

Abstract
Although age-at-injury influences chronic recovery from traumatic brain injury (TBI), the differential effects of age on early outcome remain understudied. Using a male murine model of moderate contusion injury, we investigated the underlying mechanism(s) regulating the distinct response between juvenile and adult TBI. We demonstrate similar biomechanical and physical properties of naive juvenile and adult brains. However, following controlled cortical impact (CCI), juvenile mice displayed reduced cortical lesion formation, cell death, and behavioral deficits at 4 and 14 d. Analysis of high-resolution laser Doppler imaging showed a similar loss of cerebral blood flow (CBF) in the ipsilateral cortex at 3 and 24 h post-CCI, whereas juvenile mice showed enhanced subsequent restoration at 2–4 d compared with adults. These findings correlated with reduced blood–brain barrier (BBB) disruption and increased perilesional vessel density. To address whether an age-dependent endothelial cell (EC) response affects vessel stability and tissue outcome, we magnetically isolated CD31+ ECs from sham and injured cortices and evaluated mRNA expression. Interestingly, we found increased transcripts for BBB stability-related genes and reduced expression of BBB-disrupting genes in juveniles compared with adults. These differences were concomitant with significant changes in miRNA-21-5p and miR-148a levels. Accompanying these findings was robust GFAP immunoreactivity, which was not resolved by day 35. Importantly, pharmacological inhibition of EC-specific Tie2 signaling abolished the juvenile protective effects. These findings shed new mechanistic light on the divergent effects that age plays on acute TBI outcome that are both spatial and temporal dependent.
SIGNIFICANCE STATEMENT Although a clear “window of susceptibility” exists in the developing brain that could deter typical developmental trajectories if exposed to trauma, a number of preclinical models have demonstrated evidence of early recovery in younger patients. Our findings further demonstrate acute neuroprotection and improved restoration of cerebral blood flow in juvenile mice subjected to cortical contusion injury compared with adults. We also demonstrate a novel role for endothelial cell-specific Tie2 signaling in this age-related response, which is known to promote barrier stability, is heightened in the injured juvenile vasculature, and may be exploited for therapeutic interventions across the age spectrum following traumatic brain injury.
Keywords: angiopoietin, blood–brain barrier, cerebral blood flow, juvenile, miRNA, Tie2 receptor
Introduction
Traumatic brain injury (TBI) is a growing health concern that afflicts a broad range of the population worldwide. (Schneier et al., 2006; Marin et al., 2014; Thurman, 2016). TBI initiates a multitudinous cascade of complex biochemical events including disruption of the cerebral vasculature and breakdown of the blood–brain barrier (BBB) that contribute to secondary injury such as hypoxia, ischemia, oxidative stress, and inflammation (McGinn and Povlishock, 2016; Price et al., 2016; Pearn et al., 2017). Although physical trauma to the developing brain may interrupt the maturation of key developmental processes that support higher-order cognition in adulthood, numerous studies support an age-related difference in early functional recovery. Several human studies demonstrate that increased age negatively influences outcome with the greatest amount of improvement in disability was seen in early adolescent patients (Marquez de la Plata et al., 2008), whereas children under 2 years of age represent a high-risk group with poorer outcome (Adelson et al., 1997). These findings are supported by preclinical studies in swine demonstrating greater lesion formation with increased age after acute cortical impact injury (Duhaime et al., 2003; Missios et al., 2009). The mechanism(s) underlying this age-at-injury difference in cortical lesion formation and functional outcome remains unknown.
More recently, age-specific differences in the chronic outcome from TBI have been linked to dysregulation of metabolism, oxidative stress, BBB function, unresolved inflammation, and β-amyloid accumulation after TBI (Fan et al., 2003; Giza and Prins, 2006; Claus et al., 2010; Kamper et al., 2013; Pop et al., 2013; Moretti et al., 2015). However, in addition to the well described evolution of chronic injury following juvenile TBI, there is evidence of acute tissue protection compared with adults in several swine models (Duhaime et al., 2000, 2003). Age-related differences in the cerebrovascular response has been suggested to play a role (Adelson et al., 1997; Armstead, 1999; Durham et al., 2000; Claus et al., 2010; Curvello et al., 2017). Preserving adequate vascularization, tone, and function can minimize the loss of oxygen and nutrient supply as well as prevent buildup of metabolic waste after TBI (Fitzgerald et al., 2010; Salehi et al., 2017). Moreover, numerous vascular-mediated secondary events (hemorrhage, angiogenic edema, vasospasms, hypoxia-ischemia, vascular inflammation, and BBB disruption) play a central role in regulating the injured milieu following trauma and are current targets for therapeutic intervention. Although minimizing the detrimental consequences of vascular dysfunction may aide in the restoration of homeostasis (Logsdon et al., 2015; Salehi et al., 2017), exploration into the age-specific differences and the underlying elements that mediate the vascular response after acute and chronic TBI is limited. To address this gap in knowledge, we subjected adult and juvenile (postnatal day 21, P21) CD1 mice to the same controlled cortical impact injury and evaluated the acute histological and functional outcomes. We further interrogated the cortical endothelial cell (EC)-specific response at the cellular and molecular level, which revealed differential changes in gene expression necessary for vessel stability. This is the first study that provides a comparative analysis of acute outcomes in juvenile and adult mice subjected to cortical trauma and uncovers vascular age as a major contributor to the age-at-injury response. Our novel findings also highlight a novel age-related mechanism involving Tie2 receptor signaling that may be exploited for modulating tissue stability across the age spectrum of TBI.
Materials and Methods
Animals.
All mice were housed in an AAALAC approved, virus/specific antigen-free facility with a 12 h light/dark cycle and food and water available ad libitum. CD1 male mice were purchased from Charles River Laboratories and reared until age P60–P75 for experimentation. All experiments were conducted in accordance with the NIH Guide for the Care and Use of Laboratory Animals and were conducted under the approval of the Virginia Tech Institutional Animal Care and Use Committee (IACUC; #15-063) and the Virginia Maryland Regional College of Veterinary Medicine.
Controlled cortical impact (CCI) injury and soluble Tie2-Fc administration.
Animals were prepared for CCI as described previously (Brickler et al., 2016). Male CD1 mice at P21 (average weight 14–17 g) and P60–P75 (average weight 25–35 g) were anesthetized with ketamine (100 mg/kg) and xylazine (10 mg/kg) intraperitoneal injection and positioned in a stereotaxic frame. Body temperature was monitored with a homeothermic blanket system containing a rectal probe and maintained at 37°C with an autoregulated heating pad. A 4 mm craniotomy was made using a portable drill over the right parietal-temporal cortex (−2.5 mm A/P and 2.0 mm lateral from bregma). Injury was induced by moderate CCI using the eCCI- 6.3 device (Custom Design and Fabrication, 3 mm beveled steel impact tip) at a velocity of 5.0 ± 0.3 m/s, 1.0 mm depth, and 150 ms impact duration (Theus et al., 2010; Baumann et al., 2013). Sham controls received craniotomy only. For Tie-Fc experiments, juvenile mice were injected with 1 mg/kg/d soluble Tie2-Fc or Fc control (SinoBiological) via tail vein injection starting immediately following CCI injury. Following injury, the incision was closed using Vetbond tissue adhesive (3M) and the animals were placed into a heated cage and monitored every 20 min until fully recovered from anesthesia.
Cerebral blood flow (CBF) analysis.
CBF measurement was conducted using a laser Doppler imager (LDI 5061 and software version 5.3; Moor Instruments) as described previously (Ojo et al., 2016). Mice were anesthetized and kept at 37.0 ± 0.5°C body temperature. The measurements were performed pre- and post-CCI and post-sham and then under 2–4% isoflurane at 3 h and 1–4 d post-CCI or post-sham injury. Perfusion units (PFUs) were collected using a 2.5 × 2.5 cm scanning area. A standardized region of interest (ROI) was used for each image in the contralateral and ipsilateral hemispheres to quantify PFUs. The PFUs for each injured animal (n = 10 for P21 and n = 7 for adult) were then normalized to the average sham values (n = 5 for both ages) for each time point and represented as relative change compared with the average sham PFUs in each hemisphere. This was performed to take into account any minor CBF changes that occur during sham surgery under anesthesia.
Behavioral evaluations.
For all behavioral analysis, mice were habituated in the experimental room for 1 h before testing and all equipment was cleaned with 70% ethanol between each mouse. We used the rotarod (Rotamex; Columbus Instruments) to assess gross motor function as described previously (Jones and Roberts, 1968; Bouët et al., 2007; Okyere et al., 2018). Briefly, animals were pretrained on days −4 d to −1 d and then CCI or sham injury was performed on day 0. A baseline was collected on the fourth day of training, then again at 4 d, 7 d, and 14 d post-CCI (n = 13 for P21 and n = 6 for adult) or post-sham injury (n = 6 for P21 and n = 9 for adult) for each mouse. For each session, mice were placed on the rod and the starting velocity was set at 10 rpm and accelerated at 0.1 rpm/s. Mice were given 4 trials each testing day with a 2 min intertrial interval. The time to fall was recorded for each mouse and the data were graphed as the mean individual time per day relative each animal's baseline performance (percentage of baseline). The beam walk test was used to assess fine motor coordination and balance as described previously (Okyere et al., 2018). The square wood beam was 30 cm in height, 6 mm wide, and 80 cm long. Mice were pretrained to walk to a dark goal box on days −4 d to −1 d with 4 trials each day; the baseline total number of foot slips were collected on the fourth day of training and then again at 4 d, 7 d, and14 d post-injury. Cognitive nonspatial hippocampal-mediated memory (Clark et al., 2000) was tested using novel object recognition (NOR). Long-term memory deficits were tested using the NOR test as described previously (Okyere et al., 2018). Briefly, no habituation was performed (Leger et al., 2013) and, in this case, mice were introduced to two identical objects, 14 cm high, spaced 20 cm apart, and 10 cm from the wall on day one (day 3, 6, and 13 d post-injury) and then long-term memory was tested 24 h later by replacing one familiar object with a new or “novel” object (test day or 4, 7, and 14 d post-injury; 4 h after motor assessment) in a 40 × 40 cm arena surrounded by 20 cm high walls covered by clear plexiglas. The novel object was different for each testing day and was constructed with Legos or a 50 ml plastic conical tube. Time of exploration of the familiar and novel object was recorded over 5 min by a video camera positioned over the chamber and later scored by an observer blinded to the treatment. Exploration was defined as touching the objects with the nose and/or forepaws. Preference for the novel object was calculated as a ratio of exploration time of novel object divided by the total time of exploration of both objects and represented as preference index.
Serial sectioning and staining.
For histological evaluations, fresh frozen brains were embedded in optimal cutting temperature compound (OCT; Fisher Scientific). Then, serial coronal sections were cut at 30 μm thick using CryoStar Cryostat NX70 (Thermo Fisher Scientific) at 300 μm apart for a total of 5 sections per animal spanning the lesion (−1.1 to −2.6 mm posterior to bregma) and then stored at −80°C. For histological analysis, the following numbers of animals were collected for 4 d post-CCI injury: n = 8 P21; n = 6 adult and 14 d post-CCI injury: n = 6 P21; n = 7 adult) and 35 d post-CCI injury n = 3 for both, which were used for lesion volume, vessel density, and astrogliosis. At room temperature (RT), the dried sections were postfixed with 10% formalin, washed 3 times in 1× PBS, and blocked in 2% cold water fish gelatin (Sigma-Aldrich) with 0.1% Triton X-100 for 1 h. They were then incubated with primary antibodies diluted in block at 4°C overnight as follows: goat anti-CD31: 1/500 (R&D Systems, AF-3628) or rabbit anti-GFAP (1/500, Cell Signaling Technology, 12389S). Sections were washed 5 times with 1× PBS and incubated with 1/500 Alexa Fluor 488-conjugated secondary antibodies or Alex Flour 594-conjugated secondary antibodies (Invitrogen) for 1 h at RT, washed 3× in 1× PBS, and then mounted in Pro-Long anti-fade mounting solution with DAPI (Molecular Probes). TUNEL staining was performed as described previously (Theus et al., 2010, 2014). For Figure 1, Nissl substance was stained using cresyl violet acetate 2.5% solution (Electron Microscopy Science) on serial coronal sections for 30 min, followed by water rinse, dehydration in ethanol, and mounting with xylene-based media. For Figure 2, NeuroTrace 488 fluorescent Nissl stain (1/2000, Thermo Fisher Scientific, N21482) was applied to the coronal tissue sections for 30 m following 10% formalin fixation, 3× PBS washes, and fish gelatin/0.2% Triton X-100 block for 10 min.
Figure 1.

Naive brain dimensions, water content, and viscoelasticity. a, Brain dimensions of naive juvenile and adult CD1 mice were assessed on Nissl stained coronal brain slices Cortical mantle thickness (T) is the length of the cortical layer. The width (W) and height (H) were determined as the distance from medial-lateral and dorsal-ventral, respectively. b, Juvenile mice showed a significant increase in the percentage of overall water content of the brain (**p = 0.0046) as well as between each hemisphere (right, **p = 0.0020; left **p = 0.0071) compared with adult (c). d, No significant differences were seen between the width (in millimeters) of adult and juvenile mice whether combined or analyzed by bregma levels −1.5, −1.8, and −2.1. e, f, No significant difference in combined height or by bregma levels −1.5, −1.8, and −2.1 was observed. g, h, Cortical mantle thickness (CMT) was significantly increased in juvenile compared with adult (**p = 0.0010.). i, CMT by bregma levels −1.8 and −2.1 was also increased at −1.8 and −2.1 compared with adult (**p = 0.0017 and ***p = 0.0008, respectively). j, No significant difference was observed between the elasticity (p = 0.50 sham; p = 0.50 injured) or viscosity (p = 0.27 sham; p = 0.61 injured) (k) of juvenile and adult sham-injured or 4 d injured brains.
Figure 2.

Analysis of cortical lesion volume and cell death after CCI injury. Comparison of lesion volume at 4 d and 14 d post-CCI injury on Nissl-stained serial coronal sections from adult (a and c, respectively) and juvenile (b and d, respectively) mice using Caveleri estimator. e, Quantified representation of lesion volume (in cubic millimeters) showing that juvenile mice have reduced cortical loss at 4 d (***p = 0.0004) and 14 d (***p = 0.005) post-CCI compared with adult. f, Quantification of the number of TUNEL+ cells in the cortex and dentate gyrus (DG) at 4 d post-CCI injury shows a significant reduction in the number of dying cells in the juvenile cortex (*p = 0.0164) and no significant difference in the DG (p = 0.2792). CTX, Cortex; hippo, hippocampus (adult, n = 6–7 per group; juvenile, n = 6–8 per group). Scale bar, 1 mm in a–d.
Naive brain hemispheric analysis and determination of water content and biomechanical properties.
Comparison of brain dimensions was performed on 3 coronal serial brain sections (A/P −1.5, −1.8, −2.1) from n = 5 each of naive P21 and adult CD-1 male mice. Following Nissl staining, comparison of developmental perimeters were calculated by measuring cortical mantle thickness, width, and height (all in millimeters) of each hemisphere using the line measure tool within StereoInvestigator software (MicroBrightField). Cortical hemispheric water content was compared between P21 and adult naive mice; hemispheres were freshly dissected and then weighed to obtain the wet weight (in milligrams). Brains were then dried at 70°C for 3 d to obtain the dry weight. Hemispheric water content was calculated and represented as follows: percentage hemispheric water content = (wet weight − dry weight)/(wet weight) × 100. For analysis of biomechanical properties, dynamic oscillatory shear testing was used to measure the mechanical properties of adult and P21 naive and TBI brain tissue. Fresh naive brains were coronal cut at 2 mm thickness and keep on ice in dissecting medium until testing. Brain viscoelasticity was measured using a Discovery Hybrid Rheometer (TA Instruments) with cylindrical 8 mm parallel shear plates at RT. Samples were compressed to a uniform 0.3 mm thickness and then tested using an oscillatory amplitude sweep with a frequency set at 10 Hz. The sweep began at a 0.05% strain rate and ended at 5%.
Evaluation of lesion volume.
Lesion volume (in cubic millimeters) was assessed by a blinded investigator using the Caveleri Estimator from StereoInvestigator (MicroBrightField) and an upright Olympus BX51TRF motorized microscope, as described previously (Theus et al., 2014; Brickler et al., 2016). Briefly, volume analysis was performed by estimating the area of tissue loss in the ipsilateral cortical hemisphere using five serial coronal sections (−1.1 to −2.6 mm posterior from bregma). Nissl-stained sections were viewed under fluorescent microscopy at a magnification of 4×. A random sampling scheme was used that estimates every 10th section from rostral to caudal, yielding five total sections to be analyzed. A randomly placed grid with 100 μm spaced points was placed over the ipsilateral hemisphere and the area of contusion was marked within each grid. Lesion boundaries were identified by loss of Nissl staining, pyknotic neurons, and tissue hemorrhage. The marked areas, using grid spacing, was then used to estimate total tissue volume based on section thickness, section interval, and total number of sections within the Caveleri probe in StereoInvestigator. Data are represented as volume of tissue loss or damage (in cubic millimeters) for juvenile and adult mice.
Vessel density and area analysis.
For vessel density, five CD31-immunostained serial tissue sections were viewed under epifluorescence at 10×. Using the optical fractionator probe from StereoInvestigator, a contour was drawn around the medial and lateral perilesion cortical tissue, which measured 200 μm from the border of the lesion cavity. A 150 × 150 μm counting frame was used for random nonbiased stereology over a 400 × 400 μm grid set within the drawn contour. Each probe run calculated the total number of CD31+ vessels per animal which was estimated based on these parameters. For analysis of CD31 and GFAP in the perilesion at 35d percentage area, images were first acquired on an inverted Zeiss 880 confocal microscope. The percentage of area covered by CD31+ vessels and GFAP reactivity was determined using ImageJ software on z-stacks (7 images/stack, 1 μm step size, 0.6 zoom, 3 stacks/regions per animal) from confocal images acquired at 25× and 40×, respectively; a default/auto threshold followed by color inversion was applied to each single channel image in each stack and the percentage coverage was calculated using the measurement (percentage area) tool expressed as an average percentage across images within the stack. The percentage colocalization of GFAP+ processes with CD31+ vessels was quantified using the “Coloc” threshold function in ImageJ on double-labeled confocal z-stacks acquired at 40×. The CD31 channel was used to generate bounded ROIs to restrict colocalization analysis to blood vessels. Double-channeled images were separated into single-channel images and the background was subtracted from each image (radius = 60). After running the Coloc threshold tool, the total number of colocalized pixels were normalized to the number of bounded CD31 pixels to generate a percentage of colocalized pixels expressed as GFAP+CD31+/CD31+.
Isolation of brain-derived CD31+ ECs and primary culture.
Mice were euthanized by decapitation under anesthesia and the fresh brains were removed and placed in L15 dissecting media on ice (ThermoFisher), then a 4 mm × 4 mm cortical piece of tissue from the ipsilateral hemisphere was subjected to neural dissociation (kit from Miltenyi Biotec, Auburn, CA). Five mice were pooled per group and age (sham vs CCI injury) for each isolation. Single-cell suspension from freshly dissociated brain tissue was subjected to CD31+ magnetic microbeads and column separation, as per manufacturer instructions to isolate CD31+ cell fraction following CD45 depletion (MACS; Miltenyi Biotec, Auburn, CA). The flow through and CD31-enriched fractions were then placed in Trizol and prepared for mRNA isolation. Technical triplicates were used for qRT-PCR. Cell culture of primary brain-derived ECs was performed as described previously (Okyere et al., 2016). For Tie2-Fc Experiments 100, 000 cells were plated in each well of a 0.2% gelatin coated 6-well plate in media without growth factors containing either 20 μg/ml Tie2-Fc or 10 μg/ml Fc control for 24 h then Trizol was used to collect and isolate mRNA. Biological triplicates were used for qRT-PCR.
Quantitative real-time PCR (qRT-PCR).
Total RNA from cultured ECs or freshly isolated cortex-derived CD31+ cells was isolated according to the manufacturer's instructions using TRIzol extraction (Invitrogen). RNA quantification was performed by measuring absorbance with spectrophotometer ND-1000 (NanoDrop). RNA was reverse transcribed into cDNA with Im-Prom II Reverse Transcription System (Promega). RNA samples were treated with DNase I (Thermo Fisher Scientific) before reverse transcription. Each DNase reaction (1 μg of RNA, 1 μl of 10× DNase I buffer, 1 μl of DNase I, 0.5 μl of RNase inhibitor, up to 10 μl with water) was incubated at 37°C for 60 min before inactivation by addition of 1 μl of 50 mm EDTA and incubation at 65°C for 10 min. The DNase-treated RNA samples were incubated with oligo(dT) 15 primer at 70°C for 5 min [250 ng of RNA, 1 μl oligo(dT) 15, up to 5 μl with water] and chilled on ice for 5 min to allow annealing before reverse transcription. To prepare the cDNA samples, 15 μl of reverse transcription mix (3.7 μl of water, 4 μl of 5× ImProm-II buffer, 4.8 μl of 25 mm MgCl2, 1 μl of 10 mm dNTPs, 0.5 μl of RNase inhibitor, 1 μl of ImProm-II reverse transcriptase) were added per 5 μl of RNA sample and reverse transcription was performed using the following PCR scheme: 25°C for 5 min; 42°C for 1 h; 70°C for 15 min. For qRT-PCR analysis, 7 ng of cDNA per reaction was amplified using SYBR Green PCR Master Mix (Applied Biosystems). Expression changes were calculated using ΔΔCq values with reference to Gapdh internal control gene and then calculated as relative expression compared with appropriate sham samples.
eNOS.mRNA.F: ATGAGTTCAGAGATTGGCATGA, eNOS.mRNA.R: CTTGTCTTTCCACAGGGATGAG;Bcl2.mRNA.F: CCTGTGGATGACTGAGTACCTG, Bcl2.mRNA.R: AGCCAGGAGAAATCAAACAGAG; Angpt1.mRNA.F: CGAAAGCTGACAGATGTTGAGA, Angpt1.mRNA.R: TTTGTCTGTTGGAGAAGTTGCT; Angpt2.mRNA.F: GGAAAAGCAGATTTTGGATCAG, Angpt2.mRNA.R: TTCTGCTCCTTCATGGACTGTA; Vegf.mRNA.F: GAAGTCCCATGAAGTGATCCAG, Vegf.mRNA.R: TCACCGCCTTGGCTTGTCA; Vegfr2.mRNA.F: GGGACCTGGACTGGCTTTG, Vegfr2.mRNA.R: CCGCATTCAGTCACCAATACC; Vegfr1.mRNA.F: TTCGGAAGACAGAAGTTCTCGTT, Vegfr1.mRNA.R: ACCTCGTAGTCACTGAGGTTTTG; NeuN.mRNA.F: CACTCTCTTGTCCGTTTGCTTC, NeuN.mRNA.R: CTGCTGGCTGAGCATATCTGTA; Gfap.mRNA.F: ACCAGTAACATGCAAGAGACAGAG, Gfap.mRNA.R: GATAGTCGTTAGCTTCGTGCTTG; PdgfrB.mRNA.F: GGTACGTGTGAAGGTGTCAGAA, PdgfrB.mRNA.R: ACTCTCACTCAGCTCCAGCAC; TgfB.mRNA.F: TTAGGAAGGACCTGGGTTGGA, TgfB.mRNA.R: CAGGGTCCCAGACAGAAGTT; Tie2.mRNA.F: AAATGACCCTAGTGAAGCCAGA, Tie2.mRNA.R: GTCAGGAGGTAAGACTCGGTTG; Vecad.mRNA.F: AGGACAGCAACTTCACCCTCA, Vecad.mRNA.R: AACTGCCCATACTTGACCGTG; Cx3cr1.mRNA.F: GTGAGTGACTGGCACTTCCTG, Cx3cr1.mRNA.R: AATAACAGGCCTCAGCAGAATC; Ocln.mRNA.F: TGGATCGGTATGATAAGTCCAA, Ocln.mRNA.R: CATAGTCAGATGGGGGTGGAG; Cldn5.mRNA.F: ATGCAGTGCAAGGTGTATGAAT, Cldn5.mRNA.R: CCGGTCAAGGTAACAAAGAGTG; Il6.mRNA.F: CTTCACAAGTCGGAGGCTTAAT, Il6.mRNA.R: GATTGTTTTCTGCAAGTGCATC; Nfkb.mRNA.F: GAGAGAGGCAGACAGTGGAC, Nfkb.mRNA.R: AGGCCATTGAAGTGATCCAGG; Vcam1.mRNA.F: GGTGTACGAGCCATCCACAG, Vcam1.mRNA.R: ACTTGTGGAAATGTGCCCGA; mGAPDH.F: ATTGTGTCCGTCGTGGATCTGA, mGAPDH.mRNA.R: GATGCCTGCTTCACCACCTTCTT.
Quantification of miRNA expression.
As we described previously (Dai et al., 2010, 2013), the TaqMan miRNA assay kit (Thermo Fisher Scientific) was used to quantify miRNA expression per the manufacturer's instructions. Briefly, 16.5 ng (3.3 μl) total RNA, isolated as described above, was reverse transcribed using the TaqMan MicroRNA Reverse Transcription kit (Thermo Fisher Scientific) with miRNA-specific RT primers in 10 μl of reverse transcription mix (2.8 μl of water, 1 μl of 10× RT buffer, 0.1 μl of 100 mm dNTPs, 0.13 μl of RNase inhibitor at 20 U/ μl), 0.67 μl of MutiScribe reverse transcriptase at 50 U/μl, and 2 μl of 5× RT primers). The reverse transcription was performed by incubating the reaction mixes at 16°C for 30 min; 42°C for 30 min; 85°C for 5 min. qRT-PCR was performed in 18 μl of qRT-PCR mix (6.9 μl of water, 1.2 μl of miRNA-specific RT products, 0.9 μl of 20× miRNA-specific TaqMan small RNA assay mix, 9 μl of 2× TaqMan Fast Universal PCR Master Mix) on a 7500 Fast Real-Time PCR System (Thermo Fisher Scientific) following the manufacturer's recommendation. The relative miRNA expression levels in specific treatment groups were referred to the control group by normalizing to endogenous small RNA control, snoRNA 202, and calculating with the 2−ΔΔCt formula (ΔCt = Cttarget miRNA − CtsnoRNA202, ΔΔCt = ΔCt treatment − ΔCt control).
BBB analysis.
A 2% sterile Evans blue (EB; Sigma-Aldrich, E2129) solution was prepared in 0.1 m PBS and passed through a 0.2 μm filter. Mice having undergone either sham injury (n = 4 for both P21 and adult) or CCI injury (n = 9 for P21 and n = 5 for adult) were restrained and injected with 5 μl/g EB solution via tail vein. Three hours post-injection, brains were removed and the ipsilateral and contralateral cortical hemispheres were dissected and incubated separately in 500 μl of formamide (Invitrogen, 15515-026) for 24 h at 55°C. Samples were then centrifuged to pellet the tissue and absorbance of the solution was measured at 610 nm using a NanoDrop 1000 spectrophotometer (Thermo Fisher Scientific.). Absorbance at 610 nm was quantified and graphed for each cortical hemisphere. For Tie2-Fc experimentation, the following mice were evaluated: n = 4 P21-Fc; n = 7 P21-Tie2-Fc; n = 5 adult-Fc; and n = 3 adult Tie2-Fc.
Experimental design and statistical analysis.
Data were graphed using GraphPad Prism version 7 XML software. Student's unpaired two-tailed t test was used for comparison of two experimental groups. For three or more groups, multiple comparisons were done using one-way and two-way ANOVA with repeated measures (RM) where appropriate, followed by Bonferroni post hoc test for multiple pairwise examinations. Changes were identified as significant at p < 0.05. Mean values were reported together with the SEM. Sample size was determined based on an effect size measured for each outcome by pilot or prior studies. G*Power 3 (Universitat Dusseldorf, Germany) was used to retrieve sample size using an acceptable power range between 80% and 90%. All animal and serial sections were coded and a double-blinded strategy was used in all stereological and behavioral analysis.
Results
Brain dimensions, water content, and viscoelasticity in naive juvenile and adult mice
To evaluate whether any major differences in the biophysical properties of the adult and juvenile brain would affect the outcome from CCI injury, we investigated the naive dimensions and viscoelastic properties of the adult and P21 outbred CD1 mouse brain using Nissl-stained coronal serial sections (Fig. 1a) and using rheometry on freshly dissected 2 mm brain slices, respectively. The percentage of water content was also evaluated from freshly dissected right and left hemispheres, which showed that juvenile mice (80.7 ± 0.5%, n = 5) had ∼2.6% more compared with adult (78.1 ± 0.4%; n = 4, p = 0.0046 unpaired t test) in combined hemispheres (Fig. 1b), which was similar when comparing right and left hemispheres individually (Fig. 1c). Measurement of the hemispheric width showed no statistical significance between adults (5.9 ± 0.06 mm, n = 5) and juveniles (6.1 ± 0.06 mm, n = 5, p = 0.12 unpaired t test) (Fig. 1d,e), nor was there a difference in height found between adult mice (5.9 ± 0.06 mm, n = 5) and juvenile mice (6.1 ± 0.06 mm, n = 5, p = 0.12 unpaired t test) (Fig. 1f,g). However, cortical mantle thickness was ∼10.8% larger in juvenile mice (1.23 ± 0.01 mm, n = 5) compared with adult mice (1.11 ± 0.02 mm, n = 5, p = 0.001 unpaired t test) (Fig. 1h). Interestingly, this increase was significant only in posterior bregma levels −1.8 and −2.1 (Fig. 1i). These findings are similar to those previously reported for the C57BL/6 inbred strain of mice (Fan et al., 2003). Last, we evaluated whether the 2–3% increase in water content in the juvenile brain may affect its deformation properties compared with adult following cortical impact and if any compositional change in brain elasticity and viscosity may be occurring using oscillatory shear testing. We found no significant difference in the normalized G′ storage modulus (elasticity) between naive adult mice (1.0 ± 0.10, n = 7) and juvenile mice (1.0 ± 0.20, n = 7, p = 0.50 unpaired t test) nor between 4 d injured adult mice (0.31 ± 0.03, n = 4) and juvenile mice (0.39 ± 0.04, n = 3). Likewise, no differences in the G″ loss modulus (viscosity) between naive adult mice (1.0 ± 0.09, n = 7) and juvenile mice (0.95 ± 0.18, n = 7, p = 0.27 unpaired t test) nor between 4 d injured adult mice (0.33 ± 0.13, n = 4) and juvenile mice (0.34 ± 0.17, n = 3). These findings demonstrate that the deformation properties of the adult and juvenile brain are similar and that compositional changes occurring in the juvenile brain after CCI injury does not significantly affect the change in response to applied forces compared with adult.
Juvenile mice display reduced cortical lesion volume and cell death after CCI injury
To evaluate whether age-at-injury influences the acute and subacute histological outcome following cortical trauma, we evaluated lesion formation at 4 d and 14 d post-CCI injury (Theus et al., 2010, 2014; Baumann et al., 2013), respectively, in adult and juvenile mice. Serial Nissl-stained sections were assessed for cortical tissue damage using the Caveleri estimator (one-way ANOVA, F = 16.63, p < 0.0001). We found that juvenile mice displayed smaller lesion volumes (1.6 ± 0.13 mm3; n = 8) (Fig. 2b,e) compared with adults (2.5 ± 0.08 mm3; n = 6) (Fig. 2a,e) at 4 d post-CCI injury. This acute protection was maintained at 14 d, when juvenile mice (1.2 ± 0.1 mm3; n = 6) (Fig. 2d,e) showed reduced lesion volume compared with adult mice (2.15 ± 0.20 mm3; n = 7) (Fig. 2c,e). These findings correlated with reduced TUNEL+ cells in the cortex of juvenile mice (8422 ± 1335, n = 5) compared with adult mice (22, 508 ± 2879, n = 5, p = 0.0164) using nonbiased stereological analysis (Fig. 2f). Although a trend toward reduced TUNEL+ cells was observed, no significant difference was seen in the dentate gyrus of juvenile mice (54.2 ± 8.9, n = 5) compared with adult mice (114.6 ± 51.3, n = 5, p = 0.2792). These findings suggest the reduction in cortical lesion volume found in juvenile mice may be due in part to a significant reduction in cell death.
Age-at-injury influences functional recovery following cortical impact injury
Cortical impact injury results in significant behavioral deficits in adult aged mice (Zhao et al., 2012; Theus et al., 2014; Dixon et al., 2015). To evaluate whether age-at-injury influences behavioral recovery, we assessed changes in gross and fine motor function using rotarod and beam walk tests, respectively, as well NOR for learning and memory at 4, 7, and 14 d post-sham or post-CCI injury in juvenile and adult mice. As demonstrated previously (Theus et al., 2014), we found a significant loss of motor ability in adult mice subject to CCI (F(1,13) = 17.45, p = 0.0011, two-way RM ANOVA) at 4 d (60.2 ± 3.5% baseline; n = 6), 7 d (66.8 ± 6.9% baseline; n = 6) and 14 d (74.2 ± 8.9% baseline; n = 6) compared with sham at 4 d (109.8 ± 7.5% baseline; n = 9), 7 d (122.2 ± 11.1% baseline; n = 9), and 14 d (122.8 ± 13.2% baseline; n = 9) (see Fig. 5a). However, juvenile mice showed no statistical difference (F(1,17) = 2.01, p = 0.1748, two-way RM ANOVA) in motor function between sham and CCI injury at 4 d (138.2 ± 14.5% vs 116.4 ± 5.3% baseline, respectively, n = 6 vs n = 13), 7 d (123.7 ± 20.0% vs 112.2 ± 5.7% baseline, respectively, n = 6 vs n = 13), and 14 d (135.3 ± 18.7% vs 114.2 ± 5.2% baseline; n = 6 vs n = 13) (Fig. 3b). Although no difference was found between adult and juvenile sham mice (F(1,13) = 0.638, p = 0.4388, two-way RM ANOVA), there was a significant reduction in motor deficits in juvenile compared with adult injured mice at every time point assessed (F(1,17) = 40.51, p < 0.0001, two-way RM ANOVA). Using the beam walk task to further measure fine motor coordination, pronounced deficits were observed in adult injured mice at 4 d (2.5 ± 0.5 foot slips) compared with sham (0.25 ± 0. 17 foot slips) (F(1,10) = 10.22, p = 0.0095, two-way RM ANOVA); however, statistical significance was not reached at 7 d and 14 d post-CCI injury (Fig. 3e). No significant difference was observed in the number of foot slips between sham and injured juvenile mice (F(1,18) = 1.55, p = 0.2298, two-way RM ANOVA) or between adult and juvenile sham mice (F(1,10) = 0, p > 0.9999, two-way RM ANOVA). Adult injured mice did show significance at 4 d post-CCI injury compared with sham injured mice (F(1,10) = 0, p > 0.9999, two-way RM ANOVA) (Fig. 3f).
Figure 5.

Vascular density and BBB stability is enhanced in juvenile mice following CCI injury. a, Representative low-magnification confocal image of DAPI (blue) and CD31+ vessels (green) in the perilesion cortex at 4 d post-CCI injury in adult mice. b, Representative high-magnification confocal image from image a inset. c, Low-magnification confocal image of CD31+ vessels in the perilesion cortex at 4 d post-CCI injury in adult mice. d, High-magnification confocal image from image c, inset. e, Quantification of the number of CD31+ vessels by nonbiased stereology shows a significant decrease in the number of vessels in adult and juvenile ipsilateral cortex compared with their shams (***p < 0.001 and ***p = 0.0007, respectively). Juvenile injured mice displayed an increase in CD31+ vessels compared with adult injured (#p = 0.0292). f, Representative image of Evans blue in the adult cortex at 4 d post-CCI injury. g, Representative image of Evans blue in the juvenile cortex at 4 d post-CCI injury. h, Quantification of Evans blue A610nm showing a significant increase in the adult ipsilateral injured cortex at 4 d compared with ipsilateral sham injury or contralateral (Con) hemispheres (*p = 0.0433). Juvenile mice showed a significant reduction in Evans blue absorbance in the ipsilateral cortex compared with ipsilateral adult cortex (#p = 0.0281). Scale bar, 20 μm in b and d and 100 μm in a and c.
Figure 3.

Age-at-injury influences functional recovery following CCI injury. Sensorimotor and cognitive behavior was evaluated in adult and juvenile mice following sham and CCI injury. a, Rotarod analysis in adult mice showing a significant reduction in motor function at 4 d (***p = 0.0009), 7 d (***p = 0.002), and 14 d (**p = 0.0012) post-CCI injury compared with sham. b, No significant change was observed in juvenile mice in gross motor function at 4 d (p = 2.907), 7 d (p = 0.8243), and 14 d (p = 0.3200) post-CCI injury compared with sham. #p < 0.05 compared with adult CCI injury. c, Preference index, the ratio of preference for novel object to total time exploring objects ×100, was significantly reduced in adult injured mice at 4 d (*p = 0.0189), but not 7 d (p = 0.0587) or 14 d (>0.9999), compared with sham. d, Juvenile mice showed no deficit in novel object preference (p > 0.9999; p = 0.5394; p > 0.9999, respectively). e, Beam walk testing for fine motor function showed a significant increase in the number of foot slips in adult injured mice compared with sham at 4 d (**p = 0.0019), but not at 7 d (p = 0.0842) or 14 d (p = 0.6618). f, No difference in beam walk function was observed in juvenile injured mice compared with sham (p > 0.9999; p = 0.5394; p > 0.9999, respectively). #p < 0.05 compared with adult CCI injury.
Last, selective cell loss in the dentate gyrus has been shown to correlate with learning and memory deficits following CCI injury (Blaiss et al., 2011; Theus et al., 2014). Using the NOR preference index to assess cognition, we found no difference between sham juvenile and adult mice (F(1,12) = 0.0078, p = 0.9313, two-way RM ANOVA) or between sham and injured juvenile mice (F(1,17) = 0.0078, p = 0.4583, two-way RM ANOVA) at 4, 7, and 14 d post-injury (Fig. 3c,d). There was, however, a significant reduction in the NOR preference index in adult injured mice compared with adult sham at 4 d post-injury (F(1,10) = 10.22, p = 0.0095, two-way RM ANOVA), which did not reach significance at 7 d and 14 d (Fig. 3c). These findings demonstrate that CCI injury results in pronounced gross motor deficits up to at least 14 d, whereas fine motor and cognitive dysfunction are most prominent at 4 d post-injury in adult mice. We also show that juvenile mice display reduced functional deficits compared with adult, which correlates with reduced lesion volume.
Restoration of acute CBF in juvenile mice following CCI injury
To evaluate whether the age-dependent effects on cortical injury outcome following CCI was mediated in part by enhanced CBF, juvenile and adult mice were subjected to noninvasive high-resolution laser Doppler imaging (moorLDI2-HIR) to measure perfusion in the cortex before and after CCI injury (Fig. 4). Using a standardized ROI, we quantified the PFUs in the ipsilateral and contralateral hemispheres at 0 h, 5 min, 3 h, and 1–4 d post-CCI and calculated the relative change in CBF normalized to sham injury at each time point. We found no statistical difference at 5 min, 3 h, or 1 d post-CCI injury in the ipsilateral hemisphere between juvenile mice (0.35 ± 0.03; 0.36 ± 0.02, and 0.56 ± 0.06; n = 10, respectively) and adult mice (0.46 ± 0.04; 0.30 ± 0.03, and 0.53 ± 0.06; n = 7, respectively) (Fig. 4a–g). However, juvenile mice displayed enhanced reperfusion at 2, 3, and 4 d post-CCI injury (0.58 ± 0.04; 0.67 ± 0.05 and 0.62 ± 0.05; n = 10 respectively) compared with adult mice (0.41 ± 0.06; 0.38 ± 0.06 and 0.44 ± 0.03, n = 7, respectively) (F(1,15) = 3.23; p = 0.0326, two-way RM ANOVA) (Fig. 4g). Interestingly, there was also a significant increase in relative perfusion in the contralateral hemisphere of juvenile mice compared with adult at 3 h post-CCI injury (1.03 ± 0.03 juvenile vs 0.70 ± 0.07 adult; p = 0.0177) but not 1, 2, 3, or 4 d (1.0 ± 0.07 vs 0.86 ± 0.05; 1.12 ± 0.11 vs 0.86 ± 0.05; 1.09 ± 0.1 vs 0.82 ± 0.05; and 1.02 ± 0.09 vs 0.86 ± 0.04, respectively) (F(1,15) = 4.14; p = 0.060, two-way RM ANOVA) (Fig. 4h). These data show greater restoration of CBF in the juvenile ipsilateral and contralateral hemispheres compared with adult mice, which may account for greater tissue protection following CCI injury.
Figure 4.
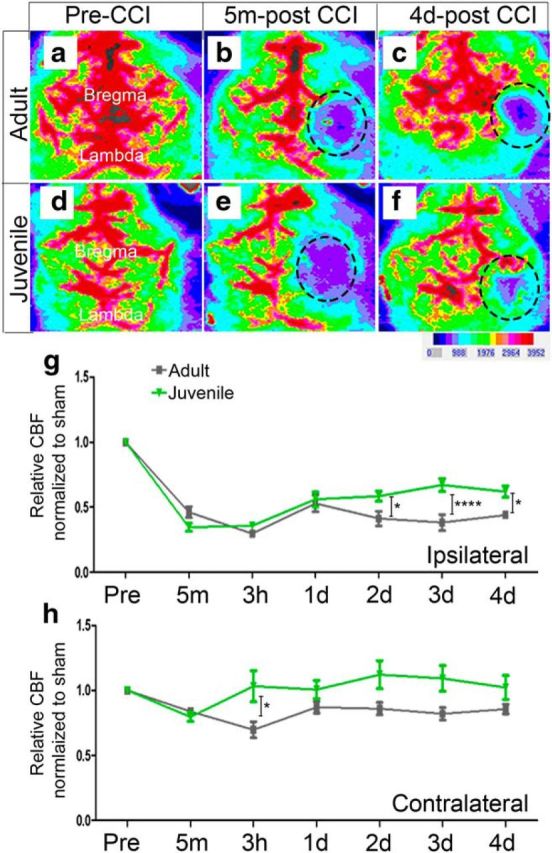
Age-at-injury influences restoration of CBF following CCI injury. CBF analysis was performed using high-resolution laser Doppler imaging, moorLDI2-HIR, before and after sham and CCI injury. Image analysis of CBF in adult and juvenile mice shows a dramatic loss in the ipsilateral cortex at 5 m post-CCI injury (dotted black circle; defined ROI) (b and e, respectively) compared with preinjury (a and d, respectively); however, at 4 d post-CCI injury, juvenile mice (f) displayed a greater restoration of CBF in the same ROI compared with adult (c). g, Quantitative analysis comparing the PFUs relative to sham at each time point shows a significant increase in the restoration of CBF in juvenile mice at 2 d (*p = 0.0340), 3 d (****p ≤ 0.0001), and 4 d (*p = 0.0210) post-injury compared with adult. h, Quantitative analysis of CBF in the contralateral hemisphere shows an increase in CBF at only 3 h juvenile injured compared with adult injured mice (*p = 0.018).
Vessel density and BBB stability are enhanced in the juvenile injured cortex
Various models of CNS injury have shown that a significant loss of microvascular structures can contribute to the breakdown of the BBB and loss of CBF (Allen and Bayraktutan, 2009; Zhang et al., 2014; Wang et al., 2016b; Liu et al., 2017; Assis-Nascimento et al., 2018). To determine whether vessel density and function correlate with the overall increase in acute CBF in juvenile mice after CCI injury, we quantified the number of CD31+ vessels in the ipsilateral perilesion cortex at 4 d postinjury. We found that the density of CD31+ vessels was comparable between adult sham mice (76,403 ± 7844; n = 3) and juvenile sham mice (80,044 ± 5477; n = 3) (Fig. 5e). However, juvenile injured mice had a significant increase in the number of CD31+ vessels (47,015 ± 3104; n = 6) (Fig. 5c–e) in the ipsilateral perilesion cortex compared with adult injured mice (29,951 ± 2731; F = 29.44, p < 0.0001, one-way ANOVA; n = 5) (Fig. 5a,b,e). In addition, the morphology of the vessels was more strikingly unhealthy and pyknotic in the adult injured perilesion (Fig. 5b) compared with the juvenile injured perilesion (Fig. 5d). Last, we found these changes to be concomitant with BBB protection. Evans blue absorbance (A610nm) analysis at 4 d post-sham or post-CCI injury revealed a significant decrease (F(3,36) = 3.817, p = 0.0179, two-way ANOVA) in EB extravasation in the ipsilateral cortical tissue of juvenile injured cortex (0.015 ± .002; n = 9) (Fig. 5g,h) compared with ipsilateral adult injured cortex (0.038 ± 0.011; n = 5) (Fig. 5f,h). These findings suggest that the observed age-dependent neuroprotection and CBF restoration may result from enhancement of vessel protection and stability.
Age-related differences in mRNA of cortical ECs following CCI injury
To evaluate whether age-dependent differences in vascular stability is regulated in part by changes in gene expression, we isolated ECs from the cortex of juvenile and adult mice at 4 d post-sham and post-CCI injury, quantified mRNA expression levels using qRT-PCR, and then analyzed relative to adult sham levels. We show the purity of the EC isolates for each group relative to Tie2. The relative mRNA expression of tie2 receptor and VE-cadherin (vecad) in the cortical-derived ECs of each group were higher compared with platelet-derived growth factor receptor β (pdgfrβ; perictes), GFAP (astrocytes), NeuN (neurons), and CX3 chemokine receptor 1 (cx3cr1; macrophages/microglia) (Fig. 6a). Expression of factors known to stabilize the BBB were significantly increased in juvenile compared with adult ECs at 4 d post-CCI injury, such as the tight junction markers occludin-1 (F = 28.20; p = 0.0017; one-way ANOVA) and claudin-5 (F = 20.36; p = 0.0004; n = 3; one-way ANOVA). Angiopoietin-1 (Ang1) (F = 11.09; p = 0.0032; one-way ANOVA) and Ang2 (F = 7.12; p = 0.012; n = 3; one-way ANOVA) were significantly increased in juvenile 4 d injured ECs compared with adult and juvenile sham; however, significance was not reached in adult injured cells (Fig. 6b). Conversely, vegf expression was significantly increased in adult and juvenile injured ECs compared with their respective sham controls, but juvenile ECs showed a reduction in relative vegf expression compared with adults after CCI injury (F = 27.88; p = 0.0001; one-way ANOVA) (Fig. 6c). Likewise, vegfr-1 (F = 8.88; p = 0.0063; n = 3; one-way ANOVA) and vegfr-2 (F = 18.87; p = 0.0005; n = 3; one-way ANOVA) were reduced in injured juvenile ECs compared with adult. Although not significant, juvenile ECs showed a trend toward increased anti-apoptotic bcl-2 mRNA expression compared with adults. Additionally, NF-κβ was significantly increased after CCI injury in both adults and juveniles compared with their respective sham controls, but no difference was seen between injured samples (F = 63.71; <p = 0.0001; n = 3; one-way ANOVA). Last, vascular cell adhesion molecule (vcam) (F = 83.77; p < 0.0001; n = 3; one-way ANOVA) and endothelial nitric oxide synthase (eNOS) (F = 69.42; p < 0.0001; n = 3; one-way ANOVA) mRNA levels were increased after CCI injury; however, juvenile ECs showed a significant reduction in response compared with adults (Fig. 6d). Overall, these findings suggest that differential changes in mRNA expression may mediate age-dependent differences in CBF, vessel density, and stability following CCI injury. Specifically, the balance between stabilization and destabilization signals may be shifted to provide greater stability through angiopoietin/Tie2 activation, which would improve the health of vascular elements in the damaged juvenile cortex.
Figure 6.
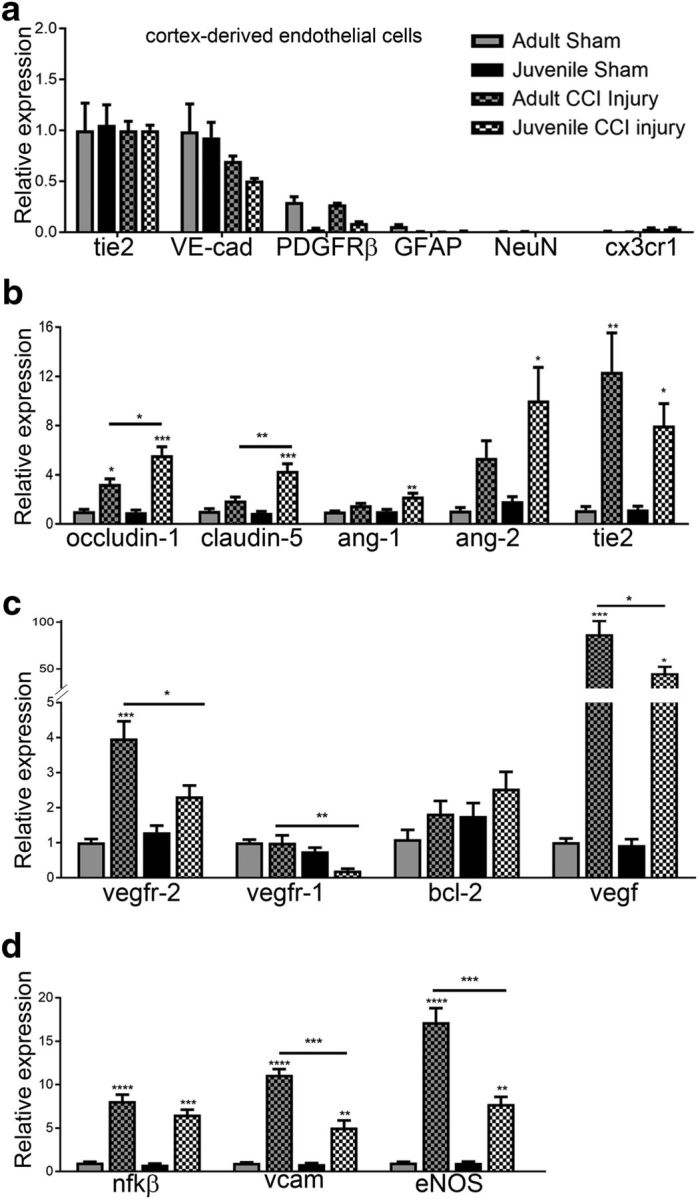
Age-dependent mRNA expression changes in cortex-derived ECs following CCI injury. ECs were isolated from the cortex of 4 d sham and injured adult and juvenile mice using magnetic bead separation. a, Expression of cell-specific genes was quantified using qRT-PCR and is represented relative to tie2 for each group. PDGFRβ, GFAP, NeuN, and cx3cr1 were absent or reduced compared with tie2 or VE-cadherin in all four groups, indicating EC enrichment following cortex isolation. b, Additional genes were analyzed and represented relative to adult sham. Occludin-1 was significantly increased in adult and juvenile ECs after CCI injury (*p = 0.0217 and ***p = 0.002, respectively); however, juvenile mice showed higher levels compared with adult (*p = 0.0017). Claudin-5 was significantly increased in juvenile injured ECs compared with sham (***p = 0.0006) and adult injured cells (***p = 0.0055). Ang1 and Ang2 were increased only in juvenile injured ECs compared with juvenile sham (**p = 0.0052 and p = 0.0223, respectively). c, Vegf and its receptors, vegfr1 and vegfr2, were significantly reduced in juvenile injured ECs compared with adult injured cells (*p = 0.0232, **p = 0.0093, and *p = 0.0219, respectively). d, No difference in NF-κβ was found; however, vcam and eNOS were significantly reduced in juvenile injured ECs compared with adult injured cells (****p = 0.00001 compared to respective sham levels).
Age-related miRNA differences in cortex-derived ECs following CCI injury
Given the differential mRNA expression changes in ECs between juvenile and adult following CCI injury, we sought to evaluate whether these differences were relate to miRNA levels. Recent findings implicate small, noncoding miRNAs as a major regulator of gene expression after TBI (Lei et al., 2009; Redell et al., 2011; Ge et al., 2014; Abdul-Muneer, 2016; Meissner et al., 2016). To address this, we analyzed the relative expression of miR-21-5p, miR-155, miR-31, miR-127, and miR-148a, which target known vascular and inflammatory genes. Interestingly, juvenile sham ECs showed significantly higher levels of miR-21–5p (F = 3132; p < 0.0001, one-way ANOVA) and miR-31 (F = 15.99; p = 0.0108, one-way ANOVA) relative to adult shams (Fig. 7a). CCI injury resulted in a significant increase in miR-21–5p and miR-155 (F = 131.1; p = 0.0002, one-way ANOVA) in adult and juvenile ECs; however, juvenile ECs had an approximately twofold increase in miR-21 compared with adult. Although miR-31 was enriched in juvenile sham ECs, it was significantly reduced after CCI injury to adult sham levels, which were unchanged after injury. Although no significant differences in miR-127 (F = 5.549; p = 0.0656, one-way ANOVA) or miR-148a were observed in adult ECs after CCI injury, juvenile ECs showed increased levels of miR-148a (F = 21.88; p = 0.0061, one-way ANOVA) compared with adult injured ECs.
Figure 7.

Age-related changes in miRNA expression from cortex-derived ECs following CCI injury. ECs were isolated from the cortex of 4 d sham and injured adult and juvenile mice using magnetic bead separation. a, Expression of miR-21–5p, miR-155, miR-31, miR-127, and miR-148a was quantified using qRT-PCR and is represented relative to adult sham. We found that miR-21–5p was increased in juvenile sham ECs (*p = 0.0270) and adult injured ECs (****p < 0.0001) compared with adult sham. We also observed that juvenile injured ECs had a greater increase in miR-21–5p compared with adult injured ECs (****p < 0.0001). Adult and juvenile injured ECs showed an increase in miR-155 over their respective sham controls (***p = 0.0009 and ***p = 0.0010, respectively) and miR-31 was increased in juvenile sham ECs compared adult (*p = 0.0461), which was reduced after injury (*p = 0.0383). No difference was seen in miR-127; however, miR-148a was increased only in juvenile injured ECs compared with adult injured ECs (**p = 0.0135). b, Using miRSystem pathway ranking scores, we identified significant biological processes predicted to be regulated by both miR-21 and miR-148a, which were enhanced in juvenile ECs compared with adult. c, We used KEGG pathway analysis to generate a graphical representation of cell signaling processes targeted by miR-21 and miR-148a in juvenile ECs, including apoptosis, vegf, and cytokine expression. Pathway components targeted by miR-21 and/or 148a are shown in red.
Next, we used miRSystem (Lu et al., 2012) to identify the biological pathways common to both miR-21–5p and miR-148a, which are elevated in juvenile injured ECs over adult. miRSystem uses seven miRNA target prediction programs and multiple gene ontology databases to generate a list of predicted miRNA targets and enriched ontological categories among the predicted targets. Target genes were shown if predicted by ≥3 algorithms and had an observed/expected ratio ≥2. Biological pathways containing 25–500 genes were included in the analysis. We found that the actin cytoskeleton and pathways associated with TGF-β, Wnt, MAPK, and JAK-STAT signaling were among the highly ranked pathways targeted by both miR-21–5p and miR-148a (Fig. 7b). Using the KEGG pathway mapping program to illustrate miRNA regulation of EC pathways (Kanehisa et al., 2017), we identified genes that are predicted to be negatively regulated by miR-21 and miR-148a and may therefore collectively suppress vegf, apoptosis, cytokine, and actin cytoskeleton signaling after TBI (Fig. 7c). These findings correlate with reduced vegf and vegfR1/2 mRNA expression in juvenile ECs (Fig. 6c), which are known to induce vessel permeability.
Soluble Tie2-Fc receptor attenuates juvenile neuroprotection following CCI injury
In addition to suppressing the VEGF pathway, recent studies demonstrate that miR-21–5p activates the Ang1/Tie2 axis and plays a protective role in TBI (Ge et al., 2014, 2016; Han et al., 2014). Our data show angiopoietin, the ligand for Tie2 that is known alleviate VEGF-induced vascular permeability (Brindle et al., 2006), is increased in juvenile ECs. To determine whether the juvenile EC response to CCI-injury is mediated by the ang/Tie2 axis, we blocked Tie2 activation by injecting 1 mg/kg/d soluble Tie2-Fc or Fc control via tail vein for 2 d following CCI injury and then analyzed histological outcome and BBB permeability. We found that blocking Tie signaling in juvenile injured mice significantly attenuated cortical tissue protection (Fig. 8a–h). Although no difference in lesion volume was observed in adult mice (3.69 ± 1.04 Tie2-Fc n = 5 vs 4.39 ± 0.72 Fc control n = 3), juvenile mice receiving Tie2-Fc showed a significant increase in lesion volume (Fig. 8e–i) compared with Fc control (Fig. 8a–d,i) (2.6 ± 0.1 mm3 vs 1.5 ± 0.11 mm3, respectively, n = 6 for both) (F(3,16) = 27.26, p < 0.0001; one-way ANOVA). These findings correlated with increased disruption of the BBB in the ipsilateral cortex (Fig. 8j). Compared with juvenile Fc controls (0.015 ± 0.001 A610, n = 4), Evans blue absorbance was increased ∼1.6-fold in juvenile Tie2-Fc-treated mice (0.024 ± 0.002 A610, n = 7) 4 d following CCI injury. Tie2-Fc values in the juvenile ipsilateral cortex were not significantly different from adult Fc control or Tie2-Fc treatment (0.026 ± 0.002 A610, Fc n = 5 vs 0.03 ± 0.006 A610, Tie2-Fc n = 3, respectively) (F(3,30) = 3.375, p = 0.0311; two-way ANOVA). No significant changes were observed in the contralateral cortex among treatment groups.
Figure 8.
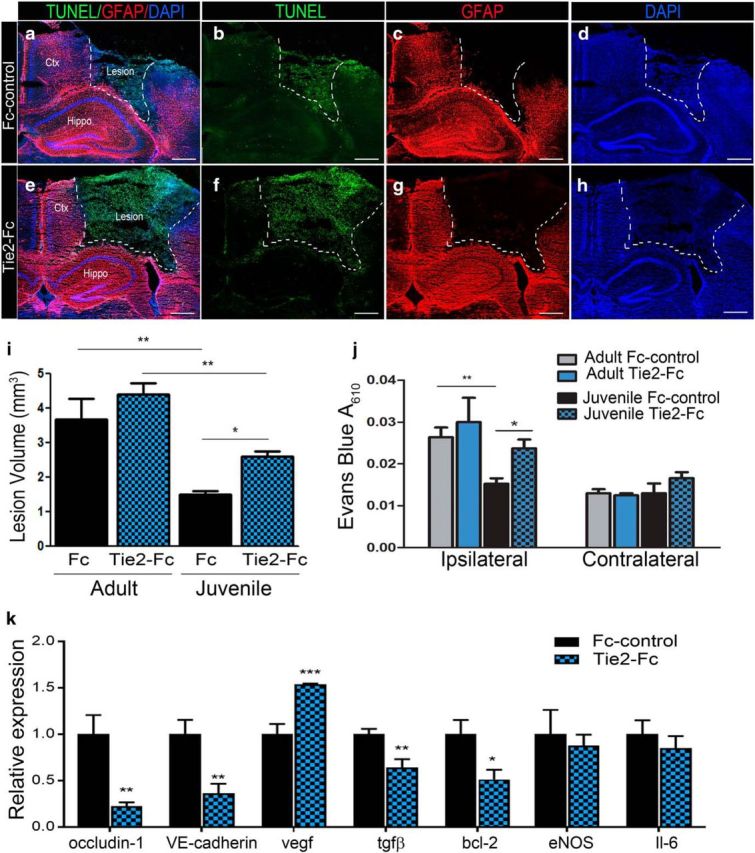
Blocking Tie2 receptor attenuates neuroprotection and BBB stability in juvenile injured mice. a, Representative merged confocal image of cortical injury at 2 d post-CCI in juvenile mice receiving 1 mg/kg/d Fc-control via tail vein injection. b–d, Serial coronal sections were stained for terminal deoxynucleotidyl transferase dUTP Nick-End Labeling (TUNEL) (b), GFAP (c), and DAPI (d). e–h, Representative confocal images of cortical injury at 2 d post-injury in juvenile mice receiving 1 mg/kg/d Tie2-Fc, which shows greater lesion area compared with Fc control. i, Quantified data analysis demonstrating that Tie2-Fc significantly increases lesion volume in juvenile mice compared with Fc-control (*p = 0.016). j, Quantification of Evans blue (EB) absorbance (A610nm) also shows Tie2-Fc increased EB absorbance in the juvenile ipsilateral cortex compared with Fc control (*p = 0.0402). k, Cultured brain-derived ECs treated with 20 μg/ml Tie2-Fc for 24 h reduced mRNA expression of BBB-associated occludin-1 (**p = 0.0088), VE-cadherin (**p = 0.0042), tgfβ (**p = 0.0059), and pro-survival bcl-2 (*p = 0.0199), whereas vegf levels were increased (***p = 0.0002) relative to Fc control. Unpaired t test (k), one-way ANOVA (i), and two-way ANOVA (j). Scale bar, 500 μm in a–h.
To address the molecular signaling affected by blocking Tie2 on ECs, we evaluated mRNA expression using qRT-PCR in primary brain-derived ECs following 24 h of treatment with 20 μg/ml soluble Tie2-Fc versus Fc control (Fig. 8i). Relative to Fc controls, we found that Tie-Fc treatment significantly reduced the vascular barrier-associated genes occludin-1 (t test, p = 0.0089, n = 6), VE-cadherin (t test, p = 0.0042, n = 8), and tgfβ (t test, p = 0.0051, n = 8), along with the anti-apoptotic gene bcl-2 (t test, p = 0.0199, n = 8). Interestingly, preventing Tie2 receptor signaling increased vegf expression (t test, p = 0.0002, n = 8), which is known to destabilize vessels and contribute to BBB permeability. No difference in eNOS (t test, p = 0.6801, n = 4) or IL-6 (t test, p = 0.4640, n = 8) expression was observed. These findings demonstrate that the Tie2 receptor mediates the age-at-injury response to CCI injury, which may diminish the severity of injury by regulating the expression of genes critical for vessel stability and survival.
Prominent GFAP immunoreactivity in the juvenile cortex at 4 d and 35 d post-CCI injury
Injury-induced astrogliosis, represented by increased astrocyte numbers and GFAP immunoreactivity, contributes to BBB function and tissue health (Bélanger and Magistretti, 2009). Moreover, astrocytes are a major source of angiopoietins, which may contribute to the stability of the cortical tissue in juvenile injured mice. To evaluate whether acute changes in astrogliosis correlate with juvenile tissue protection, we used immunohistochemistry and confocal image analysis to evaluate GFAP immunoreactivity in the juvenile and adult brain at 4 and 35 d post-CCI injury. Upregulation of GFAP expression was seen in the adult ipsilateral perilesional cortex, hippocampus, and thalamus compared with the contralateral hemisphere at 4 d (Fig. 9a). The extent of GFAP immunoreactivity, however, was greatly enhanced in these regions in juvenile mice (Fig. 9b, white arrows). GFAP immunoreactivity remained prominent in the ipsilateral perilesion of juvenile mice (Fig. 9f) at the chronic 35 d time point compared with adults (Fig. 9c). High-magnification images revealed increased GFAP reactivity on CD31+ vessels (Fig. 9h1–h3) in the juvenile perilesion compared with contralateral (Fig. 9g1–g3) or adult ipsilateral (Fig. 9e1–e3). Quantification of vessel density at 35 d post-CCI injury showed an increase in the percentage area occupied by CD31+ vessels in the juvenile perilesion (10.02 ± 0.28%, n = 3) compared with adult (7.66 ± 0.51%, n = 3; t test, p = 0.0143). Although not significant, there was a trend toward an increase in percentage area of GFAP immunoreactivity in the juvenile ipsilateral perilesion (36.68 ± 5.64%, n = 3) compared with adult (23.49 ± 3.27%, n = 3; t test, p = 0.083). Last, quantified data showed a significant increase in the percentage of GFAP and CD31 colocalization (area of GFAP + CD31/total area of CD31) in juvenile mice (61.1 ± 3.63%, n = 3) compared with adult mice (48.33 ± 1.69%, n = 3; t test, p = 0.0332) (Fig. 9k). These findings demonstrate that age at injury affects the astroglial response following acute and chronic cortical trauma, which may contribute to the vascular niche environment.
Figure 9.
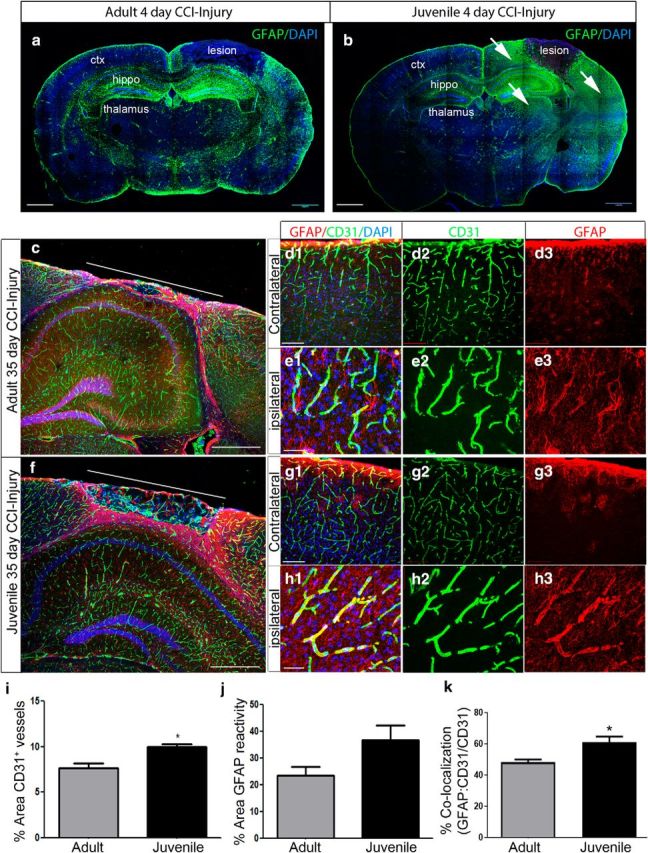
Prominent GFAP immunoreactivity accompanies neuroprotection in juvenile mice after CCI injury. a, Representative mosaic confocal image of GFAP colabeling at 4 d post-CCI injury. b, Representative mosaic confocal image of GFAP colabeling at 4 d post-CCI injury in juvenile mice. Increased immunoreactivity for GFAP is seen in the perilesion cortex, hippocampus, and thalamus (white arrows) compared with adult. c, Confocal mosaic image of the ipsilateral cortex at 35 d post-CCI injury in adult mice. Double immunolabeling for GFAP (red) and CD31 (green) shows the glial scar in the perilesion cortex. d1–d3, High-magnification confocal image of GFAP and CD31 in contralateral and ipsilateral cortex (e1–e3) after CCI injury. f, Confocal image of the juvenile ipsilateral cortex at 35 d post-CCI injury. High-magnification confocal image of GFAP and CD31 in contralateral (g1–g3) and ipsilateral (h1–h3). i, Quantification of the percentage area of CD31+ vessels in the perilesion at 35 d shows an increase in vessel density in the juvenile perilesion compared with adult mice (*p = 0.0143). No difference was seen in the percentage area of GFAP reactivity (j), but a significant increase in the percentage colocalization was seen (GFAP:CD31/CD31) (*p = 0.0332) (k). ctx, Cortex; hippo, hippocampus. Scale bar, 1 mm in a–c, and f; 500 μm in d1 and g1; and 100 μm in e1 and h1.
Discussion
TBI is increasing in prevalence across the age spectrum, leading to long-term deleterious effects on neurological function and developmental outcomes. Our understanding of the acute age-at-injury response, however, remains limited. Using a well established focal contusion model of TBI, we aimed to evaluate the acute pathophysiological, behavioral, and vascular-specific changes in adult and juvenile mice. We found that cortical lesion formation was significantly reduced at 4 d and 14 d post-CCI injury in juvenile versus adult mice, which correlated with reduced numbers of TUNEL+ cells in the cortex, reduced motor and cognitive deficits, and enhanced restoration of CBF. Juvenile neuroprotection was accompanied by increased vessel density in the perilesion region and reduced cortical BBB disruption following CCI injury. EC isolation from the cortex showed increased transcripts for BBB-associated genes occludin-1, claudin-5, and angiopoietins in injured juvenile versus adult mice. Conversely, vegf and its receptors, which contribute to BBB breakdown, and immune-mediated vascular cell adhesion molecule (vecam) were reduced in juvenile versus adult, indicative of greater vessel stability in response to cortical impact. Interestingly, expression of miRNA-21-5p and miRNA-148a were enhanced in juvenile injured ECs. Soluble inhibition of EC-specific Tie2 receptor reversed the cortical tissue and BBB protection observed in juvenile injured mice, possibly due to the negative regulation of occludin-1, VE-cadherin, vegf, and tgfβ expression. Last, we found temporospatial age-dependent differences in the astrogliotic response to CCI injury that may play a role in early outcome.
The present study shows EC age may be a critical factor in the cortical response to physical impact. Although the murine cerebral vasculature is well established by P21 (Zeller et al., 1996; Wälchli et al., 2015), we found increased levels of miRNA-21-5p and miR-31 in uninjured juvenile ECs, which may significantly affect how ECs mount a response to brain injury (Bhalala et al., 2013; Chamorro-Jorganes et al., 2013). miRNAs are short-stranded RNAs that regulate gene expression at the posttranscriptional level and are involved in a wide range of biological processes (Chamorro-Jorganes et al., 2013; Adlakha and Saini, 2014). Recently, miR-21–5p was shown to be upregulated following adult TBI and contributed to BBB protection and reduced neuronal cell death (Lei et al., 2009; Ge et al., 2014; Han et al., 2015). Additional studies revealed miR-21–5p agonists suppressed inflammation via NF-kβ signaling and promoted the Ang/Tie2 axis (Ge et al., 2016). Conversely, miR-21–3p, which is also generated following pre-miRNA-21 splicing, contributes to BBB leakage and intraventricular infusion of its antagomir alleviated BBB disruption (Ge et al., 2018). This finding suggests that miR-21 plays an indispensable role in TBI outcome; however, cell-specific expression of miR-21 variants may affect the overall response. We recently observed reduced cortical injury in miR-21 global knock-out (both −5p and −3p) adult mice compared with wild-type, indicating a more complex picture of miRNAs in TBI (data not shown). Last, the molecular mechanism(s) underlying the regulation of miRNAs in ECs at discrete stages of maturation remain ill defined but may include epigenetic modifications by DNA methylation and histone modification (Morales et al., 2017). Our data suggest that EC-specific miRNAs may be differentially regulated in the juvenile brain which may confer an acute protective advantage following TBI.
Tie2, the cognate receptor for Ang1 and Ang2, is expressed predominately on the vascular endothelium and is a potent regulator of cell survival, inhibition of vascular leakage, and suppression of inflammatory genes (Brindle et al., 2006; Fiedler and Augustin, 2006; Fukuhara et al., 2009). Ang1 is an agonist of Tie2 and Ang2 is a competitive inhibitor; however, this effect appears to be cell type and context dependent (Brindle et al., 2006). For example, at high concentrations or sustained concentrations for long periods, Ang2 can act as an agonist to stimulate Tie2 on ECs (Kim et al., 2000; Teichert-Kuliszewska et al., 2001) and may act as an agonist alongside Ang1 in the presence of TNF stimulation to attenuate Tie2 expression as part of a negative feedback system (Hashimoto et al., 2004). This may explain why we find increased Ang2 concomitant with reduced lesion volume and BBB permeability compared with adult. Interestingly, Tie2, Ang1, and Ang2 mRNA transcripts were significantly increased in juvenile ECs following 4 d CCI injury, which correlates with increased expression of BBB-associated occludin-1 and claudin-5. Conversely, adult ECs do not display changes in Ang1 or Ang2, but do show increased Tie2 expression following CCI injury. Inhibition of Tie2 receptor attenuated BBB stability in the juvenile cortex while having no effect in adult. These findings exclude the possibility that structural differences in brain size contributed to the juvenile neuroprotection and highlights Tie2 signaling as a central player in this protective response to cortical impact. Although the Ang/Tie2 axis has not been extensively evaluated following TBI (Gu et al., 2016), plasma levels of Ang1 have been shown to be decreased in patients with chronic TBI and the Ang1/Ang2 ratio may represent an important diagnostic and prognostic tool for TBI (Chittiboina et al., 2013; Reis et al., 2015). Additional studies are needed to improve our mechanistic understanding of the age-dependent response underlying Ang/Tie2 regulation following TBI.
The current study establishes a novel acute protective response to juvenile focal contusion injury. Whether this is the case for other forms of brain injury such as diffuse axonal injury (DAI) or single versus repeated mild concussion is unclear. It is plausible that the juvenile brain is more resistant to focal cortical lesions rather than DAI, which often occurs in the absence of major vessel damage following mild TBI (Oppenheimer, 1968; Povlishock et al., 1983). Although these assumptions were made based on gross pathology, histology, or conventional MRI (Jane et al., 1985; Povlishock, 1993), recent evidence suggests that cerebral microbleeds, or small brain hemorrhages, may lead to prolonged disability in mild TBI or DAI (Park et al., 2009; Liu et al., 2014; van der Horn et al., 2018). Hemorrhagic shearing has been identified in children and adolescents with post-traumatic DAI using high-spatial-resolution susceptibility-weighted (SW) MRI (Tong et al., 2003). It is unclear whether these hemorrhagic lesions are smaller in size or number compared with adult or older-aged patients. Additional studies are needed to evaluate the vascular-specific response across multiple models and brain regions.
Although EC-specific Tie2 signaling plays a significant role in the juvenile response to CCI injury by protecting the BBB and preventing cortical tissue loss, it is plausible that juvenile ECs may also modulate brain glucose metabolism to help improve outcome after TBI (Appelberg et al., 2009). Cellular metabolism in the brain relies on delivery of fuel substrate and oxygen through the vasculature. Recent findings support a critical role for the endothelium in regulating brain glucose metabolism which is mediated by HIF-1α and glucose transporter (Glut1) expression (Huang et al., 2012). Glucose transport into the brain occurs by way of Glut1 found predominately on BBB ECs (Boado et al., 1994; Devraj et al., 2011; Huang et al., 2012). To date, little is known about the regulation of Glut1 expression following TBI; however, it is well established that TBI induces glucose metabolic depression (Passineau et al., 2000). Interestingly, this phase is shortened in P17 rats following fluid percussion injury and in P35 rats after CCI injury (Thomas et al., 2000), which could be related to differences in Glut1 expression or other metabolic coping strategies such as utilization of ketones (Prins et al., 2004), which is better exploited by the juvenile brain compared with adult(Prins et al., 2005; Deng-Bryant et al., 2011). Future avenues of research warrant investigation into whether age at injury influences Glut1 expression and/or other carrier-mediated transport of lactate, pyruvate, and ketone bodies such as monocarboxylate transport (MCT1-4).
Last, we show an age-at-injury difference in acute GFAP immunoreactivity following CCI injury. Astrocytes are pivotal to the evolution of TBI sequelae (McGraw et al., 2001; Burda et al., 2016) and they respond to acute mechanical stress by regulating inflammation, limiting spread of injury, and modifying the BBB (McGraw et al., 2001; Chen and Swanson, 2003; Myer et al., 2006). It is therefore plausible that increased GFAP immunoreactivity may represent overzealous astrogliosis in the juvenile cortex following acute CCI injury, which may potentiate the positive effects of reactive astrocytes. Although astrocytes are important regulators of tissue recovery, there is a two-way exchange of signals between ECs and astrocytes (Goldstein, 1987; Estrada et al., 1990; Spoerri et al., 1997; Wagner and Gardner, 2000; Mi et al., 2001; Abbott, 2002; Engelhardt and Liebner, 2014; Wang et al., 2016a). In what way this bidirectional relationship is affected following TBI and whether the age-at-injury EC response alters EC-astrocyte communication is unclear.
Our findings demonstrate a previously unidentified acute protective response in juvenile mice to cortical impact injury that is mediated in part by Tie2 receptor signaling; a known regulator of vessel stability, anti-inflammation, and EC survival (Engelhardt and Liebner, 2014; Milam and Parikh, 2015; Gurnik et al., 2016). Our novel findings indicate that the EC response is central to maintaining tissue homeostasis after TBI and is regulated by age-dependent cellular and molecular mechanisms, including mRNA and miRNA regulation. These data improve our understanding of the differential response that age plays in TBI outcome and provide potential targets for therapeutic intervention across the age spectrum.
Footnotes
This work was supported by the National Institute of Neurological Disorders and Stroke of the National Institutes of Health (Grant R01NS096281 to M.H.T.). We thank the Regenerative Medicine–Interdisciplinary Graduate Education Program at Virginia Tech and Institute of Critical Technology and Applied Sciences Programs for student support (T.R.B., A.H.), the Virginia–Maryland Regional College of Veterinary Medicine, and Dr. William Huckle for the generous gift of qRT-PCR primers.
The authors declare no competing financial interests.
References
- Abbott NJ. (2002) Astrocyte-endothelial interactions and blood-brain barrier permeability. J Anat 200:629–638. 10.1046/j.1469-7580.2002.00064.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdul-Muneer PM. (2016) MicroRNA in the pathophysiology of CNS injury: implication in neuroregenerative medicine. CNS Neurosci Ther 22:543–545. 10.1111/cns.12579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adelson PD, Clyde B, Kochanek PM, Wisniewski SR, Marion DW, Yonas H (1997) Cerebrovascular response in infants and young children following severe traumatic brain injury: a preliminary report. Pediatr Neurosurg 26:200–207. 10.1159/000121192 [DOI] [PubMed] [Google Scholar]
- Adlakha YK, Saini N (2014) Brain microRNAs and insights into biological functions and therapeutic potential of brain enriched miRNA-128. Mol Cancer 13:33. 10.1186/1476-4598-13-33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen CL, Bayraktutan U (2009) Oxidative stress and its role in the pathogenesis of ischaemic stroke. Int J Stroke 4:461–470. 10.1111/j.1747-4949.2009.00387.x [DOI] [PubMed] [Google Scholar]
- Appelberg KS, Hovda DA, Prins ML (2009) The effects of a ketogenic diet on behavioral outcome after controlled cortical impact injury in the juvenile and adult rat. J Neurotrauma 26:497–506. 10.1089/neu.2008.0664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstead WM. (1999) Cerebral hemodynamics after traumatic brain injury of immature brain. Exp Toxicol Pathol 51:137–142. 10.1016/S0940-2993(99)80087-6 [DOI] [PubMed] [Google Scholar]
- Assis-Nascimento P, Tsenkina Y, Liebl DJ (2018) EphB3 signaling induces cortical endothelial cell death and disrupts the blood-brain barrier after traumatic brain injury. Cell Death Dis 9:7. 10.1038/s41419-017-0016-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumann G, Travieso L, Liebl DJ, Theus MH (2013) Pronounced hypoxia in the subventricular zone following traumatic brain injury and the neural stem/progenitor cell response. Exp Biol Med 238:830–841. 10.1177/1535370213494558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bélanger M, Magistretti PJ (2009) The role of astroglia in neuroprotection. Dialogues Clin Neurosci 11:281–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhalala OG, Srikanth M, Kessler JA (2013) The emerging roles of microRNAs in CNS injuries. Nat Rev Neurol 9:328–339. 10.1038/nrneurol.2013.67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaiss CA, Yu TS, Zhang G, Chen J, Dimchev G, Parada LF, Powell CM, Kernie SG (2011) Temporally specified genetic ablation of neurogenesis impairs cognitive recovery after traumatic brain injury. J Neurosci 31:4906–4916. 10.1523/JNEUROSCI.5265-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boado RJ, Wang L, Pardridge WM (1994) Enhanced expression of the blood-brain barrier GLUT1 glucose transporter gene by brain-derived factors. Brain Res Mol Brain Res 22:259–267. 10.1016/0169-328X(94)90054-X [DOI] [PubMed] [Google Scholar]
- Bouët V, Freret T, Toutain J, Divoux D, Boulouard M, Schumann-Bard P (2007) Sensorimotor and cognitive deficits after transient middle cerebral artery occlusion in the mouse. Exp Neurol 203:555–567. 10.1016/j.expneurol.2006.09.006 [DOI] [PubMed] [Google Scholar]
- Brickler T, Gresham K, Meza A, Coutermarsh-Ott S, Williams TM, Rothschild DE, Allen IC, Theus MH (2016) Nonessential role for the NLRP1 inflammasome complex in a murine model of traumatic brain injury. Mediators Inflamm 2016:6373506. 10.1155/2016/6373506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brindle NP, Saharinen P, Alitalo K (2006) Signaling and functions of angiopoietin-1 in vascular protection. Circ Res 98:1014–1023. 10.1161/01.RES.0000218275.54089.12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burda JE, Bernstein AM, Sofroniew MV (2016) Astrocyte roles in traumatic brain injury. Exp Neurol 275:305–315. 10.1016/j.expneurol.2015.03.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamorro-Jorganes A, Araldi E, Suárez Y (2013) MicroRNAs as pharmacological targets in endothelial cell function and dysfunction. Pharmacol Res 75:15–27. 10.1016/j.phrs.2013.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Swanson RA (2003) Astrocytes and brain injury. J Cereb Blood Flow Metab 23:137–149. 10.1097/01.WCB.0000044631.80210.3C [DOI] [PubMed] [Google Scholar]
- Chittiboina P, Ganta V, Monceaux CP, Scott LK, Nanda A, Alexander JS (2013) Angiopoietins as promising biomarkers and potential therapeutic targets in brain injury. Pathophysiology 20:15–21. 10.1016/j.pathophys.2012.02.004 [DOI] [PubMed] [Google Scholar]
- Clark RE, Zola SM, Squire LR (2000) Impaired recognition memory in rats after damage to the hippocampus. J Neurosci 20:8853–8860. 10.1523/JNEUROSCI.20-23-08853.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claus CP, Tsuru-Aoyagi K, Adwanikar H, Walker B, Whetstone W, Noble-Haeusslein LJ (2010) Age is a determinant of leukocyte infiltration and loss of cortical volume after traumatic brain injury. Dev Neurosci Basel 32:454–465. 10.1159/000322448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curvello V, Hekierski H, Riley J, Vavilala M, Armstead WM (2017) Sex and age differences in phenylephrine mechanisms and outcomes after piglet brain injury. Pediatr Res 82:108–113. 10.1038/pr.2017.83 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Dai R, Zhang Y, Khan D, Heid B, Caudell D, Crasta O, Ahmed SA (2010) Identification of a common lupus disease-associated microRNA expression pattern in three different murine models of lupus. PLoS One 5:e14302. 10.1371/journal.pone.0014302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai R, McReynolds S, Leroith T, Heid B, Liang Z, Ahmed SA (2013) Sex differences in the expression of lupus-associated miRNAs in splenocytes from lupus-prone NZB/WF1 mice. Biol Sex Differ 4:19. 10.1186/2042-6410-4-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng-Bryant Y, Prins ML, Hovda DA, Harris NG (2011) Ketogenic diet prevents alterations in brain metabolism in young but not adult rats after traumatic brain injury. J Neurotrauma 28:1813–1825. 10.1089/neu.2011.1822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devraj K, Klinger ME, Myers RL, Mokashi A, Hawkins RA, Simpson IA (2011) GLUT-1 glucose transporters in the blood-brain barrier: differential phosphorylation. J Neurosci Res 89:1913–1925. 10.1002/jnr.22738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon KJ, Theus MH, Nelersa CM, Mier J, Travieso LG, Yu TS, Kernie SG, Liebl DJ (2015) Endogenous neural stem/progenitor cells stabilize the cortical microenvironment after traumatic brain injury. J Neurotrauma 32:753–764. 10.1089/neu.2014.3390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duhaime AC, Margulies SS, Durham SR, O'Rourke MM, Golden JA, Marwaha S, Raghupathi R (2000) Maturation-dependent response of the piglet brain to scaled cortical impact. J Neurosurg 93:455–462. 10.3171/jns.2000.93.3.0455 [DOI] [PubMed] [Google Scholar]
- Duhaime AC, Hunter JV, Grate LL, Kim A, Golden J, Demidenko E, Harris C (2003) Magnetic resonance imaging studies of age-dependent responses to scaled focal brain injury in the piglet. J Neurosurg 99:542–548. 10.3171/jns.2003.99.3.0542 [DOI] [PubMed] [Google Scholar]
- Durham SR, Raghupathi R, Helfaer MA, Marwaha S, Duhaime AC (2000) Age-related differences in acute physiologic response to focal traumatic brain injury in piglets. Pediatr Neurosurg 33:76–82. 10.1159/000028980 [DOI] [PubMed] [Google Scholar]
- Engelhardt B, Liebner S (2014) Novel insights into the development and maintenance of the blood-brain barrier. Cell Tissue Res 355:687–699. 10.1007/s00441-014-1811-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estrada C, Bready JV, Berliner JA, Pardridge WM, Cancilla PA (1990) Astrocyte growth stimulation by a soluble factor produced by cerebral endothelial cells in vitro. J Neuropathol Exp Neurol 49:539–549. 10.1097/00005072-199011000-00001 [DOI] [PubMed] [Google Scholar]
- Fan P, Yamauchi T, Noble LJ, Ferriero DM (2003) Age-dependent differences in glutathione peroxidase activity after traumatic brain injury. J Neurotrauma 20:437–445. 10.1089/089771503765355513 [DOI] [PubMed] [Google Scholar]
- Fiedler U, Augustin HG (2006) Angiopoietins: a link between angiogenesis and inflammation. Trends Immunol 27:552–558. 10.1016/j.it.2006.10.004 [DOI] [PubMed] [Google Scholar]
- Fitzgerald A, Aditya H, Prior A, McNeill E, Pentland B (2010) Anoxic brain injury: clinical patterns and functional outcomes: a study of 93 cases. Brain Injury 24:1311–1323. 10.3109/02699052.2010.506864 [DOI] [PubMed] [Google Scholar]
- Fukuhara S, Sako K, Noda K, Nagao K, Miura K, Mochizuki N (2009) Tie2 is tied at the cell-cell contacts and to extracellular matrix by angiopoietin-1. Exp Mol Med 41:133–139. 10.3858/emm.2009.41.3.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge XT, Lei P, Wang HC, Zhang AL, Han ZL, Chen X, Li SH, Jiang RC, Kang CS, Zhang JN (2014) miR-21 improves the neurological outcome after traumatic brain injury in rats. Sci Rep 4:6718. 10.1038/srep06718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge X, Huang S, Gao H, Han Z, Chen F, Zhang S, Wang Z, Kang C, Jiang R, Yue S, Lei P, Zhang J (2016) miR-21–5p alleviates leakage of injured brain microvascular endothelial barrier in vitro through suppressing inflammation and apoptosis. Brain Res 1650:31–40. 10.1016/j.brainres.2016.07.015 [DOI] [PubMed] [Google Scholar]
- Ge X, Li W, Huang S, Yin Z, Yang M, Han Z, Han Z, Chen F, Wang H, Lei P, Zhang JN (2018) Increased miR-21–3p in injured brain microvascular endothelial cells following traumatic brain injury aggravates blood-brain barrier damage by promoting cellular apoptosis and inflammation through targeting MAT2B. J Neurotrauma. Advance online publication. Retrieved from April 26, 2018. doi: 10.1089/neu.2018.5728. [DOI] [PubMed] [Google Scholar]
- Giza CC, Prins ML (2006) Is being plastic fantastic? Mechanisms of altered plasticity after developmental traumatic brain injury. Dev Neurosci 28:364–379. 10.1159/000094163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein GW. (1987) The blood-brain barrier: interacters between endothelial cells and astrocytes. Mead Johnson Symp Perinat Dev Med 29:15–17. [PubMed] [Google Scholar]
- Gu H, Fei ZH, Wang YQ, Yang JG, Zhao CH, Cai Y, Zhong XM (2016) Angiopoietin-1 and angiopoietin-2 expression imbalance influence in early period after subarachnoid hemorrhage. Int Neurourol J 20:288–295. 10.5213/inj.1632692.346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurnik S, Devraj K, Macas J, Yamaji M, Starke J, Scholz A, Sommer K, Di Tacchio M, Vutukuri R, Beck H, Mittelbronn M, Foerch C, Pfeilschifter W, Liebner S, Peters KG, Plate KH, Reiss Y (2016) Angiopoietin-2-induced blood-brain barrier compromise and increased stroke size are rescued by VE-PTP-dependent restoration of Tie2 signaling. Acta Neuropathol 131:753–773. 10.1007/s00401-016-1551-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Z, Chen F, Ge X, Tan J, Lei P, Zhang J (2014) miR-21 alleviated apoptosis of cortical neurons through promoting PTEN-akt signaling pathway in vitro after experimental traumatic brain injury. Brain Res 1582:12–20. 10.1016/j.brainres.2014.07.045 [DOI] [PubMed] [Google Scholar]
- Han Z, Ge X, Tan J, Chen F, Gao H, Lei P, Zhang J (2015) Establishment of lipofection protocol for efficient miR-21 transfection into cortical neurons in vitro. DNA Cell Biol 34:703–709. 10.1089/dna.2015.2800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto T, Wu Y, Boudreau N, Li J, Matsumoto M, Young W (2004) Regulation of tie2 expression by angiopoietin–potential feedback system. Endothelium 11:207–210. 10.1080/10623320490512417 [DOI] [PubMed] [Google Scholar]
- Huang Y, Lei L, Liu D, Jovin I, Russell R, Johnson RS, Di Lorenzo A, Giordano FJ (2012) Normal glucose uptake in the brain and heart requires an endothelial cell-specific HIF-1alpha-dependent function. Proc Natl Acad Sci U S A 109:17478–17483. 10.1073/pnas.1209281109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jane JA, Steward O, Gennarelli T (1985) Axonal degeneration induced by experimental noninvasive minor head injury. J Neurosurg 62:96–100. 10.3171/jns.1985.62.1.0096 [DOI] [PubMed] [Google Scholar]
- Jones BJ, Roberts DJ (1968) The quantiative measurement of motor incoordination in naive mice using an acelerating rotarod. J Pharm Pharmacol 20:302–304. 10.1111/j.2042-7158.1968.tb09743.x [DOI] [PubMed] [Google Scholar]
- Kamper JE, Pop V, Fukuda AM, Ajao DO, Hartman RE, Badaut J (2013) Juvenile traumatic brain injury evolves into a chronic brain disorder: behavioral and histological changes over 6 months. Exp Neurol 250:8–19. 10.1016/j.expneurol.2013.09.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, Furumichi M, Tanabe M, Sato Y, Morishima K (2017) KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res 45:D353–D361. 10.1093/nar/gkw1092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim I, Kim JH, Moon SO, Kwak HJ, Kim NG, Koh GY (2000) Angiopoietin-2 at high concentration can enhance endothelial cell survival through the phosphatidylinositol 3′-kinase/Akt signal transduction pathway. Oncogene 19:4549–4552. 10.1038/sj.onc.1203800 [DOI] [PubMed] [Google Scholar]
- Leger M, Quiedeville A, Bouët V, Haelewyn B, Boulouard M, Schumann-Bard P, Freret T (2013) Object recognition test in mice. Nat Protoc 8:2531–2537. 10.1038/nprot.2013.155 [DOI] [PubMed] [Google Scholar]
- Lei P, Li Y, Chen X, Yang S, Zhang J (2009) Microarray based analysis of microRNA expression in rat cerebral cortex after traumatic brain injury. Brain Res 1284:191–201. 10.1016/j.brainres.2009.05.074 [DOI] [PubMed] [Google Scholar]
- Liu J, Kou Z, Tian Y (2014) Diffuse axonal injury after traumatic cerebral microbleeds: an evaluation of imaging techniques. Neural Regen Res 9:1222–1230. 10.4103/1673-5374.135330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YL, Xu ZM, Yang GY, Yang DX, Ding J, Chen H, Yuan F, Tian HL (2017) Sesamin alleviates blood-brain barrier disruption in mice with experimental traumatic brain injury. Acta Pharmacol Sin 38:1445–1455. 10.1038/aps.2017.103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logsdon AF, Lucke-Wold BP, Turner RC, Huber JD, Rosen CL, Simpkins JW (2015) Role of microvascular disruption in brain damage from traumatic brain injury. Compr Physiol 5:1147–1160. 10.1002/cphy.c140057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu TP, Lee CY, Tsai MH, Chiu YC, Hsiao CK, Lai LC, Chuang EY (2012) miRSystem: an integrated system for characterizing enriched functions and pathways of microRNA targets. PLoS One 7:e42390. 10.1371/journal.pone.0042390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marin JR, Weaver MD, Yealy DM, Mannix RC (2014) Trends in visits for traumatic brain injury to emergency departments in the united states. JAMA 311:1917–1919. 10.1001/jama.2014.3979 [DOI] [PubMed] [Google Scholar]
- Marquez de la Plata CD, Hart T, Hammond FM, Frol AB, Hudak A, Harper CR, O'Neil-Pirozzi TM, Whyte J, Carlile M, Diaz-Arrastia R (2008) Impact of age on long-term recovery from traumatic brain injury. Arch Phys Med Rehabil 89:896–903. 10.1016/j.apmr.2007.12.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGinn MJ, Povlishock JT (2016) Pathophysiology of traumatic brain injury. Neurosurg Clin N Am 27:397–407. 10.1016/j.nec.2016.06.002 [DOI] [PubMed] [Google Scholar]
- McGraw J, Hiebert GW, Steeves JD (2001) Modulating astrogliosis after neurotrauma. J Neurosci Res 63:109–115. 10.1002/1097-4547(20010115)63:2%3C109::AID-JNR1002%3E3.0.CO;2-J [DOI] [PubMed] [Google Scholar]
- Meissner L, Gallozzi M, Balbi M, Schwarzmaier S, Tiedt S, Terpolilli NA, Plesnila N (2016) Temporal profile of MicroRNA expression in contused cortex after traumatic brain injury in mice. J Neurotrauma 33:713–720. 10.1089/neu.2015.4077 [DOI] [PubMed] [Google Scholar]
- Mi H, Haeberle H, Barres BA (2001) Induction of astrocyte differentiation by endothelial cells. J Neurosci 21:1538–1547. 10.1523/JNEUROSCI.21-05-01538.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milam KE, Parikh SM (2015) The angiopoietin-Tie2 signaling axis in the vascular leakage of systemic inflammation. Tissue Barriers 3:e957508. 10.4161/21688362.2014.957508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Missios S, Harris BT, Dodge CP, Simoni MK, Costine BA, Lee YL, Quebada PB, Hillier SC, Adams LB, Duhaime AC (2009) Scaled cortical impact in immature swine: effect of age and gender on lesion volume. J Neurotrauma 26:1943–1951. 10.1089/neu.2009.0956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales S, Monzo M, Navarro A (2017) Epigenetic regulation mechanisms of microRNA expression. Biomol Concepts 8:203–212. 10.1515/bmc-2017-0024 [DOI] [PubMed] [Google Scholar]
- Moretti R, Pansiot J, Bettati D, Strazielle N, Ghersi-Egea JF, Damante G, Fleiss B, Titomanlio L, Gressens P (2015) Blood-brain barrier dysfunction in disorders of the developing brain. Front Neurosci 9:40. 10.3389/fnins.2015.00040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myer DJ, Gurkoff GG, Lee SM, Hovda DA, Sofroniew MV (2006) Essential protective roles of reactive astrocytes in traumatic brain injury. Brain 129:2761–2772. 10.1093/brain/awl165 [DOI] [PubMed] [Google Scholar]
- Ojo JO, Mouzon B, Algamal M, Leary P, Lynch C, Abdullah L, Evans J, Mullan M, Bachmeier C, Stewart W, Crawford F (2016) Chronic repetitive mild traumatic brain injury results in reduced cerebral blood flow, axonal injury, gliosis, and Increased T-Tau and Tau oligomers. J Neuropathol Exp Neurol 75:636–655. 10.1093/jnen/nlw035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okyere B, Giridhar K, Hazy A, Chen M, Keimig D, Bielitz RC, Xie H, He JQ, Huckle WR, Theus MH (2016) Endothelial-specific EphA4 negatively regulates native pial collateral formation and re-perfusion following hindlimb ischemia. PLoS One 11:e0159930. 10.1371/journal.pone.0159930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okyere B, Creasey M, Lebovitz Y, Theus MH (2018) Temporal remodeling of pial collaterals and functional deficits in a murine model of ischemic stroke. J Neurosci Methods 293:86–96. 10.1016/j.jneumeth.2017.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oppenheimer DR. (1968) Microscopic lesions in the brain following head injury. J Neurol Neurosurg Psychiatry 31:299–306. 10.1136/jnnp.31.4.299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JH, Park SW, Kang SH, Nam TK, Min BK, Hwang SN (2009) Detection of traumatic cerebral microbleeds by susceptibility-weighted image of MRI. J Korean Neurosurg Soc 46:365–369. 10.3340/jkns.2009.46.4.365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passineau MJ, Zhao W, Busto R, Dietrich WD, Alonso O, Loor JY, Bramlett HM, Ginsberg MD (2000) Chronic metabolic sequelae of traumatic brain injury: prolonged suppression of somatosensory activation. Am J Physiol Heart Circ Physiol 279:H924–H931. 10.1152/ajpheart.2000.279.3.H924 [DOI] [PubMed] [Google Scholar]
- Pearn ML, Niesman IR, Egawa J, Sawada A, Almenar-Queralt A, Shah SB, Duckworth JL, Head BP (2017) Pathophysiology associated with traumatic brain injury: current treatments and potential novel therapeutics. Cell Mol Neurobiol 37:571–585. 10.1007/s10571-016-0400-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pop V, Sorensen DW, Kamper JE, Ajao DO, Murphy MP, Head E, Hartman RE, Badaut J (2013) Early brain injury alters the blood-brain barrier phenotype in parallel with beta-amyloid and cognitive changes in adulthood. J Cereb Blood Flow Metab 33:205–214. 10.1038/jcbfm.2012.154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Povlishock JT. (1993) Pathobiology of traumatically induced axonal injury in animals and man. Ann Emerg Med 22:980–986. 10.1016/S0196-0644(05)82738-6 [DOI] [PubMed] [Google Scholar]
- Povlishock JT, Becker DP, Cheng CL, Vaughan GW (1983) Axonal change in minor head injury. J Neuropathol Exp Neurol 42:225–242. 10.1097/00005072-198305000-00002 [DOI] [PubMed] [Google Scholar]
- Price L, Wilson C, Grant G (2016) Blood-brain barrier pathophysiology following traumatic brain injury. In: Translational research in traumatic brain injury (Laskowitz D, Grant G, eds). Boca Raton, FL: CRC Press/Taylor and Francis Group. [PubMed] [Google Scholar]
- Prins ML, Lee SM, Fujima LS, Hovda DA (2004) Increased cerebral uptake and oxidation of exogenous betaHB improves ATP following traumatic brain injury in adult rats. J Neurochem 90:666–672. 10.1111/j.1471-4159.2004.02542.x [DOI] [PubMed] [Google Scholar]
- Prins ML, Fujima LS, Hovda DA (2005) Age-dependent reduction of cortical contusion volume by ketones after traumatic brain injury. J Neurosci Res 82:413–420. 10.1002/jnr.20633 [DOI] [PubMed] [Google Scholar]
- Redell JB, Zhao J, Dash PK (2011) Altered expression of miRNA-21 and its targets in the hippocampus after traumatic brain injury. J Neurosci Res 89:212–221. 10.1002/jnr.22539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reis C, Wang Y, Akyol O, Ho WM, Ii RA, Stier G, Martin R, Zhang JH (2015) What's new in traumatic brain injury: update on tracking, Monitoring and Treatment. Int J Mol Sci 16:11903–11965. 10.3390/ijms160611903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salehi A, Zhang JH, Obenaus A (2017) Response of the cerebral vasculature following traumatic brain injury. J Cereb Blood Flow Metab 37:2320–2339. 10.1177/0271678X17701460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneier AJ, Shields BJ, Hostetler SG, Xiang H, Smith GA (2006) Incidence of pediatric traumatic brain injury and associated hospital resource utilization in the united states. Pediatrics 118:483–492. 10.1542/peds.2005-2588 [DOI] [PubMed] [Google Scholar]
- Spoerri PE, Grant MB, Gomez J, Vernadakis A (1997) Endothelial cell conditioned media mediated regulation of glutamine synthetase activity in glial cells. Brain Res Dev Brain Res 104:205–208. 10.1016/S0165-3806(97)00173-9 [DOI] [PubMed] [Google Scholar]
- Teichert-Kuliszewska K, Maisonpierre PC, Jones N, Campbell AI, Master Z, Bendeck MP, Alitalo K, Dumont DJ, Yancopoulos GD, Stewart DJ (2001) Biological action of angiopoietin-2 in a fibrin matrix model of angiogenesis is associated with activation of Tie2. Cardiovasc Res 49:659–670. 10.1016/S0008-6363(00)00231-5 [DOI] [PubMed] [Google Scholar]
- Theus MH, Ricard J, Bethea JR, Liebl DJ (2010) EphB3 limits the expansion of neural progenitor cells in the subventricular zone by regulating p53 during homeostasis and following traumatic brain injury. Stem Cells 28:1231–1242. 10.1002/stem.449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theus MH, Ricard J, Glass SJ, Travieso LG, Liebl DJ (2014) EphrinB3 blocks EphB3 dependence receptor functions to prevent cell death following traumatic brain injury. Cell Death Dis 5:e1207. 10.1038/cddis.2014.165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas S, Prins ML, Samii M, Hovda DA (2000) Cerebral metabolic response to traumatic brain injury sustained early in development: a 2-deoxy-D-glucose autoradiographic study. J Neurotrauma 17:649–665. 10.1089/089771500415409 [DOI] [PubMed] [Google Scholar]
- Thurman DJ. (2016) The epidemiology of traumatic brain injury in children and youths: a review of research since 1990. J Child Neurol 31:20–27. 10.1177/0883073814544363 [DOI] [PubMed] [Google Scholar]
- Tong KA, Ashwal S, Holshouser BA, Shutter LA, Herigault G, Haacke EM, Kido DK (2003) Hemorrhagic shearing lesions in children and adolescents with posttraumatic diffuse axonal injury: improved detection and initial results. Radiology 227:332–339. 10.1148/radiol.2272020176 [DOI] [PubMed] [Google Scholar]
- van der Horn HJ, de Haan S, Spikman JM, de Groot JC, van der Naalt J (2018) Clinical relevance of microhemorrhagic lesions in subacute mild traumatic brain injury. Brain Imaging Behav 12:912–916. 10.1007/s11682-017-9743-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner S, Gardner H (2000) Modes of regulation of laminin-5 production by rat astrocytes. Neurosci Lett 284:105–108. 10.1016/S0304-3940(00)00987-3 [DOI] [PubMed] [Google Scholar]
- Wälchli T, Mateos JM, Weinman O, Babic D, Regli L, Hoerstrup SP, Gerhardt H, Schwab ME, Vogel J (2015) Quantitative assessment of angiogenesis, perfused blood vessels and endothelial tip cells in the postnatal mouse brain. Nat Protoc 10:53–74. 10.1038/nprot.2015.002 [DOI] [PubMed] [Google Scholar]
- Wang JD, Khafagy el-S, Khanafer K, Takayama S, ElSayed ME (2016a) Organization of endothelial cells, pericytes, and astrocytes into a 3D microfluidic in vitro model of the blood-brain barrier. Mol Pharm 13:895–906. 10.1021/acs.molpharmaceut.5b00805 [DOI] [PubMed] [Google Scholar]
- Wang Y, Fan X, Tang T, Fan R, Zhang C, Huang Z, Peng W, Gan P, Xiong X, Huang W, Huang X (2016b) Rhein and rhubarb similarly protect the blood-brain barrier after experimental traumatic brain injury via gp91(phox) subunit of NADPH oxidase/ROS/ERK/MMP-9 signaling pathway. Sci Rep 6:37098. 10.1038/srep37098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeller K, Vogel J, Kuschinsky W (1996) Postnatal distribution of Glut1 glucose transporter and relative capillary density in blood-brain barrier structures and circumventricular organs during development. Brain Res Dev Brain Res 91:200–208. 10.1016/0165-3806(95)00177-8 [DOI] [PubMed] [Google Scholar]
- Zhang J, Wei L, Sun WL, Wang L, Zhang WJ, You H (2014) Radiation-induced endothelial cell loss and reduction of the relative magnitude of the blood flow in the rat spinal cord. Brain Res 1583:193–200. 10.1016/j.brainres.2014.06.014 [DOI] [PubMed] [Google Scholar]
- Zhao Z, Loane DJ, Murray MG 2nd, Stoica BA, Faden AI (2012) Comparing the predictive value of multiple cognitive, affective, and motor tasks after rodent traumatic brain injury. J Neurotrauma 29:2475–2489. 10.1089/neu.2012.2511 [DOI] [PMC free article] [PubMed] [Google Scholar]