

Summary
Obstructive sleep apnea (OSA) is an increasingly prevalent sleep disorder characterized by upper airway obstruction during sleep, resulting in breathing pauses, intermittent hypoxia, and fragmented sleep. In parallel, the constellation of adverse health outcomes associated with prolonged obesity, such as insulin resistance, elevated blood pressure, triglycerides, and reduced high-density lipoprotein cholesterol – termed metabolic syndrome –raises the risk of cardiovascular morbidity and mortality, type 2 diabetes, and all-cause mortality. Affecting 35–40% of U.S. adults, risk factors for metabolic syndrome, including obesity, middle age, sedentary behavior, and genetics, share considerable overlap with those for OSA. Thus, it has been difficult to disentangle cause, effect, and whether certain treatments, such as CPAP, can improve these outcomes. In this paper, we provide an update to our 2005 review which explored the association between OSA and metabolic syndrome, highlighting visceral obesity as the common etiological factor of both conditions. This update includes (a) recent data on physiological and biochemical mechanisms, (b) new data in nonobese men and women as well as children and adolescents, (c) insight from the latest treatment studies, (d) the role of aging in understanding clinically-meaningful phenotypes of the disorder, and (e) the potential diagnostic/prognostic utility of biomarkers in identifying OSA patients with the strongest cardiometabolic risk.
Keywords: obstructive sleep apnea, metabolic syndrome, inflammation, obesity, biomarkers, phenotypes
Obstructive sleep apnea (OSA) is a prevalent sleep disorder characterized by upper airway obstruction during sleep, resulting in intermittent breathing pauses despite effort, acute reductions in hemoglobin oxygen saturation (SpO2), and fragmented sleep. Importantly, OSA is associated with a number of chronic medical conditions that contribute to poor quality of life, morbidity, and mortality, including hypertension, diabetes, coronary artery disease, and diminished cognitive ability, among others. Patients with untreated OSA have been shown to contribute significantly to increased healthcare costs and resource use, while treatment reduces healthcare use to that of the general population [1]. Population cohort studies over the last two decades have estimated the prevalence of OSA at 17–24% of men and 5–9% of women [2–4], though more recent work emphasizes how rates of OSA have increased between 14% to 55%, depending on the subpopulation studied, in conjunction with the ongoing obesity epidemic [5].
In parallel, adverse health outcomes associated with prolonged obesity – elevated blood pressure, insulin resistance, elevated triglycerides, and reduced high-density lipoprotein (HDL) cholesterol –raise the risk of cardiovascular morbidity and mortality, type 2 diabetes, some cancers, all-cause mortality, and even neurological disorders [6]. Collectively, this constellation of health risks is termed metabolic syndrome. Affecting 35–40% of U.S. adults [7], risk factors for metabolic syndrome – including middle age, sedentary behavior, poor diet, and genetics – are similar to those for OSA. The considerable overlap in biomarkers and health outcomes in obese individuals with and without OSA, thus, makes it difficult to disentangle cause, effect, and whether certain treatments, such as continuous positive airway pressure (CPAP), can improve these outcomes.
In this paper, we provide an update to our 2005 review [8], which proposed the argument that both OSA and metabolic syndrome were, largely, manifestations of central obesity, the common etiological factor of both conditions. In that review, we proposed a theoretical model based largely on cross-sectional associations – that metabolic syndrome and OSA represent, essentially, heads of two similar coins, with a bidirectional, feedforward association driven primarily by visceral obesity and insulin resistance (Figure 1). This updated review includes (a) recent data that digs into physiological and biochemical mechanisms underlying the association between OSA and metabolic syndrome, (b) new data in nonobese men and women, as well as children and adolescents, (c) insight from the latest treatment studies, (d) the important role of aging in understanding clinically-meaningful phenotypes of the disorder, and (e) the potential diagnostic and prognostic utility of biomarkers in identifying OSA patients with the strongest cardiometabolic risk.
Figure 1.
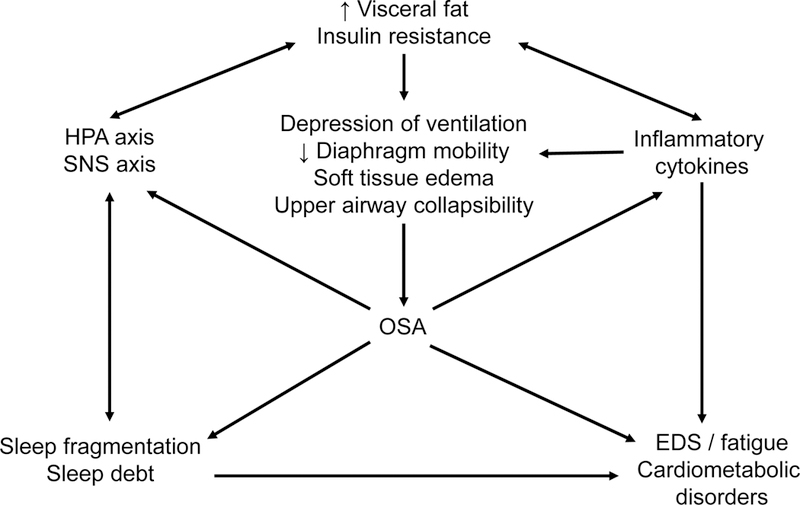
A heuristic model of the feedforward associations between visceral fat, insulin resistance, inflammatory cytokines, stress hormones, excessive daytime sleepiness (EDS) and fatigue, and obstructive sleep apnea (OSA). Adapted from Vgontzas et al., 2005 [8].
Obstructive sleep apnea and metabolic syndrome: the chicken or the egg?
Epidemiological studies have reported that metabolic syndrome is 6 to 9 times more likely to be present in individuals with OSA compared to the general population [9, 10]. The prevalence of OSA peaks around ages 50–59 years for men and 60–69 years for women (Figure 2A) [3, 4]. Particularly in women, OSA is more prevalent following hormonal changes such as menopause, which may explain the delay in peak prevalence compared to men [4]. This quadratic relationship mirrors the prevalence of the metabolic syndrome in U.S. adults according to the Third National Health and Nutrition Examination Survey (NHANES) data collected during the same time frame (1988–1994; Figure 2B) [11].
Figure 2.

The prevalence of clinically-defined OSA in men and women (A), and metabolic syndrome in U.S. men and women (B; National Health and Nutrition Examination Survey [NHANES] 1988–1994). From Ford et al., 2002 and Vgontzas et al., 2005 [8, 11].
Just as the prevalence of metabolic syndrome declines in older age (Figure 2A), the cardiometabolic comorbidities associated with OSA also diminish in parallel. Both cross-sectional [Bixler2000, 13] and prospective [14, 15] studies have demonstrated that the relationship between OSA and hypertension, coronary heart disease, heart failure, and mortality is strongest in the youngest, and that OSA is not a significant risk factor for adverse health outcomes in older men and women. Excessive daytime sleepiness (EDS), a cardinal symptom of OSA, is also strongly and independently associated with obesity and metabolic syndrome [16, 17]; similar to cardiometabolic comorbidities, EDS diminishes in frequency and intensity in older patients with OSA. [97]. Together, these findings suggest that cardiometabolic morbidities in the elderly occur independently from OSA, and that OSA in older age is not as strongly associated with the metabolic syndrome as in younger age. These studies are described in greater detail in the section below entitled Obstructive sleep apnea in the young and middle-aged is a different phenotype than in the elderly.
Visceral fat is the key predictor of both OSA and metabolic syndrome.
Increased prevalence of OSA has mirrored the surge in obesity over the last two decades. A four-year follow-up study in the Wisconsin Sleep Cohort reported that modest (10%) weight gain predicted a 32% increase in AHI and 6-fold odds of developing moderate-to-severe OSA; on the other hand, 10% weight loss predicted a 26% decrease in AHI [18].
While there are multiple definitions of metabolic syndrome featuring different cut-offs for blood pressure, lipid, and glucose levels [19], the presence of abdominal obesity, specifically – defined by either an elevated waist circumference or waist/hip ratio – is central to all definitions. Waist circumference is largely driven by intra-abdominal, or visceral, adipose tissue in the peritoneal cavity, packed between the stomach, liver, kidneys, intestines, and other internal organs. Higher levels of visceral fat have been observed in obese [20] and, importantly, non-obese [21] men with OSA, even when matched with controls for age and body mass index (BMI). Metabolic parameters including cholesterol, triglycerides, homeostatic model assessment (HOMA, a measure of insulin resistance), and inflammatory markers are also elevated in non-obese men and, for some parameters, women with OSA compared to controls [22]. Visceral fat accounts for 5–8% of total body fat in women and increases during menopause, and up to 10–20% in men [23], which may, in large part, explain the sex dimorphism in OSA prevalence. Of note, longitudinal pediatric studies have also demonstrated that incident OSA in adolescents is most strongly predicted by childhood waist circumference [24], and OSA severity in adolescence is positively associated with a greater increase in waist circumference between childhood and adolescence [25].
The mechanism linking central obesity and OSA is likely the result of interplay between various processes. Excess fat tissue, particularly in the abdominal region, is also associated with accumulation of excess fat in the neck, which contributes to upper airway narrowing [26], increased collapsibility due to caudal traction, decreased efficiency of dilator muscle contraction [27], and sarcopenia, denervation, and skeletal muscle dysfunction due to lipid accumulation [28] and increased inflammatory gene expression [29]. While the mechanisms by which obesity causes OSA remain incompletely understood, we propose that local and systemic inflammation may be a partially contributory factor, as outlined in the following sections.
Systemic inflammation in OSA is largely derived from visceral fat.
Compared to age-and BMI-matched controls, patients with OSA demonstrate significantly elevated plasma interleukin-6 (IL-6), tumor necrosis factor alpha (TNFα) [20, 30], and C-reactive protein (CRP) levels [31, 32]. Levels of CRP have been positively correlated with AHI, arousal index, and oxygen desaturation [33, 34].
OSA-associated inflammation is thought to arise from two major sources. For one, mechanical damage due to snoring, breathing effort, and upper airway obstruction is associated with elevated immune cells in nasal and oropharyngeal mucosa, breath condensate, and sputum [35–38]. A study by Sériès and colleagues [39] comparing OSA patients and age-/BMI-matched controls, however, demonstrated that inflammation in the uvula is correlated with BMI, but not with AHI, suggesting that obesity per se produces tissue inflammation in structures subject to mechanical damage. Second, OSA patients experience a unique pattern of oxygen deficiency – termed intermittent hypoxia (IH) – in which short, repetitive cycles of oxygen desaturation are followed by rapid reoyxgenation of tissue. In an in-vitro model, Ryan and colleagues [40] have described how IH, but not sustained hypoxia (SH), preferentially activates inflammatory pathways mediated by the transcription factor nuclear factor kappa B (NF-κB) which increase the transcription of genes coding for TNFα, interleukins, and other immune proteins.
While OSA is an independent predictor of elevated proinflammatory cytokines, there is considerable evidence in adult and child literature, however, suggesting that central obesity largely precedes systemic inflammation, metabolic dysfunction, and subsequent development of OSA. Obesity per se is recognized as a chronic, low-grade inflammatory state. In addition to adipocytes, fibroblasts, and vascular endothelial cells, adipose tissue contains immune cells such as mast cells, eosinophils, B cells, T cells, and macrophages [41]. Compared to subcutaneous fat, visceral fat contains proportionally more adipocytes that are capable of growing quite large before dividing [42]; as the size of these lipid droplets increases with weight gain, the adipocytes themselves begin secreting low levels of TNFα. In turn, TNFα stimulates preadipocytes and surrounding endothelial cells to produce monocyte chemoattract protein-1 (MCP-1), promoting macrophage recruitment and adhesion to endothelial cells [43]. As adipocytes continue to grow and divide, endothelial damage due to oxidative stress and cell crowding recruits additional macrophages [44], which secrete TNFα, IL-6, IL-1β, and other proinflammatory cytokines. As a result, plasma levels of acute-phase reactant proteins, such as CRP, become elevated. Taken together, obesity – particularly visceral obesity – creates a proinflammatory state that perpetuates a vicious cycle of macrophage recruitment, impaired adipocyte functioning, and activation of transcription factors for genes encoding proinflammatory proteins [44].
Examining OSA in children and adolescents presents a unique vantage point into the early pathophysiology of the disorder, as the cases are new-onset and are often not confounded by multiple diseases. In a recent study of the Penn State Child Cohort, we demonstrated that visceral fat area is significantly elevated in adolescents with new-onset OSA (AHI ≥ 5). Importantly, mediation analysis revealed that 82% of the association between visceral fat and OSA in adolescents was mediated by CRP levels, while 42% was mediated by IL-6 (Figure 3A). Although the model cannot account for other potentially important factors that were not included, our findings suggest that the release of proinflammatory cytokines by visceral adipocytes may explain, to some degree, the association between central obesity and incident OSA [45]. In a longitudinal subsample of the Cohort, we also demonstrated that greater increases in waist circumferences from childhood to adolescence (∆waist) predicted higher adolescent AHI, and that this association may be accounted for by increases in CRP from childhood to adolescence (∆CRP) [25; Figure 3B]. Interestingly, pre- and perinatal factors such as chorioamnionitis, maternal smoking, weight gain during pregnancy, gestational diabetes, and prematurity have also been associated with childhood OSA [46–48], with inflammation during this critical period hypothesized as a possible mechanism.
Figure 3.
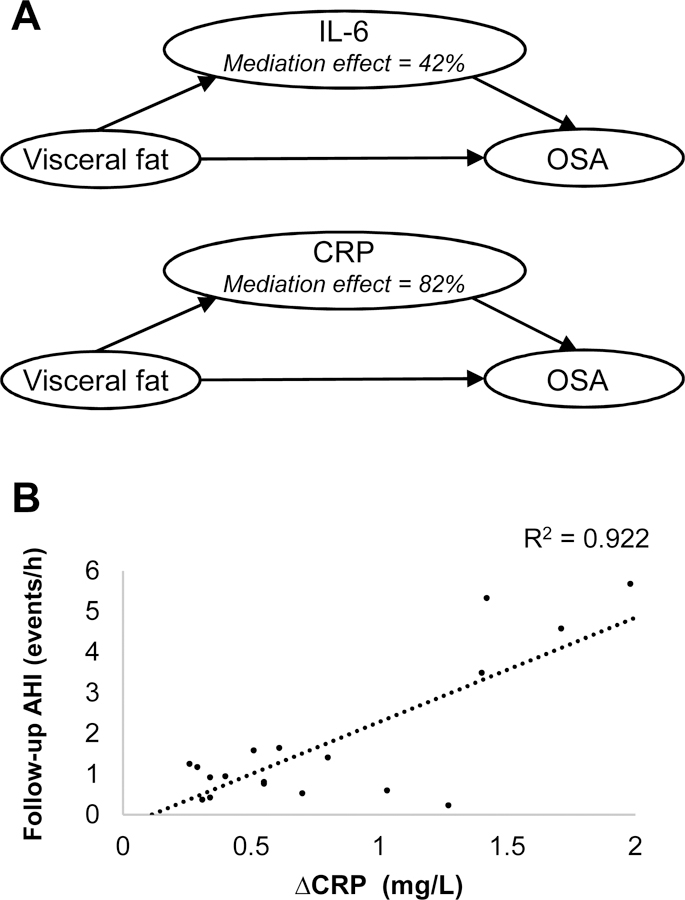
Model (A) for the association between visceral adiposity and moderate OSA, as mediated by IL-6 and CRP, in adolescents with incident OSA. Scatterplot (B) depicting the association of increased CRP from childhood to adolescence (∆CRP) with adolescent AHI in boys. Adapted from Gaines et al., 2016 and Gaines et al., 2017a [25, 45].
Obstructive sleep apnea prevalence is higher in disorders of insulin resistance.
Visceral fat tissue is rich in vasculature and readily contributes adipokines into the bloodstream and nervous system [42]; compared to subcutaneous fat, the proximity of visceral fat to vasculature, central nerves, and critical organs such as the liver make this tissue type relatively more dangerous to overall health. Visceral adipose tissue also has a relatively high concentration of glucocorticoid, androgen, and adrenergic receptors, making the tissue more susceptible to stress hormone, catecholamine, and testosterone signaling [49, 50]. Furthermore, as visceral adipocytes accumulate larger lipid droplets with weight gain, they become dysfunctional, adopting a higher rate of glucose uptake and developing insulin resistance [51]. Through these pathways, increased waist circumference and elevated visceral fat have been strongly linked to metabolic syndrome, cardiovascular events, and cardiovascular and all-cause mortality, particularly in men [52, 53].
TNFα levels have been associated with insulin resistance in individuals both with and without type 2 diabetes [54, 55]. Given the well-known association between obesity and inflammation with insulin resistance, it is unsurprising that many studies have reported associations between OSA severity and degree of insulin resistance [20, 56, 57], even in non-obese subjects [22, 58]. Several experimental [59, 60] and observational [61, 62] studies in both humans and animals have demonstrated that hypoxic stress contributes to acute and long-term elevations in glucose, insulin, and/or hemoglobin A1c (HbA1c) levels.
At the same time, while many studies have explored the hypothesis that OSA causes chronic insulin resistance and diabetes, the relative contribution of OSA per se, compared to obesity and other metabolic factors, is not as strong. For instance, in the Wisconsin Sleep Cohort, Reichmuth and colleagues demonstrated that although the prevalence of type 2 diabetes was significantly higher in subjects with OSA, the risk of developing diabetes over a four-year follow-up period was not, suggesting that OSA may be only weakly causal in the development of the disorder [63]. Furthermore, a more recent prospective study reported that baseline fasting insulin, HOMA, and triglycerides significantly predicted OSA six years later, even after adjusting for age, sex, and waist circumference; interestingly, however, baseline OSA did not predict elevated insulin levels at follow-up. Unsurprisingly, baseline waist circumference was the strongest predictor of incident OSA in this study [64]. A recent study in patients with moderate-to-severe OSA reported that CPAP withdrawal caused dynamic increases in nocturnal glucose levels; however, this was observed primarily in those who were already diabetic [60]. While the hypoxemia, sleep fragmentation, and sympathetic nervous system activation associated with OSA undoubtedly exacerbate metabolic aberrations, considerable data suggest that metabolic factors largely precede the development of the disorder.
As we and others have demonstrated, OSA is very frequent in disorders in which insulin resistance is a primary pathophysiologic abnormality. Polycystic ovarian syndrome (PCOS) is the most common endocrine disorder in women of reproductive age, characterized by oligoovulation, hyperandrogenism, obesity, and insulin resistance. Compared to pre-menopausal women without PCOS, we have reported that those with PCOS are 30.6 times more likely to also suffer from sleep-disordered breathing (SDB), including snoring, and 28.7 times more likely to have OSA (AHI ≥ 10) [65]; later studies have confirmed this finding [66–68]. Obese women with PCOS have significantly higher fasting insulin levels than non-obese women with PCOS [69]; we have reported that insulin resistance is the strongest risk factor for OSA in women with PCOS – stronger than even BMI or testosterone [65].
The association between insulin resistance states and OSA severity has even been demonstrated as young as childhood. We have previously demonstrated that the frequency of EDS increases with increasing BMI and OSA severity in children aged 5–17 years, with significant linear increases in IL-6, TNF receptor-1 (TNFR1), CRP, and leptin, and decreases in adiponectin [70]. Interestingly, just as elevations in inflammation from childhood to adolescence predict OSA in adolescence [25], a recent prospective study demonstrated that metabolic exposures in mid-childhood (age 9) – including adiposity, leptin, HOMA, CRP, and global metabolic risk – predicted SDB just four years later at the early teenage visit (mean age 13.2 years), even after adjusting for age, sex, BMI, race, and other confounders [71].
Insight from treatment studies
As the gold-standard treatment for OSA, CPAP devices apply continuous mild air pressure to keep airways open, allowing patients to breathe spontaneously on their own. CPAP use has been consistently associated with higher sleep efficiency, fewer arousals, lower AHI, improved oxygen saturation, and decreased daytime sleepiness [72].
Despite these beneficial effects, however, a recent meta-analysis concluded that CPAP does not significantly improve lipid levels, insulin resistance, inflammatory markers, or the proportion of patients with the metabolic syndrome [73]. Specifically, CPAP does not significantly reduce IL-6, TNFα, TNFR1 [74, 75], CRP [76], leptin [22, 77], nor fasting insulin and glucose [74], even after comparing groups with low versus high CPAP compliance [22]. A sham-controlled study also demonstrated no reversal of metabolic syndrome after 12 weeks of CPAP use [78], and a recent study demonstrated that CPAP did not significantly reduce HbA1 levels in individuals with Type 2 diabetes [79]. While several of these studies have been criticized for short follow-up periods (≤ three months), another recent paper demonstrates that even one year of CPAP use does not significantly alter levels of CRP, IL-8, or TNFα in patients with OSA and coronary artery disease [80]. Interestingly, in this study, even though IL-6 levels decreased with CPAP use, IL-6 levels had also decreased in parallel in the non-CPAP group, which may have been due to other lifestyle changes (e.g. diet, exercise, and weight reduction), about which the non-CPAP group received education [80]. In another recent randomized trial, improvements in inflammation, insulin resistance, and serum triglycerides were only observed in patients with OSA who combined their CPAP use with weight loss during a 24-week period [81].
On the other hand, while CPAP use largely does not improve metabolic parameters nor reverse metabolic syndrome, there is evidence suggesting that OSA is associated with dynamic increases in cortisol in proportion to OSA severity [60] and elevated 24-hour and nighttime cortisol levels [82] and are, in fact, corrected with CPAP therapy [82–84]. The intermittent hypoxia, subsequent autonomic arousal, and frequent nocturnal awakenings are hypothesized to create a state of sympathetic hyperarousal. Despite little to no effect on other metabolic parameters, longer-term CPAP use (three months) has been associated with modest reductions in blood pressure [74, 85].
Taken together, these treatment studies suggest that metabolic factors (including inflammatory markers, lipids, and markers of insulin resistance) are not reversed by improved AHI and sleep efficiency unless accompanied by long-term weight loss. Sympathetic activation and increased blood pressure as a consequence of intermittent hypoxia, however, may be improved by CPAP therapy. Of note, the relatively high failure rate of upper airway surgeries for patients with OSA, such as uvulopalatopharyngoplasty (UPPP) [86], is even higher in obese patients [87], further suggesting that obesity and metabolic factors may play a contributing role in the pathophysiology of the disorder.
In line with the hypothesis that OSA stems from a systemic cause, several pilot treatment studies employing anti-inflammatory therapy have modestly reduced OSA severity in both children and adults without CPAP treatment or surgical intervention. In a placebo-controlled, double-blind study of eight obese men with severe OSA (AHI = 52.8 ± 9.1 events/h), a three-week trial of the TNFα antagonist etanercept significantly reduced AHI (to 44.3 ± 10.3 events/h), IL-6 levels, and objective daytime sleepiness (as measured by the Multiple sleep latency test [MSLT]) [88]. Several randomized controlled trials in children, spanning across several weeks to months, also report reduced AHI and relative normalization of sleep parameters with intranasal corticosteroid treatment in combination with oral anti-inflammatory medication, despite no significant reductions in adenotonsillar size [89–93]. An emerging body of literature also reports an association between OSA and autoimmune and rheumatic disease populations, particularly rheumatoid arthritis [94, 95]. Common to both conditions are increased levels of circulating proinflammatory cytokines; however, the potential direction of this association has not been explicitly examined. Interestingly, however, a recent study reported a significantly lower prevalence of OSA in patients with spondyloarthritis who take TNF inhibitors compared to those who do not [96].
There are certainly limitations to drawing physiological conclusions from these studies given a lack of rigorous data, mostly due to methodological limitations in being able to conduct the type of cellular- and molecular-level analyses necessary in human patients with OSA. Future clinical trials, as well as animal studies of obesity-derived OSA, will better shed light on these complex mechanisms. Taken together, however, these studies do lend evidence that systemic inflammation may play some role in the development of OSA, and that anti-inflammatory therapies, as well as therapies designed to reduce visceral adiposity – compared to CPAP, upper airway surgery, or other mechanical treatments – may address the systemic root cause of OSA for many patients.
Obstructive sleep apnea in the young and middle-aged is a different phenotype than in the elderly
As mentioned earlier in this review, while the prevalence of OSA based on clinical criteria tends to peak in middle age, the cardiometabolic comorbidities associated with OSA tend to diminish. We have previously reported in the Penn State Adult Cohort that, cross-sectionally, the prevalence of OSA based on AHI criteria alone (i.e. AHI ≥ 10, with or without accompanying symptoms) continues to increase linearly after 65 years of age; however, the peak prevalence based on comorbid AHI and clinical criteria is around age 55 years for men and 65 years for women (Figure 2A) [3]. Furthermore, oxygen desaturation in older individuals was significantly less in the elderly, suggesting the possibility that OSA in the young and middle-aged is more severe [3].
Subsequent cross-sectional [12, 13] and prospective [14, 15] studies have reported that the associations between AHI and hypertension [12, 13], incident coronary heart disease and heart failure [15], and even mortality [14] are not as strong, or are even non-significant, in the elderly compared to the young and middle-aged. In a recent 10-year follow-up of the Penn State Adult Cohort, we report that both mild (OR = 4.35) and moderate (OR = 3.80) OSA are associated with increased odds for developing incident hypertension; however, this association is only observed in those < 60 years (p = 0.01) and lost in the elderly. Furthermore, young and middle-aged subjects with OSA also have higher rates of obesity, increased BMI, and higher fasting blood glucose and triglyceride levels compared to those ≥ 60 years with incident OSA [97]. Together, these findings suggest that cardiometabolic morbidities in the elderly occur independently from OSA, and that OSA in older age is not as strongly associated with the metabolic syndrome as in younger age.
Several physiological mechanisms have been hypothesized to explain this observation. A decade ago, Kirkness and colleagues studied the effects of age on pharyngeal pressure under hypotonic conditions (passive Pcrit) during sleep in men and women with and without OSA. The authors reported that OSA increased in severity when passive Pcrit rose above −5 cmH2O, with a predictive power of 0.73. Passive Pcrit was significantly higher in men than women, and increased with age [98]. Edwards et al. expanded on these findings in 2014 in a small study of ten young (20–40 years) and old (≥60 years) patients with OSA matched for sex and BMI, measuring collapsibility, loop gain, upper airway muscle responsiveness, and respiratory arousal threshold during drops in positive airway pressure. Compared to younger patients with OSA, older patients had, on average, a more collapsible airway, lower ventilation prior to the pressure drop, and a lower loop gain. The authors concluded that airway anatomy and collapsibility play a relatively greater role in the pathogenesis of OSA in older adults [99]. In line with these studies, a recent longitudinal study of elderly patients with OSA showed no association between dual X-ray absorptiometry (DXA)-measured central obesity and changes in AHI across an 8-year period [100]. Taken together, these findings are in line with the notion that OSA in older age is a distinctly different phenotype than in the young – due to an increasingly independent association with cardiovascular and metabolic comorbidities – and differential diagnosis, prognosis, and treatment options should be considered in this population.
In search of a clinically-useful biomarker of cardiometabolic risk
As detailed throughout this review, (a) markers of metabolic dysfunction, such as inflammation, are highly prevalent in OSA, and (b) OSA treatments, while successful at reducing AHI, do not significantly improve metabolic parameters unless combined with weight loss. While it is clear that treatment should be pursued in more severe cases of OSA (AHI ≥ 30), OSA in the mild (5 ≤ AHI < 15) to moderate (15 ≤ AHI < 30) range presents a challenge, as AHI alone cannot satisfactorily predict the development of OSA-associated comobidities – particularly when patients do not complain of excessive daytime sleepiness or other troubling symptoms. Furthermore, the gold-standard treatment, CPAP, has relatively poor compliance; 31% of patients with OSA who are prescribed CPAP never commence therapy after diagnosis and titration, and an additional 15% abandon their machines after 10 months of use [101]. CPAP devices can also be highly disturbing to both the patient and their bed partner due to discomfort and noise [102, 103].
A recent joint biomarker workshop sponsored by the National Heart Lung and Blood Institute, National Institute on Aging, and the Sleep Research Society advised that the sleep field lacks a “unique biomarker signature that is context -relevant, simple and easy to use” that can help clinicians determine a patient’s long-term prognosis [104]. Furthermore, a recent editorial highlights the importance of identifying precise, cost-effective biomarkers for cardiometabolic risk that allow clinicians to identify OSA patients who are most likely to benefit from therapy [105]. We have previously demonstrated that inflammation (specifically, CRP) is elevated in a dose-response manner across OSA patients with hypertension, OSA patients without hypertension, and controls, suggesting that inflammation may be a clinically useful marker of OSA severity and comorbid cardiovascular problems [106]. We recently demonstrated that objectively-measured daytime sleepiness (via MSLT), but not subjectively-measured sleepiness (Epworth sleepiness scale), correlates with 24-hour IL-6 [107]. Furthermore, CRP has been shown to partially mediate the association between AHI and incident type 2 diabetes [108], suggesting its potential efficacy in discriminating which OSA patients may have a worse prognosis.
In a new study [109], we compared the relative utility of AHI versus CRP in identifying the presence and severity of hypertension and hyperglycemia in 60 middle-aged adults with mild-to-moderate OSA (5 ≤ AHI < 30). We found that CRP levels were associated with greater odds for having hypertension (OR = 1.42, p = 0.08) and hyperglycemia (OR = 1.83, p = 0.01) compared to AHI (both p > 0.12). Furthermore, receiver-operating characteristics (ROC) curves demonstrated that adding a measure of CRP to standard clinical factors (age, sex, and BMI) yielded moderately good to strong risk prediction models for the disorders (AUC = 0.72 and AUC = 0.81, respectively), while adding AHI did not (both AUC < 0.69). According to these models, a “typical” obese (BMI = 30 kg/m 2), middle‐aged (50 years) man with AHI = 15 has a 36.6% probability of having hypertension and 25.7% probability of having hyperglycemia if their CRP = 0.5 mg/L (healthy levels); these probabilities increase to 58.1% and 60.2%, respectively, if their CRP levels are increased to 3.0 mg/L (“at risk” levels; Figure 4A). Similarly, an obese, middle‐aged woman with AHI = 10 has a 15.3% and 8.3% probability of hypertension and hyperglycemia, respectively, if their CRP = 0.5 mg/L; these probabilities increase to 30.2% and 28.4% if their CRP = 3.0 mg/L (Figure 4B). Our findings are also interesting given the results of another recent study [110] showing that a combination of elevated CRP, hemoglobin A1c (HbA1c, a measure of plasma glucose levels), and erythropoietin (a cytokine released in response to cellular hypoxia) levels provided the greatest predictive power for the presence of moderate-to-severe OSA (AUC = 0.84), with high sensitivity (85%) and specificity (79%). No current test for detecting OSA, however, can replace the gold-standard polysomnography.
Figure 4.

Probabilities of hypertension and hyperglycemia in “typical” obese, middle-aged men (A) and women (B) with mild-to-moderate OSA when CRP levels are in the healthy (0.5 mg/L) vs. “at-risk” (3.0 mg/L) range. From Gaines et al., 2017b [108].
Although CRP represents a nonspecific marker of inflammation, we propose that incorporating a patient’s CRP levels through routine bloodwork – along with other standard clinical sleep measures – may enhance the ability f or clinicians to detect cases of mild-to-moderate OSA with true cardiometabolic risk, thus improving the accuracy of prognosis and clarifying which treatment option may be most beneficial to the patient. Future studies should follow up patients over a period of several years to explore whether CRP levels at baseline predict the severity of future cardiometabolic sequelae, more so than AHI alone.
Conclusions
At the time that our 2005 review [8] was published, our proposed heuristic model (Figure 1) was largely theoretical – based primarily on cros s-sectional associations and astute clinical observation of patients with OSA who did and did not present with comorbid metabolic syndrome. Our proposed research agenda at the time detailed the need to examine the association of OSA and metabolic syndrome in non-obese populations and children; explore the potential for anti-inflammatory agents in reducing OSA severity; and a need for prospective studies on the mechanisms underlying the consequences of lifestyle changes, such as weight gain and weight loss, on OSA prevalence, incidence, and severity.
In the past decade, we and others have been able to better unravel these complexmechanisms via in-vitro and animal models, studies in children and adolescents, and prospective observational and treatment studies. And our conclusions largely remain the same: there is a bi-directional, feedforward association between metabolic syndrome and OSA.
Visceral obesity and insulin resistance – determine d by genetic, constitutional, and environmental factors – are the principal culprits leading to OSA, and these associations may be driven by a chronic, low-grade inflammatory state. In turn, central obesity and inflammation may lead to upper airway narrowing, respiratory muscle fatigue, and decreased dilator muscle contraction. Furthermore, these mechanisms may explain why mechanical treatments, such as CPAP or upper airway surgeries, are often unsuccessful at reversing metabolic syndrome unless combined with weight loss. Given our significant progress in better understanding these mechanisms over the last decade, it is certainly a worthwhile endeavor for future studies to focus on the use of biomarkers for improving the accuracy of OSA diagnosis and prognosis, and for clarifying which treatment option may be most beneficial to the patient.
But in the meantime, our recommendations remain the same: for the average patient with OSA, weight management, healthy eating habits, and regular exercise should obvious, complement to treatment for any patient with OSA.
Practice Points:
The relationship between age and OSA across the lifespan mirrors that of age and metabolic syndrome.
The increased prevalence of OSA has followed the surge in obesity over the last two decades.
Visceral fat is the key predictor for both metabolic syndrome and OSA, even in non-obese and younger populations.
Increased systemic inflammation and insulin resistance in patients with OSA are largely derived from visceral adiposity.
Continuous positive airway pressure does not reverse inflammation, insulin resistance, or elevated lipid levels, but lowers cortisol and has a modest effect on blood pressure.
OSA in the elderly is more strongly associated with upper airway collapsibility, not metabolic abnormalities, and may represent a different phenotype.
C-reactive protein may be a useful biomarker for improving the accuracy of diagnosis and prognosis of mild-to-moderate OSA, and for clarifying which treatment option may be most beneficial to the patient.
Research Agenda:
Future studies should:
Follow up patients of a wide age spectrum over a period of several years to explore whether biomarker levels at baseline, such as CRP, can predict the severity of future cardiometabolic sequelae.
Explore the characteristics and mechanisms that delineate OSA phenotypes (for example, in young and middle-aged vs. older individuals).
Study how inflammatory and metabolic molecules affect central and peripheral breathing mechanisms in animals and humans.
Assess the impact of lifestyle or pharmacological therapies that reduce inflammation or metabolic syndrome (alone, or in combination with existing treatments such as CPAP), on OSA and associated adverse health outcomes.
Acknowledgments:
Several research studies discussed in this review were funded in part by the National Institutes of Health grants R01 51931, R01 40916, R01 HL64415, R01 HL-63772, R01 HL-97165, UL1 RR033184, and C06 RR6499.
Abbreviations
- AHI
apnea-hypopnea index
- BMI
body mass index
- CPAP
continuous positive airway pressure
- CRP
C-reactive protein
- DXA
dual X-ray absorptiometry
- EDS
excessive daytime sleepiness
- HbA1c
hemoglobin A1c
- HDL
high-density lipoprotein
- HOMA
homeostatic model assessment
- IH
intermittent hypoxia
- IL-6
interleukin-6
- MSLT
Multiple sleep latency test
- OSA
obstructive sleep apnea
- SDB
sleep-disordered breathing
- SpO2
blood oxygen saturation
- TNFα
tumor necrosis factor alpha
- TNFR1
tumor necrosis factor receptor-1
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest: The authors have no conflicts of interest to disclose.
References
- 1.Leger D, Bayon V, Laaban JP, Philip P. Impact of sleep apnea on economics. Sleep Med Rev 2012, 16: 455–62. [DOI] [PubMed] [Google Scholar]
- 2.Young T, Palta M, Dempsey J, Skatrud J, Weber S, Badr S. The occurrence of sleep-disordered breathing among middle-aged adults. N Engl J Med 1993, 328: 1230–5. [DOI] [PubMed] [Google Scholar]
- 3.*.Bixler EO, Vgontzas AN, Ten Have T, Tyson K, Kales A. Effects of age on sleep apnea in men: I. Prevalence and severity. Am J Respir Crit Care Med 1998; 157: 144–8. [DOI] [PubMed] [Google Scholar]
- 4.*.Bixler EO, Vgontzas AN, Lin HM, Ten Have T, Rein J, Vela-Bueno A,et al. Prevalence of sleep-disordered breathing in women: effects of gender. Am J Respir Crit Care Med 2001; 163: 608–13. [DOI] [PubMed] [Google Scholar]
- 5.Peppard PE, Young T, Barnet JH, Palta M, Hagen EW, Hla KM. Increased prevalence of sleep-disordered breathing in adults. Am J Epidemiol 2013, 177: 1006–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ford ES. Risks for all-cause mortality, cardiovascular disease, and diabetes associated with the metabolic syndrome: a summary of the evidence. Diabetes Care 2005; 28: 1769–78. [DOI] [PubMed] [Google Scholar]
- 7.Ford ES. Prevalence of the metabolic syndrome defined by the International Diabetes Federation among adults in the U.S. Diabetes Care 2005, 28: 2745–9. [DOI] [PubMed] [Google Scholar]
- 8.*.Vgontzas AN, Bixler EO, Chrousos GP. Sleep apnea is a manifestation of the metabolic syndrome. Sleep Med Rev 2005, 9: 211–24. [DOI] [PubMed] [Google Scholar]
- 9.Coughlin SR, Mawdsley L, Mugarza JA, Calverley PM, Wilding JP. Obstructive sleep apnoea is independently associated with an increased prevalence of metabolic syndrome. Eur Heart J 2004; 25: 735–41. [DOI] [PubMed] [Google Scholar]
- 10.Gruber A, Horwood F, Sithole J, Ali NJ, Idris I. Obstructive sleep apnoea is independently associated with the metabolic syndrome but not insulin resistance state. Cardiovasc Diabetol 2006; 5: 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ford ES, Giles WH, Dietz WH. Prevalence of the metabolic syndrome among US adults: findings from the third National Health and Nutrition Examination Survey. JAMA 2002; 287: 356–9. [DOI] [PubMed] [Google Scholar]
- 12.*.Bixler EO, Vgontzas AN, Lin HM, Ten Have T, Leiby BE, Vela-Bueno A, et al. Association of hypertension and sleep-disordered breathing. Arch Intern Med 2000; 160: 2289–95. [DOI] [PubMed] [Google Scholar]
- 13.Sjöström C, Lindberg E, Elmasry A, Hägg A, Svärdsud d K, Janson C. Prevalence of sleep apnoea and snoring in hypertensive men: a population based study. Thorax 2002; 57: 602–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lavie P, Lavie L. Unexpected survival advantage in elderly people with moderate sleep apnoea. J Sleep Res 2009; 18: 397–403. [DOI] [PubMed] [Google Scholar]
- 15.Gottlieb DJ, Yenokyan G, Newman AB, O’Connor GT, Punjabi NM, Quan SF, et al. Prospective study of obstructive sleep apnea and incident coronary heart disease and heart failure: the Sleep Heart Health Study. Circulation 2010; 122: 352–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bixler EO, Vgontzas AN, Lin HM, Calhoun SL, Vela-Bueno A, Kales A. Excessive daytime sleepiness in a general population sample: the role of sleep apnea, age, obesity, diabetes, and depression. J Clin Endocrinol Metab 2005; 90: 4510–5. [DOI] [PubMed] [Google Scholar]
- 17.Vgontzas AN. Excessive daytime sleepiness in sleep apnea: it is not just apnea hypopnea index. Sleep Med 2008; 9: 712–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.*.Peppard PE, Young T, Palta M, Dempsey J, Skatrud J. Longitudinal study of moderate weight change and sleep-disordered breathing. JAMA 2000; 284: 3015–21. [DOI] [PubMed] [Google Scholar]
- 19.Huang PL. A comprehensive definition for metabolic syndrome. Dis Model Mech 2009; 2: 231–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vgontzas AN, Papanicolaou DA, Bixler EO, Hopper K, Lotsikas A, Lin HM, et al. Sleep apnea and daytime sleepiness and fatigue: relation to visceral obesity, insulin resistance, and hypercytokinemia. J Clin Endocrinol Metab 2000; 85: 1151–8. [DOI] [PubMed] [Google Scholar]
- 21.Kritikou I, Basta M, Tappouni R, Pejovic S, Fernandez-Mendoza J, Nazir R, et al. Sleep apnoea and visceral adiposity in middle-aged male and female subjects. Eur Respir J 2013; 41: 601–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kritikou I, Basta M, Vgontzas AN, Pejovic S, Liao D, Tsaoussoglou M, et al. Sleep apnoea, sleepiness, inflammation and insulin resistance in middle-aged males and females. Eur Respir J 2014; 43: 145–55. [DOI] [PubMed] [Google Scholar]
- 23.Wajchenberg BL. Subcutaneous and visceral adipose tissue: their relation to the metabolic syndrome. Endocr Rev 2000; 21: 697–738. [DOI] [PubMed] [Google Scholar]
- 24.Bixler EO, Fernandez-Mendoza J, Liao D, Calhoun SL, Rodriguez-Colon SM, Gaines J, He F, Vgontzas AN. Natural history of sleep disordered breathing in prepubertal children transitioning to adolescence. Eur Respir J 2016; 47: 1402–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.*.Gaines J, Vgontzas AN, Fernandez-Mendoza J, He F, Calhoun SL, Liao D, et al. Increased inflammation from childhood to adolescence predicts sleep apnea in boys: A preliminary study. Brain Behav Immun 2017; 64: 259–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lubrano C, Saponara M, Barbaro G, Specchia P, Addessi E, Costantini D, et al. Relationships between body fat distribution, epicardial fat and obstructive sleep apnea in obese patients with and without metabolic syndrome. PLoS One 2012; 7: e47059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Deegan PC, McNicholas WT. Pathophysiology of obstructive sleep apnoea. Eur Respir J 1995; 8: 1161–78. [DOI] [PubMed] [Google Scholar]
- 28.Gueugneau M, Coudy-Gandilhon C, Théron L, Meunier B, Barboiron C, Combaret L, et al. Skeletal muscle lipid content and oxidative activity in relation to muscle fiber type in aging and metabolic syndrome. J Gerontol A Biol Sci Med Sci 2015; 70: 566–76. [DOI] [PubMed] [Google Scholar]
- 29.Poelkens F, Lammers G, Pardoel EM, Tack CJ, Hopman MT. Upregulation of skeletal muscle inflammatory genes links inflammation with insulin resistance in women with the metabolic syndrome. Exp Physiol 2013; 98: 1485–94. [DOI] [PubMed] [Google Scholar]
- 30.Yokoe T, Minoguchi K, Matsuo H, Oda N, Minoguchi H, Yoshino G, et al. Elevated levels of C-reactive protein and interleukin-6 in patients with obstructive sleep apnea syndrome are decreased by nasal continuous positive airway pressure. Circulation 2003; 1129–34. [DOI] [PubMed] [Google Scholar]
- 31.Shamsuzzaman AS, Winnicki M, Lanfranchi P, Wolk R, Kara T, Accurso V, et al. Elevated C-reactive protein in patients with obstructive sleep apnea. Circulation 2002; 2462–4. [DOI] [PubMed]
- 32.Chien MY, Lee P, Tsai YF, Yang PC, Wu YT. C-reactive protein and heart rate recovery in middle-aged men with severe obstructive sleep apnea. Sleep Breath 2012; 16: 629–37. [DOI] [PubMed] [Google Scholar]
- 33.Tauman R, Ivanenko A, O’Brien LM, Gozal D. Plasma C-reactive protein levels among children with sleep-disordered breathing. Pediatrics 2004; 113: e564–9. [DOI] [PubMed] [Google Scholar]
- 34.Larkin EK, Rosen CL, Kirchner HL, Storfer-Isser A, Emancipator JL, Johnson NL, et al. Variation of C-reactive protein levels in adolescents: association with sleep-disordered breathing and sleep duration. Circulation 2005; 111: 1978–84. [DOI] [PubMed] [Google Scholar]
- 35.Rubinstein I Nasal inflammation in patients with obstructive sleep apnea. Laryngoscope 1995; 105: 175–7. [DOI] [PubMed] [Google Scholar]
- 36.Sekosan M, Zakkar M, Wenig BL, Olopade CO, Rubinstein I. Inflammation in the uvula mucosa of patients with obstructive sleep apnea. Laryngoscope 1996; 106: 1018–20. [DOI] [PubMed] [Google Scholar]
- 37.Olopade CO, Christon JA, Zakkar M, Hua C, Swedler WI, Scheff PA, Rubinstein I. Exhaled pentane and nitric oxide levels in patients with obstructive sleep apnea. Chest 1997; 111: 1500–4. [DOI] [PubMed] [Google Scholar]
- 38.Goldbart AD, Krishna J, Li RC, Serpero LD, Gozal D. Inflammatory mediators in exhaled breath condensate of children with obstructive sleep apnea syndrome. Chest 2006; 130: 143–8. [DOI] [PubMed] [Google Scholar]
- 39.Sériès F, Chakir J, Boivin D. Influence of weight and sleep apnea status on immunologic and structural features of the uvula. Am J Resp Crit Care Med 2004; 170: 1114–9. [DOI] [PubMed] [Google Scholar]
- 40.Ryan S, Taylor CT, McNicholas WT. Selective activation of inflammatory pathways by intermittent hypoxia in obstructive sleep apnea syndrome. Circulation 2005; 112: 2660–7. [DOI] [PubMed] [Google Scholar]
- 41.Schipper HS 1, Prakken B, Kalkhoven E, Boes M Adipose tissue-resident immune cells: key players in immunometabolism. Trends Endocrinol Metab 2012; 23: 407–15. [DOI] [PubMed] [Google Scholar]
- 42.Ibrahim MM. Subcutaneous and visceral adipose tissue: structural and functional differences. Obes Rev 2010; 11: 11–8. [DOI] [PubMed] [Google Scholar]
- 43.Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest 2003; 112: 1821–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wellen KE, Hotamisligil GS. Obesity-induced inflammatory changes in adipose tissue. J Clin Invest 2003; 112: 1785–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.*.Gaines J, Vgontzas AN, Fernandez-Mendoza J, Calhoun SL, He F, Liao D, et al. Inflammation mediates the association between visceral adiposity and obstructive sleep apnea in adolescents. Am J Physiol Endocrinol Metab 2016; 311: E851–E858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hibbs AM, Johnson NL, Rosen CL, Kirchner HL, Martin R, Storfer-Isser A, et al. Prenatal and neonatal risk factors for sleep disorder breathing in school-aged children born preterm. J Pediatr 2008; 153: 176–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Calhoun SL, Vgontzas AN, Mayes SD, Tsaoussoglou M, Sauder K, Mahr F, et al. Prenatal and perinatal complications: is it the link between race and SES and childhood sleep disordered breathing? J Clin Sleep Med 2010; 6: 264–9. [PMC free article] [PubMed] [Google Scholar]
- 48.Tapia IE, Shults J, Doyle LW, Nixon GM, Cielo CM, Traylor J, et al. Prenatal risk factors associated with the obstructive sleep apnea syndrome in school-aged children born preterm. Sleep 2016; 39: 737–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rebuffé-Scrive M, Lundholm K, Björntorp P. Glucocorticoid hormone binding to human adipose tissue. Eur J Clin Invest 1985; 15: 267–71. [DOI] [PubMed] [Google Scholar]
- 50.Arner P, Hellström L, Wahrenberg H, Brönnegård M. B eta-adrenoceptor expression in human fat cells from different regions. J Clin Invest 1990; 86: 1595–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mårin P, Andersson B, Ottosson M, Olbe L, Chowdhury B, Kvist H, et al. The morphology and metabolism of intraabdominal adipose tissue in men. Metabolism 1992; 41: 1242–8. [DOI] [PubMed] [Google Scholar]
- 52.Dobbelsteyn CJ, Joffres MR, MacLean DR, Flowerdew G. A comparative evaluation of waist circumference, waist-to-hip ratio and body mass index as indicators of cardiovascular risk factors. The Canadian Heart Health Surveys. Int J Obes Relat Metab Disord 2001; 25: 652–61. [DOI] [PubMed] [Google Scholar]
- 53.Kuk JL, Katzmarzyk PT, Nichaman MZ, Church TS, Blair SN, Ross R. Visceral fat is an independent predictor of all-cause mortality in men. Obesity (Silver Spring) 2006; 14: 336–41. [DOI] [PubMed] [Google Scholar]
- 54.Hotamisligil GS, Spiegelman BM. Tumor necrosis factor alpha: a key component of the obesity-diabetes link. Diabetes 1994; 43: 1271–8. [DOI] [PubMed] [Google Scholar]
- 55.Kern PA, Ranganathan S, Li C, Wood L, Ranganathan G. Adipose tissue tumor necrosis factor and interleukin-6 expression in human obesity and insulin resistance. Am J Physiol Endocrinol Metab 2001; 280: E745–51. [DOI] [PubMed] [Google Scholar]
- 56.Punjabi NM, Sorkin JD, Katzel LI, Goldberg AP, Schwartz AR, Smith PL. Sleep-disordered breathing and insulin resistance in middle-aged and overweight men. Am J Respir Crit Care Med 2002; 165: 677–82. [DOI] [PubMed] [Google Scholar]
- 57.Punjabi NM, Shahar E, Redline S, Gottlieb DJ, Givelber R, Resnick HE, et al. Sleep-disordered breathing, glucose intolerance, and insulin resistance: the Sleep Heart Health Study. Am J Epidemiol 2004; 160: 521–30. [DOI] [PubMed] [Google Scholar]
- 58.Ip MS, Lam B, Ng MM, Lam WK, Tsang KW, Lam KS. Obstructive sleep apnea is independently associated with insulin resistance. Am J Respir Crit Care Med 2002; 165: 670–6. [DOI] [PubMed] [Google Scholar]
- 59.Polak J, Shimoda LA, Drager LF, Undem C, McHugh H, Polotsky VY, et al. Intermittent hypoxia impairs glucose homeostasis in C57BL6/J mice: partial improvement with cessation of the exposure. Sleep 2013; 36: 1483–90; 1490A-1490B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chopra S, Rathore A, Younas H, Pham LV, Gu C, Beselman A, et al. Obstructive sleep apnea dynamically increases nocturnal plasma free fatty acids, glucose, and cortisol during sleep. J Clin Endocrinol Metab 2017; in press. [DOI] [PMC free article] [PubMed]
- 61.Polotsky VY, Patil SP, Savransky V, Laffan A, Fonti S, Frame LA, et al. Obstructive sleep apnea, insulin resistance, and steatohepatitis in severe obesity. Am J Respir Crit Care Med 2009; 179: 228–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Aronsohn RS, Whitmore H, Van Cauter E, Tasali E. Impact of untreated obstructive sleep apnea on glucose control in type 2 diabetes. Am J Respir Crit Care Med 2010; 181: 507–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Reichmuth KJ, Austin D, Skatrud JB, Young T. Association of sleep apnea and type II diabetes: a population-based study. Am J Respir Crit Care Med 2005; 172: 1590–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Balkau B, Vol S, Loko S, Andriamboavonjy T, Lantieri O, Gusto G, et al. High baseline insulin levels associated with 6-year incident observed sleep apnea. Diabetes Care 2010; 1044–9 [DOI] [PMC free article] [PubMed]
- 65.Vgontzas AN, Legro RS, Bixler EO, Grayev A, Kales A, Chrousos GP. Polycystic ovary syndrome is associated with obstructive sleep apnea and daytime sleepiness: role of insulin resistance. J Clin Endocrinol Metab 2001; 86: 517–20. [DOI] [PubMed] [Google Scholar]
- 66.Fogel RB, Malhotra A, Pillar G, Pittman SD, Dunaif A, White DP. Increased prevalence of obstructive sleep apnea syndrome in obese women with polycystic ovary syndrome. J Clin Endocrinol Metab 2001; 86:1175–80. [DOI] [PubMed] [Google Scholar]
- 67.Gopal M, Duntley S, Uhles M, Attarian H. The role of obesity in the increased prevalence of obstructive sleep apnea in patients with polycystic ovarian syndrome. Sleep Med 2002; 3: 401–4. [DOI] [PubMed] [Google Scholar]
- 68.Tasali E, Van Cauter E, Ehrmann DA. Relationships between sleep disordered breathing and glucose metabolism in polycystic ovary syndrome. J Clin Endocrinol Metab 2006; 36–42. [DOI] [PubMed]
- 69.Mokhlesi B, Scoccia B, Mazzone T, Sam S. Risk of obstructive sleep apnea in obese and non-obese women with polycystic ovary syndrome and healthy reproductively normal women. Fertil Steril 2012; 97: 786–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tsaoussoglou M, Bixler EO, Calhoun S, Chrousos GP, Sauder K, Vgontzas AN. Sleep-disordered breathing in obese children is associated with prevalent excessive daytime sleepiness, inflammation, and metabolic abnormalities. J Clin Endocrinol Metab 2010;143–50. [DOI] [PMC free article] [PubMed]
- 71.Farr OM, Rifas-Shiman SL, Oken E, Taveras EM, Mantzoros CS. Current child, but not maternal, snoring is bi-directionally related to adiposity and cardiometabolic risk markers: A cross-sectional and a prospective cohort analysis. Metabolism 2017; S0026–0495(17)30180–4. [DOI] [PMC free article] [PubMed]
- 72.Ha SC, Hirai HW, Tsoi KK. Comparison of positional therapy versus continuous positive airway pressure in patients with positional obstructive sleep apnea: a meta-analysis of randomized trials. Sleep Med Rev 2014; 18: 19–24. [DOI] [PubMed] [Google Scholar]
- 73.*.Jullian-Desayes I, Joyeux-Faure M, Tamisier R, Launois S, Borel AL, Levy P, et al. Impact of obstructive sleep apnea treatment by continuous positive airway pressure on cardiometabolic biomarkers: a systematic review from sham CPAP randomized controlled trials. Sleep Med Rev 2015; 21: 23–38. [DOI] [PubMed] [Google Scholar]
- 74.Vgontzas AN, Zoumakis E, Bixler EO, Lin HM, Collins B, Basta M, et al. Selective effects of CPAP on sleep apnoea-associated manifestations. Eur J Clin Invest 2008; 38: 585–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Arias MA, García-Río F, Alonso-Fernández A, Hernanz A, Hidalgo R, Martínez-Mateo V, et al. CPAP decreases plasma levels of soluble tumour necrosis factor-alpha receptor 1 in obstructive sleep apnoea. Eur Respir J 2008; 32: 1009–15. [DOI] [PubMed] [Google Scholar]
- 76.Kohler M, Ayers L, Pepperell JC, Packwood KL, Ferry B, Crosthwaite N, et al. Effects of continuous positive airway pressure on systemic inflammation in patients with moderate to severe obstructive sleep apnoea: a randomised controlled trial. Thorax 2009; 64: 67–73. [DOI] [PubMed] [Google Scholar]
- 77.Hoyos CM, Killick R, Yee BJ, Phillips CL, Grunstein RR, Liu PY. Cardiometabolic changes after continuous positive airway pressure for obstructive sleep apnoea: a randomised sham-controlled study. Thorax 2012; 67: 1081–9. [DOI] [PubMed] [Google Scholar]
- 78.Hoyos CM, Sullivan DR, Liu PY. Effect of CPAP on the metabolic syndrome: a randomised sham-controlled study. Thorax 2013; 68: 588–9. [DOI] [PubMed] [Google Scholar]
- 79.Shaw JE, Punjabi NM, Naughton MT, Willes L, Bergenstal RM, Cistulli PA, et al. The effect of treatment of obstructive sleep apnea on glycemic control in Type 2 diabetes. Am J Respir Crit Care Med 2016; 194: 486–92. [DOI] [PubMed] [Google Scholar]
- 80.Thunström E, Glantz H, Yucel-Lindberg T, Lindberg K , Saygin M, Peker Y. CPAP does not reduce inflammatory biomarkers in patients with coronary artery disease and nonsleepy obstructive sleep apnea: a randomized controlled trial. Sleep 2017; 40. [DOI] [PubMed] [Google Scholar]
- 81.*.Chirinos JA, Gurubhagavatula I, Teff K, Rader DJ, Wadden TA, Townsend R, et al. CPAP, weight loss, or both for obstructive sleep apnea. N Engl J Med 2014; 370: 2265–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kritikou I, Basta M, Vgontzas AN, Pejovic S, Fernandez-Mendoza J, Liao D, et al. Sleep apnoea and the hypothalamic-pituitary-adrenal axis in men and women: effects of continuous positive airway pressure. Eur Respir J 2016; 47: 531–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Vgontzas AN, Pejovic S, Zoumakis E, Lin HM, Bentley CM, Bixler EO, et al. Hypothalamic-pituitary-adrenal axis activity in obese men with and without sleep apnea: effects of continuous positive airway pressure therapy. J Clin Endocrinol Metab 2007; 92: 4199–207. [DOI] [PubMed] [Google Scholar]
- 84.Schmoller A, Eberhardt F, Jauch-Chara K, Schweiger U, Zabel P, Peters A, et al. Continuous positive airway pressure therapy decreases evening cortisol concentrations in patients with severe obstructive sleep apnea. Metabolism 2009; 58: 848–53. [DOI] [PubMed] [Google Scholar]
- 85.Dimsdale JE, Loredo JS, Profant J. Effect of continuous positive airway pressure on blood pressure: a placebo trial. Hypertension 2000; 35: 144–7. [DOI] [PubMed] [Google Scholar]
- 86.Enoz M UPPP failure due to lingual tonsils and epiglottal laxity. Sleep Med 2006; 7(8):660–1. [DOI] [PubMed] [Google Scholar]
- 87.Riley RW, Powell NB, Guilleminault C. Obstructive sleep apnea syndrome: a review of 306 consecutively treated surgical patients. Otolaryngol Head Neck Surg 1993; 108: 117–25. [DOI] [PubMed] [Google Scholar]
- 88.Vgontzas AN, Zoumakis E, Lin HM, Bixler EO, Trakada G, Chrousos GP. Marked decrease in sleepiness in patients with sleep apnea by etanercept, a tumor necrosis factor-alpha antagonist. J Clin Endocrinol Metab 2004; 89: 4409–13. [DOI] [PubMed] [Google Scholar]
- 89.Brouillette RT, Manoukian JJ, Ducharme FM, Oudjhane K, Earle LG, Ladan S, et al. Efficacy of fluticasone nasal spray for pediatric obstructive sleep apnea. J Pediatr 2001; 838–44. [DOI] [PubMed]
- 90.Goldbart AD, Goldman JL, Veling MC, Gozal D. Leukotriene modifier therapy for mild sleep-disordered breathing in children. Am J Respir Crit Care Med 2005; 172: 364–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kheirandish-Gozal L, Gozal D. Intranasal budesonide treatment for children with mild obstructive sleep apnea syndrome. Pediatrics 2008; 122: e149–55. [DOI] [PubMed] [Google Scholar]
- 92.Kuhle S, Urschitz MS. Anti-inflammatory medications for obstructive sleep apnea in children. Cochrane Database Syst Rev 2011; 19: CD007074. [DOI] [PubMed] [Google Scholar]
- 93.Kheirandish-Gozal L, Bhattacharjee R, Bandla HPR, Gozal D. Antiinflammatory therapy outcomes for mild OSA in children. Chest 2014; 146: 88–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Reading SR, Crowson CS, Rodeheffer RJ, Fitz-Gibbon PD, Maradit-Kremers H, Gabriel SE. Do rheumatoid arthritis patients have a higher risk for sleep apnea? J Rheumatol 2009; 36: 1869–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Taylor-Gjevre RM, Nair BV, Gjevre JA. Obstructive sleep apnoea in relation to rheumatic disease. Rheumatology (Oxford) 2013; 52: 15–21. [DOI] [PubMed] [Google Scholar]
- 96.Walsh JA, Duffin KC, Crim J, Clegg DO. Lower frequency of obstructive sleep apnea in spondyloarthritis patients taking TNF-inhibitors. J Clin Sleep Med 2012; 8: 643–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Vgontzas AN, Li Y, He F, Fernandez-Mendoza J, Gaines J, Basta M, et al. Mild-to-moderate obstructive sleep apnea is associated with incident hypertension: a longitudinal, population-based study. Sleep 2016; 40: A158. [Google Scholar]
- 98.Kirkness JP, Schwartz AR, Schneider H, Punjabi NM, Maly JJ, Laffan AM, et al. Contribution of male sex, age, and obesity to mechanical instability of the upper airway during sleep. J Appl Physiol 2008; 104: 1618–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Edwards BA, Wellman A, Sands SA, Owens RL, Eckert DJ, White DP, et al. Obstructive sleep apnea in older adults is a distinctly different physiological phenotype. Sleep 2014; 1227–36. [DOI] [PMC free article] [PubMed]
- 100.Saint Martin M, Roche F, Thomas T, Collet P, Barthélémy JC, Sforza E. Association of body fat composition and obstructive sleep apnea in the elderly: A longitudinal study. Obesity 2015; 23: 1511–6. [DOI] [PubMed] [Google Scholar]
- 101.Wolkove N, Baltzan M, Kamel H, Dabrusin R, Palayew M. Long-term compliance with continuous positive airway pressure in patients with obstructive sleep apnea. Can Respir J 2008; 15: 365–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wozniak DR, Lasserson TJ, Smith I. Education, supportive and behavioural intervensions to improve usage of continuous positive airway pressure machines in adults with obstructive sleep apnea. Cochrane Database Syst Rev 1:CD007736. [DOI] [PubMed] [Google Scholar]
- 103.McArdle N, Kingshott R, Engleman HM, Mackay TW, Douglas NJ. Partners of patients with sleep apnoea/hypopnoea syndrome: effect of CPAP treatment on sleep quality and quality of life. Thorax 56: 513–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Mullington JM, Abbott SM, Carroll JE, Davis CJ, Dijk DJ, Dinges DF, et al. Developing biomarker arrays predicting sleep and circadian-coupled risks to health. Sleep 2016; 39: 727–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Mehra R Building evidence implicating novel cardiovascular biomarkers in obstructive sleep apnea. J Clin Sleep Med 2017; 13: 361–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Gaines J, Vgontzas AN, Fernandez-Mendoza J, Kritikou I, Basta M, Bixler EO. Gender differences in the association of sleep apnea and inflammation. Brain Behav Immun 2015; 47: 211–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Li Y, Vgontzas AN, Fernandez-Mendoza J, Kritikou I, Basta M, Pejovic S, et al. Objective, but not subjective, sleepiness is associated with inflammation in sleep apnea. Sleep 2017; 40(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Nagayoshi M, Punjabi NM, Selvin E, Pankow JS, Shahar E, Iso H, et al. Obstructive sleep apnea and incident type 2 diabetes. Sleep Med 2016; 25: 156–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.*.Gaines J, Kong L, Li M, Fernandez-Mendoza J, Bixler EO, Basta M, et al. C-reactive protein improves the ability to detect cardiometabolic risk in mild-to-moderate obstructive sleep apnea. Physiol Rep 2017; 5: e13454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Fleming WE, Ferouz-Colborn A, Samoszuk MK, Azad A, Lu J, Riley JS, et al. Blood biomarkers of endocrine, immune, inflammatory, and metabolic systems in obstructive sleep apnea. Clin Biochem 2016; 49: 854–61. [DOI] [PubMed] [Google Scholar]