

Abstract
Abnormalities in α-synuclein are implicated in the pathogenesis of Parkinson's disease (PD). Because α-synuclein is highly concentrated within presynaptic terminals, presynaptic dysfunction has been proposed as a potential pathogenic mechanism. Here, we report novel, tau-dependent, postsynaptic deficits caused by A53T mutant α-synuclein, which is linked to familial PD. We analyzed synaptic activity in hippocampal slices and cultured hippocampal neurons from transgenic mice of either sex expressing human WT, A53T, and A30P α-synuclein. Increased α-synuclein expression leads to decreased spontaneous synaptic vesicle release regardless of genotype. However, only those neurons expressing A53T α-synuclein exhibit postsynaptic dysfunction, including decreased miniature postsynaptic current amplitude and decreased AMPA to NMDA receptor current ratio. We also found that long-term potentiation and spatial learning were impaired by A53T α-synuclein expression. Mechanistically, postsynaptic dysfunction requires glycogen synthase kinase 3β-mediated tau phosphorylation, tau mislocalization to dendritic spines, and calcineurin-dependent AMPA receptor internalization. Previous studies reveal that human A53T α-synuclein has a high aggregation potential, which may explain the mutation's unique capacity to induce postsynaptic deficits. However, patients with sporadic PD with severe tau pathology are also more likely to have early onset cognitive decline. Our results here show a novel, functional role for tau: mediating the effects of α-synuclein on postsynaptic signaling. Therefore, the unraveled tau-mediated signaling cascade may contribute to the pathogenesis of dementia in A53T α-synuclein-linked familial PD cases, as well as some subgroups of PD cases with extensive tau pathology.
SIGNIFICANCE STATEMENT Here, we report mutation-specific postsynaptic deficits that are caused by A53T mutant α-synuclein, which is linked to familial Parkinson's disease (PD). The overexpression of WT, A53T, or A30P human α-synuclein leads to decreased spontaneous synaptic vesicle release. However, only those neurons expressing A53T α-synuclein exhibit tau phosphorylation-dependent postsynaptic dysfunction, which is characterized by decreased miniature postsynaptic current amplitude and decreased AMPA to NMDA receptor current ratio. The mutation-specific postsynaptic effects caused by human A53T α-synuclein will help us better understand the neurobiological basis of this specific form of familial PD. The differential effects of exogenous human WT, A53T, A30P, and E46K α-synuclein on glutamatergic synaptic responses will help to explain the clinical heterogeneity of sporadic and familial PD.
Keywords: AMPA receptor, frontotemporal dementia, Parkinson's disease, synaptic plasticity, synuclein, tau
Introduction
Parkinson's disease (PD) is the second most common late-onset neurodegenerative disease. It is characterized by both motor symptoms and the convergence of α-synuclein (αS), tau, and amyloid-β pathology (Irwin et al., 2013). Sporadic PD is clinically heterogeneous. Statistical cluster analysis suggests the existence of distinct subgroups of patients in the early stages of idiopathic PD, perhaps due to different etiopathologies (Lewis et al., 2005; Kehagia et al., 2010; van Balkom et al., 2016). Genetic abnormalities including αS gene (SNCA) amplification (Singleton et al., 2003; Chartier-Harlin et al., 2004; Ibáñez et al., 2004), as well as A53T (Polymeropoulos et al., 1997), A30P (Krüger et al., 2001), and E46K (Zarranz et al., 2004) αS point mutations, are linked to familial PD. These inherited forms of PD are also heterogeneous. Each features a different time of onset, clinical presentation, and histopathology (Petrucci et al., 2016). Particularly, tau and αS pathologies frequently coexist in the Contursi kindred who carry the A53T αS point mutation (Duda et al., 2002). Here, we investigated whether A53T mutant αS activates additional signaling pathways that are distinct from those activated by WT αS and other mutants.
αS is a cytosolic protein that is enriched in the presynaptic terminals of neurons and can associate with the plasma membrane. The role of αS in presynaptic function has been intensely studied over the past 25 years (Burré, 2015). αS promotes SNARE assembly (Burré et al., 2010) and interacts with multiple presynaptic proteins including SNARE (Burré et al., 2010), synapsin III (Zaltieri et al., 2015), and VMAT2 (Guo et al., 2008). Under physiological conditions, αS has been implicated as both a negative and positive regulator of synaptic transmission. αS and other synuclein homologs support synaptic function and the release of synaptic vesicles by chaperoning the SNARE complex in presynaptic terminals (Burré et al., 2014; Burré, 2015). Conversely, loss of αS function has been linked to enhanced vesicle release in dopaminergic terminals (Abeliovich et al., 2000; Nemani et al., 2010), suggesting that αS normally functions to suppress vesicle release. Consistent with this view, overexpression of WT or mutant human αS suppresses presynaptic transmission by depleting the recycling and readily releasable pools of synaptic vesicles (Nemani et al., 2010). Because both mutant and WT human αS as well as homologous β-synuclein have similar effects on vesicular release (Nemani et al., 2010), it is difficult to attribute the clinical heterogeneity of PD to effects of αS on presynaptic function alone. To identify the cellular mechanisms underlying the clinical and pathological diversity of PD, we have compared changes in the synaptic function of neurons expressing multiple αS variants. We found that A53T αS induces postsynaptic deficits that require glycogen synthase kinase 3β (GSK3β)-dependent tau missorting to dendritic spines and calcineurin-dependent loss of postsynaptic surface AMPA receptors.
Materials and Methods
Animals.
The four transgenic mouse lines used in the present study are listed in Figure 1A. The method for the generation of transgenic (Tg) mice that express human WT (line I2-2), A53T mutant (lines G2-3 and H5), and A30P mutant (line O2) αS under the control of a mouse prion protein promoter have been described previously (Lee et al., 2002). The mice from line G2-3 develop progressive neurological dysfunction at 12–16 months of age, which rapidly progress to end stage paralysis within 14–21 d following initial onset of symptoms (Lee et al., 2002). For this study, Tg mice were bred to establish neuronal cultures, acute slice electrophysiology, biochemical analysis, and behavioral analysis. Mouse genotype was confirmed by Northern blot and RT-PCR analysis as described previously (Lee et al., 2002). For all experiments, data were collected from animals of both sexes. All experimental protocols involving mice and rats were in strict adherence to the National Institutes of Health's Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee at the University of Minnesota.
Figure 1.

A53T αS causes mutation-specific postsynaptic deficits in AMPAR signaling, whereas overexpression of human αS variants, regardless of genotype, causes presynaptic suppression in acute hippocampal slices. A, List of transgenic mice used in the present study. B, C, Immunoblots and quantification of human αS (HuSyn-1 antibody) and total (mouse and human) αS (BD Biosciences antibody 610787) in hippocampal lysates from 4- to 6-month-old MoPrP-Hu-αS transgenic and TgNg mice. Each lane represents an individual animal. αS levels are normalized to tubulin. I2-2 and H5 have comparable expression levels but have lower expression levels than G2-3 and O2. D, Input–output relationships of EPSCs (TgNg, n = 9; I2-2, n = 10; H5, n = 11; G2-3, n = 9; O2, n = 9; two-way ANOVA, F = 0.29, p = 1.0). E, Paired-pulse ratio induced by two consecutive stimuli delivered at different time intervals (TgNg, n = 15, I2-2, n = 9, H5, n = 11, G2-3, n = 10, O2, n = 11); two-way ANOVA, F = 0.56, p = 0.96. Representative traces are illustrated as insets, scale bars: 20 pA, 30 ms. F, Synaptic fatigue induced by 15 consecutive stimuli at 25 ms interpulse intervals (TgNg, n = 13; I2-2, n = 8; H5, n = 10; G2-3, n = 13; O2, n = 9; two-way ANOVA, F = 0.57, p = 0.99). Representative traces are illustrated as insets. Scale bars, 40 pA, 70 ms. For D–F, two-way ANOVA with Fisher LSD post hoc analysis. G, Representative AMPA and NMDA receptor-mediated synaptic response traces and AMPA to NMDA receptor current ratio (TgNg, n = 11; I2-2, n = 7; H5, n = 10; G2-3, n = 8; O2, n = 11). Scale bars, 20 pA, 100 ms. Kruskal–Wallis test with Dunn's method post hoc analysis H = 21.53, df = 4; H5. H–J, Representative traces, mean amplitude, and mean frequency of mEPSCs obtained in the presence of TTX (1 μm) (TgNg, n = 7; I2-2, n = 10; H5, n = 11; G2-3, n = 8; O2, n = 10). Scale bar, 5 pA, 2 s. One-way ANOVA with Fisher LSD post hoc analysis, F = 8.23, p < 0.001 (amplitude); F = 5.54, p = 0.001 (frequency). All data are expressed as mean ± SEM; *p < 0.05, **p < 0.01, and ***p < 0.001 compared with TgNg, #p < 0.05 and ##p < 0.01 compared with I2-2. TgNg control was taken from littermates of I2-2 mice. For all, n-values represent neurons, at least 3 3- to 6-month-old mice were used for every experimental condition.
Biochemistry: gel electrophoresis and immunoblotting.
Hippocampi from Tg mice were suspended and mechanically homogenized in 10 volumes of ice-cold TNE buffer (50 mm Tris-HCl, 150 mm NaCl, 5 mm EDTA, complete mini protease inhibitor cocktail, and phosphatase inhibitor cocktails 2 and 3 inhibitors, 1:100, Sigma-Aldrich) in a polystyrene tube. Homogenized tissue was aliquoted and diluted with equal volumes of ice-cold Complete TNE (TNE, 1% SDS, 0.5% Nonidet P-40, 0.5% sodium deoxycholate). Estimation of protein concentration for protein correction and dilution was performed using the bicinchoninic acid (BCA) assay (Thermo Fisher Scientific). Concentration-corrected protein samples were diluted in reducing sample buffer (Boston BioProducts), electrophoresed on 4–20% Criterion TGX gels (Bio-Rad), and transferred onto 0.45 μm nitrocellulose membranes (GE Healthcare). Membranes were probed with primary antibodies of total αS (BD Biosciences, 610787), human αS (HuSyn1; Lee et al., 2002), and α-tubulin (Abcam, ab4074) and visualized using enhanced chemiluminescent reagents (Thermo Fisher Scientific) via the ImageQuant LAS 4000 detection system (GE Healthcare). Densitometry analysis was performed using ImageQuant TL version 8.1 software (GE Healthcare).
Acute slice electrophysiology.
We obtained acute coronal hippocampal slices (350 μm thick) from 3- to 6-month-old nontransgenic (TgNg) and Tg mice from lines I2-2 (WT), H5 (A53T), G2-3 (A53T), and O2 (A30P). We kept slices in ice-cold artificial CSF (ACSF) containing the following (in mm): 124 NaCl, 5 KCl, 1.25 NaH2PO4, 2 MgSO4, 26 NaHCO3, 2 CaCl2, and 10 glucose, gassed with 95% O2/5% CO2, pH 7.3–7.4. Slices were incubated in ACSF at room temperature for at least 1 h before use and then transferred to an immersion recording chamber, superfused at 2 ml/min with gassed ACSF, and visualized under an Olympus BX50WI microscope. Picrotoxin (50 μm) and CGP54626 (1 μm) were added to the solution to block the GABAa and GABAb receptors, respectively. Whole-cell electrophysiological recordings were obtained from CA1 pyramidal neurons. Patch electrodes (3–10 MΩ) were filled with internal solution containing the following (in mm): 117 cesium-gluconate, 20 HEPES, 0.4 EGTA, 2.8 NaCl, 5 TEA-Cl, 2 ATP-Mg2+, and 0.3 GTP-Na+, pH 7.3. Recordings were obtained with PC-ONE amplifiers (Dagan Instruments). Membrane potential was held at −70 mV. Signals were filtered at 1 kHz and acquired at 10 kHz sampling rate and fed to a Pentium-based PC through a DigiData 1440A interface board. pCLAMP version 10.4 (Axon Instruments) software was used for stimulus generation, data display, acquisition, and storage. To record evoked EPSCs, theta capillaries filled with ACSF were used for bipolar stimulation and placed in the stratum radiatum to stimulate Schaffer collaterals (SCs). Input–output curves of EPSCs were made by increasing stimulus intensities from 20 to 80 μA. Paired pulses (2 ms duration) were applied in the SC with 25, 50, 75, 100, 200, 300, and 500 ms interpulse intervals and the paired-pulse ratio was calculated as follows: second EPSC/first EPSC. Synaptic fatigue was assessed with 15 consecutive stimuli with a 25 ms interval. AMPA currents were obtained at a holding potential of −70 mV and NMDA currents at +30 mV. To ascertain the AMPA to NMDA receptor current ratio, we measured the NMDA component 50 ms after the stimulus, when the AMPA component had decayed. For LTP induction, a tetanic stimulation (4 trains at 100 Hz for 1 s; 30 s intervals) was applied in the SC. EPSC amplitude was normalized to 10 min of baseline recording. After LTP induction, neurons were recorded for 45 min. The presence of LTP was determined by comparing the last 5 min of baseline with the last 5 min of recording. For miniature EPSC recordings, TTX (1 μm) was also included in the solution. Normality was verified with a Kolmogorov–Smirnov test in analyses of cumulative curves and groups were compared using a one-way ANOVA with Fisher's LSD post hoc analysis. When data did not meet normality, a one-way Kruskal–Wallis test with Dunn's method post hoc was applied.
Plasmid constructs.
All eGFP, tau, αS, and DsRed constructs were expressed in the pRK5 vector and driven by a cytomegalovirus promoter (Clontech). All tau and αS constructs were tagged with eGFP on the N terminus. The WT tau construct encodes human four-repeat tau lacking the N-terminal sequences (0N4R) and contained exons 1, 4 and 5, 7, and 9–13, intron 13, and exon 14. Using WT tau as a template, we used QuikChange site-directed mutagenesis (Agilent Technologies) to generate two tau constructs termed AP tau and E14 tau. We mutated all 14 S/P or T/P amino acid residues (T111, T153, T175, T181, S199, S202, T205, T212, T217, T231, S235, S396, S404, and S422) to alanine (AP) or glutamate (E14). Numbering is based on the longest (2N4R) 441-aa adult brain isoform of human tau. All tau constructs have been characterized in our previous study (Hoover et al., 2010). We used site-directed mutagenesis to generate A30P, E46K, and A53T αS from WT αS. All sequences were confirmed with Sanger sequencing (UMN Genomics, Minneapolis).
High-density neuronal cultures and neuronal transfection.
A 25 mm glass polylysine-coated coverslip (thickness, 0.08 mm) was glued to the bottom of a 35 mm culture dish with a 22 mm hole using silicone sealant as described previously (Lin et al., 2004). Dissociated neuronal cultures from mouse and rat hippocampi at postnatal day 1 (P1) were prepared as described previously (Hoover et al., 2010). Briefly, hippocampi were dissected and stored in ice-cold Earl's balanced medium supplemented with 1 mm d-glucose. Rat hippocampal neurons from each litter were pooled before plating, whereas mouse hippocampal neurons were separated by pup before plating. Neurons were plated onto prepared 35 mm culture dishes at a density of 1 × 106 cells per dish. The age of cultured neurons was counted from the day of plating as 1 d in vitro (DIV). All experiments were performed on neurons from at least three independent cultures. Neurons at 6–8 DIV were transfected with appropriate plasmids using the standard calcium phosphate precipitation method as described previously (Liao et al., 2005). After transfection, neurons were placed in a tissue culture incubator (37°C, 5% CO2) and allowed to mature and develop until 3 weeks in vitro, a time at which neurons express high numbers of dendritic spines with mature morphologies. Mouse culture genotype was ascertained by Northern blot and RT-PCR analysis of ex vivo tail clippings (see above).
Low-density neuronal cultures.
To detect the distribution of endogenous synaptic proteins with high resolution, low-density neuronal cultures were prepared as described previously with some modifications (Lin et al., 2009). Dissociated neuronal cultures from Tg mouse hippocampi at P1–P2 were plated into 12-well culture plates at a density of 50,000–100,000 cells per well. Each well contained a polylysine-coated 12 mm glass coverslip. The 12 mm coverslips with 7 DIV low-density cultured neurons were transferred to high-density neuronal cultures in 60 mm dishes (4 coverslips per dish to encourage survival).
In vitro electrophysiology.
Miniature EPSCs (mEPSCs) were recorded from cultured dissociated rat hippocampal neurons at 21–25 DIV with a glass pipette (resistance of ∼5 MΩ) at holding potentials of −55 mV and filtered at 1 kHz with an output gain, α, of 0.5 (mouse culture) and 1 (rat culture) as described previously (Miller et al., 2014). Briefly, neurons were bathed in ACSF at room temperature (25°C) with 100 μm APV (an NMDAR antagonist), 1 μm TTX (a sodium channel blocker), and 100 μm picrotoxin (a GABAa receptor antagonist) gassed with 95% O2–5% CO2. The ACSF contained the following (in mm): 119 NaCl, 2.5 KCl, 5.0 CaCl2, 2.5 MgCl2, 26.2 NaHCO3, 1 NaH2PO4, and 11 glucose. The internal solution in the patch pipette contained the following (in mm): 100 cesium gluconate, 0.2 EGTA, 0.5 MgCl2, 2 ATP, 0.3 GTP, and 40 HEPES, pH 7.2 with cesium hydroxide. mEPSC traces were recorded using an Axopatch 200B amplifier and pClamp 11 (Molecular Devices). Recordings ranged from 5–20 min and stable traces longer than 2 min in duration were analyzed. All mEPSCs >3 pA were manually counted with MiniAnalysis (Synaptosoft). Each mEPSC event was visually inspected and only events with a distinctly fast-rising phase and a slow-decaying phase were accepted. Relative cumulative frequencies were derived from individual events and the averaged parameters from each neuron were treated as single samples in any further statistical analyses.
In vitro neuronal imaging and analysis.
The 35 mm culture dishes fit tightly in a custom holding chamber on a fixed platform above an inverted Nikon microscope sitting on an X–Y translation stage (Burleigh). A 60× oil lens was used for all imaging experiments. Original images were 157.3 μm wide (x-axis) and 117.5 μm tall (y-axis). The z-axis was composed of 15 images taken at 0.5 μm intervals. All digital images were analyzed with the MetaMorph Imaging System (Universal Imaging). Unless stated otherwise, live image stacks were processed by 2D deconvolution of nearest planes and averaged into a single image. Dendritic protrusions, with an expanded head that was >50% wider than its neck, were defined as spines. The spines from each dendrite were manually counted and normalized per 100 μm of dendritic length.
Immunocytochemistry in fixed tissues.
Cultured neurons were fixed and permeabilized successively with 4% paraformaldehyde, 100% methanol, and 0.2% Triton X-100 (Lin et al., 2009). For all immunocytochemical staining, primary antibodies were diluted at 1:50 or 1:100 in 10% donkey serum in PBS and rhodamine (red)- or FITC (green)-labeled secondary antibodies were diluted at 1:100 or 1:200, respectively. Mouse anti-synaptophysin (Thermo Fisher Scientific) antibodies (1:100 dilution) were used to detect presynaptic terminals. Commercial antibodies against PSD-95 were used as a postsynaptic marker to stain dendritic spines (rabbit polyclonal, Invitrogen; mouse monoclonal, Millipore; 1:100 dilution) as described previously (Lin et al., 2009). The rabbit polyclonal antibodies against the N terminus of GluA1 subunits were generous gifts from Dr. Richard Huganir (Johns Hopkins University Medical School). The fixed neurons were incubated with primary antibodies at 4°C overnight and subsequently incubated with secondary antibodies for 1–2 h at room temperature (PSD-95) or a 37°C incubator (synaptophysin). The fluorescent images of antibody staining and transfected exogenous proteins (eGFP-αS or eGFP alone) were taken with an inverted Nikon microscope (see “In vitro neuronal imaging and analysis” section above). In Figure 4, A and B, the number of synaptophysin clusters and their colocalization with boutons of neurons expressing eGFP or eGFP-labeled αS were automatically counted using ImageJ software (https://imagej.nih.gov/ij/). For DAPI-stained neurons, we incubated paraformaldehyde-fixed neurons in 10 mm DAPI dilactate (Thermo Fisher Scientific) at 23°C for 5 min before imaging. DAPI-stained nuclei were manually counted from wide-field photomicrographs.
Figure 4.

A53T αS-induced postsynaptic deficits are cell autonomous. A, Wide-field fluorescence photomicrographs from cultured rat hippocampal DAPI-stained neurons expressing eGFP-tagged WT and A53T αS plasmids via calcium-phosphate transfection. The percentage of untransfected cells was tabulated. B, Photomicrographs of fixed neurons that had been transfected with eGFP, eGFP-WT αS, or eGFP-A53T αS plasmids (left) and subsequently stained with a mouse anti-synaptophysin antibody (middle; with overlay on the right). Axons were visually traced and defined as thin, long neurites emerging from the soma with occasional perpendicular branch points. Arrows represent nonoverlapping synaptophysin clusters; arrowheads point to synaptophysin-filled, eGFP-expressing synaptic boutons. The percentage of synaptophysin clusters free of exogenous αS expression was calculated. C, Whole-cell AMPAR mEPSCs were recorded from cultured rat hippocampal neurons transfected with eGFP alone or eGFP-fused αS species. Scale bar, 10 pA, 100 ms. D, Relative cumulative frequency of mEPSC amplitudes. Kolmogorov–Smirnov test, D = 0.39; ***p < 0.0001. E, F, Mean mEPSC amplitudes and frequencies. One-way ANOVA with Bonferroni post hoc analysis, F(4,32) = 3.65; *p = 0.012. All data are expressed as mean ± SEM.
Barnes maze learning and memory test.
Spatial learning and memory were evaluated using the Barnes maze as described previously with some modifications (http://www.nature.com/protocolexchange/protocols/349). A Barnes maze with a video-tracking system was purchased from San Diego Instrument (http://www.sandiegoinstruments.com/). ANY-maze video-tracking software (Stoelting) was used for behavioral analysis. Briefly, the maze consists of 20 exploration holes with only one hole leading to a recessed escape box during task acquisition on an elevated platform (see Fig. 2). In each trial, an 11- to 12-month-old mouse was first placed under a box in the center of the maze for ∼15 s and then allowed to freely explore the maze to search for the escape hole (target) for 3 min after the removal of the box. An escape from the maze was defined as the movement of the mouse completely through the escape hole into the recessed box. In the acquisition period (learning phase), the mouse underwent 4 trials per day with an intertrial interval of 25–30 min for 4 consecutive days. The retention of memory (probe test) was performed 24 h following the fourth day of acquisition by covering all holes and occupancy plots as the exploration pattern for each group of mice was determined. Retention of memory was measured by quantifying the time that the mouse spent in each zone and the distance from the animal to the position of the removed escape hole (the target) during this 90 s probe test.
Figure 2.
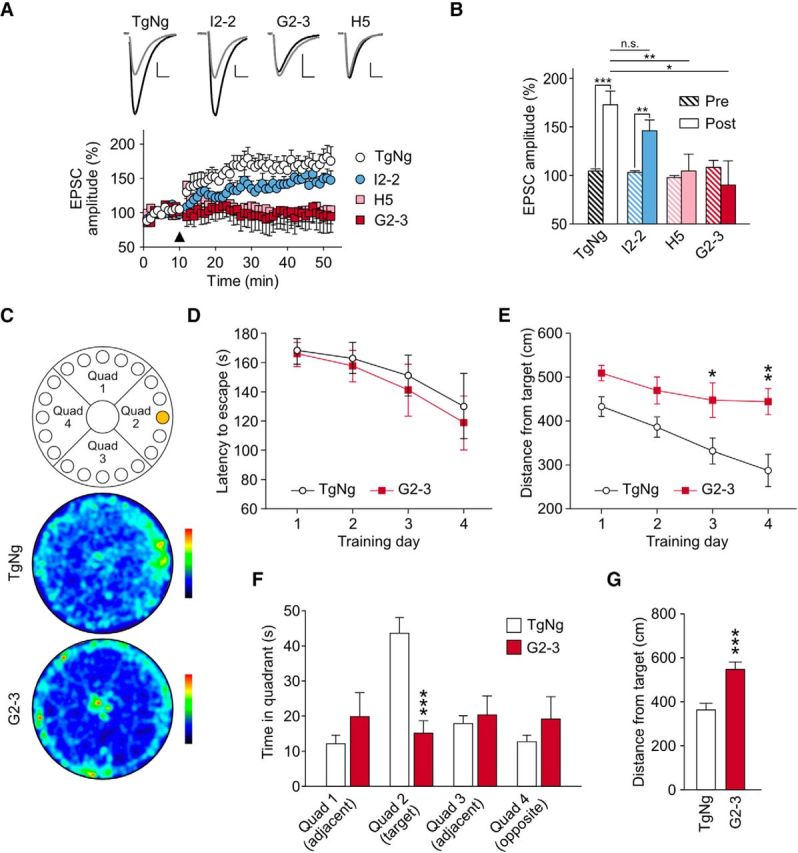
A53T αS causes deficits in LTP and spatial learning and memory. A, Top, Representative EPSC traces before (gray) and after (black) a high-frequency stimulation (HFS) of the Schaffer collaterals recorded from TgNg, I2-2, H5, and G2-3 mice. Scale bars, 10 pA, 15 ms. Bottom, EPSC amplitude versus time obtained from TgNg, I2-2, H5, and G2-3 mice (n = 9, n = 9, n = 9, and n = 8, respectively). Arrowhead indicates HFS application. TgNg controls were taken from I2-2 littermates. B, EPSC amplitude before and 45 min after stimulation in the different mouse models. Within-group analysis: two-tailed paired t test: t/df/P = −5.07/8/0.0010; −3.56/0.0074; −0.41/8/0.70; 0.39/6/0.71; for TgNg, I2-2, H5, and G2-3, respectively. Between-group analysis: one-way ANOVA with a Fisher LSD post hoc analysis F = 3.54, p = 0.027. At least 3 3- to 6-month-old mice were used for every experimental condition. n-values represent neurons. C, Diagram of the Barnes circular maze and representative occupancy plots from TgNg and G2-3 probe trials (color gradient bar plot, black: least occupied region, red: highest occupancy). D, Latency time to escape the maze during 4 consecutive training days. Two-way ANOVA, F(3,51) = 0.093. E, Mean distance from target measured on each training day. Two-way ANOVA with Bonferroni post hoc analysis, F(3,51) = 1.056; *p = 0.030, **p = 0.0015. F, Mean time 11- to 12-month-old TgNg and G2-3 animals spent in each quadrant of the maze during the probe trial. Two-way ANOVA with Bonferroni post hoc analysis, F(3,51) = 5.34; ***p = 0.0002. G, Average distance between the animals and the target during the probe trial. G2-3 mice were significantly more distant from the target than their TgNg littermates (TgNg, n = 9; G2-3, n = 10). Analyzed by Student's t test, t = 4.50, df = 17; ***p = 0.0003. All data are expressed as mean ± SEM.
Flow cytometry.
Rat hippocampal neurons at 21 DIV transfected with eGFP-tagged exogenous αS species were suspended, stained for viability, and analyzed via flow cytometry (see Fig. 5). Briefly, neurons were washed in 37°C PBS and then incubated for 6 min in 0.05% Trypsin/EDTA (Thermo Fisher Scientific) at 25°C with gentle shaking to detach cells. Suspended cells were manually triturated and MEM + 10% fetal bovine serum (FBS) 1× GlutaMAX (Thermo Fisher Scientific) was added to inactivate trypsin. Cells were pelleted by centrifugation (1000 × g, 4°C, 3 min), resuspended in PBS + 2% FBS, and then passed through a 70 μm strainer (Fisher Scientific) and reserved at 4°C. Cells were pelleted as before, washed once with 1 ml of PBS, and then resuspended in 50:1 staining buffer (BioLegend, 420201) and Ghost Dye Red 780 (Tonbo Biosciences). Cells were incubated on ice for 30 min, pelleted, and washed twice with staining buffer. Finally, cells were resuspended in PBS + 0.1% bovine serum albumin, passed through a 35 μm strainer and analyzed on a BD LSR II Flow Cytometer (BD Biosciences). Data were analyzed in FlowJo version 7.6.5 software, with gating parameters represented in Figure 5.
Figure 5.
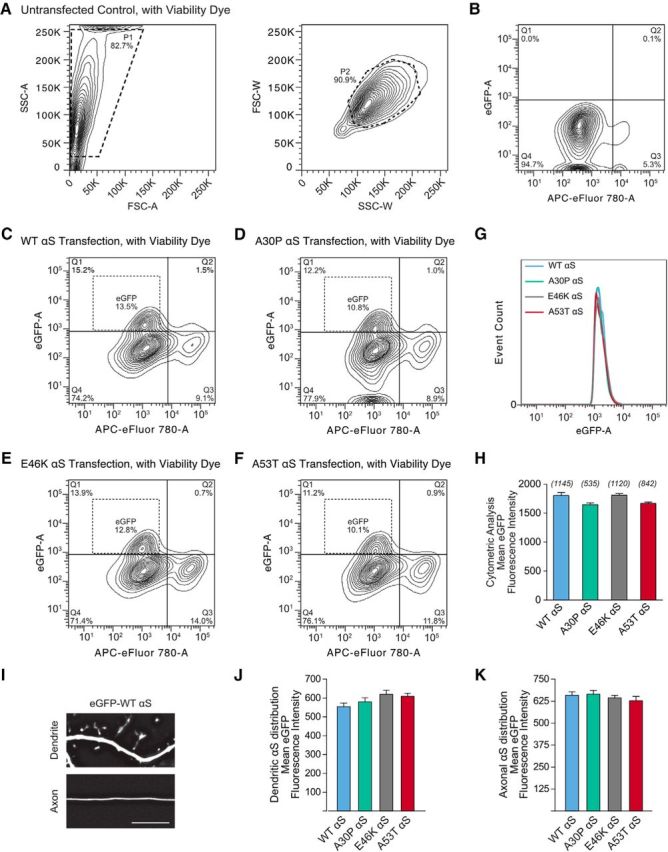
Stable and consistent expression of eGFP-fused human αS across constructs. A, Contour plots of flow cytometry gating parameters from the nontransfected group. B, Contour plots of two populations of cells in the nontransfected group: eGFP-negative, living cells (Q4) and eGFP-negative, dead cells (Q3). C–F, Contour plots of neurons transfected with eGFP-fused WT, A30P, E46K, and A53T mutant human αS respectively. A small population of cells emerged that is both living and eGFP-positive (Q1). G, Histogram comparison of fluorescence in eGFP cell population. H, Mean eGFP fluorescence intensity from flow cytometer detection. Data were analyzed by one-way ANOVA, F(3,3638) = 2.38. n-values are eGFP-positive events and shown parenthetically. There was no difference between the cellular distributions of αS variants. I, Deconvoluted example micrographs of an axon and a dendrite of a neuron expressing eGFP-WT αS. Scale bar, 10 μm. J, K, Fifteen-image Z-series of dendrites and axons were analyzed to estimate cellular distribution of αS using linear analysis perpendicular to the shaft. Total area under the curve of dendritic or axonal fluorescence in each image series was averaged and normalized to background fluorescence. One-way ANOVA, F(3,25) = 0.45 (dendrite), F(3,14) = 0.90 (axon); n > 4. All data are expressed as mean ± SEM.
Pharmacology and common reagents.
All common reagents used in this report were purchased from Sigma-Aldrich-Millipore unless otherwise noted. CHIR-99021 and FK506 were purchased from Sigma-Aldrich. Both drugs were prepared as stock solutions (CHIR-99021 5 mm and FK506 1 mm) in fresh DMSO and stored at −20°C in aliquots. Either drug or DMSO vehicle was applied to cultured cells at 16 DIV with appropriate dilutions 5 d before imaging or electrophysiology experiments.
Experimental design and statistical analysis.
All statistics were performed in Prism 6 (GraphPad) or Origin (OriginLab) software. Except where discussed above, we used one- and two-way ANOVA for univariate and two-variable analysis, respectively. If ANOVA revealed significant variance between all groups, then post hoc analysis was performed using Bonferroni's analysis adjusted for multiple groups. Univariate cumulative frequency distributions were compared using the unmodified Kolmogorov–Smirnov goodness of fit test. For all, statistical significance was set at α = 0.05. Data representations are described in figure legends.
Results
Human A53T αS induces mutation-specific synaptic deficits and spatial memory dysfunction
Although most PD cases are sporadic, familial PD can be caused by the duplication (Chartier-Harlin et al., 2004; Ibáñez et al., 2004) or triplication (Singleton et al., 2003) of the WT αS gene (SNCA) as well as point mutations, including the A53T (Polymeropoulos et al., 1997) or A30P mutation (Krüger et al., 2001). We used Tg mouse lines expressing WT and mutant αS at various levels to test effects of these genetic mutations on synaptic responses. We first used Western blots to determine the expression levels of both mouse and human αS in four mouse lines: I2-2 mice expressing WT human αS, H5 mice expressing A53T human αS at a lower level, G2-3 mice expressing A53T human αS at a higher level, and O2 mice expressing A30P human αS (Fig. 1A–C, Lee et al., 2002). Transgenic negative (TgNg) littermates of I2-2 mice were used for control.
Next, we performed whole-cell patch-clamp recordings of CA1 pyramidal neurons in acute hippocampal slices from 3- to 6-month-old mice from each line (Fig. 1D–J). Analysis of evoked synaptic responses showed that the input–output curve of all Tg neurons was comparable to that of TgNg neurons (Fig. 1D) and there was no significant difference in paired-pulse facilitations (Fig. 1E) and synaptic fatigue (Fig. 1F), suggesting that there was no overt degeneration. However, there was a significant reduction in AMPA to NMDA receptor current ratios in hippocampi of H5 and G2-3 mice (Fig. 1G). Overexpression of either WT or A30P αS had no significant effect on the AMPA to NMDA receptor current ratios (Fig. 1G). To further characterize the presynaptic and postsynaptic changes, we recorded mEPSCs in acute slices (Fig. 1I,J). Consistent with the potential loss of AMPA receptor response, expression of A53T αS, but not WT or A30P αS, significantly decreased the amplitude of AMPA receptor-mediated mEPSCs recorded in hippocampal slices (Fig. 1I). These results indicate that A53T αS expression is unique in its ability to produce postsynaptic deficits. By contrast, the expression of all three forms of αS (WT, A53T, and A30P) significantly decreased the frequency of mEPSCs (Fig. 1J), suggesting a decrease in the release probability of presynaptic vesicles. The specific postsynaptic deficits caused by A53T αS expression and nonspecific presynaptic deficits associated with all αS variants imply that presynaptic and postsynaptic deficits are mediated through two separate intracellular mechanisms.
Synaptic plasticity such as LTP is known to increase the synaptic recruitment of AMPA receptors to dendritic spines (Liao et al., 1995; Malinow and Malenka, 2002). Therefore, the results shown in Figure 1 may be associated with deficits in LTP and memory. We induced LTP in acute hippocampal slice from 3- to 6-month-old mice expressing WT αS or A53T αS at two expression levels (Fig. 2). Compared with TgNg littermates, lower or higher levels of A53T αS expression suppressed LTP, whereas the expression of WT αS had no significant effect (Fig. 2A,B). Consistent with prior studies using another Tg A53T αS mouse line, M83) (Paumier et al., 2013), we also found that expression of A53T αS impaired spatial memory at 11–12 months of age (Fig. 2C–G).
The above effects observed in acute hippocampal slices could result from differences in neural circuit development rather than neuron-autonomous differences in postsynaptic responses. Therefore, we recorded glutamatergic mEPSCs in cultured hippocampal neurons from TgNg, I2-2, H5, G2-3, and O2 mice (Fig. 3A). We found that the amplitude of mEPSCs was significantly decreased in neurons from the H5 and G2-3 neurons but was unchanged in I2-2 and O2 neurons (Fig. 3B,C). By contrast, the frequency of mEPSCs was significantly decreased in I2-2, H5, G2-3, and O2 neurons (Fig. 3D), confirming that presynaptic deficits are not mutation specific and are induced by hyperexpression of any of the synuclein species (Nemani et al., 2010). Furthermore, the reduced mEPSC frequency and amplitude is not due to loss of postsynaptic structures because there was no alteration in dendritic spine density (Fig. 3E,F). Again, the human αS expression level was similar between the I2-2 and H5 mouse lines and between the G2-3 and O2 mouse lines (Fig. 1A). We therefore conclude that differences in the expression level cannot explain the postsynaptic deficits induced by A53T αS expression.
Figure 3.

A53T αS-induced postsynaptic deficits are independent of expression levels. A, Representative traces of events represented in C. Scale bars, 5 pA, 2 s. B, Relative cumulative frequency of whole-cell mEPSC amplitudes from cultured transgenic mouse hippocampal neurons. Kolmogorov–Smirnov test; D = 0.30, *p = 0.048; D = 0.34, **p = 0.0086; D = 0.43, ***p = 0.0002. C, D, Amplitude and frequency of mEPSCs. One-way ANOVA with Bonferroni post hoc analysis. For C: F(4,47) = 4.48; H5:*p = 0.014; G2-3: *p = 0.026; **p = 0.0036. For D: F(4,47) = 3.54; H5: p = 0.032; G2-3: p = 0.031; O2: p = 0.016. E, Representative images of eGFP-illuminated dendrites and spines from cultured Tg mouse hippocampal neurons. Scale bar, 5 μm. F, Spine density of neurons represented in E. One-way ANOVA, F(4,47) = 2.42. Data are expressed mean ± SEM. n-values are represented parenthetically.
Human A53T αS induces postsynaptic deficits in a cell-autonomous manner
In neuronal cultures established from Tg mice (Fig. 3), αS is present in presynaptic structures, which could lead to secondary postsynaptic deficits. To rule out a presynaptic influence on postsynaptic transmission, we used calcium phosphate neuronal transfection to express exogenous plasmid αS DNA in a small proportion of cells (<5%; Fig. 4A) in rat primary neuronal hippocampal cultures. In this model, neurons expressing the transfected proteins receive presynaptic inputs almost exclusively from terminals that express endogenous proteins alone; that is, no transfected eGFP or eGFP-αS (Fig. 4B). Therefore, when patching eGFP-expressing cells, any observed postsynaptic changes are cell autonomous and are not associated with the expression of mutant protein in presynaptic neurons. We patched neurons transfected with eGFP control and eGFP-tagged human WT, A30P, E46K, and A53T αS and recorded glutamatergic mEPSCs (Fig. 4C). We found that only those neurons expressing A53T αS showed a significant reduction in the amplitude of mEPSCs and expression of the other variants of αS had no significant postsynaptic effect (Fig. 4D,E). By contrast, consistent with the lack of transfected αS expression in presynaptic terminals, postsynaptic expression of the transfected αS variants had no significant effect on mEPSC frequency compared with control (Fig. 4F). Together, these results indicate that the expression of A53T αS leads to mutation-specific, cell-autonomous postsynaptic dysfunction (Fig. 4). As further control studies, we used flow cytometry and fluorescence microscopy to compare the expression (Fig. 5A–H) and cellular distribution (Fig. 5I–K) of αS variants in transfected rat neurons, respectively. There was no significant difference between neurons expressing WT, A30P, E46K, and A53T αS in the above analyses (Fig. 5), excluding the potential complication that the A53T αS-induced cell-autonomous postsynaptic deficits are due to nonspecific changes in expression level.
A53T αS induces phosphorylation-dependent tau mislocalization to dendritic spines and associated postsynaptic deficits
We showed that only the A53T mutation caused postsynaptic deficits even though both A30P and E46K mutations are linked to autosomal-dominant PD (Krüger et al., 2001; Zarranz et al., 2004). Unlike other kindred with familial PD, one unique pathological feature of PD brains from Contursi kindred, who carry the A53T mutation, is the frequent concurrence of both αS and tau pathology (Duda et al., 2002). Previously, we have shown that tau missorting to dendritic spines is associated with memory loss and postsynaptic AMPA receptor signaling in FTDP-17 and Alzheimer's disease (Hoover et al., 2010; Miller et al., 2014). Therefore, we tested whether there was a mechanistic relationship among A53T αS, tau, and postsynaptic deficits. We cultured hippocampal neurons from H5 and G2-3 mice, which express A53T αS, as well as their TgNg littermates. We cotransfected these neurons with DsRed and three eGFP-tagged tau constructs (Fig. 6A–C): WT human tau, AP tau (in which the 14 proline-directed serine and threonine residues were converted to unphosphorylatable alanine residues), and E14 tau (in which the 14 residues were converted to phosphomimetic glutamate) (Hoover et al., 2010). The proportion of dendritic spines containing eGFP-tau proteins versus total number of spines labeled by DsRed, was quantified (Hoover et al., 2010; Miller et al., 2014). Results show that the fraction of dendritic spines containing eGFP-WT tau is significantly higher in the neurons expressing both levels of A53T αS (G2-3 and H5 mice) compared with that in neurons from the TgNg littermates (Fig. 6A,B). By contrast, AP tau does not mislocalize to dendritic spines even when A53T αS is expressed (fifth bar in Fig. 6B), indicating that tau phosphorylation is necessary for A53T αS-induced mislocalization to dendritic spines. As a positive control, expression of E14 tau causes maximal mislocalization of tau into dendritic spines in both TgNg and G2-3 neurons (bottom two rows in Fig. 6A; right-most two bars in Fig. 6B). The mislocalization of tau is not due to alterations in the neuronal health because the spine density, a sensitive indicator of neurotoxicity, is comparable between all groups (Fig. 6C).
Figure 6.

A53T αS at two expression levels induces phosphorylation-dependent mislocalization of tau to dendritic spines. Neurons were cultured from TgNg, H5, and G2-3 hippocampi and transfected with DsRed to visualize cellular architecture and eGFP-fused human tau to visualize subcellular location of tau. A, Representative photomicrographs of cultured TgNg, G2-3, and H5 hippocampal neurons expressing WT tau, AP tau (phosphorylation-blocking), or E14 tau (phosphomimetic). Scale bar, 10 μm. B, Quantification of percentage of total dendritic spines containing tau. C, Spine density. For all, TgNg, n = 8; H5, n = 6; G2-3, n = 8; one-way ANOVA with Bonferroni post hoc analysis, F(6,47) = 1.52; ***p < 0.0001. All data are expressed as mean ± SEM.
Tau missorting is known to cause functional deficits in dendritic spines, which also depend upon tau phosphorylation (Hoover et al., 2010; Ittner et al., 2010; Miller et al., 2014). Therefore, we also tested whether A53T αS-induced synaptic dysfunction is mediated by tau phosphorylation (Fig. 7). As before, we used calcium phosphate transfection to cotransfect cultured rat hippocampal neurons with an αS construct (WT or A53T) and a tau construct (eGFP-tagged WT or AP tau). eGFP-expressing neurons were patched at 20–23 DIV in whole-cell voltage-clamp configuration to record AMPA receptor-mediated mEPSCs. The amplitudes of mEPSCs were significantly lower in neurons cotransfected with WT tau + A53T αS than those in neurons cotransfected with WT tau + WT αS (Fig. 7B,C). However, coexpression of AP tau + A53T αS rescued the mEPSC amplitudes to control levels (Fig. 7B,C), suggesting that tau phosphorylation is required for the A53T αS-induced deficits given that human AP tau may establish a dominant-negative block of endogenous tau. Again, no significant differences in mEPSC frequency were found between the groups (Fig. 7D), confirming that the low transfection rates limit the effect of exogenous protein expression on presynaptic terminals innervating the patched neurons. These results provide a mechanistic link among tau phosphorylation, missorting, and A53T αS-induced postsynaptic deficits.
Figure 7.

A53T αS induces tau phosphorylation-dependent, cell-autonomous postsynaptic deficits. A, Representative traces of whole-cell mEPSCs recorded from cultured rat hippocampal neurons cotransfected with tau and αS variants. Scale bar, 5 pA, 100 ms. B, Relative cumulative frequency plot of mEPSC amplitude. C, D, Quantification of mean mEPSC amplitude and mEPSC frequency of cotransfected neurons. For all, n = 12; two-way ANOVA with Bonferroni post hoc analysis, F(1,44) = 4.86; **p = 0.0095, ***p < 0.001. All data are expressed as mean ± SEM.
A53T αS-induced tau missorting and synaptic dysfunction require the activation of GSK3β
To further clarify the postsynaptic roles of αS, we tested whether pharmacological blockade of αS-initiated tau mislocalization can rescue deficits in AMPA receptor signaling (Figs. 8, 9, 10). Many kinases have been reported to phosphorylate tau (Liao et al., 2014). Among these, GSK3β is the most-studied tau kinase in PD pathogenesis and previous reports indicated that αS can cause GSK3β-mediated tau hyperphosphorylation (Haggerty et al., 2011; Kawakami et al., 2011; Wills et al., 2011). Therefore, we first sought to determine whether GSK3β activity is necessary for tau mislocalization in neurons expressing A53T αS (Fig. 8A–C). As above, cultured neurons from G2-3 and TgNg mice were cotransfected with eGFP-WT tau and DsRed. We treated the neurons with 3 μm CHIR-99021 (CHIR), a GSK3β inhibitor, or vehicle at 16 DIV and then imaged the neurons at 21 DIV (Fig. 8A). We found that the increase in dendritic spines containing mislocalized eGFP-tau in G2-3 neurons was blocked by the presence of CHIR (Fig. 8A,B). Therefore, GSK3β activity is necessary for A53T αS-dependent mislocalization of tau to dendritic spines. Furthermore, inhibition of GSK3β can completely reverse the postsynaptic deficits caused by A53T αS transfection (Fig. 8D–F). Collectively, these results indicate that the functional deficits in AMPA receptor-mediated synaptic responses caused by A53T αS require the activity of GSK3β, a major tau kinase.
Figure 8.

GSK3β activation is required for tau mislocalization and synaptic deficits in A53T αS-expressing neurons. A, Representative photomicrographs from cultured TgNg and G2-3 neurons that were either untreated or treated with the GSK3β-specific inhibitor CHIR-99021 (CHIR). Scale bar, 5 μm. B, C, Quantification of spines containing tau and spine density. Two-way ANOVA with Bonferroni post hoc analysis, F(2,42) = 27.27. D, Representative mEPSC traces from untreated (top) and CHIR-treated neurons (bottom) expressing eGFP alone, eGFP-WT αS, and eGFP-A53T αS. Scale bar, 10 pA, 100 ms. E, Relative cumulative frequency of mEPSC amplitudes from neurons represented in D. Kolmogorov–Smirnov comparison with eGFP; D = 0.53 ***p < 0.0001. F, G, Quantification of mEPSC amplitude and frequency. Two-way ANOVA with Bonferroni post hoc analysis. F(2,66) = 3.214; *p = 0.041. For all, n = 12. All data are expressed as mean ± SEM.
Figure 9.

GluA1 surface expression in dendritic spines is decreased by A53T αS expression in a GSK3β-dependent fashion. A, Photomicrographs of neurons from G2-3 mice and their TgNg littermates without (top two panels) and with treatment of CHIR-99021 (bottom two panels). As described previously (Liao et al., 1999), live neurons were stained for N-GluA1 antibodies (green), fixed, permeablized, and stained for PSD-95 (red). Arrows indicate tightly clustered surface N-GluA1 colocalized with PSD-95, whereas weak, nonspecific N-GluR1 immunoreactivity appeared along the dendritic shafts as diffuse staining rather than distinct clusters in G2-3 neurons (arrowheads). The diffuse staining is likely due to the presence of extrasynaptic AMPA receptors (Newpher and Ehlers, 2008). Treatment with CHIR-99021 restored surface N-GluA1 synaptic localization in G2-3 mice. Scale bar, 10 μm. B, GluA1 surface fluorescence in PSD-95 immunoreactive spines was normalized to dendritic fluorescence. Two-way ANOVA with Bonferroni post hoc analysis, F(1,28) = 5.69; **p = 0.0029. For all, n = 8. All data are expressed as mean ± SEM.
Figure 10.

Calcineurin activation is required for tau- and A53T αS-induced postsynaptic deficits. A, Representative mEPSC traces recorded from cultured rat hippocampal neurons transfected with WT αS treated with DMSO vehicle or with A53T αS treated with DMSO vehicle or FK506. Scale bar, 10 pA, 100 ms. B, Relative cumulative frequency of mEPSC amplitudes from neurons represented in A. Kolmogorov–Smirnov comparison with vehicle-treated neurons expressing eGFP-WT αS, D = 0.43 ***p < 0.0001. C, D, Quantification of mEPSC amplitude and frequency. One-way ANOVA with Bonferroni post hoc analysis, F(4,41) = 3.84; **p = 0.0025. For all, n = 12. All data are expressed as mean ± SEM. E, Hypothetical pathways for αS-induced changes in neuronal transmission: In pathway #1, A53T αS induces mutation-specific, GSK3β-dependent phosphorylation of tau, leading to tau missorting to dendritic spines. Here, tau leads to calcineurin (CaN)-mediated endocytosis of GluA1-containing AMPA receptors, leading to postsynaptic deficits. However, we cannot rule out tau-mediated inhibition of AMPA receptor insertion into the synaptic membrane. In pathway #2, hyperexpression of WT or mutant αS (A53T, A30P) leads to presynaptic release suppression through an unknown mechanism regardless of genotypes. The differential effects of αS on these two separate pathways may contribute to PD heterogeneity.
Mislocalization of phospho-tau to dendritic spines leads to reduced mEPSC amplitude by reducing the surface levels of AMPARs (Hoover et al., 2010; Miller et al., 2014). To determine whether this also occurs with αS-dependent postsynaptic deficits, we treated low-density cultures of hippocampal neurons from G2-3 and TgNg mouse lines with CHIR or vehicle and stained the live neurons with a FITC-conjugated antibody against the N terminus of GluA1 subunits (N-GluA1; Liao et al., 1999). We next fixed and permeabilized the neurons and stained with an antibody against PSD-95 to reveal the location of dendritic spines (see representative images in Fig. 9). Surface GluA-1 signal is normally clustered with PSD-95 at the synapses in mature neurons (>3 weeks in vitro); however, in G2-3 neurons, this colocalization is significantly diminished, leaving only nonspecific extrasynaptic staining in the dendritic shaft. When GSK3β activity was blocked with CHIR, strong colocalization of PSD-95 and N-GluA1 clusters is restored (Fig. 9). Together, these results suggest that A53T αS expression causes a GSK3β-dependent decrease in AMPA receptor signaling via postsynaptic internalization of GluA1 subunits or inhibition of synaptic recruitment of these subunits.
A53T αS-induced synaptic dysfunction also requires the activation of calcineurin
Given that A53T αS causes a loss of surface GluA1, we hypothesized that calcineurin-mediated AMPAR internalization may play a role in synaptic deficits caused by A53T αS. Calcineurin is a Ca2+-dependent protein phosphatase (Klee et al., 1979) that mediates AMPAR internalization under multiple conditions including LTD (Esteban et al., 2003; Dell'Acqua et al., 2006; He et al., 2011), morphine treatment (Kam et al., 2010; Miller et al., 2012), neuronal toxicity, and exposure to Aβ (Hsieh et al., 2006; Wu et al., 2012; Miller et al., 2014). We examined calcineurin involvement by recording mEPSCs from neurons expressing A53T αS in the presence of the calcineurin inhibitor FK506 (tacrolimus) or vehicle (Fig. 10). mEPSC amplitudes in neurons expressing A53T αS treated with vehicle were significantly smaller than those in neurons expressing A53T αS treated with FK506, indicating that calcineurin activation is required for A53T αS-induced synaptic dysfunction (Fig. 10A–C). Again, mEPSC frequency in this low-efficiency transfection was unaffected (Fig. 10D), further supporting that the A53T αS-induced calcineurin-mediated changes are mostly postsynaptic and cell autonomous.
Discussion
Although hyperexpression of WT, A30P, and A53T human αS results in presynaptic deficits, we demonstrate here that A53T αS causes unique additional deficits in postsynaptic neuronal function (Figs. 1, 3, 4). The presence or absence of postsynaptic deficits may contribute to the clinical and pathological heterogeneity of PD (Lewis et al., 2005; Kehagia et al., 2010; van Balkom et al., 2016). Unlike presynaptic deficits, postsynaptic deficits are not due to a simple increase in αS expression level. Rather, postsynaptic deficits require expression of αS with the specific A53T mutation (Figs. 1, 3, 4). These postsynaptic deficits are likely mediated by a mechanism distinct from that underlying presynaptic deficits. We propose two separate signaling cascades that can be activated by changes in αS. A53T αS causes postsynaptic deficits by inducing tau missorting to dendritic spines (Fig. 10E, pathway #1), whereas an abnormal increase in the expression level of human WT or mutant αS induces presynaptic deficits by suppressing the release probability of neurotransmitter vesicles (Fig. 10E, pathway #2). The absence of paired pulse ratio changes between TgNg and Tg mouse lines does not exclude presynaptic alterations. Subtle presynaptic changes may be concealed by the variability inherent to EPSCs and paired pulses. Measuring mEPSC frequency is a more sensitive test for changes in vesicle release probability. Additionally, it is unclear whether the vesicles recruited by an evoked action potential and a spontaneous release are from the same reserve pool (for review, see Schneggenburger and Rosenmund, 2015).
It remains unresolved why only A53T αS activates the postsynaptic signaling cascade presented here. In a recent study, Dhavale et al. (2017) used seed growth by monomer association assays to measure fibril growth of αS with mutations linked to familial PD including A30P, E46K and A53T. Among these three mutations, only the A53T mutation significantly increases the growth of αS fibrils over an observation window of 3 h. Moreover, only this mutation increases de novo fibril formation of αS over 72 h compared with the WT αS control (Dhavale et al., 2017). It has been shown that certain forms of αS fibrils (“strain B”) significantly increase the aggregation of pathological forms of tau proteins (Guo et al., 2013). An in-depth future study of how various αS mutations affect toxic oligomer and fibril formation and the effects of these oligomers and fibril species on presynaptic and postsynaptic deficits is warranted.
We have also characterized a postsynaptic signaling cascade that directly links the A53T αS mutation to tau-dependent pathophysiology (Fig. 10E, pathway #1). Our results support the involvement of GSK3β-dependent tau phosphorylation and calcineurin-mediated suppression of AMPA receptor currents in this cascade (Figs. 8, 10). However, further studies are required to fully characterize the signaling cascade involved. No previous study has shown that changes in αS can induce tau missorting to dendritic spines and subsequent loss of postsynaptic AMPA receptors. There is strong evidence of frequent concurrence of tau and αS pathologies in the Contursi kindred (Duda et al., 2002). Additionally, a recent clinical study found that frontotemporal dementia is the presenting phenotype in some A53T carriers with atrophy of prefrontal cortex and elevated tau concentration in CSF (Bougea et al., 2017). Together with our present findings, it is possible that the A53T mutation has a unique pathogenic association with tau, leading to frontotemporal dementia and parkinsonism. However, tau and αS histopathology overlap in many non-A53T αS PD etiologies. In a recent pathological analysis of 63 cases of Lewy body disorders, neurofibrillary tangles at Braak stage III or greater were found in 31.3% of PD cases, 53.6% of PD dementia cases, and 89.5% of dementia with Lewy body cases (Colom-Cadena et al., 2017). Because WT αS can also form fibrils with features of “strain B” under certain conditions (Guo et al., 2013), it is possible that the postsynaptic link between αS and tau revealed here also plays a role in the pathogenesis of some cases of sporadic PD.
Consistent with a previous report (Paumier et al., 2013), we also found that aged G2-3 mice exhibit deficits in synaptic plasticity and spatial memory tests. However, our studies extend the previous analysis by describing a novel molecular basis for synaptic deficits caused by A53T αS (i.e., tau-missorting and postsynaptic deficits). Tau missorting to dendritic spines has been shown to be associated with cognitive deficits in models of Alzheimer's disease (Ittner et al., 2010; Miller et al., 2014), FTDP-17 (Hoover et al., 2010), and stress (Lopes et al., 2016). Our results suggest that A53T mutation-induced tau missorting may contribute to dementia observed in the Contursi kindred. However, A53T-linked familial PD is not uniquely associated with the dementia seen in humans with SNCA multiplications (Fuchs et al., 2007; Olgiati et al., 2015) and E46K (Lucas-Jiménez et al., 2016) mutations as well. It is possible that αS abnormalities can cause dementia via multiple mechanisms, which would be consistent with the well documented clinical heterogeneity of PD (Lewis et al., 2005; Kehagia et al., 2010; van Balkom et al., 2016). A recent retrospective study of 213 sporadic synucleinopathy patients demonstrates that the severity of tau pathologies is strongly correlated with the early onset of both motor and cognitive symptoms (Irwin et al., 2017). Tau, it seems, is important to the development of dementia in various synucleinopathies. Our present study illuminates one possible pathway connecting tau pathology to familial A53T αS PD, but may also be relevant to other synucleinopathies with concurrent tauopathic dementia.
In summary, here we have characterized a postsynaptic signaling cascade that is initiated by αS and mediated by tau. Among the three autosomal-dominant PD-linked αS mutations that we tested (A30P, E46K, and A53T), only the A53T mutation activates this postsynaptic cascade. Although the unraveled signal cascade will help us better understand the neuropathopysiology of familial PD cases specifically linked to A53T mutation in αS, its role in dementia seen in sporadic α-synucleinopathy remains to be determined. Given that tau pathology is strongly correlated with early onset dementia in parkinsonism (Irwin et al., 2017), that the A53T mutation uniquely increases the de novo formation of αS fibrils (Dhavale et al., 2017), and that WT αS monomers can also be transformed to pathological strains of αS fibrils (Guo et al., 2013), the novel functional link between αS and tau unraveled here will also help our understanding of the cellular basis for the pathogenesis of some subgroups of sporadic PD dementia.
Footnotes
This work was supported by the National Institutes of Health [Grants R21-NS084007-01, R21NS096437-01, R01-NS NS086074, R01-092093, R01 NS108686-01 (to M.K.L., D.L., and A.A.), National Institute on Drug Abuse Training Grant T32 DA07234, Predoctoral Training Grant P32-GM008471 (to E.C.M.); and National Institute on Aging-funded predoctoral fellowship to T32-AG029796 to C.G.]; the Michael J. Fox Foundation; a UMN–Mayo partnership grant and a grant from Minnesota Higher Education (D.L.); the Susan and David Plimpton Fund (M.K.L.). Behavioral studies were performed in the Mouse Behavioral Phenotyping Core at the University of Minnesota, which is supported by National Institute of Neurological Disorders and Stroke–NIH (Center Grant P30 NS062158). We thank Drs. Sylvain Lesne, Karen Hsiao Ashe, Harry Orr, Rachel Penrod, and Daniel Miller for thoughtful discussion; Drs. Ben Smith and Xiaohui Zhao for thoughtful discussion and technical expertise; Susan Bushek, Samantha Sydow, and Ben Dummer for technical assistance; and special thanks to Clifford Csizmar for advice and technical assistance concerning flow cytometry and analysis.
The authors declare no competing financial interests.
References
- Abeliovich A, Schmitz Y, Fariñas I, Choi-Lundberg D, Ho WH, Castillo PE, Shinsky N, Verdugo JM, Armanini M, Ryan A, Hynes M, Phillips H, Sulzer D, Rosenthal A (2000) Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron 25:239–252. 10.1016/S0896-6273(00)80886-7 [DOI] [PubMed] [Google Scholar]
- Bougea A, Koros C, Stamelou M, Simitsi A, Papagiannakis N, Antonelou R, Papadimitriou D, Breza M, Tasios K, Fragkiadaki S, Geronicola Trapali X, Bourbouli M, Koutsis G, Papageorgiou SG, Kapaki E, Paraskevas GP, Stefanis L (2017) Frontotemporal dementia as the presenting phenotype of p.A53T mutation carriers in the alpha-synuclein gene. Parkinsonism Relat Disord 35:82–87. 10.1016/j.parkreldis.2016.12.002 [DOI] [PubMed] [Google Scholar]
- Burré J. (2015) The synaptic function of α-synuclein. J Parkinsons Dis 5:699–713. 10.3233/JPD-150642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burré J, Sharma M, Tsetsenis T, Buchman V, Etherton MR, Südhof TC (2010) Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 329:1663–1667. 10.1126/science.1195227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burré J, Sharma M, Südhof TC (2014) α-synuclein assembles into higher-order multimers upon membrane binding to promote SNARE complex formation. Proc Natl Acad Sci U S A 111:E4274–E4283. 10.1073/pnas.1416598111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chartier-Harlin MC, Kachergus J, Roumier C, Mouroux V, Douay X, Lincoln S, Levecque C, Larvor L, Andrieux J, Hulihan M, Waucquier N, Defebvre L, Amouyel P, Farrer M, Destée A (2004) Alpha-synuclein locus duplication as a cause of familial Parkinson's disease. Lancet 364:1167–1169. 10.1016/S0140-6736(04)17103-1 [DOI] [PubMed] [Google Scholar]
- Colom-Cadena M, Grau-Rivera O, Planellas L, Cerquera C, Morenas E, Helgueta S, Muñoz L, Kulisevsky J, Martí MJ, Tolosa E, Clarimon J, Lleó A, Gelpi E (2017) Regional overlap of pathologies in Lewy body disorders. J Neuropathol Exp Neurol 76:216–224. 10.1093/jnen/nlx002 [DOI] [PubMed] [Google Scholar]
- Dell'Acqua ML, Smith KE, Gorski JA, Horne EA, Gibson ES, Gomez LL (2006) Regulation of neuronal PKA signaling through AKAP targeting dynamics. Eur J Cell Biol 85:627–633. 10.1016/j.ejcb.2006.01.010 [DOI] [PubMed] [Google Scholar]
- Dhavale DD, Tsai C, Bagchi DP, Engel LA, Sarezky J, Kotzbauer PT (2017) A sensitive assay reveals structural requirements for alpha-synuclein fibril growth. J Biol Chem 292:9034–9050. 10.1074/jbc.M116.767053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duda JE, Giasson BI, Mabon ME, Miller DC, Golbe LI, Lee VM, Trojanowski JQ (2002) Concurrence of alpha-synuclein and tau brain pathology in the contursi kindred. Acta Neuropathol 104:7–11. 10.1007/s00401-002-0563-3 [DOI] [PubMed] [Google Scholar]
- Esteban JA, Shi SH, Wilson C, Nuriya M, Huganir RL, Malinow R (2003) PKA phosphorylation of AMPA receptor subunits controls synaptic trafficking underlying plasticity. Nat Neurosci 6:136–143. 10.1038/nn997 [DOI] [PubMed] [Google Scholar]
- Fuchs J, Nilsson C, Kachergus J, Munz M, Larsson EM, Schüle B, Langston JW, Middleton FA, Ross OA, Hulihan M, Gasser T, Farrer MJ (2007) Phenotypic variation in a large Swedish pedigree due to SNCA duplication and triplication. Neurology 68:916–922. 10.1212/01.wnl.0000254458.17630.c5 [DOI] [PubMed] [Google Scholar]
- Guo JL, Covell DJ, Daniels JP, Iba M, Stieber A, Zhang B, Riddle DM, Kwong LK, Xu Y, Trojanowski JQ, Lee VM (2013) Distinct α-synuclein strains differentially promote tau inclusions in neurons. Cell 154:103–117. 10.1016/j.cell.2013.05.057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JT, Chen AQ, Kong Q, Zhu H, Ma CM, Qin C (2008) Inhibition of vesicular monoamine transporter-2 activity in alpha-synuclein stably transfected SH-SY5Y cells. Cell Mol Neurobiol 28:35–47. 10.1007/s10571-007-9227-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haggerty T, Credle J, Rodriguez O, Wills J, Oaks AW, Masliah E, Sidhu A (2011) Hyperphosphorylated tau in an α-synuclein-overexpressing tg model of Parkinson's disease. Eur J Neurosci 33:1598–1610. 10.1111/j.1460-9568.2011.07660.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- He K, Lee A, Song L, Kanold PO, Lee HK (2011) AMPA receptor subunit GluR1 (GluA1) serine-845 site is involved in synaptic depression but not in spine shrinkage associated with chemical long-term depression. J Neurophysiol 105:1897–1907. 10.1152/jn.00913.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoover BR, Reed MN, Su J, Penrod RD, Kotilinek LA, Grant MK, Pitstick R, Carlson GA, Lanier LM, Yuan LL, Ashe KH, Liao D (2010) Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron 68:1067–1081. 10.1016/j.neuron.2010.11.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh H, Boehm J, Sato C, Iwatsubo T, Tomita T, Sisodia S, Malinow R (2006) AMPAR removal underlies abeta-induced synaptic depression and dendritic spine loss. Neuron 52:831–843. 10.1016/j.neuron.2006.10.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibáñez P, Bonnet AM, Débarges B, Lohmann E, Tison F, Pollak P, Agid Y, Dürr A, Brice A (2004) Causal relation between alpha-synuclein gene duplication and familial Parkinson's disease. Lancet 364:1169–1171. 10.1016/S0140-6736(04)17104-3 [DOI] [PubMed] [Google Scholar]
- Irwin DJ, Lee VM, Trojanowski JQ (2013) Parkinson's disease dementia: convergence of α-synuclein, tau and amyloid-β pathologies. Nat Rev Neurosci 14:626–636. 10.1038/nrn3549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin DJ, Grossman M, Weintraub D, Hurtig HI, Duda JE, Xie SX, Lee EB, Van Deerlin VM, Lopez OL, Kofler JK, Nelson PT, Jicha GA, Woltjer R, Quinn JF, Kaye J, Leverenz JB, Tsuang D, Longfellow K, Yearout D, Kukull W, et al. (2017) Neuropathological and genetic correlates of survival and dementia onset in synucleinopathies: a retrospective analysis. Lancet Neurol 16:55–65. 10.1016/S1474-4422(16)30291-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J, Wölfing H, Chieng BC, Christie MJ, Napier IA, Eckert A, Staufenbiel M, Hardeman E, Götz J (2010) Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer's disease mouse models. Cell 142:387–397. 10.1016/j.cell.2010.06.036 [DOI] [PubMed] [Google Scholar]
- Kam AY, Liao D, Loh HH, Law PY (2010) Morphine induces AMPA receptor internalization in primary hippocampal neurons via calcineurin-dependent dephosphorylation of GluR1 subunits. J Neurosci 30:15304–15316. 10.1523/JNEUROSCI.4255-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawakami F, Suzuki M, Shimada N, Kagiya G, Ohta E, Tamura K, Maruyama H, Ichikawa T (2011) Stimulatory effect of α-synuclein on the tau-phosphorylation by GSK-3β. FEBS J 278:4895–4904. 10.1111/j.1742-4658.2011.08389.x [DOI] [PubMed] [Google Scholar]
- Kehagia AA, Barker RA, Robbins TW (2010) Neuropsychological and clinical heterogeneity of cognitive impairment and dementia in patients with Parkinson's disease. Lancet Neurol 9:1200–1213. 10.1016/S1474-4422(10)70212-X [DOI] [PubMed] [Google Scholar]
- Klee CB, Crouch TH, Krinks MH (1979) Calcineurin: a calcium- and calmodulin-binding protein of the nervous system. Proc Natl Acad Sci U S A 76:6270–6273. 10.1073/pnas.76.12.6270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krüger R, Kuhn W, Leenders KL, Sprengelmeyer R, Müller T, Woitalla D, Portman AT, Maguire RP, Veenma L, Schröder U, Schöls L, Epplen JT, Riess O, Przuntek H (2001) Familial parkinsonism with synuclein pathology: clinical and PET studies of A30P mutation carriers. Neurology 56:1355–1362. 10.1212/WNL.56.10.1355 [DOI] [PubMed] [Google Scholar]
- Lee MK, Stirling W, Xu Y, Xu X, Qui D, Mandir AS, Dawson TM, Copeland NG, Jenkins NA, Price DL (2002) Human alpha-synuclein-harboring familial Parkinson's disease-linked ala-53 –> thr mutation causes neurodegenerative disease with alpha-synuclein aggregation in tg mice. Proc Natl Acad Sci U S A 99:8968–8973. 10.1073/pnas.132197599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis SJ, Foltynie T, Blackwell AD, Robbins TW, Owen AM, Barker RA (2005) Heterogeneity of Parkinson's disease in the early clinical stages using a data driven approach. J Neurol Neurosurg Psychiatry 76:343–348. 10.1136/jnnp.2003.033530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao D, Hessler NA, Malinow R (1995) Activation of postsynaptically silent synapses during pairing-induced LTP in CA1 region of hippocampal slice. Nature 1995 375:400–404. 10.1038/375400a0 [DOI] [PubMed] [Google Scholar]
- Liao D, Lin H, Law PY, Loh HH (2005) Mu-opioid receptors modulate the stability of dendritic spines. Proc Natl Acad Sci U S A 102:1725–1730. 10.1073/pnas.0406797102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao D, Miller EC, Teravskis PJ (2014) Tau acts as a mediator for Alzheimer's disease-related synaptic deficits. Eur J Neurosci 39:1202–1213. 10.1111/ejn.12504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao D, Zhang X, O'Brien R, Ehlers MD, Huganir RL (1999) Regulation of morphological postsynaptic silent synapses in developing hippocampal neurons. Nat Neurosci 2:37–43. 10.1038/4540 [DOI] [PubMed] [Google Scholar]
- Lin H, Huganir R, Liao D (2004) Temporal dynamics of NMDA receptor-induced changes in spine morphology and AMPA receptor recruitment to spines. Biochem Biophys Res Commun 316:501–511. 10.1016/j.bbrc.2004.02.086 [DOI] [PubMed] [Google Scholar]
- Lin H, Higgins P, Loh HH, Law PY, Liao D (2009) Bidirectional effects of fentanyl on dendritic spines and AMPA receptors depend upon the internalization of mu opioid receptors. Neuropsychopharmacology 34:2097–2111. 10.1038/npp.2009.34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes S, Vaz-Silva J, Pinto V, Dalla C, Kokras N, Bedenk B, Mack N, Czisch M, Almeida OF, Sousa N, Sotiropoulos I (2016) Tau protein is essential for stress-induced brain pathology. Proc Natl Acad Sci U S A 113:E3755–E3763. 10.1073/pnas.1600953113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas-Jiménez O, Del Pino R, Acera M, Gabilondo I, Gómez-Esteban JC, Tijero B, Díez-Cirarda M, Peña J, Ibarretxe-Bilbao N, Ojeda N (2016) Neuropsychological profile in carriers of E46K mutation of the alpha-synuclein gene. Neurology 86:P5.200. [Google Scholar]
- Malinow R, Malenka RC (2002) AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci 25:103–126. 10.1146/annurev.neuro.25.112701.142758 [DOI] [PubMed] [Google Scholar]
- Miller EC, Zhang L, Dummer BW, Cariveau DR, Loh H, Law PY, Liao D (2012) Differential modulation of drug-induced structural and functional plasticity of dendritic spines. Mol Pharmacol 82:333–343. 10.1124/mol.112.078162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller EC, Teravskis PJ, Dummer BW, Zhao X, Huganir RL, Liao D (2014) The role of tau phosphorylation and tau mislocalization in soluble A-beta oligomer-induced deficits in AMPA glutamate receptor signaling. Eur J Neurosci 39:1214–1224. 10.1111/ejn.12507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemani VM, Lu W, Berge V, Nakamura K, Onoa B, Lee MK, Chaudhry FA, Nicoll RA, Edwards RH (2010) Increased expression of alpha-synuclein reduces neurotransmitter release by inhibiting synaptic vesicle reclustering after endocytosis. Neuron 65:66–79. 10.1016/j.neuron.2009.12.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newpher TM, Ehlers MD (2008) Glutamate receptor dynamics in dendritic microdomains. Neuron 58:472–497. 10.1016/j.neuron.2008.04.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olgiati S, Thomas A, Quadri M, Breedveld GJ, Graafland J, Eussen H, Douben H, de Klein A, Onofrj M, Bonifati V (2015) Early-onset parkinsonism caused by alpha-synuclein gene triplication: clinical and genetic findings in a novel family. Parkinsonism Relat Disord 21:981–986. 10.1016/j.parkreldis.2015.06.005 [DOI] [PubMed] [Google Scholar]
- Paumier KL, Sukoff Rizzo SJ, Berger Z, Chen Y, Gonzales C, Kaftan E, Li L, Lotarski S, Monaghan M, Shen W, Stolyar P, Vasilyev D, Zaleska M, D Hirst W, Dunlop J (2013) Behavioral characterization of A53T mice reveals early and late stage deficits related to Parkinson's disease. PLoS One 8:e70274. 10.1371/journal.pone.0070274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrucci S, Ginevrino M, Valente EM (2016) Phenotypic spectrum of alpha-synuclein mutations: new insights from patients and cellular models. Parkinsonism Relat Disord 22:S16–S20. 10.1016/j.parkreldis.2015.08.015 [DOI] [PubMed] [Google Scholar]
- Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL (1997) Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science 276:2045–2047. 10.1126/science.276.5321.2045 [DOI] [PubMed] [Google Scholar]
- Schneggenburger R, Rosenmund C (2015) Molecular mechanisms governing Ca(2+) regulation of evoked and spontaneous release. Nat Neurosci 18:935–941. 10.1038/nn.4044 [DOI] [PubMed] [Google Scholar]
- Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, Lincoln S, Crawley A, Hanson M, Maraganore D, Adler C, Cookson MR, Muenter M, Baptista M, Miller D, Blancato J, et al. (2003) Alpha-synuclein locus triplication causes Parkinson's disease. Science 302:841. 10.1126/science.1090278 [DOI] [PubMed] [Google Scholar]
- van Balkom TD, Vriend C, Berendse HW, Foncke EM, van der Werf YD, van den Heuvel OA, Klein M (2016) Profiling cognitive and neuropsychiatric heterogeneity in Parkinson's disease. Parkinsonism Relat Disord 28:130–136. 10.1016/j.parkreldis.2016.05.014 [DOI] [PubMed] [Google Scholar]
- Wills J, Credle J, Haggerty T, Lee JH, Oaks AW, Sidhu A (2011) Tauopathic changes in the striatum of A53T alpha-synuclein mutant mouse model of Parkinson's disease. PLoS One 6:e17953. 10.1371/journal.pone.0017953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu HY, Hudry E, Hashimoto T, Uemura K, Fan ZY, Berezovska O, Grosskreutz CL, Bacskai BJ, Hyman BT (2012) Distinct dendritic spine and nuclear phases of calcineurin activation after exposure to amyloid-β revealed by a novel fluorescence resonance energy transfer assay. J Neurosci 32:5298–5309. 10.1523/JNEUROSCI.0227-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaltieri M, Grigoletto J, Longhena F, Navarria L, Favero G, Castrezzati S, Colivicchi MA, Della Corte L, Rezzani R, Pizzi M, Benfenati F, Spillantini MG, Missale C, Spano P, Bellucci A (2015) α-synuclein and synapsin III cooperatively regulate synaptic function in dopamine neurons. J Cell Sci 128:2231–2243. 10.1242/jcs.157867 [DOI] [PubMed] [Google Scholar]
- Zarranz JJ, Alegre J, Gómez-Esteban JC, Lezcano E, Ros R, Ampuero I, Vidal L, Hoenicka J, Rodriguez O, Atarés B, Llorens V, Gomez Tortosa E, del Ser T, Muñoz DG, de Yebenes JG (2004) The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol 55:164–173. 10.1002/ana.10795 [DOI] [PubMed] [Google Scholar]