RPE: A Mouse Model of Progressive Retinal Degeneration. Retin. Degener. Dis. Mech. Exp. Ther 31–37. [DOI] [PubMed] [Google Scholar]
- Bonawitz ND, Chatenay-Lapointe M, Pan Y, and Shadel GS (2007). Reduced TOR Signaling Extends Chronological Life Span via Increased Respiration and Upregulation of Mitochondrial Gene Expression. Cell Metab 5, 265–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand MD (2010). The sites and topology of mitochondrial superoxide production. Exp. Gerontol 45, 466–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case AJ, and Domann FE (2012). Manganese superoxide dismutase is dispensable for post-natal development and lactation in the murine mammary gland. Free Radic. Res 46, 1361–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandel NS (2015). Evolution of Mitochondria as Signaling Organelles. Cell Metab 22, 204–206. [DOI] [PubMed] [Google Scholar]
- Dillin A, Hsu A, Arantes-oliveira N, Lehrer-graiwer J, Hsin H, Fraser AG, Kamath RS, Ahringer J, and Kenyon C (2002). Rates of Behavior and Aging Specified by Mitochondrial Function During Development. 298, 2398–2402. [DOI] [PubMed] [Google Scholar]
- Dow LE, Premsrirut PK, Zuber J, Fellmann C, McJunkin K, Miething C, Park Y, Dickins RA, Hannon GJ, and Lowe SW (2012). A pipeline for the generation of shRNA transgenic mice. Nat. Protoc. 7, 374–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durieux J, Wolff S, and Dillin A (2011). The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell 144, 79–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaton JS, Lin ZP, Sartorelli AC, Bonawitz ND, and Shadel GS (2007). Ataxia-telangiectasia mutated kinase regulates ribonucleotide reductase and mitochondrial homeostasis. J. Clin. Invest. 117, 2723–2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J, Bussiére F, and Hekimi S (2001). Mitochondrial electron transport is a key determinant of life span in Caenorhabditis elegans. Dev. Cell 1, 633–644. [DOI] [PubMed] [Google Scholar]
- Flynn JM, Choi SW, Day NU, Gerencser AA, Hubbard A, and Melov S (2011). Impaired spare respiratory capacity in cortical synaptosomes from Sod2 null mice. Free Radic Biol Med. 50, 866–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frezza C, Cipolat S, and Scorrano L (2007). Organelle isolation: functional mitochondria from mouse liver, muscle and cultured filroblasts. Nat. Protoc 2, 287–295. [DOI] [PubMed] [Google Scholar]
- Jang YC, and Van Remmen H (2009). The mitochondrial theory of aging: Insight from transgenic and knockout mouse models. Exp. Gerontol. 44, 256–260. [DOI] [PubMed] [Google Scholar]
- Kim J, Kim SK, Kim HK, Mattson MP, and Hyun DH (2013). Mitochondrial Function in Human Neuroblastoma Cells Is Up-Regulated and Protected by NQO1, a Plasma Membrane Redox Enzyme. PLoS One 8, e69030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokoszka JE, Coskun P, Esposito L. a, and Wallace DC (2001). Increased mitochondrial oxidative stress in the Sod2 (+/−) mouse results in the age-related decline of mitochondrial function culminating in increased apoptosis. Proc. Natl. Acad. Sci. U. S. A. 98, 2278–2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebovitz R, Zhang H, Vogel H, Cartwright J, Dionne L, Lu N, Huang S, and Matzuk MM (1996). Neurodegeneration, myocardial injury, and perinatal death in mitochondrial superoxide dismutase-deficient mice. Protc. Natl. Acad. Sci. USA 93, 9782–9787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Hwang AB, and Kenyon C (2010). Report Inhibition of Respiration Extends C . elegans Life Span via Reactive Oxygen Species that Increase HIF-1 Activity. Curr. Biol. 20, 2131–2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Huang T-T, Carlson EJ, Melov S, Ursell PC, Olson JL, Noble LJ, Yoshimura MP, Berger C, Chan PH, et al. (1995). Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nat Genet. 11, 376–381. [DOI] [PubMed] [Google Scholar]
- Liang LP, and Patel M (2004). Mitochondrial oxidative stress and increased seizure susceptibility in Sod2−/+ mice. Free Radic. Biol. Med. 36, 542–554. [DOI] [PubMed] [Google Scholar]
- Ma Q (2013). Role of Nrf2 in Oxidative Stress and Toxicity. Annu. Rev. Pharmacol. Toxicol. 53, 401–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merkwirth C, Jovaisaite V, Durieux J, Matilainen O, Jordan SD, Quiros PM, Steffen KK, Williams EG, Mouchiroud L, Tronnes SU, et al. (2016). Two Conserved Histone Demethylases Regulate Mitochondrial Stress-Induced Longevity. Cell 165, 1209–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunnari J, and Suomalainen A (2012). Mitochondria: In sickness and in health. Cell 148, 1145–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owusu-Ansah E, Song W, and Perrimon N (2013). Muscle Mitohormesis Promotes Longevity via Systemic Repression of Insulin Signaling. Cell 155, 1286–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Y, Schroeder EA, Ocampo A, Barrientos A, and Shadel GS (2011). Regulation of yeast chronological life span by TORC1 via adaptive mitochondrial ROS signaling. Cell Metab 13, 668–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Premsrirut PK, Dow LE, Kim SY, Camiolo M, Malone CD, Miething C, Scuoppo C, Zuber J, Dickins RA, Kogan SC, et al. (2011). A rapid and scalable system for studying gene function in mice using conditional RNA interference. Cell 145, 145–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Raamsdonk JM, and Hekimi S (2009). Deletion of the mitochondrial superoxide dismutase sod-2 extends lifespan in Caenorhabditis elegans. PLoS Genet. 5, e1000361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Remmen H, Ikeno Y, Hamilton M, Pahlavani M, Wolf N, Thorpe SR, Alderson NL, Baynes JW, Epstein CJ, Huang T-T, et al. (2003). Life-long reduction in MnSOD activity results in increased DNA damage and higher incidence of cancer but does not accelerate aging. Physiol. Genomics 16, 29–37. [DOI] [PubMed] [Google Scholar]
- Riera CE, Merkwirth C, De Magalhaes Filho CD, and Dillin A (2016). Signaling Networks Determining Life Span. Annu. Rev. Biochem. 85, 35–64. [DOI] [PubMed] [Google Scholar]
- Ristow M, and Zarse K (2010). How increased oxidative stress promotes longevity and metabolic health: The concept of mitochondrial hormesis ( mitohormesis ). Exp. Gerontol. 45, 410–418. [DOI] [PubMed] [Google Scholar]
- Ross D, and Siegel D (2017). Functions of NQO1 in cellular protection and CoQ10 metabolism and its potential role as a redox sensitive molecular switch. Front. Physiol. 8, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder EA, Raimundo N, and Shadel GS (2013). Epigenetic silencing mediates mitochondria stress-induced longevity. Cell Metab. 17, 954–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz TJ, Zarse K, Voigt A, Urban N, Birringer M, and Ristow M (2007). Glucose Restriction Extends Caenorhabditis elegans Life Span by Inducing Mitochondrial Respiration and Increasing Oxidative Stress. Cell Metab. 6, 280–293. [DOI] [PubMed] [Google Scholar]
- Shadel GS, and Horvath TL (2015). Mitochondrial ROS Signaling in Organismal Homeostasis. Cell 163, 560–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunagawa T, Shimizu T, Matsumoto A, Tagashira M, Kanda T, Shirasawa T, and Nakaya H (2014). Cardiac Electrophysiological Alterations in Heart/Muscle-Specific Manganese-Superoxide Dismutase-Deficient Mice: Prevention by a Dietary Antioxidant Polyphenol. Biomed Res. Int. 2014, 704291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian Y, Garcia G, Bian Q, Wolff S, Meyer BJ, Tian Y, Garcia G, Bian Q, Steffen KK, Joe L, et al. (2016). Mitochondrial Stress Induces Chromatin Reorganization to Promote Longevity and UPR mt Article Mitochondrial Stress Induces Chromatin Reorganization to Promote Longevity and UPR mt. Cell 165, 1197–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velarde MC, Flynn JM, Day NU, Melov S, and Campisi J (2012). Mitochondrial oxidative stress caused by Sod2 deficiency promotes cellular senescence and aging phenotypes in the skin. Aging (Albany. NY). 4, 3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergnes L, Phan J, Stolz Aand Reue K (2003). A cluster of eight hydroxysteroid dehydrogenase genesbelonging to the aldo-keto reductase supergene family on mouse chromosome 13. J. Lipid Res. 44, 503–511. [DOI] [PubMed] [Google Scholar]
- Wang T, Shah YM, Matsubara T, Zhen Y, Tanabe T, Nagano T Fotso S, Krausz KW, Zabriskie TM, Idle JR, and Gonzales FJ (2010) Control of steroid 21-oic acid synthesis by peroxisome proliferator-activated receptor alpha and role of the hypothalamic-pituitary-adrenal axis. J. Biol. Chem. 285, 7670–7685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AP, Shadel GS, and Ghosh S (2011). Mitochondria in innate immune responses. Nat. Rev. Immunol. 11, 389–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AP, Khoury-Hanold W, Staron M, Tal MC, Pineda CM, Lang SM, Bestwick M, Duguay BA, Raimundo N, MacDuff DA, et al. (2015). Mitochondrial DNA stress primes the antiviral innate immune response. Nature 520, 553–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, and Hekimi S (2010). A mitochondrial superoxide signal triggers increased longevity in caenorhabditis elegans. PLoS Biol. 8, e1000556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yee C, Yang W, and Hekimi S (2014). The Intrinsic Apoptosis Pathway Mediates the Pro-Longevity Response to Mitochondrial ROS in C . elegans. Cell 157, 897–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun J, and Finkel T (2014). Mitohormesis. Cell Metab 19, 757–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Ikeno Y, Qi W, Chaudhuri A, Li Y, Bokov A, Thorpe SR, Baynes JW, Epstein C, Richardson A, et al. (2009). Mice deficient in both Mn superoxide dismutase and glutathione peroxidase-1 have increased oxidative damage and a greater incidence of pathology but no reduction in longevity. Journals Gerontol.-Ser. A Biol. Sci. Med. Sci 64, 1212–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Differentially Expressed Genes in Adapted Liver, Related to Figure 2
Data Availability Statement
The microarray data has been deposited under the GEO accession number GSE116676.