

Abstract
In higher plants, a P-type proton-pumping ATPase generates the proton-motive force essential for the function of all other transporters and for proper growth and development. X-ray crystallographic studies of the plant plasma membrane proton pump have provided information on amino acids involved in ATP catalysis but provided no information on the structure of the C-terminal regulatory domain. Despite progress in elucidating enzymes involved in the signaling pathways that activate or inhibit this pump, the site of interaction of the C-terminal regulatory domain with the catalytic domains remains a mystery. Genetic studies have pointed to amino acids in various parts of the protein that may be involved, but direct chemical evidence for which ones are specifically interacting with the C terminus is lacking. In this study, we used in vivo cross-linking experiments with a photoreactive unnatural amino acid, p-benzoylphenylalanine, and tandem MS to obtain direct evidence that the C-terminal regulatory domain interacts with amino acids located within the N-terminal actuator domain. Our observations are consistent with a mechanism in which intermolecular, rather than intramolecular, interactions are involved. Our model invokes a “head-to-tail” organization of ATPase monomers in which the C-terminal domain of one ATPase molecule interacts with the actuator domain of another ATPase molecule. This model serves to explain why cross-linked peptides are found only in dimers and trimers, and it is consistent with prior studies suggesting that within the membrane the protein can be organized as homopolymers, including dimers, trimers, and hexamers.
Keywords: H+-ATPase, protein cross-linking, mass spectrometry (MS), protein-protein interaction, Arabidopsis thaliana
Introduction
The plasma membrane proton pump (H+-ATPase) is the primary energy-transducing transporter in fungi and plants. By analogy to the sodium pump in animals, the plant/fungal protein creates an electrical potential and proton chemical gradient that powers secondary transporters to move solutes such as ions, amino acids, glucose, and other metabolites in and out of the cell. The H+-ATPase belongs to the family of ATPases known as “P-type” because they form a covalently modified intermediate, i.e. a phosphoaspartate, as they change their conformational states during the catalytic cycle (1). Because the H+-ATPase is often a major consumer of ATP, e.g. during rapid cell division in yeast, and in plant cells that are specialized for performing large amounts of transport (e.g. root hairs, phloem companion cells, and guard cells surrounding the leaf stomata), its catalytic activity must be tightly controlled (2, 3). The structure of H+-ATPase consists of 10 transmembrane domains and three catalytic domains facing the cytoplasm: the phosphorylation domain containing the conserved phosphoaspartate, the nucleotide binding domain, and the actuator domain (4). In addition, the ∼100 amino acid C-terminal domain is thought to have an autoinhibitory effect on the enzyme because when it is removed by genetic deletion mutagenesis (5) or proteolytic digestion (6, 7), the enzyme activity increases. The C-terminal domain may inhibit the enzyme via interactions with the actuator domain and the top of transmembrane domains 1 and 2 because point mutations in these regions activate the enzyme (8, 9). Similarly, point mutations in the C-terminal domain that displace its autoinhibition are clustered in two regions: region I from Lys-863 to Leu-885, and region II from Ser-904 to Leu-919 (8–11). This suggests the interaction of these two regions with the rest of enzyme. However, another study suggested that regions I and II do not inhibit enzyme in a direct manner, and three other regions corresponding to three critical phosphorylated threonines (region A, Thr-861; region B, Thr-881; and region C, Thr-947) seem to function directly as autoinhibitors (12). Changes in the degree of phosphorylation of these threonines as well as other serines in the C-terminal domain correlate with changes in catalytic activity. For example, phosphorylation of Ser-899, which is induced by binding of a peptide hormone rapid-alkalinizing factor, to the receptor-like protein kinase FERONIA inhibits the enzyme. The precise molecular mechanism of how this reduces the catalytic activity of the enzyme is unknown (13). In contrast, phosphorylation of AHA2 at Thr-881 and the penultimate Thr-947 activates the enzyme (14–16); phosphorylation of Thr-947 also induces the binding of the 14-3-3 protein to the C-terminal domain and eliminates the inhibitory effect on the rest of the enzyme. In addition, recent studies suggest that phosphorylation of Nicotiana plumbaginifolia PMA2 (a tobacco counterpart of AHA2) at the penultimate threonine of the C terminus results in binding of a 14-3-3 protein. This event ultimately drives the conversion of pre-existing H+-ATPase dimers into a hexamer complexed with six 14-3-3 molecules (17, 18). Intriguingly, it is thought that plasma membrane H+-ATPase exists as a homo-oligomer in vivo, yet the pump retains activity as a monomer when it is reconstituted into liposomes (19) or produced in nanodiscs (20); this suggests that oligomerization is not required for plasma membrane H+-ATPase to function. Unfortunately, oligomerization studies of the pump have been impeded by the inherent difficulties to identifying the exact size of the protein in the presence of detergent. For example, Saccharomyces cerevisiae ScPMA1 behaves as a trimer on SDS/Triton X-100 velocity gradients (21). However, it can behave as a trimer, hexamer, and dodecamer when run on blue native gels under different detergent conditions (22). Schizosaccharomyces pombe SpPMA1 also forms higher-order homo-oligomers (8–10 monomers) when solubilized in lysophosphatidylcholine (23). Neurospora crassa PMA1 exists as a hexamer when isolated by gradient centrifugation with Tween 20 (24), and this hexamer form is also observed by EM of 2D crystals (25). In contrast, radiation inactivation studies suggest that H+-ATPase of N. crassa and red beet form dimers in the native membrane environment (26, 27). In summary, it is clear that the quaternary structure of plasma membrane H+-ATPase in intact cells remains to be determined. Unlike in fungi, the H+-ATPase enzyme is not observed as a hexamer when it is expressed in plants or heterologously expressed in yeast unless the 14-3-3 protein is bound to its penultimate phosphorylated threonine in the regulatory carboxyl domain. This observation has led to the suggestion that in plant phosphorylation followed by binding of 14-3-3 locks the enzyme into a hyperactive state that exists primarily as a hexamer (18).
The crystal structure (Protein Data Bank code 5KSD) of the C-terminally truncated version of the AHA2 lacking the last 73 residues reveal the three-dimensional locations for amino acids within the 10 transmembrane domains as well as three catalytic domains (4). In this study, the authors also attempted to crystallize the full-length AHA2 with the C-terminal domain but failed to obtain the density for the large segment from 858 to 948, presumably because it is flexible and/or intrinsically disordered. Thus, the long-standing questions of where the C-terminal domain interacts in the rest of the protein and what conformational changes are associated with its function remain unanswered. To address these questions, we have applied a chemical genomic method using an engineered tRNA/tRNA-synthetase pair to incorporate the unnatural photolabile amino acid, p-benzoylphenylalanine (BPA)3 (28–30), into AHA2. We then expressed BPA–AHA2 in yeast and used tandem MS to identify cross-linked products after photoactivation. We call this scanning cross-linking mutagenesis because one can introduce the modified residue at many different positions and then after treatment with UV light use SDS-PAGE and tandem MS to identify regions of the protein with which the BPA interacts. This method is also advantageous over other cross-linking approaches because we performed these experiments in living cells, allowing the pump to retain its native three-dimensional structure without introducing variables due to potential denaturation or other changes caused by solubilization with detergents. The results of the experiments presented herein demonstrate that the C-terminal domain interacts with amino acids within the N-terminally located actuator domain and that this interaction is an intermolecular rather than intramolecular event. These results provide the first direct evidence for the location of peptides that interact with the C-terminal regulatory domain and support a model in which oligomerization plays a role in enzyme regulation. This result may be entirely separate from that concerning how oligomers are formed and what their role is, if any, in regulating the enzyme in vivo. Given the many important roles that the pump plays in plant physiology, acting as the nexus for responses to many environmental changes (e.g. light, gravity, and pathogens), it is not surprising that its manner of regulation may be complicated. Clearly, this question remains an important subject for scrutiny at the level of a protein's three-dimensional structure in order to determine the conformational changes associated with various states of activation and inhibition of the pump.
Results
Site-specific incorporation of BPA in AHA2
Although genetic truncation and proteolytic experiments indicate that the C-terminal domain inhibits H+-ATPase catalytic function, the exact location where it interacts remains a mystery. To address this question, we exploited amber codon recoding to site-specifically incorporate the unnatural photolabile amino acid BPA, which can cross-link to alkyl carbons of nearby amino acid residues (31), into His6–HA-tagged AHA2 using the S. cerevisiae RS72 strain as the expression system (32). Amber mutant AHA2 constructs were coexpressed with the mutant tRNA synthetase/tRNASup pair, and in the presence of BPA, full-length proteins were synthesized for subsequent studies. We initially attempted to incorporate BPA directly into the very end of the protein within the C terminus, but unfortunately, we obtained a very low yield of BPA–AHA2 at these sites, although major products were truncated proteins (cf., Fig. 2A and Table S1). To circumvent this problem, we placed BPA at several dozen locations scattered throughout the protein (Fig. 1), especially within the N-terminal actuator domain that was predicted to be involved in interaction with the C-terminal domain in previous genetic studies (8, 9). Also, to minimize the effect of BPA on protein structure and enzyme function, most of our point mutations were positioned in loops, i.e. locations not found in β-sheets or α-helices, based on the 3.5-Å crystal structure of AHA2 (4). These sites were located in the first cytoplasmic domain (Val-12, Leu-14, Phe-23, Lys-27, Leu-33, Lys-57, and Leu-59), in the first transmembrane domain (Phe-61, Phe-64, and Trp-71), in the intracellular loops between transmembrane domain 2 and transmembrane domain 3 (Leu-169, His-217, His-225, and Ser-229), in the transmembrane domain 4 (Pro-286), in the phosphorylation domain (Lys-330), and in the nucleotide-binding domain (Asn-506, Met-531, Tyr-532, and Ala-547) (Fig. 1). As expected, when BPA was not present in the culture medium, very few or no full-length proteins were detected, whereas upon addition of BPA, full-length AHA2 was produced (Fig. 2A). Although we were able to generate BPA mutant proteins for each position, it was not clear whether AHA2 function was compromised by the BPA substitution. To study the effect of these individual amino acid substitutions, we tested the ability of each variant to rescue the RS72 mutant (32). In this yeast strain, expression of the nuclear gene encoding an essential isoform of H+-ATPase, PMA1, is under control of the galactose-inducible promoter. Thus, when the cells are switched to glucose medium, PMA1 expression is inhibited, and yeast growth depends on the activity of transformed AHA2s. Among 22 sites tested, 18 of them can complement the yeast pma1 mutant (Fig. 2B), indicating that most of these BPA–AHA2 variants can produce active enzymes. Interestingly, two mutants at the C-terminal domain, N856TAG and R880TAG, activate the enzyme because the growth rate at 0.05 optical density (OD) of these two sites is similar to that at 0.5 OD of other sites (Fig. 2B). This is probably due to the presence of the truncated versions of AHA2 lacking the C-terminal domain. In contrast, BPA insertion at several conserved residues such as Leu-33, Pro-286, and Lys-330 cannot complement the yeast growth in glucose medium (Fig. 2B). This is not surprising because these amino acids are conserved in rabbit Ca2+-ATPase, plant H+-ATPase AHA2, and yeast H+-ATPase PMA1 (4). In addition, corresponding mutations at these residues in yeast PMA1, such as P335A (Pro-286 in AHA2) and K379Q (Lys-330 in AHA2), were dominant-lethal to yeast (33). Notably, L169BPA also failed to complement pma1, which is consistent with a previous study (34). Note that the expression efficiency varied among the nonlethal BPA mutants (Fig. S1A), but this did not appear to affect their ability to complement pma1, suggesting that expression of the active pump in these experiments is sufficient to support growth.
Figure 2.

BPA was incorporated at different positions of AHA2 and cross-linked in vivo with UV irradiation. A, whole-cell extract from yeast cells expressing His6-HA–tagged AHA2 amber mutants and the tRNA synthetase/tRNASup in the absence (−) or presence of 2 mm BPA(+) were analyzed by Western blotting with anti-HA antibody. B, drop test for the growth of yeast strains expressing BPA–AHA2s on plates containing either 2% galactose or 2% glucose + 2 mm BPA at pH ∼6.5. Growth was recorded after 4 days. C, cells expressing BPA-containing AHA2 were kept in the dark (−) or exposed to UV 365 nm (+), and AHA2 was purified from cell extracts using nickel-Sepharose resin, followed by Western blotting with anti-HA antibody. The cross-linked species are observed as higher molecular bands in the upper part of the gel above the monomeric 110-kDa ATPase polypeptide. D, Strep-HA–tagged AHA2s containing BPA was purified with StrepTactin-Sepharose (GE Healthcare) resin and analyzed by NuPAGE Tris acetate gel. On this gel, the cross-link bands are indicated as a dimer and a trimer of AHA2.
Figure 1.

Two-dimensional structure of AHA2 with BPA incorporated at different positions (indicated with purple stars) in AHA2, including the actuator domain (orange), phosphorylation domain (blue), nucleotide-binding domain (red), and the regulatory domain (last 105 amino acids at the C terminus) that includes the three regulatory regions R-I (black), R-B (white), and R-II (black).
BPA substitutions in the actuator domain form cross-links in a UV-dependent manner
To cross-link the protein, we exposed the cells to long-wavelength UV light and purified protein from whole-cell lysates using nickel-Sepharose resin. Proteins were resolved by SDS-PAGE and detected by Western blotting, with an equal amount of sample from cells not exposed to UV loaded side-by-side to compare the pattern of UV-dependent cross-linking at different positions. Among the tested amino acid positions, F64BPA, H225BPA, and S229BPA showed significant high molecular weight bands upon UV irradiation (Fig. 2C, boxed lanes). Strikingly, these three residues belong to the actuator domain (F64BPA is also to be part of the first transmembrane domain) (4). S229BPA showed the highest cross-linking efficiency as most of its monomer band disappeared concomitantly with the formation of high-weight bands after UV irradiation. To accurately assess the high-weight species that were forming in response to irradiation, we also ran our samples on an acetate gel for better resolution of the higher molecular weight proteins. The results of these experiments showed that the cross-linked proteins migrated as dimers and trimers of AHA2, indicating that the cross-linked products consisted of multiple AHA2 monomers (Fig. 2D). It is worth noting that we also observed a faint dimer band in most of the samples regardless of the UV irradiation, and this is probably due to the preformed AHA2 dimer (18) containing strong noncovalent interactions that cannot always be broken by the SDS detergent treatment during gel electrophoresis.
Identification of peptides cross-linked to S229BPA by tandem MS
We found that a His6 tag was inferior to a Strep–HA-tagged AHA2 for purifying BPA–AHA2 in the high yields required for subsequent MS analysis. Therefore, studies on identifying the precise cross-linked amino acid residues were performed using Strep–HA-tagged AHA2. This system provided us with sufficient quantities of pure protein with BPA incorporation at Phe-64, His-225, and Ser-229 for MS (Fig. 2D and Fig. S1B). Initially, we focused our analysis on AHA2-S229BPA, because this residue showed the highest cross-linking efficiency (Fig. 2C). In these experiments, the intact cells containing AHA2-S229BPA were either kept in the dark as a negative control or exposed to long-wavelength UV light. We then enriched for the microsomal fraction, added detergent, and purified the solubilized Strep-AHA2 with StrepTactin-Sepharose. The eluted proteins were subjected to SDS-PAGE (Fig. S1B), followed by in-gel digestion with trypsin and AspN, and analysis via nanoLC-MS/MS on an Orbitrap Elite hybrid mass spectrometer (ThermoFisher Scientific). We used StavroX software (35) to search for cross-linked dipeptides in the datasets. Only cross-linked peptides with a precursor mass difference from the expected of less than 5 ppm and containing fragment ions detected from both cross-linked peptides (36) were selected; these are summarized in Table 1 and Table S2. In these experiments, we can identify the peptides that cross-link to the BPA site, but because one spectrum can often be assigned to several sites in the same peptide, we cannot always pinpoint with certainty which amino acids within those peptides are being cross-linked to the BPA residue.
Table 1.
Summary of photo-cross-links identified in AHA2-S229BPA after in-gel digestion
Precursor area and spectral count were manually calculated from raw data using Xcalibur and StavroX.
| Peptides with a UV-induced cross-link to S229BPA (MS1) | Possible amino acids cross-link to BPA | Spectral count | Precursor area | |
|---|---|---|---|---|
| S229BPA_trimer | ||||
| 228DXTNQVGHFQK238 | 889EAVNIFPEKGSYR901 | Val-892/Ile-893/Phe-894/Pro-895/Glu-896/Lys-897/Ser-899/Tyr-900 | 19 | 8.22E + 06 |
| 228DXTNQVGHFQK238 | 889EAVNIFPEK897 | Val-892/Ile-893/Phe-894/Pro-895/Glu-896 | 25 | 1.50E + 06 |
| 228DXTNQVGHFQK238 | 858TAFTMKK864 | Thr-861/Met-862/Lys-863 | 7 | 2.44E + 06 |
| 228DXTNQVGHFQK238 | 865DYGKEEREAQWALAQR880 | Lys-868/Glu-869/Glu-872/Arg-873 | 4 | 1.47E + 06 |
| 228DXTNQVGHFQK238 | 865DYGKEER871 | Lys-868/Glu-869/Glu-870 | 2 | 2.10E + 05 |
| 228DXTNQVGHFQK238 | 176VDQSALTGESLPVTK190 | Asp-177/Ser-179/Ala-180/Leu-181/Gly-183 | 4 | 3.45E + 05 |
| 228DXTNQVGHFQK238 | 177DQSALTGESLPVTK190 | Ser-179/Ala-180/Leu-181/Gly-183 | 2 | 5.59E + 05 |
| S229BPA_dimer | ||||
| 228DXTNQVGHFQK238 | 889EAVNIFPEKGSYR901 | Val-892/Ile-893/Phe-894/Pro-895/Glu-896 | 13 | 8.22E + 06 |
| 228DXTNQVGHFQK238 | 889EAVNIFPEK897 | Val-892/Ile-893/Phe-894/Pro-895/Glu-896/Lys-897/Ser-899/Tyr-900 | 17 | 9.76E + 06 |
| 228DXTNQVGHFQK238 | 858TAFTMKK864 | Met-862/Lys-863 | 1 | 8.48E + 05 |
| 228DXTNQVGHFQK238 | 865DYGKEEREAQWALAQR880 | Lys-868/Glu-869/Glu-872/Arg-873 | 3 | 8.33E + 05 |
| 228DXTNQVGHFQK238 | 177DQSALTGESLPVTK190 | Ser-179/Ala-180/Leu-181/Gly-183 | 2 | 8.03E + 05 |
| 228DXTNQVGHFQK238 | 176VDQSALTGESLPVTK190 | Asp-177/Ser-179/Ala-180/Leu-181/Gly-183 | 2 | 7.76E + 05 |
| S229BPA_monomer | ||||
| 228DXTNQVGHFQK238 | 176VDQSALTGESLPVTK190 | Asp-177/Ser-179/Ala-180/Leu-181/Gly-183 | 4 | 1.02E + 06 |
| 228DXTNQVGHFQK238 | 177DQSALTGESLPVTK190 | Ser-179/Ala-180/Leu-181/Gly-183 | 3 | 5.50E + 05 |
| 228DXTNQVGHFQK238 | 172DPLKVDQSALTGESLPVTK190 | Leu-181/Ala-180/Gln-178 | 1 | 1.65E + 05 |
Notably, we found that S229BPA interacts with several peptides from the first 50 amino acids spanning region I and region B (R-I and R-B on Fig. 1A) of the C-terminal domain. In general, S229BPA interacts most efficiently with amino acids within peptide 889EAVNIFPEKGSYR901 in region B of the C-terminal domain of AHA2 as their precursor area and spectral counts are both more abundant compared with other cross-links (Table 1). Strikingly, this cross-linked product was detected only in the dimer and trimer bands but not in the monomer band of S229BPA treated with UV (Fig. 3A), suggesting that this is more likely an intermolecular cross-link between AHA2 monomers rather than an intramolecular one. In Fig. 3, B and C, examples of the MS/MS analysis of a cross-linked product between S229BPA and peptide 889–901 detected in the trimer and dimer bands of AHA2-S229BPA are shown. We also found that S229BPA interacts with two other peptides in region I of the C-terminal domain, peptide 858TAFTMKK864 (Fig. S2) and peptide 865DYGKEEREAQWALAQR880 (Fig. S3). Interestingly, both of these cross-link products are again observed only in the trimer and dimer bands but not in the monomer band. We also never observed these cross-linked products in the non-UV–treated samples, indicating that the S229BPA cross-links are dependent on UV irradiation. From the monomer band of S229BPA plus UV, we identified an intramolecular cross-link between S229BPA and a nearby peptide 176VDQSALTGESLPVTK190, within the actuator domain, and this intramolecular cross-linking was also detected in the dimer and trimer bands of Ser-229 treated with UV but not in the non-UV irradiated sample (Fig. S4).
Figure 3.

In UV-irradiated samples, MS and MS/MS analysis show a cross-linked product between S229BPA and peptide 889EAVNIFPEKGSYR901 in the trimer and dimer bands but not in the monomer band of AHA2-S229BPA. A, extracted ion chromatogram and mass spectrum showing a quadruply charged ion at m/z 734.109 in the trimer and dimer bands. B and C, fragment ion mass spectrum (MS/MS) of the precursor at m/z 734.109 in the trimer band (B) and dimer band (C). The fragment ions from peptide α are in red and from peptide β are in blue.
Identification of peptides cross-linked to H225BPA
Because H225BPA forms the same pattern of cross-linked dimers and trimers on SDS-PAGE as S229BPA and it is just four amino acids away from the S229BPA residue, we performed LC-MS/MS experiments to determine whether a similar interacting domain was observed. This time, two different double proteolytic digestions were performed, either with trypsin and chymotrypsin or trypsin and AspN. We found that H225BPA also cross-linked with several peptides in the C-terminal domain of AHA2. From the precursor area and spectral counts of the cross-linked peptide, we also found that H225BPA reacts most efficiently with peptide 889EAVNIFPEK897 in region B of the C-terminal domain (Table 2 and Table S2), and again the cross-linked peptides were only detected in the dimer and trimer bands of H225BPA treated with UV but not in the monomer band (Fig. 4). MS/MS fragmentation for H225BPA interacting with peptide 889–897 in the trimer band and the dimer band are shown in Fig. 4, B and C. In addition, we found H225BPA cross-linked to peptide 865DYGKEEREAQWALAQR880 in the trimer band only (Fig. S5) and to peptide 872EAQWALAQRTLHGLQPK888 to a greater extent in the trimer band compared with the dimer band (Fig. S6). These two peptides are in region I of the C-terminal domain. The cross-link of H225BPA to peptides in the C-terminal domain is observed exclusively in either trimer or dimer bands but not in the monomer, indicating that they are most likely the intermolecular cross-links between AHA2 subunits rather than an intramolecular link within the AHA2 monomer. We questioned whether the ability to identify a cross-link may depend on the ionization properties of peptides involved in the cross-link. However, from the spectral counts for several peptides in the C-terminal domain involved in cross-linking with S229BPA and H225BPA, we found that there is no significant difference in their observed abundance (Tables S3 and S4). Therefore, the specific cross-link of BPA to these peptides is more or less independent of ionization efficiency.
Table 2.
Summary of photo-cross-links identified in AHA2-H225BPA after in-gel digestion
Precursor area and spectral count were manually calculated from raw data using Xcalibur and StavroX.
| Peptides with a UV induced cross-link to H225BPA (MS1) | Possible amino acids cross-link to BPA | Spectral count | Precursor area | |
|---|---|---|---|---|
| H225BPA_trimer | ||||
| 223AAXLV227 | 889EAVNIFPEK897 | Phe-894/Pro-895/Glu-896/Ile-893 | 9 | 1.49E + 07 |
| 223AAXLV227 | 872EAQWALAQRTLHGLQPK888 | Ala-878/Leu-882/Pro-887 | 4 | 9.99E + 05 |
| 223AAXLV227 | 865DYGKEEREAQWALAQR880 | Gln-874/Glu-869/Lys-868 | 2 | 5.25E + 05 |
| 223AAXLVDSTNQVGHFQK238a | 889EAVNIFPEK897 | Phe-894/Pro-895/Glu-896/Ile-893 | 14 | 8.01E + 06 |
| 223AAXLVDSTNQVGHFQK238a | 881TLHGLQPK888 | Pro-887/Gln-886/Leu-885/Glu-884 | 3 | 2.32E + 06 |
| H225BPA_dimer | ||||
| 223AAXLV227 | 889EAVNIFPEK897 | Ile-893/Phe-894/Pro-895/Glu-896 | 6 | 9.98E + 06 |
| 223AAXLV227 | 872EAQWALAQRTLHGLQPK888 | Ala-878/Leu-882/Pro-887 | 1 | 9.35E + 04 |
| 223AAXLVDSTNQVGHFQK238 | 889EAVNIFPEK897 | Phe-894/Pro-895/Glu-896/Ile-893 | 11 | 5.30E + 06 |
a Samples were digested with trypsin and chymotrypsin, all others were digested with trypsin and AspN.
Figure 4.

MS and MS/MS analysis show a cross-linked product between H225BPA and peptide 889EAVNIFPEK897 in the trimer and dimer bands but not in the monomer band of AHA2-H225BPA upon UV irradiation. A, extracted ion chromatogram and mass spectrum showing a quadruply charged ion at m/z 728.621 in the trimer and dimer bands. B and C, fragment ion mass spectrum (MS/MS) of the precursor at m/z 728.621 in the trimer band (B) and dimer band (C). The fragment ions from peptide α are in red and from peptide β are in blue.
We attempted to analyze the cross-linked AHA peptides with cells containing F64BPA with various combinations of enzyme digestions and reaction conditions; however, we did not identify any cross-linked products in these samples. Because we have never been able to observe the peptide containing the F64BPA substitution itself and Phe-64 is also part of the first transmembrane domain, it is reasonable to conclude that the lack of success in pinpointing the cross-link with F64BPA is due to the technical challenges of working with hydrophobic peptides that behave poorly during the chromatographic and mass spectrometric procedures.
Discussion
In this work, we present the first evidence that the C-terminal domain of AHA2 directly interacts with the N-terminal actuator domain in vivo by using an unnatural amino acid as a photoaffinity cross-linker and light delivered to the enzyme in intact cells. Because the reactive volume of a benzophenone group is approximated as a sphere with a radius of 3.1 Å centered on the ketone oxygen (31), we believe that our results indicate that there is a direct and close contact of the N-terminal actuator domain with the first half of the C-terminal domain of AHA2 in the native enzyme in living cells. Importantly, our observation that the C-terminal domain interacts with two residues (H225BPA and S229BPA) that are both in the actuator loop corroborates previous genetic studies in which mutation of several residues in the actuator domain disrupts the inhibitory effect of the regulatory domain (8, 9). Furthermore, a cross-link between these two amino acids in the N-terminal actuator domain with both region B and region I of the C-terminal domain is consistent with previous genetic studies suggesting that the first 50 amino acids in this regulatory domain are directly involved in the interaction with the rest of AHA2, whereas the other half of C-terminal domain is involved in the interaction with the 14-3-3 protein (10, 12, 37). Interestingly, both H225BPA and S229BPA cross-link most efficiently with peptide 889–901 in region B, which contains a phosphorylation site Ser-899. As discussed previously, increased phosphorylation of this serine inhibits the enzyme with an unknown mechanism, and our observation underscores the potential importance of this residue for understanding pump regulation. The wide range of interactions of H225BPA and S229BPA with multiple amino acids in close proximity in these regions also reflects the highly dynamic nature of the C-terminal domain that may be the cause of failure to obtain information from X-ray crystallography experiments. It is also relevant to note that our cross-linking experiments are performed in live cells, and thus, any aberrant effect of denaturing detergents during protein extraction and purification is eliminated. Most importantly, our observation that the cross-link between S229BPA or H225BPA with the C-terminal domain is only found in the dimer and trimer bands but not in the monomer band supports a model in which the C-terminal domain's inhibitor actions are mediated by an intermolecular interaction with the actuator domain, as opposed to an intramolecular interaction. Our model for inhibition by intermolecular interaction is not meant to imply that this interaction mediates or is involved with oligomerization because there may be many points of contact and conformational changes involved in forming this higher-order structure.
Note that when samples were digested with trypsin and only ions with +1 charge state were rejected for MS/MS fragmentation, we were able to obtain 60% protein coverage that, while high, is less than 100%. A lower coverage (∼30%) was obtained when the samples were digested with trypsin/AspN or trypsin/chymotrypsin, and only ions with ≥+3 charge state were selected for fragmentation (for reference, the raw data were submitted to PRIDE). We excluded the lower charge ions to increase the ability to identify cross-linked dipeptides that are usually very low in abundance but have higher charge states than the unlinked peptides. Further studies using additional chemical cross-linking reagents, different proteolytic digestion, and different chromatographic procedures may provide evidence of putative intramolecular interactions that were missed in our study, but there is no evidence for this in our current work. If the polypeptides within a hexamer can exchange with polypeptides in a different hexamer molecule in vitro, isotopically labeled protein mixtures may help to confirm or refute our model in which intermolecular interactions between polypeptides predominate (38). Finally, our model predicts that genetic experiments in which cells contain both WT and mutant ATPase polypeptides should reveal dominant-negative mutations, i.e. “poison” subunits, that destabilize the hexamer.
We propose a model in which H+-ATPase can exist in two active states: active and hyper-active (Fig. 5). In the active state, AHA2 may exist as a mixture of homo-oligomers that includes dimeric, trimeric, tetrameric, or even hexameric species. However, these higher-order oligomers may be very labile in vivo in the absence of 14-3-3 protein bound to the phosphorylated penultimate threonine (16). In this normal active state, the C-terminal domain of one monomer interacts with and prevents the movement of actuator domains of the neighboring monomer in a “head-to-tail” fashion (Fig. 5). This interaction acts as an on and off switch during normal physiological conditions. Actuator domain movement is known to be important for dephosphorylation of the conserved catalytic aspartate in the phosphorylation domain (39, 40), and this movement may be inhibited when the first half of the C-terminal domain is interacting with the actuator domain. Once the other half of the C-terminal domain is phosphorylated, for example at the penultimate threonine (Thr-947) and then bound to 14-3-3, it will enter the hyper-active state perhaps via conformational changes in the first half of the C-terminal domain that cause the release of the actuator domain from all of the monomers. In this hyperactive state, the H+-ATPase forms a more stable and different type of hexamer complexed with six 14-3-3 proteins. This model accounts for the observation that in order to hyperactivate the H+-ATPase, six molecules of 14-3-3 are required to coordinate and completely release six C-terminal autoinhibitory domains of H+-ATPase from each monomer. Our model is a derivative of and is consistent with a previous study of the structure of the 14-3-3 protein complexed with 52 amino acids containing region II and bearing the penultimate threonine in the C terminus of N. plumbaginifolia PMA2. In this crystal structure, they observed the intermolecular interaction of region II of C-terminal peptides between two PMA2 monomers (17). This observation is consistent with our model in which 14-3-3's main role is to prevent the intermolecular head-to-tail interaction of the regulatory domain and the actuator domain, and when this interaction is blocked, other intermolecular interactions, e.g. of region II in two PMA2 monomers, may occur.
Figure 5.
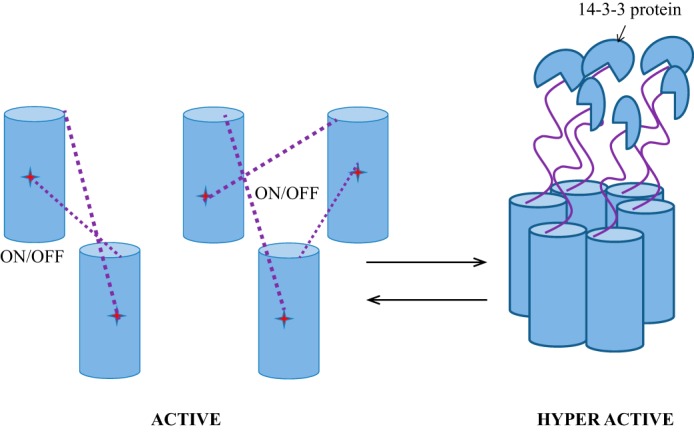
Model describing a head-to-tail interaction of the N-terminal actuator domain and the C-terminal regulatory domain of the 100,000 Da H+-ATPase in oligomeric AHA2. In the active state, the protein can exist as a dimer, as a trimer, or as a higher order oligomer in which the C-terminal domain of one monomer interacts with and inhibits the actuator movement of the neighboring unit. Under normal conditions, this inhibition may be switched on or off as the dynamic C-terminal domains swing back and forth to the N-terminal actuator domains. The enzyme can also enter a stable hyperactive state, as shown in the hexamers on the right, in which phosphorylation of the penultimate threonine results in the binding of a 14-3-3 protein to the C-terminal domain of each monomer, locking it into place so that it can no longer swing back to bind the actuator domain. It remains unclear whether the interaction between the actuator domain and the regulatory domain specifically mediates oligomerization, although hexamer formation may be required for hyperactivation.
In conclusion, the results of our experiments with a photolyzable cross-linking reagent incorporated and cross-linked in intact cells provide insights on the specific sites of interaction between regions of the AHA2 with the protein in its functional, native active state. Our results do not rule out the possibility that the C-terminal domain could interact with other parts of AHA2, such as the phosphorylation domain, which has been suggested to occur based on genetic studies (8, 9), but they do not provide evidence in its favor either. The hydrophobic domains in this protein present challenges that are not insurmountable for bottom up chemical studies such as ours that rely on traditional chromatographic techniques. With these same protocols for protein expression, purification, and digestion, there are many other mass spectrometric-based tools available, including hydrogen–deuterium exchange and hydroxy-radical footprinting, for studying protein conformation in solution, although in contrast to our approach with a photolyzable amino acid, these cannot yet be applied as readily to the enzyme in its native form in intact cells. Together with orthogonal technologies, such as site-directed mutagenesis and high-resolution EM, future experiments will provide additional means of testing this model.
Experimental procedures
Yeast strains, plasmid, and genetic techniques
All in vivo experiments were carried out in S. cerevisiae strain RS-72 (MATα, ade1-100, his4-519, leu2-3, 312, pPMA1:pGAL1) (32). The pPMA1:AHA2 and PMA1 terminator cassette from pMP1745 (41) was digested with HindIII and ligated to pRSII428 vector containing an ADE1 marker (Addgene). The His6-HA tag or Strep-HA tag was inserted at the N terminus of AHA2 using the QuikChange Lightning kit (Agilent). The QuikChange Lightning kit was also used to place the amber codon (TAG) in the coding sequence of AHA2, and all the constructs were subsequently confirmed by sequencing. ptRNA-BPA plasmid with LEU2 marker containing the nonsense suppressor tRNA/tRNA synthetase system was generously gifted by Anna Mapp (30).
In vivo photocross-linking
Yeast transformed with different mutant AHA2s containing the amber stop codon and the nonsense suppressor tRNA/tRNA synthetase were grown in selective medium (YNB + 100 mg/liter histidine + 2% galactose) for 2 days and then transferred to YPD medium (1% yeast extract, 2% peptone, 2% d-glucose) supplemented with 2 mm BPA (Chemimpex) and grown overnight. Cells were kept in the dark for the entire time. Yeast cells were harvested, suspended, and divided in two. One-half was irradiated with UV 365 nm for 30 min at 4 °C at a distance of 2 cm (Stratalinker 1800 UV lamp), and the other half was kept on ice in the dark as a negative control for no cross-linking. Approximately 0.15 g wet weight of yeast were resuspended in 1 ml of cell lysis buffer (50 mm MOPS, pH 7.4, 5% glycerol, 150 mm KCl, and protease inhibitor mixture was freshly added before experiment). Cell lysates were prepared using 0.5-mm glass beads with a vortex mixer with six to eight cycles of agitation (1 min on and 1 min off) in a cold room. The cell lysates were then spun at 16, 000 rcf at 4 °C using a tabletop centrifuge for 5 min. The supernatant was purified with the nickel-Sepharose purification system (GE Healthcare) and analyzed by immunoblotting with anti-HA antibody (Covance).
A large-scale preparation was used for MS using Strep-HA–tagged AHA2 constructs instead of His6-HA tag system (5–8 g of cells, wet weight for each sample was used). The cells were first grown in selective medium for 4 days and then transferred to YPD medium containing 2 mm BPA. Yeast cells were then harvested, resuspended in MilliQ water, and then divided into two parts: one part was kept in darkness and the other half was irradiated with UV at 365 nm for 45 min in a cold box at a distance of 2 cm, and the cells were agitated for 1 min for every 10 min during irradiation. The cells were then pelleted and broken using a cell disruptor (PBI) in the cell lysis buffer (20 mm HEPES, pH 7.6, 150 mm KCl, 1 mm EDTA, and protease inhibitor mixture was added before using). Cell debris was then removed by spinning down at 10,000 × g for 10 min in an SA800 rotor. The supernatant was then ultracentrifuged at 33,000 rpm for 3 h with a type 45Ti rotor (Beckman) to collect the microsomal fraction containing BPA–AHA2. The microsome was then solubilized and incubated with streptactin-Sepharose resin (GE Healthcare) overnight in the presence of 0.25% DDM (Anatrace) in the lysis buffer. The resin was then washed with lysis buffer in the presence of 0.025% DDM four times and eluted with 2.5 mm desthiobiotin in the lysis buffer supplemented with 0.025% DDM. The elutions were then run on a Bolt 4–12% BisTris gel or a NuPAGE 3–8% Tris acetate gel (Invitrogen) and stained with GelCode Blue stain (Invitrogen) for 30 min or overnight and distained in MilliQ water.
Drop test
Yeast was grown for 3 days at 28 °C in liquid selective medium (YNB + 100 mg/liter histidine + 2% galactose). Cells were then diluted to an optical density of 0.5 and 0.05 in the selective medium. 5 μl of cells were then spotted onto selective medium containing either 2% galactose at pH 6.5 or 2% glucose with 2 mm BPA at pH ∼6.5 (adjust pH after adding BPA with a pH-indicator strip). Growth was recorded after incubation for 4 days at 28 °C.
In-gel digestion
Bands corresponding to monomer, dimer, and trimer of BPA–AHA2 were excised after gel electrophoresis and GelCode Blue staining. The gel pieces were then de-stained with 50% MeOH, 100 mm NH4HCO3, dehydrated for 2 min in CH3CN/H2O/NH4HCO3 (50:50:25 mm) and then once more time for 30 s in 100% CH3CN, and finally dried in vacuum for 2 min. The gel pieces were then hydrated and reduced with 25 mm DTT in 25 mm NH4HCO3 for 15 min at 56 °C, alkylated with 55 mm iodoacetamide in 25 mm NH4HCO3 in darkness for 30 min, and dehydrated as in the previous step. Protein was digested with 200 ng of trypsin (Promega) at 42 °C for 3 h and then with 100 ng of AspN (Roche Applied Science) at 37 °C or 200 ng of chymotrypsin (Roche Applied Science) at room temperature overnight for double digestion. All the digestions were performed in 25 mm NH4HCO3 and 0.01% ProteaseMaxTM detergent (Promega). The digestion reactions were stopped with 0.3% trifluoroacetic acid (TFA), and the supernatant was transferred to a Protein LoBind tubes (∼60 μl volume). The residual proteolytic peptides were then extracted by addition of 75% CH3CN, 0.1% TFA and vortexed gently for 10 min. An additional extraction step was performed with 96% CH3CN, 0.1% TFA. All extractions were then combined, vacuum dried, and then resuspended in 30 μl of 0.05% TFA. Degraded ProteaseMAXTM was removed via centrifugation (maximum speed, 10 min), and the peptides were cleaned with a solid-phase support (Bond Elute OMIX C18 pipette tips from Agilent). Peptides were eluted off the C18 column with 15 μl of 96% CH3CN, 0.1% TFA, dried to a minimum volume, then brought up to a 12-μl total volume with 0.1% formic acid, and 3 μl was loaded onto the instrument. Three independent experiments were performed for the S229BPA sample, whereas two replicates were performed for H225BPA sample (Table S2).
NanoLC-MS/MS
Peptides were analyzed by nanoLC-MS/MS using the Agilent 1100 nanoflow system (Agilent) connected to a hybrid linear ion trap–orbitrap mass spectrometer (LTQ-Orbitrap EliteTM, ThermoFisher Scientific) equipped with an EASY-SprayTM electrospray source. Chromatography of peptides prior to mass spectral analysis was accomplished using a capillary emitter column (PepMap® C18, 3 μm, 100 Å, 150 × 0.075 mm, ThermoFisher Scientific) onto which 3 μl of extracted peptides was automatically loaded. The nanoHPLC system delivered solvents A, 0.1% (v/v) formic acid, and B, 99.9% (v/v) acetonitrile, 0.1% (v/v) formic acid at 0.50 μl/min to load the peptides (over a 30-min period) and 0.3 μl/min to elute peptides directly into the nano-electrospray with a gradual gradient from 3% (v/v) B to 20% (v/v) B over 18 min and concluded with a 5-min fast gradient from 20% (v/v) B to 50% (v/v) B, at which time a 4-min flash-out from 50 to 95% (v/v) B took place. As peptides eluted from the HPLC-column/electrospray source survey, MS scans were acquired in the Orbitrap with a resolution of 120,000 followed by MS2 for the 20 most intense peptides detected in the MS1 scan from 350 to 1800 m/z; redundancy was limited by dynamic exclusion with a repeat count of 2 and 15 s duration. Collision-induced dissociation-based MS/MS fragmentation in the ion trap portion of the instrument used an activation time of 10 ms, normalized collision energy of 35, and isolation width of 2 atomic mass units. Monoisotopic precursor selection and charge state screening were enabled, and +2 and lower charge states were rejected.
Data processing
To obtain identifications of uncross-linked peptides, raw MS/MS data were converted to mgf file format using MSConvert (ProteoWizard: Open Source Software for Rapid Proteomics Tools Development). Resulting mgf files were used to search against S. cerevisiae S288c RefSeq amino acid sequence database (5,913 protein entries) containing AHA2-BPA user construct plus a list of common contaminants using our in-house Mascot search engine 2.2.07 (Matrix Science) with variable methionine oxidation and asparagine and glutamine deamidation plus fixed cysteine carbamidomethylation. Peptide mass tolerance was set at 10 ppm and fragment mass at 0.6 Da. To determine the identity of cross-linked products, a targeted database search of raw mass spectrometry data was conducted using only the sequence of AHA2 proteins used in the cross-linking experiment plus the most abundant yeast proteins identified in the samples. Cross-linked candidates were generated using StavroX software (StavroX Freeware version 3.5.1 from University of Halle-Wittenberg) with BPA chosen as a cross-linker, trypsin, AspN, and chymotrypsin with two missed cleavages as protease digestion sites with static cysteine carbamidomethylation plus variable methionine oxidation as possible modifications. The candidates were manually verified based on the quality of MS1 and MS/MS data, matches with their theoretical isotopic and fragmentation distribution, and finally cross-checked against the non-UV–treated peptide mass spectrometry data from proteins used in the cross-linking experiment. Precursor area and spectral counts of cross-linked peptides were manually calculated from Xcalibur and StavroX correspondingly. The MS proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (42) partner repository with the dataset identifier PXD010892.
Author contributions
T. T. N. and M. R. S. conceptualization; T. T. N. formal analysis; T. T. N. investigation; T. T. N. visualization; T. T. N. methodology; T. T. N. writing-original draft; T. T. N., G. S., and M. R. S. writing-review and editing; G. S. and M. R. S. resources; M. R. S. supervision; M. R. S. funding acquisition; M. R. S. project administration.
Supplementary Material
Acknowledgments
We thank Jamison D. Wolfer and Jeremy Volkening for technical support; Pei Liu and Greg Barrett-Wilt for MS assistance; Amanda Laseke for assistance with yeast cell culture; Brian J. Conti and Matthew R. Blackburn for critical reading of the manuscript; and all other laboratory members for their helpful discussion and support.
This work was supported in part by Basic Energy Sciences Grant DEFG02-88ER13938 (to M. R. S.) from the Department of Energy and by National Science Foundation Grant MCB-1713899 (to M. R. S.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S6, Tables S1–S4 and supporting Refs. 1–9.
The MS proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier accession no. PXD010892.
- BPA
- p-benzoylphenylalanine
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- DDM
- n-dodecyl β-d-maltopyranoside.
References
- 1. Kühlbrandt W. (2004) Biology, structure and mechanism of P-type ATPases. Nat. Rev. Mol. Cell Biol. 5, 282–295 10.1038/nrm1354 [DOI] [PubMed] [Google Scholar]
- 2. Morsomme P., and Boutry M. (2000) The plant plasma membrane H+-ATPase: structure, function and regulation. Biochim. Biophys. Acta 1465, 1–16 10.1016/S0005-2736(00)00128-0 [DOI] [PubMed] [Google Scholar]
- 3. Palmgren M. G. (2001) Plant plasma membrane H+-ATPases: powerhouses for nutrient uptake. Annu. Rev. Plant Physiol. Plant Mol. Biol. 52, 817–845 10.1146/annurev.arplant.52.1.817 [DOI] [PubMed] [Google Scholar]
- 4. Pedersen B. P., Buch-Pedersen M. J., Morth J. P., Palmgren M. G., and Nissen P. (2007) Crystal structure of the plasma membrane proton pump. Nature 450, 1111–1114 10.1038/nature06417 [DOI] [PubMed] [Google Scholar]
- 5. Regenberg B., Villalba J. M., Lanfermeijer F. C., and Palmgren M. G. (1995) C-terminal deletion analysis of plant plasma membrane H+-ATPase: yeast as a model system for solute transport across the plant plasma membrane. Plant Cell 7, 1655–1666 10.1105/tpc.7.10.1655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Palmgren M. G., Larsson C., and Sommarin M. (1990) Proteolytic activation of the plant plasma membrane H+-ATPase by removal of a terminal segment. J. Biol. Chem. 265, 13423–13426 [PubMed] [Google Scholar]
- 7. Palmgren M. G., Sommarin M., Serrano R., and Larsson C. (1991) Identification of an autoinhibitory domain in the C-terminal region of the plant plasma membrane H+-ATPase. J. Biol. Chem. 266, 20470–20475 [PubMed] [Google Scholar]
- 8. Morsomme P., de Kerchove d'Exaerde A., De Meester S., Thinès D., Goffeau A., and Boutry M. (1996) Single point mutations in various domains of a plant plasma membrane H+-ATPase expressed in S. cerevisiae increase H+-pumping and permit yeast growth at low pH. EMBO J. 15, 5513–5526 10.1002/j.1460-2075.1996.tb00936.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Morsomme P., Dambly S., Maudoux O., and Boutry M. (1998) Single point mutations distributed in 10 soluble and membrane regions of the Nicotiana plumbaginifolia plasma membrane PMA2 H+-ATPase activate the enzyme and modify the structure of the C-terminal region. J. Biol. Chem. 273, 34837–34842 10.1074/jbc.273.52.34837 [DOI] [PubMed] [Google Scholar]
- 10. Axelsen K. B., Venema K., Jahn T., Baunsgaard L., and Palmgren M. G. (1999) Molecular dissection of the C-terminal regulatory domain of the plant plasma membrane H+-ATPase AHA2: mapping of residues that when altered give rise to an activated enzyme. Biochemistry 38, 7227–7234 10.1021/bi982482l [DOI] [PubMed] [Google Scholar]
- 11. Baunsgaard L., Venema K., Axelsen K. B., Villalba J. M., Welling A., Wollenweber B., and Palmgren M. G. (1996) Modified plant plasma membrane H+-ATPase with improved transport coupling efficiency identified by mutant selection in yeast. Plant J. 10, 451–458 10.1046/j.1365-313X.1996.10030451.x [DOI] [PubMed] [Google Scholar]
- 12. Speth C., Jaspert N., Marcon C., and Oecking C. (2010) Regulation of the plant plasma membrane H +-ATPase by its C-terminal domain: what do we know for sure? Eur. J. Cell Biol. 89, 145–151 10.1016/j.ejcb.2009.10.015 [DOI] [PubMed] [Google Scholar]
- 13. Haruta M., Sabat G., Stecker K., Minkoff B. B., and Sussman M. R. (2014) A peptide hormone and its receptor protein kinase regulate plant cell expansion. Science 343, 408–411 10.1126/science.1244454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Piette A. S., Derua R., Waelkens E., Boutry M., and Duby G. (2011) A phosphorylation in the C-terminal auto-inhibitory domain of the plant plasma membrane H+-ATPase activates the enzyme with no requirement for regulatory 14-3-3 proteins. J. Biol. Chem. 286, 18474–18482 10.1074/jbc.M110.211953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Niittylä T., Fuglsang A. T., Palmgren M. G., Frommer W. B., and Schulze W. X. (2007) Temporal analysis of sucrose-induced phosphorylation changes in plasma membrane proteins of Arabidopsis. Mol. Cell. Proteomics 6, 1711–1726 10.1074/mcp.M700164-MCP200 [DOI] [PubMed] [Google Scholar]
- 16. Kinoshita T., and Shimazaki K. (1999) Blue light activates the plasma membrane H+-ATPase by phosphorylation of the C terminus in stomatal guard cells. EMBO J. 18, 5548–5558 10.1093/emboj/18.20.5548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ottmann C., Marco S., Jaspert N., Marcon C., Schauer N., Weyand M., Vandermeeren C., Duby G., Boutry M., Wittinghofer A., Rigaud J. L., and Oecking C. (2007) Structure of a 14-3-3 coordinated hexamer of the plant plasma membrane H+-ATPase by combining X-ray crystallography and electron cryomicroscopy. Mol. Cell 25, 427–440 10.1016/j.molcel.2006.12.017 [DOI] [PubMed] [Google Scholar]
- 18. Kanczewska J., Marco S., Vandermeeren C., Maudoux O., Rigaud J. L., and Boutry M. (2005) Activation of the plant plasma membrane H+-ATPase by phosphorylation and binding of 14-3-3 proteins converts a dimer into a hexamer. Proc. Natl. Acad. Sci. U.S.A. 102, 11675–11680 10.1073/pnas.0504498102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Goormaghtigh E., Chadwick C., and Scarborough G. A. (1986) Monomers of the Neurospora plasma membrane H+-ATPase catalyze efficient proton translocation. J. Biol. Chem. 261, 7466–7471 [PubMed] [Google Scholar]
- 20. Justesen B. H., Hansen R. W., Martens H. J., Theorin L., Palmgren M. G., Martinez K. L., Pomorski T. G., and Fuglsang A. T. (2013) Active plasma membrane P-type H+-ATPase reconstituted into nanodiscs is a monomer. J. Biol. Chem. 288, 26419–26429 10.1074/jbc.M112.446948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bagnat M., Chang A., and Simons K. (2001) Plasma membrane proton ATPase Pma1p requires raft association for surface delivery in yeast. Mol. Biol. Cell 12, 4129–4138 10.1091/mbc.12.12.4129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lee M. C., Hamamoto S., and Schekman R. (2002) Ceramide biosynthesis is required for the formation of the oligomeric H+-ATPase Pma1p in the yeast endoplasmic reticulum. J. Biol. Chem. 277, 22395–22401 10.1074/jbc.M200450200 [DOI] [PubMed] [Google Scholar]
- 23. Dufour J. P., and Goffeau A. (1980) Molecular and kinetic properties of the purified plasma membrane ATPase of the yeast Schizosaccharomyces pombe. Eur. J. Biochem. 105, 145–154 10.1111/j.1432-1033.1980.tb04484.x [DOI] [PubMed] [Google Scholar]
- 24. Chadwick C. C., Goormaghtigh E., and Scarborough G. A. (1987) A hexameric form of the N. crassa plasma membrane H+-ATPase. Arch. Biochem. Biophys. 252, 348–356 10.1016/0003-9861(87)90041-5 [DOI] [PubMed] [Google Scholar]
- 25. Auer M., Scarborough G. A., and Kühlbrandt W. (1999) Surface crystallisation of the plasma membrane H+-ATPase on a carbon support film for electron crystallography. J. Mol. Biol. 287, 961–968 10.1006/jmbi.1999.2652 [DOI] [PubMed] [Google Scholar]
- 26. Bowman B. J., Berenski C. J., and Jung C. Y. (1985) Size of the plasma membrane H+-ATPase from N. crassa determined by radiation inactivation and comparison with the sarcoplasmic reticulum Ca2+-ATPase from skeletal muscle. J. Biol. Chem. 260, 8726–8730 [PubMed] [Google Scholar]
- 27. Briskin D. P., Thornley W. R., and Roti-Roti J. L. (1985) Target molecular size of the red beet plasma membrane ATPase. Plant Physiol. 78, 642–644 10.1104/pp.78.3.642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chin J. W., and Schultz P. G. (2002) In vivo photocrosslinking with unnatural amino acid mutagenesis. Chembiochem 3, 1135–1137 10.1002/1439-7633(20021104)3:11%3C1135::AID-CBIC1135%3E3.0.CO%3B2-M [DOI] [PubMed] [Google Scholar]
- 29. Chin J. W., Cropp T. A., Anderson J. C., Mukherji M., Zhang Z., and Schultz P. G. (2003) An expanded eukaryotic genetic code. Science 301, 964–967 10.1126/science.1084772 [DOI] [PubMed] [Google Scholar]
- 30. Krishnamurthy M., Dugan A., Nwokoye A., Fung Y. H., Lancia J. K., Majmudar C. Y., and Mapp A. K. (2011) Caught in the act: covalent cross-linking captures activator-coactivator interactions in vivo. ACS Chem. Biol. 6, 1321–1326 10.1021/cb200308e [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dormán G., and Prestwich G. D. (1994) Benzophenone photophores in biochemistry. Biochemistry 33, 5661–5673 10.1021/bi00185a001 [DOI] [PubMed] [Google Scholar]
- 32. Cid A., Perona R., and Serrano R. (1987) Replacement of the promoter of the yeast plasma membrane ATPase gene by a galactose-dependent promoter and its physiological consequences. Curr. Genet. 12, 105–110 10.1007/BF00434664 [DOI] [PubMed] [Google Scholar]
- 33. Portillo F. (1997) Characterization of dominant lethal mutations in the yeast plasma membrane H+-ATPase gene. FEBS Lett. 402, 136–140 10.1016/S0014-5793(96)01515-3 [DOI] [PubMed] [Google Scholar]
- 34. Merlot S., Leonhardt N., Fenzi F., Valon C., Costa M., Piette L., Vavasseur A., Genty B., Boivin K., Müller A., Giraudat J., and Leung J. (2007) Constitutive activation of a plasma membrane H+-ATPase prevents abscisic acid-mediated stomatal closure. EMBO J. 26, 3216–3226 10.1038/sj.emboj.7601750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Götze M., Pettelkau J., Schaks S., Bosse K., Ihling C. H., Krauth F., Fritzsche R., Kühn U., and Sinz A. (2012) StavroX–a software for analyzing crosslinked products in protein interaction studies. J. Am. Soc. Mass Spectrom. 23, 76–87 10.1007/s13361-011-0261-2 [DOI] [PubMed] [Google Scholar]
- 36. Iacobucci C., and Sinz A. (2017) To be or not to be? Five guidelines to avoid misassignments in cross-linking/mass spectrometry. Anal. Chem. 89, 7832–7835 10.1021/acs.analchem.7b02316 [DOI] [PubMed] [Google Scholar]
- 37. Fuglsang A. T., Borch J., Bych K., Jahn T. P., Roepstorff P., and Palmgren M. G. (2003) The binding site for regulatory 14-3-3 protein in plant plasma membrane H+-ATPase: involvement of a region promoting phosphorylation-independent interaction in addition to the phosphorylation-dependent C-terminal end. J. Biol. Chem. 278, 42266–42272 10.1074/jbc.M306707200 [DOI] [PubMed] [Google Scholar]
- 38. Pettelkau J., Thondorf I., Theisgen S., Lilie H., Schröder T., Arlt C., Ihling C. H., and Sinz A. (2013) Structural analysis of guanylyl cyclase-activating protein-2 (GCAP-2) homodimer by stable isotope-labeling, chemical cross-linking, and mass spectrometry. J. Am. Soc. Mass Spectrom. 24, 1969–1979 10.1007/s13361-013-0734-6 [DOI] [PubMed] [Google Scholar]
- 39. Clausen J. D., Vilsen B., McIntosh D. B., Einholm A. P., and Andersen J. P. (2004) Glutamate-183 in the conserved TGES motif of domain A of sarcoplasmic reticulum Ca2+-ATPase assists in catalysis of E2/E2P partial reactions. Proc. Natl. Acad. Sci. U.S.A. 101, 2776–2781 10.1073/pnas.0400122101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Toyoshima C. (2009) How Ca2+-ATPase pumps ions across the sarcoplasmic reticulum membrane. Biochim. Biophys. Acta 1793, 941–946 10.1016/j.bbamcr.2008.10.008 [DOI] [PubMed] [Google Scholar]
- 41. Fuglsang A. T., Guo Y., Cuin T. A., Qiu Q., Song C., Kristiansen K. A., Bych K., Schulz A., Shabala S., Schumaker K. S., Palmgren M. G., and Zhu J. K. (2007) Arabidopsis protein kinase PKS5 inhibits the plasma membrane H+-ATPase by preventing interaction with 14-3-3 protein. Plant Cell 19, 1617–1634 10.1105/tpc.105.035626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Vizcaío J. A., Csordas A., del-Toro N., Dianes J. A., Griss J., Lavidas I., Mayer G., Perez-Riverol Y., Reisinger F., Ternent T., Xu Q. W., Wang R., and Hermjakob H. (2016) 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 44, D447–D456 10.1093/nar/gkv1145 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.