

Abstract
cGMP signaling elicited by activation of the transmembrane receptor guanylyl cyclase Npr2 (also known as guanylyl cyclase B) by the ligand CNP controls sensory axon bifurcation of DRG and cranial sensory ganglion (CSG) neurons entering the spinal cord or hindbrain, respectively. Previous studies have shown that Npr2 is phosphorylated on serine and threonine residues in its kinase homology domain (KHD). However, it is unknown whether phosphorylation of Npr2 is essential for axon bifurcation. Here, we generated a knock-in mouse line in which the seven regulatory serine and threonine residues in the KHD of Npr2 were substituted by alanine (Npr2-7A), resulting in a nonphosphorylatable enzyme. Real-time imaging of cGMP in DRG neurons with a genetically encoded fluorescent cGMP sensor or biochemical analysis of guanylyl cyclase activity in brain or lung tissue revealed the absence of CNP-induced cGMP generation in the Npr27A/7A mutant. Consequently, bifurcation of axons, but not collateral formation, from DRG or CSG in this mouse mutant was perturbed at embryonic and mature stages. In contrast, axon branching was normal in a mouse mutant in which constitutive phosphorylation of Npr2 is mimicked by a replacement of all of the seven serine and threonine sites by glutamic acid (Npr2–7E). Furthermore, we demonstrate that the Npr27A/7A mutation causes dwarfism as described for global Npr2 mutants. In conclusion, our in vivo studies provide strong evidence that phosphorylation of the seven serine and threonine residues in the KHD of Npr2 is an important regulatory element of Npr2-mediated cGMP signaling which affects physiological processes, such as axon bifurcation and bone growth.
SIGNIFICANCE STATEMENT The branching of axons is a morphological hallmark of virtually all neurons. It allows an individual neuron to innervate different targets and to communicate with neurons located in different regions of the nervous system. The natriuretic peptide receptor 2 (Npr2), a transmembrane guanylyl cyclase, is essential for the initiation of bifurcation of sensory axons when entering the spinal cord or the hindbrain. By using two genetically engineered mouse lines, we show that phosphorylation of specific serine and threonine residues in juxtamembrane regions of Npr2 are required for its enzymatic activity and for axon bifurcation. These investigations might help to understand the regulation of Npr2 and its integration in intracellular signaling systems.
Keywords: axonal branching, cGMP signaling, Npr2, phosphorylation, sensory axons
Introduction
Axonal branching is one of the key processes that determine the formation of complex neuronal circuits during embryonic and postnatal stages of nervous system development. Branching enables an individual axon to establish multiple contacts, and it allows an individual neuron to communicate with other neurons that might be located in different regions. Therefore, branching provides the physical basis for the distribution and integration of information. However, the molecular mechanisms that control branch formation in vivo are not fully understood (Armijo-Weingart and Gallo, 2017). The projections of sensory axons within the spinal cord or the hindbrain represent an attractive system to characterize components implicated in branching of axons (Schmidt and Rathjen, 2010; Gibson and Ma, 2011; Dumoulin et al., 2018). Upon entering the spinal cord or hindbrain, the incoming axons from neurons of DRGs or cranial sensory ganglia (CSGs), respectively, split up into a T-like bifurcation, giving rise to two daughter axons that extend in opposite directions. The natriuretic peptide receptor 2 (Npr2), also termed guanylyl cyclase B, plays a central role in the process of sensory axon bifurcation. In loss-of-function mutations of the receptor Npr2, sensory axons no longer form these T-like branches and instead turn only in either ascending or descending direction. Identical results were obtained when the cGMP-dependent kinase I (cGKI) (Schmidt et al., 2002; Zhao et al., 2009) or the Npr2 ligand C-type natriuretic peptide (CNP) were inactivated in mice (Schmidt et al., 2007, 2009; Zhao and Ma, 2009; Ter-Avetisyan et al., 2014). More recently, this signaling pathway was shown to be of equivalent importance for the bifurcation of afferents of mesencephalic trigeminal neurons within the developing hindbrain (Ter-Avetisyan et al., 2018). A critical missing link of this signaling pathway are the downstream targets of cGKIα in somatosensory growth cones. However, by analyzing collateral branching or growth cone behavior in cell culture, experiments indicated that cGMP signaling might regulate microtubule dynamics (Xia et al., 2013; Akiyama et al., 2016). Furthermore, although the guidance receptors robo1 and robo2 and their ligands slit1 and slit2 did not perturb bifurcation itself, their absence caused sensory axons to enter the spinal cord prematurely (Ma and Tessier-Lavigne, 2007). The nitric oxide-sensitive guanylyl cyclases are not found in DRG neurons at early embryonic stages and thus are not implicated in sensory axon branching in vivo (Schmidt et al., 2009). The lack of axon bifurcation in DRG neurons results in altered termination fields of primary afferents from the skin in the spinal cord. Furthermore, behavioral testing indicated that noxious heat perception and nociception induced by chemical irritants and the maximal biting force are impaired in mouse mutants with a conditional inactivation of Npr2 in DRG or mesencephalic trigeminal neurons, whereas responses to mechanical stimulation and motor coordination are not affected (Ter-Avetisyan et al., 2018; Tröster et al., 2018).
Npr2 is a homodimeric transmembrane receptor composed of an amino-terminal extracellular ligand-binding domain, a transmembrane region, a so-called kinase homology domain (KHD), a dimerization segment, and a carboxyl-terminal guanylyl cyclase domain. Activation of Npr2 by binding of its ligand CNP results in the generation of cGMP from GTP (Potter, 2011; Kuhn, 2016). The mechanisms regulating Npr2 activity are incompletely understood. Previously published studies showed that phosphorylation of the KHD of Npr2 regulates its guanylyl cyclase activity. Mutational and mass spectrometric studies identified six chemically determined phosphorylation sites (S513, T516, S518, S523, S526, and T529) (Potter and Hunter, 1998a; Potter, 1998; Yoder et al., 2010), and a seventh site (S489) was characterized in alanine substitutions experiments using transfected cells (Yoder et al., 2012). Five of these sites are identical between Npr2 and Npr1. Dephosphorylation of Npr2 mediates meiotic resumption in response to luteinizing hormone (Egbert et al., 2014; Shuhaibar et al., 2016) and inhibition of bone growth in response to fibroblast growth factor (Robinson et al., 2017; Shuhaibar et al., 2017). However, it is unknown whether these phosphorylation sites in Npr2 are required for regulation of axon bifurcation.
Here, we applied CRISPR/Cas9 genome editing to generate a mouse line in which the seven previously identified serine and threonine residues of Npr2 were mutated to alanine (Npr2-7A), thereby preventing its phosphorylation. Biallelic abolition of Npr2 phosphorylation sites yielded a receptor that is unresponsive to the ligand CNP. We show that Npr27A/7A mutants phenocopy the dwarfed appearance of mice lacking functional alleles of Npr2 (Tamura et al., 2004) and display a loss of sensory axon bifurcation in the spinal cord and hindbrain. Interestingly, the analysis of axonal branching in a genetically modified mouse, in which the seven juxtamembrane serine and threonine residues in Npr2 are mutated to glutamic acid to mimic a constantly phosphorylated enzyme (Shuhaibar et al., 2016), demonstrated that this constitutively active form of Npr2 did not cause any miss-branching, indicating that negatively charged residues at this position are essential for full Npr2 activity. Our data show that phosphorylation of the KHD is an important regulatory element of Npr2.
Materials and Methods
Mice and generation of the Npr2-7A transgenic line
Generation of the Npr2-7A mouse line (B6-Npr2tm4/Fgr) expressing globally 7 alanine substitutions in the kinase homology domain of Npr2 was obtained by Crispr/Cas9 techniques. The following sites located in exons 8 and 9 that were previously identified by mass spectrometry to be phosphorylated in HEK293 cells (Yoder et al., 2010) were mutated to alanine: S489, S513, T516, S518, S523, S526, and T529. A donor vector covering the genomic DNA sequence from nucleotide position 43,640,567 to 43,645,697 of chromosome 4 of the C57BL/6J mouse strain (GenBank accession number NC_000070.6) was constructed in which the sequences coding for S489, S513, T516, S518, S523, S526, and T529 of mouse Npr2 were changed from AGC to GCC, from AGT to GCT, from ACG to GCG, from TCG to GCG, from AGT to GCT, from TCG to GCG, and from ACA to GCA, respectively, to alternatively encode alanine (see Fig. 1A,B). Two double-strand breaks were induced by specific 20-nt-long guide RNAs (upstream either sgRNA#1a, 5′-TACTGACTTTGGGTAGAGTT-3′; or sgRNA#1b, 5′-CTACCCAAAGTCAGTACTCC-3′ was combined with the downstream sgRNA#2, 5′-TATCACTGATTCCCAGTTGC-3′), which guide the Cas9 nuclease to the desired Npr2 sites upstream of exon 8 and downstream of exon 10. To prevent cleavage of the donor template by Cas9, the protospacer adjacent motifs in the DNA target sequence of the donor were mutated to NAA. Donor vector, guide RNA (sgRNA), and the Cas9 mRNA were coinjected into mouse zygotes and implanted into foster mouse females by the transgenic core facility of the Max Delbrück Center for Molecular Medicine (Berlin), as described previously (Wefers et al., 2017). Briefly, Cas9 mRNA was prepared in a single step by in vitro transcription from plasmid pCAG-Cas9-162A linearized with AsiSI, AscI, and XbaI, using the mMessage mMachine T7 Ultra kit (Invitrogen, Ambion, AM1345), omitting the polyadenylation step. Cas9-162A mRNA was isolated using the Oligotex mRNA mini kit (catalog #70022, QIAGEN). To produce the templates for sgRNA in vitro transcription, sgNpr2-1/2/4 were amplified by PCR from guide RNA-containing plasmids. A 1 μg template DNA was used for in vitro transcription using the Megashortscript kit (Ambion, catalog #AM1354) followed by the MEGAclear kit (Ambion, catalog #AM1908) for RNA purification. RNAs and targeting vectors were diluted in microinjection buffer (10 mm Tris, 0.1 mm EDTA, pH 7.2) to the indicated working concentrations, filtrated through a centrifugal filter (Ultrafree, PFTE, Millipore, catalog #UFC30LG25), and stored in single-use aliquots at −80°C. For microinjections, zygotes were obtained by mating of C57BL/6N males with superovulated C57BL/6N females (Charles River). Injected zygotes were transferred into pseudo-pregnant NMRI female mice to obtain live pups. Genotyping of Npr2-7A mice was done by PCR using primer Npr2-7A-GT-UP 5′-TCC TGG AAG CTT AGT GGG TG-3′ and Npr2-7A-GT-LP 5′-GGT GGT GCA AGG GTC TAT CA-3′ followed by digestion with restriction endonuclease NheI. The mutant and wild-type allele generated an amplification product of 288 bp; and after digestion with NheI, the wild-type allele splits into two fragments of 198 and 90 bp. Among 28 offspring obtained by microinjection, three independent founder mutants were obtained. Two of the founders generated progenies with identical phenotypes as reported in Figure 1 and bred according to the mendelian rules. The third founder showed a dwarfed phenotype and was therefore considered to be a homozygous Npr2 mutant. Replacement of specific nucleotides in exons 8 and 9 of the murine Npr2 gene was verified by sequencing of PCR products resulting from amplification of genomic DNA from wild-type and Npr27A/7A mice using primer Npr2-7A-Ins-UP 5′-GGC TGA GAT GTG CAG TCC TT-3′ and Npr2-7A-Ins-LP 5′-CTT GAA GTG ACC GGT GTT GG-3′ (see Fig. 1B).
Figure 1.

Generation and validation of the Npr2-7A mouse mutant. A, Targeting strategy for the replacement of seven serine and threonine phosphorylation sites that are encoded in exons 8 and 9 of the murine Npr2 gene by Crispr/Cas9. PAM, Protospacer adjacent motif; sgRNA, synthetic guide RNA. P1 and P2 indicate primer locations for genotyping. B, Sequencing of genomic DNA of the mutated locus in Npr27A/7A and in wild-type, demonstrating the replacement of specific nucleotides in the kinase homology domain. These replacements result in codons, which encode alanine residues instead of serine or threonine residues as indicated below the sequencing information. C, PCR genotyping using primer P1 and P2 followed by digestion with restriction endonuclease NheI, which specifically splits the wild-type sequence but not the mutated segment. D–F, Depiction and quantification of wild-type and Npr27A/7A littermates at postnatal day 25, demonstrating a severely reduced body length and body weight due to impaired endochondral ossification as described previously for other Npr2 loss-of-function mutants. G, Survival rate of Npr27A/7A, Npr2wt/7A, and Npr2wt/wt mice at different ages. Numbers of animals inspected are given in parentheses. H, Naso-anal length of Npr27A/7A, Npr2wt/7A, and Npr2wt/wt male mice at different ages. I, Body weight of Npr27A/7A, Npr2wt/7A, and Npr2wt/wt male mice at different ages. Females are not shown because they were underrepresented in these cohorts, but the same tendency was observed. *p < 0.05; **p < 0.01.
The following mouse lines were genotyped as detailed elsewhere: R26-CAG-cGi500(L1) cGMP-fluorescence resonance energy transfer (FRET) sensor mouse line (Gt(ROSA)26Sortm1.1(CAG-ECFP/EYFP)/Feil) (Thunemann et al., 2013b), Npr2-CreERT2 (B6.129S7-Npr2tm1(CreERT2)/Fgr) (Ter-Avetisyan et al., 2014), Thy1-YFP-H (Tg(Thy1-YFP)HJrs) (Feng et al., 2000), and Npr27E/7E (Shuhaibar et al., 2016). In this Npr27E/7E mouse, substitution of the above-mentioned serine and threonine residues by glutamate to mimic the negative charge of phosphate resulted in a variant of Npr2 that cannot be inactivated by dephosphorylation.
Animals were housed on a 12/12 h light/dark cycle with free access to water and food. The animal procedures were performed according to the guidelines from directive 2010/63/EU of the European Parliament on the protection of animals used for scientific purposes. All experiments were approved by LAGeSo Berlin (#T0313/97, X9007/16, 0143/07, G0162/12, and G0370/13).
FRET-based cGMP imaging in DRG neurons
DRGs were dissected from embryonic day (E) 12.5 mice that resulted from mating of Npr2-7A heterozygous females with males heterozygous for Npr2-7A and homozygous for R26-CAG-cGi500(L1). FRET/cGMP imaging was performed on the somata of dissociated neurons 24 h after plating using an epifluorescence imaging setup as described previously (Thunemann et al., 2013a,b; Schmidt et al., 2016). Littermates of Npr27A/7A, Npr2wt/7A, and Npr2wt/wt served as controls.
For the analysis of FRET data, ImageJ (Schneider et al., 2012), Excel (Microsoft), and OriginPro (Origin Lab Corp) software were used. The mean intensities from selected ROIs of the CFP and YFP channel images were corrected for background fluorescence and referred to as F480 and F535, respectively, before calculating the cGMP-representing F480/F535 ratio R. ΔF480/F480, ΔF535/F535, and ΔR/R traces were obtained by normalization to the respective baseline recorded for 2–3 min at the beginning of each experiment. To estimate peak areas of ΔR/R traces, the Peak Analyzer Module of Origin was used.
Axon tracing, antibodies, and immunohistology
1,1-Dioctadecyl-3,3,3,3-tetramethyl-indocarbocyanine perchlorate (DiI) axon tracing of sensory axons was done as described previously (Schmidt and Rathjen, 2011). A genetic method for sparse labeling of Npr2-positive axons involving a tamoxifen-inducible variant of Cre-recombinase under control of the Npr2 promoter (Npr2-CreERT2) and a conditional reporter allele Z/AP mice has been described previously (Lobe et al., 1999; Schmidt et al., 2013; Ter-Avetisyan et al., 2014). Tamoxifen application released reporter gene expression in a subset of Npr2-expressing cells; 0.1 mg tamoxifen per g body weight was applied by oral gavaging as described previously (Ter-Avetisyan et al., 2014). Analysis of axon branching at postnatal stages was done by crossing the Thy1-GFP-M reporter into the Npr2-7A mouse.
PFA-fixed cryostat sections (16 μm thick) were stained with the following antibodies: rabbit anti-cGKI (Ter-Avetisyan et al., 2014), guinea pig antiserum to the extracellular domain of Npr2 (dilution: 1:5000) (Ter-Avetisyan et al., 2014), guinea pig antiserum to GFAP (1:500; Synaptic Systems, #173004), or mAb anti-neurofilament (2H3, Developmental Hybridoma Bank). Secondary antibodies were applied at the following dilutions: 1:1000 goat-anti-rabbit-Cy3, 1:1000 goat-anti-rabbit-Alexa-647, 1:1000 goat-anti-mouse-Alexa-647 (Dianova), and 1:1000 donkey anti-guinea pig-Cy3 (Dianova).
All microscopic images were obtained at room temperature using an Axiovert 135 inverted microscope equipped with Neofluar/Acroplan objectives (5×, 10×, or 40× magnification with numerical apertures 0.15, 0.25, or 0.75, respectively), a charge-coupled device camera (Axiocam HRC) and acquisition software (Axiovision 3.1), or by confocal imaging using an Axio Observer.Z1 inverted microscope equipped with ZEN 2011 software and the following lenses: a Plan-Neofluar 10×/0.30 NA objective or a Plan-Achromat 63×/1.40 NA oil objective (all from Carl Zeiss). Images were imported into Photoshop CS6 (Adobe) for adjustment of contrast and brightness. Figures were assembled using Illustrator CS6 (Adobe).
Biochemical experiments
Analysis of cell surface expression of Npr2-7A on astrocytes and DRGs.
Astrocytes were prepared from postnatal day 1.5–3.5 wild-type or Npr27A/7A cortices by standard procedures and cultivated for 2–3 weeks in DMEM/10% FCS (McCarthy and de Vellis, 1980). These cultures contained GFAP-positive and -negative cells. Confluent monolayer cultures were washed three times with cold PBS, and labeling of cell surface proteins was obtained by incubation with 2 mm EZ-Link-Sulfo-NHS-LC-Biotin (Thermo Fisher Scientific) in PBS at 8°C for 1 h followed by quenching with 100 mm glycine in PBS. Cells were solubilized with 1% CHAPS in TBS, pH 7.4, supplemented with protease blockers (aprotinin, leupeptin, pepstatin, PMSF), and unsolubilized material was removed by centrifugation at 100,000 × g for 10 min. In four independent experiments, solubilized proteins in the range of 40–70 μg for each genotype were incubated with 30 μl streptavidin agarose (Thermo Fisher Scientific) for 4 h or overnight followed by 4 washing steps with solubilization buffer and elution in SDS-PAGE sample buffer.
DRGs pooled from 3 E13.5 wild-type or Npr27A/7A embryos were washed in cold PBS followed by incubation with 2 mm NHS-sulfo-biotin for 1 h and quenching with 100 mm glycine in PBS. Proteins were solubilized with 1% SDS in TBS supplemented with protease blockers, heated, and centrifuged at 100,000 × g for 10 min; 70 μg of solubilized proteins for each genotype was used to isolate biotinylated proteins as described above.
Detergent extracts of cultivated cortical astrocytes from wild-type or Npr27A/7A were treated by endoglycosidase H or endoglycosidase F as detailed by the supplier (New England BioLabs), except that 1% CHAPS, instead of NP-40, was used as detergent. Equal amounts of proteins were analyzed by Western blotting using guinea pig anti-Npr2. The intensity of bands from Western blots was calculated with Quantity One (Bio-Rad).
Preparation of membranes for guanylyl cyclase activity.
Frozen tissue samples were homogenized with a pestle and microcentrifuge tube in ice-cold phosphatase inhibitor buffer (PIB) composed of 25 mm HEPES, pH 7.4, 50 mm NaCl, 20% glycerol, 50 mm NaF, 2 mm EDTA, 0.5 μm microcystin (Cayman Chemical Company), and twofold concentrated protease inhibitor mixture (Roche Diagnostics). Following centrifugation at 20,000 × g at 4°C, the crude membranes were washed twice with PIB before final resuspension. Brain and lung samples were resuspended in 1 ml of PIB. Total protein concentrations were determined by the Bradford protein assay.
Measurement of guanylyl cyclase activity.
Crude membrane fractions containing approximately equal amounts of protein per treatment were assayed for guanylyl cyclase activity in the presence or absence of increasing concentrations of CNP (Sigma-Aldrich) to activate Npr2 or increasing concentrations of ANP (Sigma-Aldrich) to activate Npr1 (Dickey et al., 2016); 20 μl of an activator solution containing 5 mm ATP with or without the indicated concentrations of ANP or CNP was added to 20 μl of 14–75 μg of membranes resuspended in 20 μl of phosphatase inhibitor buffer (Flora and Potter, 2010). The guanylyl cyclase reaction was stimulated by addition of 60 μl of prewarmed mixture containing 25 mm HEPES, pH 7.4, 50 mm NaCl, 0.1% BSA, 500 μm isobutylmethylxanthine, 1 mm GTP, 5 mm MgCl2, 5 mm creatine phosphate, and 0.1 mg/ml creatine kinase. Membranes were assayed for 3 min at 37°C. The reactions were terminated by adding 0.4 ml of ice-cold 50 mm sodium acetate solution containing 5 mm EDTA. One-fifth of the reaction was removed and assayed for cGMP concentrations by radioimmunoassay as previously described (Abbey and Potter, 2002). The cGMP radioimmunoassay kit was purchased from PerkinElmer. Guanylyl cyclase activity in brains from Npr27E/7E mice were assayed by an ELISA-based method. GTP and ATP concentrations were 0.1 and 1 mm, respectively. Assays were stopped by addition of 110 mm zinc acetate, followed by 110 mm sodium carbonate to precipitate GTP. Samples were then purified over an alumina column (Domino et al., 1991), and cyclic GMP content of the eluate was measured by ELISA following the manufacturer's instructions (Direct cGMP ELISA kit, Enzo Life Sciences).
Transfection of the 293T cells, preparation of cellular membranes, and measurement of guanylyl cyclase activity in cells.
Twenty-four hours before transfection, cells were split to 10 cm plates. Fifty percent confluent cells were transiently transfected with 2 μg total plasmid by the HEPES-calcium-phosphate precipitation method. The medium was replaced after 16–20 h. Transfection efficiency was 50%–80% based on GFP expression. Two days after transfection, transfected cells were placed in serum-free medium for 4 h. The cells were then rinsed twice with ice-cold PBS before scraping in PIB. The cells were lysed by sonication. Following centrifugation at 20,000 × g for 15 min at 4°C, the crude membranes were washed with PIB before final resuspension in PIB. Total protein concentrations were determined by the Bradford protein assay.
Membrane fractions containing approximately equal amounts of protein per tissue were assayed for guanylyl cyclase activity by addition of ATP/Mg2+ alone (for basal determination) or with ATP/Mg2+ plus 1 μm CNP or Triton/Mn2+. The receptor was stimulated and assayed for cGMP levels as described above.
Immunoprecipitation.
Following protein determination and guanylyl cyclase assays, 0.03 g of membrane protein from the PIB lysates was fractionated on an 8% gel by SDS-PAGE and transferred to a PVDF membrane. For immunoprecipitation, the remaining PIB lysates were centrifuged and the pellets resuspended in 1 ml RIPA lysis buffer. Lysates were precleared with IPA300 (immobilized protein A from RepliGen) for 20 min. The precleared lysates were immunoprecipitated overnight with anti-FLAG M2 affinity gel or anti-HA conjugated agarose. The beads were washed three times with PBS and boiled in SDS sample buffer before fractionation on an 8% gel by SDS-PAGE and transfer to PVDF membrane. The PVDF membranes were blocked with Odyssey Li-COR blocking buffer for 1 h at room temperature and then incubated overnight at 4°C with rabbit polyclonal antiserum #6327 (against the C terminus of rat guanylyl cyclase B) and anti-FLAG M2 monoclonal antibody (Dickey et al., 2017). Following 4 × 5 min washes with TBST, the membranes were coincubated with 1:10,000 dilutions IRDye 680 goat anti-rabbit and IRDye800CW goat anti-mouse secondary antibodies for 1 h at room temperature. The membranes were then washed and imaged on a Li-Cor Odyssey imaging device. The blots were then stripped for 30 min at room temperature with 25 mm glycine, pH 2, 1% SDS, and rinsed with PBS before reblocking and reprobing with anti-HA antibody. Anti-FLAG-M2 monoclonal antibody, anti-HA monoclonal antibody (HA-7), and anti-FLAG M2 affinity gel were purchased from Sigma-Aldrich and were diluted according to the manufacturer's instructions. Odyssey blocking buffer, IRDye 680 goat anti-rabbit antibody, and IRDye 800CW goat anti-mouse antibody were from Li-Cor Biosciences.
Experimental design and statistical analysis
Mann–Whitney test was applied for comparison of body weight, naso-anal length, and cGMP/FRET peak amplitudes of different genotypes using GraphPad Prism. Results are shown as mean ± SD.
Sample size for FRET measurements and analysis of axon branching was deduced from previously published studies. No further statistical methods were used to predetermine sample size. Sample size are given in the figures or legends. Littermates were used as controls.
Unpaired t tests were conducted to determine the p values of the mean differences between tissues from mouse samples using GraphPad Prism for the MacIntosh. Data are represented as mean ± SEM, where the SEM is represented by the vertical bars in the columns or symbols.
Results
The overall phenotype of Npr27A/7A mutant mice matches the global constitutive Npr2 knock-out phenotype
Previous studies in overexpressing cell culture systems have shown that phosphorylation and dephosphorylation are required for the cellular activation and deactivation of Npr2, respectively. To test whether phosphorylation of Npr2 is a potential mechanism for the regulation of axon bifurcation, we developed a genetically modified mouse in which the seven regulatory serine and threonine residues in the kinase homology domain of Npr2 encoded by exons 8 and 9 were changed to alanines by Crispr/Cas9 technology (Fig. 1A). Three independent founders were obtained and their modified region was verified by sequencing (Fig. 1B,C). Homozygous Npr27A/7A were dwarfs as demonstrated by a nasal to anal body length and body weight as shown for P25 (Fig. 1D–F), which became visible from the second postnatal week on (Fig. 1H–I). The tail of the Npr27A/7A mice was short; the skull showed a dome-shaped appearance. Furthermore, a reduced survival rate was observed (Fig. 1G). This overall phenotype has also been described for the constitutive Npr2 knock-outs (Tamura et al., 2004; Ter-Avetisyan et al., 2014) and mice with missense mutations spontaneously arisen in the Npr2 gene (Tsuji and Kunieda, 2005; Sogawa et al., 2007; Shapiro et al., 2014). The higher mortality rate found for Npr27A/7A mice compared with the constitutive Npr2 knock-out might result from a different genetic background (Bl6 vs a mixed Bl6-SV129 background).
Npr2-7A protein is expressed on the cell surface
A number of Npr2-point mutations found in human patients (for a synopsis of human NPR2 mutations, see Vasques et al., 2014) have been reported not to reach the cell surface and were retarded in the endoplasmic reticulum (Hume et al., 2009; Vasques et al., 2013; Amano et al., 2014; Wang et al., 2016), whereas others reached the cell surface (Dickey et al., 2016). Therefore, we analyzed expression of Npr2-7A protein in transverse sections of the spinal cord, on the surface of DRGs, and in addition on cultivated astrocytes. The latter have also been found to express Npr2 in culture (Deschepper and Picard, 1994) and which can be obtained in larger quantities for biochemical experiments than DRG neurons. Staining of transversal sections with antibodies to neurofilament or cGKI demonstrated a normal overall layering of the spinal cord at the embryonic stages when bifurcation of sensory axons occurs (Fig. 2A). Western blot analysis of protein extracts revealed similar amounts of wild-type or mutated Npr2 in DRGs, which are both composed of two major bands: a slower-migrating band and a second faster-migrating band (Fig. 2B). Staining of spinal cord cross sections using an antibody to the extracellular part of Npr2 shows localization of Npr2-7A protein in the somata of DRGs, in the dorsal roots as well as at the dorsal root entry zone, and developing dorsal funiculus of the spinal cord, indicating that mutated Npr2 is transported from the cell somata along the axons (Fig. 2C,D). To demonstrate cell surface localization, intact DRGs or cultivated astrocytes were labeled by sulfo-NHS-biotin followed by precipitation of biotinylated proteins by streptavidin-agarose. Western blotting of these fractions demonstrated that similar amounts of Npr2 from wild-type or mutated cells were precipitated, which are both composed of a single major band (Fig. 2E,F). For a further comparison of both Npr2-7A and of Npr2 expressed at the surface, we analyzed the biotinylated eluate (cell surface exposed) and the nonbiotinylated unbound fraction (not expressed at the cell surface or blocked for biotinylation) from astrocyte cultures of both genotypes. In Western blots, the unbiotinylated, unbound Npr2 revealed a slower-migrating band and a second faster-migrating band from both wild-type and the mutant cultures (Fig. 2G; for comparison, see also Fig. 2B). Both forms differ in their glycosylation pattern as indicated by endoglycosidase F and H treatment (Fig. 2H). The latter cleaves the covalent bond of the high-mannose and some hybrid type oligosaccharides to asparagine but not oligosaccharides of the complex type (Maley et al., 1989). The high-mannose-type sugar, which is added to nascent polypeptides in the ER, becomes later modified in the Golgi network to the so-called complex type of oligosaccharides. The faster-migrating component shifts significantly by endoglycosidase H treatment and therefore represents an immature glycosylated form of Npr2. The upper band shifts marginally. Both bands are sensitive to endoglycosidase F, indicating that the upper band contains primarily oligosaccharides of the complex type and therefore represents the mature form of Npr2 (Fig. 2H). These data are in line with observations on Npr2 in transfected HEK293 cells treated with tunicamycin (Dickey et al., 2016). Interestingly, in both genotypes, the streptavidin-bound, biotinylated component is only composed of the slower-migrating Npr2 band (slightly higher than the upper Npr2 band from unbound Npr2 due to the incorporation of biotin) (Fig. 2G). Because the faster-migrating component was never found to be biotinylated and sensitive to endoglycosidase H treatment, we concluded that it represents intracellular Npr2, and the slower-running, biotinylated component, which is slightly sensitive to endoglycosidase H treatment, represents mature forms of Npr2. Therefore, we calculated the ratio for lower to upper migrating component for both wild-type and mutant to address the question whether lower amounts of mutated Npr2 reach the cell surface in DRGs from Npr27A/7A (Fig. 2I). The intensity ratios of the two bands were very similar for DRGs between both genotypes (1.42 and 1.44 for wild-type and mutant, respectively). In astrocytes, the lower (intracellular) component was increased (1.04 and 1.37 for wild-type and mutant, respectively), demonstrating a shift toward the intracellular, immature component of Npr2-7A in astrocyte cultures but not in embryonic DRGs.
Figure 2.

Npr2-7A protein is expressed on the cell surface of DRGs. A, Cross sections of the spinal cord from Npr27A/7A and Npr2wt/7A E12.5 embryos stained with anti-neurofilament, anti-cGKI, and DAPI reveal a normal overall structure of the spinal cord in Npr27A/7A mutants. Scale bar, 100 μm. B, Western blots of detergent extracts of DRGs from E13.5 Npr27A/7A, Npr2wt/7A, and Npr2wt/wt embryos, indicating similar amounts of Npr2 protein in all three genotypes. Npr2 and Npr2-7A are composed of two major bands. The bottom served as loading control using a monoclonal antibody to the heavy chain of clathrin. Right, Molecular mass markers. C, D, Transversal sections of the spinal cord from Npr27A/7A, Npr2LacZ/LacZ (representing Npr2−/−), and Npr2wt/wt stained with anti-Npr2, demonstrating expression of Npr2-7A-mutated protein in the somata of DRG neurons, in the dorsal root and dorsal root entry zone and the developing dorsal funiculus axons. Hyphenated line indicates the border of the spinal cord. Scale bar, 50 μm. D, Dorsal; DR, dorsal root; DREZ, dorsal root entry zone; V, ventral. E, F, Expression of Npr2 variants on the surface of DRGs and of cortical astrocyte cultures from wild-type or Npr27A/7A mutants. Cell surface proteins on freshly picked DRGs or cultivated astrocytes were labeled by sulfo-NHS-biotin followed by isolation with streptavidin agarose. Bottom, Equal protein loading was controlled by streptavidin-HRP. Right, Molecular mass markers. G, Western blot of Npr2 and Np2–7A from cortical astrocyte cultures that were cell surface biotinylated by sulfo-NHS-biotin followed by purification using streptavidin beads. The biotinylated fraction is composed of one major band in both genotypes, whereas the unbound fraction contains two bands (for DRGs, see also Fig. 2B). EL, Eluate of streptavidin beads (bound, biotinylated fraction of Npr2); FT, flow through of streptavidin beads (unbound fraction of Npr2). H, Western blot of Npr2 from astrocyte cultures treated with endoglycosidase F (EnF) or endoglycosidase H (EnH). EnF treatment resulted in one major band, whereas EnH treatment resulted in a significant shift of the lower Npr2 band. A weak stained band between both major bands might represent not fully endoglycosidase H-dependent deglycosylated Npr2. I, Quantification of Western blots of the ratio of the two major components from DRGs or astrocyte cultures from Npr27A/7A or Npr2wt/wt. Upper band is considered as mature Npr2 expressed at the cell surface. Lower band is considered as immature Npr2 expressed intracellularly. Number of blots analyzed: 9 for DRGs of each genotype, 12 for wild-type, and 9 for mutant astrocytes. Error bar indicates SEM. *p = 0.0197, for astrocytes (t test).
In sum, our data indicate that Npr2-7A mutant proteins reach the cell surface of DRG neurons as wild-type forms but might be slightly retarded in astrocytes. Therefore, we conclude that Npr2-7A does not have intracellular trafficking defects in DRGs.
In Npr27A/7A mutant mice, cGMP levels fail to increase in DRGs or in brain or lung tissue upon application of CNP
To analyze CNP-induced cGMP generation, the Npr2-7A line was crossed with transgenic mice that ubiquitously express the genetically encoded FRET-based cGMP sensor, cGi500 (Russwurm et al., 2007; Thunemann et al., 2013b). Real-time visualization of cGMP was performed by FRET measurements in cultivated E12.5 DRG neurons as previously reported (Schmidt et al., 2016). Upon application of 200 nm CNP, the intracellular cGMP level was increased immediately in neurons from wild-type embryos, whereas no change in fluorescence intensities was observed in DRG neurons from Npr27A/7A embryos (Fig. 3A–C). Fluorescence intensity values in the wild-type returned to basal levels when CNP was removed from the cell supernatants. The rise in CNP-induced cGMP concentration in DRG neurons from Npr2wt/7A embryos reached on average approximately half of the wild-type level (Fig. 3D). The FRET/cGMP measurements indicated that mutant Npr2 cannot be stimulated by CNP to generate cGMP in DRG neurons.
Figure 3.

FRET-based cGMP imaging of individual DRG neurons from E12.5 Npr2wt/wt, Npr2wt/7A, and Npr27A/7A embryos expressing the genetically encoded cGi500 cGMP biosensor. A, Representative images of cGi500 biosensor fluorescence in dissociated E12.5 DRG neurons from Npr27A/7A, Npr2wt/7A, and Npr2wt/wt 24 h after plating. Scale bar, 10 μm. B, C, Changes in fluorescence of the genetically encoded cGi500 cGMP biosensor elicited by application of 200 nm CNP (horizontal bar) recorded from individual DRG neurons from wild-type (B) and Npr27A/7A (C) mice. Traces indicate CFP emission (F480, blue), YFP emission (F535, yellow), and the CFP/YFP emission ratio (r = F480/F535, black). Emission intensities and ratios were normalized to average baseline signals and given as ΔF/F and ΔR/R, respectively. Alterations of ΔR/R indicate changes of the intracellular cGMP concentration. Shown are representative results from five experiments with independent cell cultures. D, Scatter blot of the peak amplitudes of normalized CFP/YFP emission ratios recorded from DRG neurons perfused with 200 nm CNP from wild-type, Npr2wt/7A, and Npr27A/7A mice. Data are mean ± SD (wild-type, n = 18; Npr2wt/7A, n = 25; Npr27A/7A, n = 32). ***p < 0.0001 (Mann–Whitney test).
To further support these observations on cultivated DRG neurons, guanylyl cyclase activity was measured in brain and lung tissues from Npr2wt/wt, Npr2wt/7A, and Npr27A/7A mice to determine the effect of Npr2 phosphorylation sites on Npr2 activity in response to stimulation by CNP (Shuhaibar et al., 2016). In addition, Npr1 (GC-A) activity was measured by application of ANP to determine whether the loss of Npr2 activity resulted in compensatory increases in Npr1 activity. Clear reductions in CNP-stimulated cGMP production were observed in brain tissue from Npr27A/7A mice (Fig. 4A). The slight increase in cGMP observed in the presence of 1000 nm CNP is likely due to cross-activation of Npr1 as previously reported for Npr1 in rodent tissues (Dickey et al., 2008). A similar lack of CNP-dependent activity was observed in lung tissue from Npr27A/7A mice as well (Fig. 4B). Total membrane guanylyl cyclase activity measured in response to application of Triton X-100 and manganese, which does not require phosphorylation for activity, was found to be similar in all three genotypes, which is in line with the above data that showed that Npr2 variants are equally expressed (Fig. 2B,D,E). Furthermore, no differences in guanylyl cyclase activity were observed upon application of ANP between wild-type and Npr2-7A heterozygotes and homozygous mutants.
Figure 4.
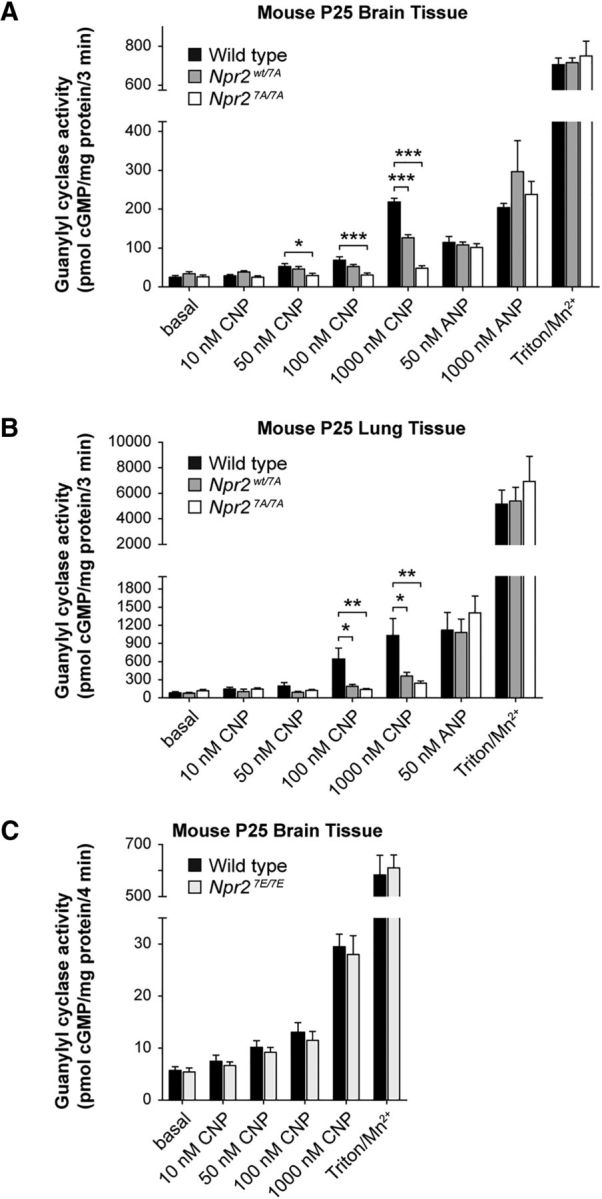
Guanylyl cyclase measurements of tissues from Npr2-7A and Npr-7E mutants. A, Changes in ANP- and CNP-dependent guanylyl cyclase activities in brains from Npr2wt/wt, Npr2wt/7A, and Npr27A/7A mice. Crude membranes were prepared from flash-frozen brains, and 60–75 μg was assayed in duplicate for each condition; n = 5 or 6. Error bars indicate mean ± SEM. *p ≤ 0.05, statistical significance from wild-type samples. **p ≤ 0.01, statistical significance from wild-type samples. ***p ≤ 0.001. B, Changes in ANP- and CNP-dependent guanylyl cyclase activities in lungs from Npr2wt/wt, Npr2wt/7A, and Npr27A/7A mice. Crude membranes were prepared from flash-frozen lungs, and 10–17 μg was assayed in duplicate for each condition; n = 5 or 6. Error bars indicate mean ± SEM. *p ≤ 0.05, statistical significance from WT samples. **p ≤ 0.01, statistical significance from WT samples. C, CNP-stimulated guanylyl cyclase activity measured in brains from Npr2wt/wt and Npr27E/7E mice does not differ. Guanylyl cyclase activity was measured in crude membranes from postnatal day 25 male and female Npr2wt/wt and Npr27E/7E mice in the presence of excess MgCl2, 0.1 mm GTP, 1 mm ATP, and the indicated concentrations of CNP or with 0.1 mm GTP and excess Mn2+ and 1% Triton X-100 (Mn/Triton). n = 6 mice for each genotype with 3 males and 3 females for each treatment. A, B, cGMP measurements were done by a highly sensitive radioimmunoassay without removal of cross-reacting GTP. C, cGMP was determined by an ELISA-based method after removal of GTP by sodium carbonate precipitation and purification of cGMP over an alumina column.
Interestingly, the amount of activity in heterozygous Npr2wt/7A mice differed between the brain versus lung tissues. In brain tissue from Npr2wt/7A mice, CNP-dependent guanylyl activity was approximately half of that observed for the Npr2wt/wt mice as expected based solely on genetic dosage. However, CNP-dependent activity in lung tissue from the Npr2wt/7A mice was less than half of the activity measured in lung tissue from the Npr2wt/wt mice (Fig. 4). One possible explanation for this discrepancy is that Npr2-7A is serving a dominant-negative function to inhibit wild-type Npr2 as previously reported (Langenickel et al., 2006; Hachiya et al., 2007; Vasques et al., 2013).
In Npr27E/7E mouse mutants, which mimic constitutive phosphorylation, guanylyl cyclase activation requires the ligand CNP
The biochemical as well as the FRET data on Npr2-7A indicated that phosphorylation is required for the sensitization of the guanylyl cyclase activity of Npr2. To ask whether phosphorylation alone is sufficient for activation, we measured the guanylyl cyclase activity from Npr27E/7E transgenic mice in which the above-described regulatory phosphorylation sites in the KHD of Npr2 are replaced by glutamic acid residues that mimic phosphorylation (Shuhaibar et al., 2016). Neither basal activity nor activation at various concentrations of CNP was different between wild-type and Npr27E/7E (Fig. 4C), which is consistent with previous reports of Npr27E/7E activity or cellular cGMP concentration measured in ovarian follicles or bones (Shuhaibar et al., 2016, 2017) or using transfected HEK293 expressing Npr2–7E (Robinson et al., 2013). Additionally, activity in the presence of Mn2+/Triton, a synthetic activator of particulate guanylyl cyclases and an indirect measure of protein content, was unchanged between groups, which demonstrates similar Npr2 protein levels between genotypes (Fig. 4C). In conclusion, these observations indicate that constitutive mimicking of phosphorylation of Npr2 alone does not activate and does not improve CNP-mediated activation of the guanylyl cyclase. In line with our data on Npr2-7A, we conclude that the negative charges of the above-mentioned sites are essential for the transduction of the CNP binding signal.
Cellular evidence of a dominant-negative effect of Npr2-7A on Npr2
To determine whether Npr2 and Npr2-7A associate in cells, an N-terminally FLAG-tagged version of mouse Npr2 and an N-terminally HA-tagged version of human Npr2-7A were expressed alone or together in the 293T cells lacking endogenous Npr2. Subsequently, the receptors were immunoprecipitated with antibodies against either the FLAG or HA epitopes, fractionated by SDS-PAGE, blotted to a PVDF membrane, and probed with antibodies to FLAG, HA, or the C terminus of Npr2 (Fig. 5A). Importantly, when receptors were immunoprecipitated with antibodies against the FLAG-tag linked to Npr2 and blotted with antibodies to the HA-tag on Npr2-7A, a positive signal was observed. The converse scenario was also observed. These data indicate that Npr2 and Npr2-7A physically associate in cells.
Figure 5.

Npr2-7A inactivates Npr2. A, Npr2 and Npr2-7A coimmunoprecipitate. The 293T cells were transfected with 1 μg GFP plasmid, 2 μg FLAG-mouse-Npr2-WT, 1 μg FLAG-mouse-Npr2-WT, and 1 μg HA-human-Npr2-7A or 2 μg HA-human-Npr2-7A. After 48 h, crude membranes were prepared and assayed for guanylyl cyclase activity. The remaining membranes were lysed, and then the Npr2 proteins were immunoprecipitated with the indicated antibodies. The proteins were separated by SDS-PAGE before transfer to PVDF. The blot was probed for Npr2 and FLAG and then stripped and reprobed for HA. B, Npr2-7A has a dominant-negative effect on Npr2. The . The 293T cells were transfected with 1 μg GFP plasmid, 2 μg FLAG-mouse-Npr2-WT, 1 μg FLAG-mouse-Npr2, and 1 μg HA-human-Npr2-7A or 2 μg HA-human-Npr2-7A. After 48 h, crude membranes were prepared and assayed for guanylyl cyclase activity. Values represent mean ± SEM from three experiments assayed in duplicate for n = 6.
When guanylyl cyclase assays were performed on membranes from the 293T cells transfected with the same receptors, a large dominant-negative effect was observed (Fig. 5B).
Bifurcation of sensory axons is impaired in Npr27A/7A mutant mice
To follow the path of sensory axons when entering the spinal cord or the hindbrain in Npr27A/7A mutants, we applied DiI tracing to embryonic DRGs and used a genetic approach for sparse labeling of Npr2-expressing axons (Schmidt et al., 2013; Ter-Avetisyan et al., 2014). Npr27A/7A mice completely lacked T-shaped bifurcation of DRG axons (Fig. 6A–C). Similarly, axons of cranial sensory ganglia entering the hindbrain turned in either rostral or caudal direction (Fig. 6D). Consistent with these observations at embryonic ages, a lack of bifurcated axons in Npr27A/7A transgenes was also evident at juvenile stages as demonstrated by studying crosses with Thy-1-YFP-H reporter mice at postnatal day (P) 15 (Fig. 6E,F). Therefore, compensatory mechanisms for the correction of the branching error at the level of the entry zone have not been developed at postnatal stages. Despite the dominant-negative effect of Npr2-7A, heterozygous Npr2wt/7A mice do not reveal bifurcations errors (Fig. 6A), which is in line with our previous investigations that gross changes in cGMP levels do not affect bifurcation (Schmidt et al., 2007, 2016).
Figure 6.

Axon bifurcation in Npr27A/7A mutants is impaired, whereas axonal branching in Npr27E/7E mutants is normal. A, DiI tracing of DRG axons from Npr27A/7A mutants reveals the absence of bifurcation in the dorsal root entry zone at E13.5. Arrows indicate bifurcation. Arrowhead indicates turn without bifurcation. Scale bar, 25 μm. B, Quantification of branching errors by DiI tracing. Numbers indicate number of axons counted and number of embryos analyzed. C, Alkaline phosphatase stained DRG axons in whole-mount spinal cord preparations from tamoxifen-stimulated Npr2wt/CreERT2;Z/APtg and Npr27A/CreERT2;Z/APtg mice demonstrate loss of axon bifurcation in the spinal cord (SC). Scale bar, 100 μm. D, Lack of bifurcation of axons from cranial sensory ganglion neurons in Npr27A/7A mutant at E13.5 using a genetic approach for sparse labeling of Npr2-positive axons as described previously (Ter-Avetisyan et al., 2014). gV, Ganglion trigeminale; gVII, ganglion geniculi; gVIII, ganglion vestibulare; gIX, ganglion glossopharyngeal superior or inferior; Hb, hindbrain. Scale bar, 200 μm. E, Lack of DRG axon bifurcation in P15 Thy1-YFP-H reporter crossed into Npr27A/7A mutant mice, indicating that compensation does not occur at postnatal stages. Scale bar, 100 μm. F, Quantification of bifurcation errors in the spinal cord using Thy-1-YFP-H reporter mice. G, H, DiI tracing of DRG axons from Npr27E/7E mutants reveals normal sensory axon bifurcation in the dorsal root entry zone at E13.5. Ectopic or repeated bifurcation was not observed, suggesting that permanent responsive Npr2 does not impair bifurcation. Scale bar, 50 μm. I, Collateral formation is not impaired in E14.5 Npr27A/7A mutant mice. Spinal cord cross sections were stained with anti-peripherin. Arrowheads indicate collaterals. MN, Motoneurons. Scale bar, 100 μm.
After bifurcation of the growth cone, the two resulting daughter axons proceed growing in opposite directions within the dorsal funiculus. After a waiting period, collaterals are generated from these stem axons by interstitial branching that extends ventrally into the cord where they terminate in specific layers of the dorsal or ventral horn. Staining of transverse sections by anti-peripherin indicated that collateral formation of nociceptive as well as proprioceptive neurons at appropriate mediolateral positions is not impaired in Npr27A/7A (Fig. 6I). In addition, the overall structure of the spinal cord was not affected by the Npr2 mutation (Fig. 2A).
In Npr27E/7E mouse mutants, which mimic constitutive phosphorylation, sensory axon bifurcation is not impaired
Previous studies using recombinant Npr2 in HEK293 cells have shown that phosphomimetic glutamate-substituted versions of transmembrane guanylyl cyclases are resistant to dephosphorylation-dependent inactivation and that their CNP-dependent guanylyl cyclase activity corresponds with that of the wild-type phosphorylated enzyme (Yoder et al., 2012; Shuhaibar et al., 2016). So far, it has not been demonstrated whether this Npr2 variant modifies axon bifurcation or extension in the spinal cord. In the absence of dephosphorylation of Npr2, a higher number of T-branches or a premature, ectopic branching already in dorsal roots before axons are entering the spinal cord might be envisioned because, in ovarian follicles, dephosphorylation of Npr2 is a physiological mediator of hormone signaling (Shuhaibar et al., 2016, 2017; Robinson et al., 2017). However, analysis of sensory axon bifurcation by DiI tracing at E13.5 demonstrated that Npr27E/7E mutants did not reveal any branching errors. DRG axons showed normal T-like bifurcations and did not form multiple branches in the dorsal root entry zone (Fig. 6G,H). Sensory axons extended at lateral regions as in wild-type spinal cords, suggesting that the process of bifurcation and extension is resilient to an impaired desensitization of Npr2.
Discussion
In this study, we showed that removal of known phosphorylation sites in the kinase homology domain of the receptor guanylyl cyclase Npr2 completely mimics the axonal bifurcation error of DRG and CSG neurons (Schmidt et al., 2007; Ter-Avetisyan et al., 2014) and the dwarfed phenotype observed in mice lacking functional alleles of Npr2 (Tamura et al., 2004). The absence of regulatory phosphorylation sites abolished CNP-dependent guanylyl cyclase activity of Npr2 and kept the enzyme unresponsive to CNP. Thus, our data demonstrated that phosphorylation of the seven regulatory serine and threonine residues in this intracellular segment of Npr2 is required for physiological activation of Npr2 in somatosensory neurons to induce bifurcation of axons and ossification of chondrocytes during long bone growth in vivo. In contrast to the Npr27A/7A mouse, no errors in axonal branching or extension were observed in DRG neurons that express a glutamate-substituted form of Npr2 (Npr27E/7E mouse mutant), indicating that negatively charged residues at these positions appear to be required for the transmission of CNP binding to the extracellular domain to the activation of the guanylyl cyclase (Fig. 7). Together with our studies on the CNP knock-out (Schmidt et al., 2009), we conclude that both binding of CNP to and phosphorylation of Npr2 are required to induce bifurcation, indicating that Npr2 is a coincidence selector in somatosensory neurons. In the skeletal system, the Npr27E/7E mouse exhibits slight bone overgrowth most likely due to the continuous activation by CNP (Shuhaibar et al., 2017) and is resistant to luteinizing hormone inhibition of Npr2 in the ovarian follicle (Shuhaibar et al., 2016), a process shown to be correlated with Npr2 dephosphorylation in rats (Egbert et al., 2014). Dephosphorylation of these serine and threonine residues is essential for the hormonal regulation of the guanylyl cyclase activity in the ovarian follicle and therefore meiotic resumption was delayed.
Figure 7.

Scheme depicting CNP-induced generation of cGMP by Npr2 in the wild-type and the effect of the Npr2-7A and Npr2–7E mutants (A–C) and the corresponding axon branching phenotype (D) of sensory neurons. Although phosphorylation is mimicked by glutamic acid substitutions, Npr2–7E does not generate cGMP in the absence of the ligand CNP. Npr2 dimers display only one catalytic site (Robinson and Potter, 2012).
Regarding the dominant-negative effects observed by measurements of guanylyl cyclase activity in lung tissue, cotransfection studies provided support for a model where Npr2-7A inactivates Npr2 by forming a physical association in cells. Similar studies were reported for the ability for dephosphorylated versions of Npr1 (GC-A) to inhibit wild-type Npr1 (Potter and Hunter, 1998b). However, a putative dominant-negative effect of Npr2-7A did not play a role in axon bifurcation because, in heterozygous Npr2wt/7A mice, a disruption of T-like branches was never observed. Because the dominant-negative effect was also only observed in lung but not in brain tissue, it appears to be a cell type-specific phenomenon. Moreover, bifurcation of axons appears to be insensitive to gross changes of cGMP levels because, in mutants, double heterozygous for Npr2 and cGKI normal T-like branches are found (Schmidt et al., 2007). Up to now, only loss-of-function mutations of the components of the cGMP pathway in DRG neurons have been shown to cause defects in sensory axon bifurcation in mice. This is remarkable because changes (either positive or negative) in the levels of CNP or Npr2 activity have been reported to affect human body height (Olney et al., 2006; Vasques et al., 2013; Hisado-Oliva et al., 2015), which was also found for mice heterozygous for Npr2 (Tamura et al., 2004).
Npr2 was first shown to be highly phosphorylated almost 20 years ago (Potter, 1998), and subsequent studies identified seven chemically determined phosphorylation sites (Potter and Hunter, 1998a; Yoder et al., 2010, 2012). Mutation of the first six chemically identified phosphorylation sites to alanine dramatically decreased the ability of CNP to stimulate Npr2 from 49-fold over basal levels for the phosphorylated wild-type receptor to 2.6-fold over basal levels for Npr2-7A (Potter and Hunter, 1998a).
It is interesting by what means individual sensory axons of DRG or CSG neurons bifurcate with high precision only at the entry zone of the spinal cord or hindbrain, respectively, and, importantly, only once so that two, and not multiple, stem axons are generated. Although Npr2 is located on sensory axons long before bifurcation takes place and still remains expressed after bifurcation, an ectopic branching or an additional T-branch deeper in the spinal cord has never been observed. T-branching therefore requires a tight control in space and time. This might be secured by regulating different set screws of CNP-induced cGMP signaling, including timing and localization of CNP, removal of CNP from the extracellular milieu by the clearance receptor Npr3, the intracellular degradation of cGMP by phosphodiesterases (PDEs), and regulation of the guanylyl cyclase activity of Npr2 by phosphorylation.
While the scavenger receptor Npr3 is negligible for T-branching (Schmidt et al., 2016), synthesis of CNP is restricted to cells located in the dorsal horn of the spinal cord or in specific rhombomers when axons enter the spinal cord or the hindbrain, respectively. CNP then becomes strongly reduced in these regions, suggesting that the timing and place of CNP expression provide some degree of specificity for the localized branching at the DREZ (Schmidt et al., 2009). Furthermore, regulation of cGMP signaling might be controlled by the degradation of cGMP by PDEs (Bender and Beavo, 2006; Maurice et al., 2014). However, bifurcation of sensory axons in the absence of PDE2A, the major PDE for the degradation of CNP-induced cGMP in embryonic DRG neurons, is normal (Schmidt et al., 2016), suggesting that intracellular degradation of cGMP might not be an essential mechanism that intervenes with bifurcation. In such a scenario where PDE2A and the scavenger receptor Npr3 seem to be dispensable, direct regulation of the guanylyl cyclase activity of Npr2 might be a central regulatory mechanism to specify and shut down bifurcation.
Several reagents have been primarily identified in cell culture systems to interfere with the phosphorylation status of Npr2, including components that modulate the activity of kinases, phosphatases, or the level of intracellular calcium (Potthast et al., 2004; Abbey-Hosch et al., 2005; Potter, 2005; Robinson et al., 2017; Shuhaibar et al., 2017). This phenomenon, referred to as heterologous desensitization, provides the basis for cross talks between Npr2 and kinase- or phosphatase-dependent signaling cascades and might establish links to axonal guidance or other branching factors. The kinases or phosphatases that phosphorylate or dephosphorylate Npr2 in DRG neurons have not yet been identified. Also, it is unknown whether phosphorylation of Npr2 is constitutive or regulated in DRG neurons. Regulated phosphorylation could make DRGs competent for CNP-induced branching, and dephosphorylation might guarantee that further T-branching is blocked. In this context, it is important to recall that axon bifurcation cannot be induced by CNP in in vitro systems, suggesting, as previously discussed, that additional molecular signals are missing in cell culture to make DRG neurons competent for bifurcation. It is notable that, in the skeletal system, a link between Npr2 and fibroblast factor receptor 3 signaling has been found, resulting in the dephosphorylation of Npr2 (Robinson et al., 2017; Shuhaibar et al., 2017).
In conclusion, this study demonstrates, for the first time, that phosphorylation of the kinase homology domain of Npr2 is of physiological relevance for axon bifurcation. Further investigation on the cross talk between CNP signaling and components implicated in heterologous desensitization of Npr2 may yield additional insights into the process of axon branching. The identification of the enzymes that phosphorylate and dephosphorylate Npr2 in sensory neurons will be one of the major steps in the dissection of the regulation of Npr2.
Footnotes
This work was supported by Deutsche Forschungsgemeinschaft Grant SFB 665/B2 to F.G.R. and Grant FOR 2060 (FE 438/5-2 and FE 438/6-2 to R.F., and SCHM 2371/1 to H.S.), National Institutes of Health Grant R01GM098309 and Hormone Receptor Fund to L.R.P., and Fund for Science Grants to L.R.P. and R.F. We thank Madlen Driesner, Mechthild Henning, Anne Banerjee, and Karola Bach for technical help; Stefanie Peters for help with FRET/cGMP measurements of embryonic DRG neurons; and Siu-Pok Yee, Leia Shuhaibar, and Laurinda Jaffe (University of Connecticut) for providing Npr27E/7E embryos and mice.
The authors declare no competing financial interests.
References
- Abbey SE, Potter LR (2002) Vasopressin-dependent inhibition of the C-type natriuretic peptide receptor, NPR-B/GC-B, requires elevated intracellular calcium concentrations. J Biol Chem 277:42423–42430. 10.1074/jbc.M206686200 [DOI] [PubMed] [Google Scholar]
- Abbey-Hosch SE, Smirnov D, Potter LR (2005) Differential regulation of NPR-B/GC-B by protein kinase c and calcium. Biochem Pharmacol 70:686–694. 10.1016/j.bcp.2005.04.034 [DOI] [PubMed] [Google Scholar]
- Akiyama H, Fukuda T, Tojima T, Nikolaev VO, Kamiguchi H (2016) Cyclic nucleotide control of microtubule dynamics for axon guidance. J Neurosci 36:5636–5649. 10.1523/JNEUROSCI.3596-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amano N, Mukai T, Ito Y, Narumi S, Tanaka T, Yokoya S, Ogata T, Hasegawa T (2014) Identification and functional characterization of two novel NPR2 mutations in japanese patients with short stature. J Clin Endocrinol Metab 99:E713–E718. 10.1210/jc.2013-3525 [DOI] [PubMed] [Google Scholar]
- Armijo-Weingart L, Gallo G (2017) It takes a village to raise a branch: cellular mechanisms of the initiation of axon collateral branches. Mol Cell Neurosci 84:36–47. 10.1016/j.mcn.2017.03.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender AT, Beavo JA (2006) Cyclic nucleotide phosphodiesterases: molecular regulation to clinical use. Pharmacol Rev 58:488–520. 10.1124/pr.58.3.5 [DOI] [PubMed] [Google Scholar]
- Deschepper CF, Picard S (1994) Effects of C-type natriuretic peptide on rat astrocytes: regional differences and characterization of receptors. J Neurochem 62:1974–1982. 10.1046/j.1471-4159.1994.62051974.x [DOI] [PubMed] [Google Scholar]
- Dickey DM, Burnett JC, Potter LR (2008) Novel bifunctional natriuretic peptides as potential therapeutics. J Biol Chem 283:35003–35009. 10.1074/jbc.M804538200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickey DM, Edmund AB, Otto NM, Chaffee TS, Robinson JW, Potter LR (2016) Catalytically active guanylyl cyclase B requires endoplasmic reticulum-mediated glycosylation, and mutations that inhibit this process cause dwarfism. J Biol Chem 291:11385–11393. 10.1074/jbc.M115.704015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickey DM, Otto NM, Potter LR (2017) Skeletal overgrowth-causing mutations mimic an allosterically activated conformation of guanylyl cyclase-B that is inhibited by 2,4,6,-trinitrophenyl ATP. J Biol Chem 292:10220–10229. 10.1074/jbc.M117.780536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domino SE, Tubb DJ, Garbers DL (1991) Assay of guanylyl cyclase catalytic activity. Methods Enzymol 195:345–355. 10.1016/0076-6879(91)95179-N [DOI] [PubMed] [Google Scholar]
- Dumoulin A, Ter-Avetisyan G, Schmidt H, Rathjen FG (2018) Molecular analysis of sensory axon branching unraveled a cGMP-dependent signaling cascade. Int J Mol Sci 19:E1266. 10.3390/ijms19051266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egbert JR, Shuhaibar LC, Edmund AB, Van Helden DA, Robinson JW, Uliasz TF, Baena V, Geerts A, Wunder F, Potter LR, Jaffe LA (2014) Dephosphorylation and inactivation of NPR2 guanylyl cyclase in granulosa cells contributes to the LH-induced decrease in cGMP that causes resumption of meiosis in rat oocytes. Development 141:3594–3604. 10.1242/dev.112219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng G, Mellor RH, Bernstein M, Keller-Peck C, Nguyen QT, Wallace M, Nerbonne JM, Lichtman JW, Sanes JR (2000) Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron 28:41–51. 10.1016/S0896-6273(00)00084-2 [DOI] [PubMed] [Google Scholar]
- Flora DR, Potter LR (2010) Prolonged atrial natriuretic peptide exposure stimulates guanylyl cyclase: a degradation. Endocrinology 151:2769–2776. 10.1210/en.2009-1239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson DA, Ma L (2011) Developmental regulation of axon branching in the vertebrate nervous system. Development 138:183–195. 10.1242/dev.046441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hachiya R, Ohashi Y, Kamei Y, Suganami T, Mochizuki H, Mitsui N, Saitoh M, Sakuragi M, Nishimura G, Ohashi H, Hasegawa T, Ogawa Y (2007) Intact kinase homology domain of natriuretic peptide receptor-B is essential for skeletal development. J Clin Endocrinol Metab 92:4009–4014. 10.1210/jc.2007-1101 [DOI] [PubMed] [Google Scholar]
- Hisado-Oliva A, Garre-Vázquez AI, Santaolalla-Caballero F, Belinchón A, Barreda-Bonis AC, Vasques GA, Ramirez J, Luzuriaga C, Carlone G, González-Casado I, Benito-Sanz S, Jorge AA, Campos-Barros A, Heath KE (2015) Heterozygous NPR2 mutations cause disproportionate short stature, similar to Leri-Weill dyschondrosteosis. J Clin Endocrinol Metab 100:E1133–E1142. 10.1210/jc.2015-1612 [DOI] [PubMed] [Google Scholar]
- Hume AN, Buttgereit J, Al-Awadhi AM, Al-Suwaidi SS, John A, Bader M, Seabra MC, Al-Gazali L, Ali BR (2009) Defective cellular trafficking of missense NPR-B mutants is the major mechanism underlying acromesomelic dysplasia-type Maroteaux. Hum Mol Genet 18:267–277. 10.1093/hmg/ddn354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn M. (2016) Molecular physiology of membrane guanylyl cyclase receptors. Physiol Rev 96:751–804. 10.1152/physrev.00022.2015 [DOI] [PubMed] [Google Scholar]
- Langenickel TH, Buttgereit J, Pagel-Langenickel I, Lindner M, Monti J, Beuerlein K, Al-Saadi N, Plehm R, Popova E, Tank J, Dietz R, Willenbrock R, Bader M (2006) Cardiac hypertrophy in transgenic rats expressing a dominant-negative mutant of the natriuretic peptide receptor B. Proc Natl Acad Sci U S A 103:4735–4740. 10.1073/pnas.0510019103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobe CG, Koop KE, Kreppner W, Lomeli H, Gertsenstein M, Nagy A (1999) Z/AP, a double reporter for cre-mediated recombination. Dev Biol 208:281–292. 10.1006/dbio.1999.9209 [DOI] [PubMed] [Google Scholar]
- Ma L, Tessier-Lavigne M (2007) Dual branch-promoting and branch-repelling actions of Slit/Robo signaling on peripheral and central branches of developing sensory axons. J Neurosci 27:6843–6851. 10.1523/JNEUROSCI.1479-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maley F, Trimble RB, Tarentino AL, Plummer TH Jr (1989) Characterization of glycoproteins and their associated oligosaccharides through the use of endoglycosidases. Anal Biochem 180:195–204. 10.1016/0003-2697(89)90115-2 [DOI] [PubMed] [Google Scholar]
- Maurice DH, Ke H, Ahmad F, Wang Y, Chung J, Manganiello VC (2014) Advances in targeting cyclic nucleotide phosphodiesterases. Nat Rev Drug Discov 13:290–314. 10.1038/nrd4228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy KD, de Vellis J (1980) Preparation of separate astroglial and oligodendroglial cell cultures from rat cerebral tissue. J Cell Biol 85:890–902. 10.1083/jcb.85.3.890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olney RC, Bükülmez H, Bartels CF, Prickett TC, Espiner EA, Potter LR, Warman ML (2006) Heterozygous mutations in natriuretic peptide receptor-B (NPR2) are associated with short stature. J Clin Endocrinol Metab 91:1229–1232. 10.1210/jc.2005-1949 [DOI] [PubMed] [Google Scholar]
- Potter LR. (1998) Phosphorylation-dependent regulation of the guanylyl cyclase-linked natriuretic peptide receptor B: dephosphorylation is a mechanism of desensitization. Biochemistry 37:2422–2429. 10.1021/bi972303k [DOI] [PubMed] [Google Scholar]
- Potter LR. (2005) Domain analysis of human transmembrane guanylyl cyclase receptors: implications for regulation. Front Biosci 10:1205–1220. 10.2741/1613 [DOI] [PubMed] [Google Scholar]
- Potter LR. (2011) Guanylyl cyclase structure, function and regulation. Cell Signal 23:1921–1926. 10.1016/j.cellsig.2011.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potter LR, Hunter T (1998a) Identification and characterization of the major phosphorylation sites of the B-type natriuretic peptide receptor. J Biol Chem 273:15533–15539. 10.1074/jbc.273.25.15533 [DOI] [PubMed] [Google Scholar]
- Potter LR, Hunter T (1998b) Phosphorylation of the kinase homology domain is essential for activation of the A-type natriuretic peptide receptor. Mol Cell Biol 18:2164–2172. 10.1128/MCB.18.4.2164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potthast R, Abbey-Hosch SE, Antos LK, Marchant JS, Kuhn M, Potter LR (2004) Calcium-dependent dephosphorylation mediates the hyperosmotic and lysophosphatidic acid-dependent inhibition of natriuretic peptide receptor-B/guanylyl cyclase-B. J Biol Chem 279:48513–48519. 10.1074/jbc.M408247200 [DOI] [PubMed] [Google Scholar]
- Robinson JW, Dickey DM, Miura K, Michigami T, Ozono K, Potter LR (2013) A human skeletal overgrowth mutation increases maximal velocity and blocks desensitization of guanylyl cyclase-B. Bone 56:375–382. 10.1016/j.bone.2013.06.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JW, Egbert JR, Davydova J, Schmidt H, Jaffe LA, Potter LR (2017) Dephosphorylation is the mechanism of fibroblast growth factor inhibition of guanylyl cyclase-B. Cell Signal 40:222–229. 10.1016/j.cellsig.2017.09.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JW, Potter LR (2012) Guanylyl cyclases A and B are asymmetric dimers that are allosterically activated by ATP binding to the catalytic domain. Sci Signal 5:ra65. 10.1126/scisignal.2003253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russwurm M, Mullershausen F, Friebe A, Jäger R, Russwurm C, Koesling D (2007) Design of fluorescence resonance energy transfer (FRET)-based cGMP indicators: a systematic approach. Biochem J 407:69–77. 10.1042/BJ20070348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt H, Peters S, Frank K, Wen L, Feil R, Rathjen FG (2016) Dorsal root ganglion axon bifurcation tolerates increased cyclic GMP levels: the role of phosphodiesterase 2A and scavenger receptor Npr3. Eur J Neurosci 44:2991–3000. 10.1111/ejn.13434 [DOI] [PubMed] [Google Scholar]
- Schmidt H, Rathjen FG (2010) Signalling mechanisms regulating axonal branching in vivo. Bioessays 32:977–985. 10.1002/bies.201000054 [DOI] [PubMed] [Google Scholar]
- Schmidt H, Rathjen FG (2011) DiI-labeling of DRG neurons to study axonal branching in a whole mount preparation of mouse embryonic spinal cord. J Vis Exp 2011:3667. 10.3791/3667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt H, Stonkute A, Jüttner R, Koesling D, Friebe A, Rathjen FG (2009) C-type natriuretic peptide (CNP) is a bifurcation factor for sensory neurons. Proc Natl Acad Sci U S A 106:16847–16852. 10.1073/pnas.0906571106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt H, Stonkute A, Jüttner R, Schäffer S, Buttgereit J, Feil R, Hofmann F, Rathjen FG (2007) The receptor guanylyl cyclase Npr2 is essential for sensory axon bifurcation within the spinal cord. J Cell Biol 179:331–340. 10.1083/jcb.200707176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt H, Ter-Avetisyan G, Rathjen FG (2013) A genetic strategy for the analysis of individual axon morphologies in cGMP signalling mutant mice. Methods Mol Biol 1020:193–204. 10.1007/978-1-62703-459-3_12 [DOI] [PubMed] [Google Scholar]
- Schmidt H, Werner M, Heppenstall PA, Henning M, Moré MI, Kühbandner S, Lewin GR, Hofmann F, Feil R, Rathjen FG (2002) cGMP-mediated signaling via cGKIalpha is required for the guidance and connectivity of sensory axons. J Cell Biol 159:489–498. 10.1083/jcb.200207058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW (2012) NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9:671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro F, Barone L, Johnson A, Flynn E (2014) The cn/cn dwarf mouse: histomorphometric, ultrastructural, and radiographic study in mutants corresponding to human acromesomelic dysplasia maroteaux type (AMDM). BMC Musculoskelet Disord 15:347. 10.1186/1471-2474-15-347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuhaibar LC, Egbert JR, Edmund AB, Uliasz TF, Dickey DM, Yee SP, Potter LR, Jaffe LA (2016) Dephosphorylation of juxtamembrane serines and threonines of the NPR2 guanylyl cyclase is required for rapid resumption of oocyte meiosis in response to luteinizing hormone. Dev Biol 409:194–201. 10.1016/j.ydbio.2015.10.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuhaibar LC, Robinson JW, Vigone G, Shuhaibar NP, Egbert JR, Baena V, Uliasz TF, Kaback D, Yee SP, Feil R, Fisher MC, Dealy CN, Potter LR, Jaffe LA (2017) Dephosphorylation of the NPR2 guanylyl cyclase contributes to inhibition of bone growth by fibroblast growth factor. Elife 6:e31343. 10.7554/eLife.31343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sogawa C, Tsuji T, Shinkai Y, Katayama K, Kunieda T (2007) Short-limbed dwarfism: slw is a new allele of Npr2 causing chondrodysplasia. J Hered 98:575–580. 10.1093/jhered/esm065 [DOI] [PubMed] [Google Scholar]
- Tamura N, Doolittle LK, Hammer RE, Shelton JM, Richardson JA, Garbers DL (2004) Critical roles of the guanylyl cyclase B receptor in endochondral ossification and development of female reproductive organs. Proc Natl Acad Sci U S A 101:17300–17305. 10.1073/pnas.0407894101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ter-Avetisyan G, Rathjen FG, Schmidt H (2014) Bifurcation of axons from cranial sensory neurons is disabled in the absence of Npr2-induced cGMP signaling. J Neurosci 34:737–747. 10.1523/JNEUROSCI.4183-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ter-Avetisyan G, Dumoulin A, Herrel A, Schmidt H, Strump J, Afzal S, Rathjen FG (2018) Loss of axon bifurcation in mesencephalic trigeminal neurons impairs the maximal biting force in Npr2-deficient mice. Front Cell Neurosci 12:153. 10.3389/fncel.2018.00153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thunemann M, Fomin N, Krawutschke C, Russwurm M, Feil R (2013a) Visualization of cGMP with cGi biosensors. Methods Mol Biol 1020:89–120. 10.1007/978-1-62703-459-3_6 [DOI] [PubMed] [Google Scholar]
- Thunemann M, Wen L, Hillenbrand M, Vachaviolos A, Feil S, Ott T, Han X, Fukumura D, Jain RK, Russwurm M, de Wit C, Feil R (2013b) Transgenic mice for cGMP imaging. Circ Res 113:365–371. 10.1161/CIRCRESAHA.113.301063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tröster P, Haseleu J, Petersen J, Drees O, Schmidtko A, Schwaller F, Lewin GR, Ter-Avetisyan G, Winter Y, Peters S, Feil S, Feil R, Rathjen FG, Schmidt H (2018) The absence of sensory axon bifurcation affects nociception and termination fields of afferents in the spinal cord. Front Mol Neurosci 11:19. 10.3389/fnmol.2018.00019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuji T, Kunieda T (2005) A loss-of-function mutation in natriuretic peptide receptor 2 (Npr2) gene is responsible for disproportionate dwarfism in cn/cn mouse. J Biol Chem 280:14288–14292. 10.1074/jbc.C500024200 [DOI] [PubMed] [Google Scholar]
- Vasques GA, Amano N, Docko AJ, Funari MF, Quedas EP, Nishi MY, Arnhold IJ, Hasegawa T, Jorge AA (2013) Heterozygous mutations in natriuretic peptide receptor-B (NPR2) gene as a cause of short stature in patients initially classified as idiopathic short stature. J Clin Endocrinol Metab 98:E1636–E1644. 10.1210/jc.2013-2142 [DOI] [PubMed] [Google Scholar]
- Vasques GA, Arnhold IJ, Jorge AA (2014) Role of the natriuretic peptide system in normal growth and growth disorders. Horm Res Paediatr 82:222–229. 10.1159/000365049 [DOI] [PubMed] [Google Scholar]
- Wang W, Song MH, Miura K, Fujiwara M, Nawa N, Ohata Y, Kitaoka T, Kubota T, Namba N, Jin DK, Kim OH, Ozono K, Cho TJ (2016) Acromesomelic dysplasia, type Maroteaux caused by novel loss-of-function mutations of the NPR2 gene: three case reports. Am J Med Genet A 170A:426–434. 10.1002/ajmg.a.37463 [DOI] [PubMed] [Google Scholar]
- Wefers B, Bashir S, Rossius J, Wurst W, Kühn R (2017) Gene editing in mouse zygotes using the CRISPR/Cas9 system. Methods 121:55–67. 10.1016/j.ymeth.2017.02.008 [DOI] [PubMed] [Google Scholar]
- Xia C, Nguyen M, Garrison AK, Zhao Z, Wang Z, Sutherland C, Ma L (2013) CNP/cGMP signaling regulates axon branching and growth by modulating microtubule polymerization. Dev Neurobiol 73:673–687. 10.1002/dneu.22078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoder AR, Robinson JW, Dickey DM, Andersland J, Rose BA, Stone MD, Griffin TJ, Potter LR (2012) A functional screen provides evidence for a conserved, regulatory, juxtamembrane phosphorylation site in guanylyl cyclase a and B. PLoS One 7:e36747. 10.1371/journal.pone.0036747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoder AR, Stone MD, Griffin TJ, Potter LR (2010) Mass spectrometric identification of phosphorylation sites in guanylyl cyclase A and B. Biochemistry 49:10137–10145. 10.1021/bi101700e [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z, Ma L (2009) Regulation of axonal development by natriuretic peptide hormones. Proc Natl Acad Sci U S A 106:18016–18021. 10.1073/pnas.0906880106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z, Wang Z, Gu Y, Feil R, Hofmann F, Ma L (2009) Regulate axon branching by the cyclic GMP pathway via inhibition of glycogen synthase kinase 3 in dorsal root ganglion sensory neurons. J Neurosci 29:1350–1360. 10.1523/JNEUROSCI.3770-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]