

Abstract
Pseudomonas aeruginosa is an opportunistic human pathogen that causes nosocomial infections. The P. aeruginosa outer membrane contains specific porins that enable substrate uptake, with the outer membrane protein OprG facilitating transport of small, uncharged amino acids. However, the pore size of an eight-stranded β-barrel monomer of OprG is too narrow to accommodate even the smallest transported amino acid, glycine, raising the question of how OprG facilitates amino acid uptake. Pro-92 of OprG is critically important for amino acid transport, with a P92A substitution inhibiting transport and the NMR structure of this variant revealing that this substitution produces structural changes in the barrel rim and restricts loop motions. OprG may assemble into oligomers in the outer membrane (OM) whose subunit interfaces could form a transport channel. Here, we explored the contributions of the oligomeric state and the extracellular loops to OprG's function. Using chemical cross-linking to determine the oligomeric structures of both WT and P92A OprG in native outer membranes and atomic force microscopy, and single-molecule fluorescence of the purified proteins reconstituted into lipid bilayers, we found that both protein variants form oligomers, supporting the notion that subunit interfaces in the oligomer could provide a pathway for amino acid transport. Furthermore, performing transport assays with loop-deleted OprG variants, we found that these variants also can transport small amino acids, indicating that the loops are not solely responsible for substrate transport. We propose that OprG functions as an oligomer and that conformational changes in the barrel–loop region might be crucial for its activity.
Keywords: outer membrane, membrane protein, Pseudomonas aeruginosa (P. aeruginosa), transporter, atomic force microscopy (AFM), single-molecule biophysics, fluorescence, chemical modification
Introduction
Pseudomonas aeruginosa (Pa) is an opportunistic human pathogen. P. aeruginosa infections account for 10–15% of total nosocomial infections worldwide (1) and pose a major threat to cystic fibrosis and immunocompromised patients (2). Its ability to form biofilms on various surfaces results in antibiotic resistance (3–5). Multidrug resistance usually originates partially in the outer membrane (OM),2 which is the first line of defense against noxious molecules (6–8). The OM is a highly complex structural assembly with an asymmetric lipid bilayer (8, 9). The composition of the inner leaflet of the OM is similar to that of the cytoplasmic membrane, i.e. it consists mainly of phosphatidylethanolamine and phosphatidylglycerol phospholipids. However, the outer leaflet of the OM is composed of a network of complex lipopolysaccharides (LPS) (9, 10). The LPS layer provides a major barrier for hydrophobic compounds. The OM of P. aeruginosa lacks general diffusion porins that are found in other Gram-negative bacteria (3). Instead, the OM of P. aeruginosa hosts ∼30 specific porins that facilitate the strictly controlled uptake of various substrates (3, 11). This specificity of porins makes the OM of P. aeruginosa less permeable to antibiotics than the OMs of other Gram-negative bacteria (6, 11, 12).
Porins are β-barrels with channels either located within monomers or formed by oligomers (13). OprG is one of the smallest porins in the outer membrane of P. aeruginosa (14, 15). OprG belongs to the widely distributed but poorly studied OmpW porin family. OprG has been hypothesized to transport hydrophobic compounds (15), cations (16), and iron (Fe2+) across the OM in aerobic growth environments (17). In support of this hypothesis, a recent report confirmed a role of OmpW from Acinetobacter baumanii in iron homeostasis (18). In addition, OprG has been shown to transport small amino acids including glycine, alanine, valine, serine, and threonine (19).
Both crystal and solution NMR structures showed OprG to be an eight-stranded β-barrel with four extracellular loops (15, 19). The NMR structure revealed that these extracellular loops are highly dynamic (19). Based on the crystal structure it has been proposed that, after binding to the loop region, hydrophobic compounds escape into the OM through a lateral gate formed by three conserved proline residues, Pro-66, Pro-91, and Pro-92, on the extracellular side of the β-barrel (15). However, in the NMR structure, this lateral gate does not exist. In addition, Pro-92 was in the transition region from loop 3 to the barrel, whereas Pro-66 and Pro-91 were part of the dynamic extracellular loops in the NMR structure (19). Mutation of the conserved Pro-92 to Ala resulted in inhibition of amino acid transport as assessed by a liposome swelling assay (19). Comparison of the NMR structures of the P92A mutant and WT showed that the barrel rim is asymmetric and loop 3 is more ordered in the mutant (19). Based on these observations, it has been suggested that these structural differences could be the reason for the inability of P92A to transport small amino acids (19). However, it is still not clear how OprG provides a pathway for amino acid transport. All structures show that the lumen within a single OprG monomer is too small to accommodate the passage of even the smallest amino acid, glycine (Fig. 1A). Therefore, the major question of how OprG is able to facilitate the uptake of small molecules remains. A plausible hypothesis is that OprG may assemble into oligomers in the OM and that the interfaces between subunits could form a channel facilitating amino acid transport. In addition, the extracellular loops and/or conformational changes in the β-barrel region may aid in substrate recognition and transport.
Figure 1.
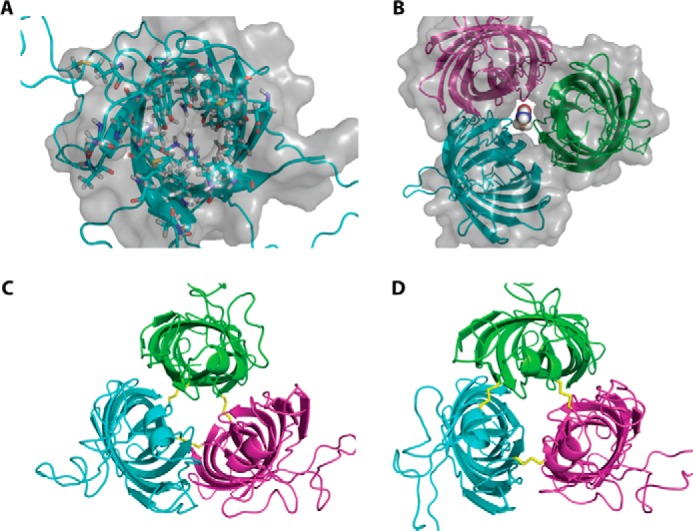
Structure models of monomeric and hypothetical trimeric OprG. A, top view of the NMR structure of OprG (PDB code 2N6L). Residues pointing inside the barrel are shown as ball and stick models and the solvent accessible surface of the barrel as a gray translucent volume. This clearly indicates that there is no luminal channel large enough to transport small amino acids. B, homology model of a hypothetical OprG trimer based on the crystal structure of OmpW (PDB code 2F1V). The solvent accessible surface shows a channel along the trimeric interface large enough to accommodate the amino acid glycine. C, disulfide-linked trimer of T65C–L90C OprG based on the homology model of B. D, disulfide-linked trimer of S128C–S136C OprG based on the homology model of B.
In this work, we explored two questions: 1) what is the quaternary structure of OprG in native outer membranes of P. aeruginosa and when reconstituted into artificial lipid bilayers; and 2) do the extracellular loops play a role in facilitating amino acid transport? Our results indicate that OprG forms monomers and dimers with a small population of higher order oligomers. Furthermore, we did not observe any differences in quaternary structure between WT and P92A OprG. Deletion of individual loops did not affect the transport of amino acids as assessed by liposome swelling. Therefore, we speculate that whereas the quaternary structure of OprG may play a crucial role, conformational changes in the barrel region could have an additional important function in facilitating amino acid transport.
Results
Cross-linking studies of OprG in outer membranes of P. aeruginosa
Because the lumen of the OprG barrel is completely blocked by the inside-facing side chains (Fig. 1A) and cannot accommodate even the smallest translocated amino acid, glycine, we hypothesized that transport might occur along subunit interfaces of an oligomer. The crystal structure of the Escherichia coli homolog OmpW (PDB code 2F1V) shows a dimer of trimers in its asymmetric unit. Although the dimer with its perpendicularly arranged subunits is clearly a crystallographic artifact, the OmpW trimer could have physiological relevance. We hence created an OprG homology model based on this OmpW trimer (Fig. 1B). This assembly features a central pore that is large enough to accommodate small amino acids. To check if OprG could form similar oligomers in its native environment, we engineered double cysteine mutants, which would allow us to trap such assemblies by thiol-reactive cross-linkers.
For an initial screen, we expressed OprG bearing an N-terminal StrepTag II for detection and cysteines in positions 65 and 90, 65 and 120, 44 and 142, 128 and 136, or 128 and 137, respectively, in P. aeruginosa PAO1. These pairs of cysteines were chosen based on proximity of neighboring subunits in the homology model. Expected Cys–Cys cross-links between T65C–L90C and S128C–S136C are shown in Fig. 1, C and D, respectively. Expected cross-links between T65C–L120C, Q44C–D142C, and S128C–S137C are shown in Fig. S1, A–C, respectively. We then isolated the outer membranes from these cells and reacted them with cross-linkers of varying length, rigidity, and hydrophobicity. Western blotting revealed the presence of monomers, dimers, and higher order oligomers of OprG (Fig. 2A). Remarkably, most samples showed a band for folded, monomeric OprG, although all samples had been boiled in SDS-PAGE sample buffer for 15 min. This indicates that OprG is considerably more stable in its native lipid environment as compared with refolded samples in micelles or proteoliposomes. Although all double cysteine mutants showed faint bands for cross-linked dimers, we focused on the 128Cys–136Cys construct, which yielded very strong dimer bands and faint bands for higher order oligomers. As we could not observe striking differences between the tested cross-linkers, we concentrated on CuP as a reversible, zero-length, catalytic cross-linker and oPDM as an irreversible, stoichiometric cross-linker of approximately 5–8 nm length (20).
Figure 2.

Cross-linking of OprG oligomers in native membranes. A, Western blotting against StrepTag II of outer membrane vesicles from P. aeruginosa PAO1 expressing five different double cysteine mutants of OprG as isolated (2nd lanes) and cross-linked with CuP (1st lanes), BM(PEG)3 (3rd lanes), or oPDM (4th lanes), respectively. Outer membrane vesicles were treated with cross-linker, quenched, boiled in nonreducing, denaturing sample buffer before SDS-PAGE and Western blot analysis. The expected position of unfolded OprG monomers to tetramers are indicated by arrows. B, Coomassie Brilliant Blue stained SDS-PAGE of purified, cross-linked OprG double cysteine mutants. Outer membrane vesicles from P. aeruginosa PAO1 expressing the 128Cys–Cys136 mutant of WT or P92A OprG were cross-linked with oPDM or CuP, respectively, and solubilized in SB 3–14. Cross-linked OprG oligomers were then purified over StrepTactin XT resin and separated by size exclusion chromatography over Superdex 200. Samples were run boiled (+) and non-boiled (−) to assess folding states of the β-barrel. The expected positions of unfolded OprG monomers to tetramers are indicated by arrows. For the reversible cross-linker CuP, samples were also run after reduction to monomers by β-mercaptoethanol (β-ME). Coomassie-stained SDS-PAGE gels are representatives of experiments that were replicated three (independent purifications) to 10 (gels) times. The labels for molecular weight markers (MW) apply to all markers in the rows of A and B.
We proceeded to check whether there is a difference in oligomerization behavior between the active WT and the inactive P92A mutant by cross-linking the respective 128Cys–136Cys double cysteine constructs in isolated outer membranes. To rule out that we observed arbitrary cross-linking to other OMPs, we isolated OprG after cross-linking in outer membrane vesicles by affinity chromatography using its StrepTag II and fractionated the eluate by size exclusion chromatography into theoretical monomer, dimer, and oligomer fractions (Fig. 2B). Cleavage of the reversible CuP cross-link by β-mercaptoethanol led to a single band corresponding to monomeric OprG. This indicates that OprG does not interact with other Cys-containing proteins and that we indeed trapped oligomeric OprG assemblies. As there were no apparent differences between the cross-linkers CuP and oPDM, we conclude that the interaction in the OprG oligomers is mostly due to flexible and exposed regions such as the dynamic extracellular loops.
Even though our homology model was a trimer, we observed mostly monomers and dimers with a very small population of trimers and higher order oligomers. This would be expected for transient, short-lived, and weak subunit interaction. Given that OmpW and OprG usually purify as monomers when extracted from native membranes (15, 21), these trapped oligomeric states are most likely held together by transient and weak interactions. Additionally, our experiments were performed in P. aeruginosa PAO1 in the background of endogenous WT OprG, which would not cross-link when being part of an oligomeric assembly. Because we also induced only low expression levels of our double cysteine constructs to avoid artificially producing oligomers by overcrowding the outer membrane, the predominance of monomers and dimers over higher order oligomers as observed in our experiments was expected. We also note that if our homology model derived from E. coli OmpW is imperfect and the actual molecular interfaces between subunits of OprG are slightly different from the model, some bifunctional Cys mutant molecules of OprG could also form higher order oligomers with more than three subunits as observed as minority species under some conditions of Fig. 2.
Atomic force microscopy (AFM) of OprG in lipid bilayers
To further probe the topography and oligomeric state of OprG in lipid bilayers, we purified WT and P92A mutant OprG and reconstituted them separately into liposomes. These proteoliposomes were deposited onto freshly cleaved mica surfaces for AFM imaging. The presence of the membrane was confirmed by observation of the characteristic ∼4 nm thickness of the lipid bilayer at the edges of membrane patches (Fig. 3, A and B). Pits with ∼4 nm depths were also observed to confirm the presence of the membrane. Mica-supported membranes containing WT or P92A OprG displayed numerous punctuate protrusions (light brown objects in Fig. 3, A and B). In contrast, supported membranes lacking proteins show no such protrusions (Fig. S2), confirming that the observed protrusions represent molecules of OprG. These protrusions were uniformly distributed in the membranes.
Figure 3.

AFM of OprG in lipid bilayers. A, representative image of WT OprG in lipid bilayer. B, representative image of P92A OprG in lipid bilayer. Lipid bilayers were identified by measuring step heights at the edges, which can be seen in both images. Protrusions of the individual OprG particles above the membrane were visualized by AFM (light brown color). The full color scale is shown on the right. C and D, height-volume distributions of membrane protrusions of WT (C) and P92A (D) OprG in lipid bilayers. E and F, modeling of the experimental volume distributions (red) of WT (E) and P92A (F) OprG as superpositions of Gaussians (dashed blue lines). The sums of these Gaussians are shown in black.
Analyses of several hundred protrusions from different membrane areas and different proteoliposome preparations were performed using software that determines the maximal height and volume of each feature. These results are summarized in two-dimensional heat maps in Fig. 3, C and D. The majority of the heights were in the range of 0.5 to 2.5 nm, with a prominent mode at ∼1 nm. Height distributions of membrane protrusions of WT and P92A OprG were very similar, indicating a similar topography for both proteins. OprG displays topographic asymmetry between the maximum heights of the protrusion on the periplasmic face compared with the extracellular face of the membrane. Based on the crystal structure of OprG, we roughly estimated the height of the extracellular region above the membrane as ∼1.3 nm (Fig. S3). Therefore we attribute the heights ∼1 to 2.5 nm to the extracellular region of OprG. This broad range of heights most likely results from the prominent conformational dynamic of the loops of OprG (19). As AFM images were collected in membranes in aqueous solutions and at room temperature, considerable conformational dynamics of the proteins are expected to be preserved (22, 23).
The majority of volumes of membrane protrusions were in the range of 0.3 to 4 × 105 Å3 with some volumes up to about 6 × 105 Å3 for both WT and P92A. The largest infrequently observed volumes were likely protein aggregates (not shown). In general, volumes measured by AFM are convoluted by the tip geometry. However, because we used similar AFM probes (biolever mini) and experimental conditions to collect all data, the effects of tip geometry should be comparable in all cases. Moreover, data collected on many different samples and different reconstitutions are very similar in terms of heights and volumes. Therefore, a comparison of the volumes of both proteins side by side is justified and should yield valuable information. The monomeric volume of OprG was estimated by dilating the extracellular region of the crystal structure (PDB entry 2X27) with a standard AFM tip geometry, producing an estimated volume of 0.8 × 105 Å3 (Fig. S4). In addition, manual inspection of several features revealed that protrusions with single Gaussian-like protrusions (Fig. S5, A and C) occupied volumes in the range of 0.5 to 1.8 × 105 Å3 (Fig. 3, C–F). Therefore, we attribute this population to monomeric OprG.
Although overall volume distributions were very similar for both WT and P92A OprG, modeling of volumes revealed small differences (Fig. 3, E and F) between the principal peaks of WT and P92A mutant OprG. For instance, WT OprG has two similar populations within the first major peak. In contrast, P92A has one major peak and a small secondary peak on the shoulder of the major peak. These data suggest that OprG likely exists in two different monomeric conformations and that the P92A mutant prefers one conformation over the other. Interestingly, the solution NMR structure of WT and P92A mutant OprG in DPC micelles showed that P92A OprG features an asymmetric barrel rim and a more rigid loop 3 (19). Therefore, we speculate that the observed small differences in volumes could be due to the different conformations of OprG that were revealed by NMR. A third distribution with a prominent peak around ∼2.3 × 105 Å3 was observed for both WT and P92A OprG. These volumes likely represent the dimeric state of OprG. Some features in this size range exhibited two Gaussian-like protrusions (Fig. S5, B and D). We also observed a minor fourth fraction with volumes ∼4 to 6 × 105 Å3 in the distributions of both proteins. This fraction, which could represent trimers or tetramers, is slightly more populated in the case of the P92A mutant than in the case of WT OprG. Taken together, the analysis of the protrusions observed by AFM is consistent with the cross-linking data of OprG in native OMs (Fig. 2), indicating that monomers, dimers, and higher order oligomers of OprG were observed by both methods.
Single molecule photobleaching of OprG in lipid bilayers
To further analyze the stoichiometry of OprG oligomers, we fluorescently labeled single cysteine mutants of OprG with Alexa 647 and reconstituted them into lipid bilayers. Using total internal reflection fluorescence microscopy, we determined the number of steps in which Alexa 647 labeled OprG photobleaches (24, 25). Two representative intensity traces of individual molecules with one and two photobleaching steps are shown in Fig. 4, A and B. We analyzed a total of 541 individual particles from 10 different sample areas and two different reconstitutions. The percentages of particles with one, two, and three steps are summarized in Fig. 4C. The maximum number of bleaching steps observed in our samples was three. Because part of the sample bleached during focusing of the microscope, the measured bleaching step distribution does not represent the distribution of oligomeric states. We determined that 30% of the fluorophores were bleached before image acquisition (see “Experimental procedures”). We calculated the binomial distributions based on the observed steps (three, two, and one) and the probability that 70% of all fluorophores were unbleached at the beginning of time-lapse imaging. From this, we determined the fraction of dimers (green area in first bar of Fig. 4C) and trimers (red area in first bar of Fig. 4C) within the observed population of particles with one step, and the fraction of dimers and trimers within the observed population of particles with two steps (green and red areas, respectively, in second bar of Fig. 4C). The total percentages of particles in monomeric, dimeric, and trimeric states are shown in Fig. 4D: 48% of the observed particles were monomers, 44% were dimers, and 8% were trimers. These results clearly indicate that OprG forms monomers, dimers, and a small population of trimers in membranes, which is consistent with and supports the results obtained by chemical cross-linking and AFM.
Figure 4.

Single molecule photobleaching of OprG in lipid bilayers. A and B, bleaching steps of single fluorescent protein complexes in lipid bilayers reveal the numbers of Alexa 647-labeled subunits. Examples of single-step (A) and two-step (B) bleaching traces are shown. The black lines represent mean intensity values from 5 × 5 pixel2 regions. The red lines represent the Chung-Kennedy nonlinear filtered versions of the data. C, average fractions of observed bleaching steps from 10 movies of two different protein reconstitutions. Division of bars into blue, green, and red illustrate the calculated fractions of monomers, dimers, and trimers within one observed population, as explained under “Experimental procedures.” D, total oligomeric fractions of monomers (blue), dimers (green), and trimers (red) are calculated from data in C.
Amino acid transport by loop deletion mutants of OprG
As the inactivating P92A mutation led to reduced dynamics of the extracellular loops, particularly of loop 3, we expressed in E. coli OprG mutants where single extracellular loops were replaced by short turns. We reconstituted these loop deletion mutants into liposomes and tested their ability to translocate small, uncharged amino acids (Fig. 5, A–D). Surprisingly, the deletion of single loops had no effect on the translocation of all tested amino acids (alanine, glycine, serine, and valine). This likely indicates that the flexible extracellular loops are either not directly involved in transport or that individual loops can mechanistically substitute for each other in facilitating amino acid transport. Therefore, the observed stiffening of loop 3 in the P92A mutant is probably not the cause for the lack of activity of this mutant. Unfortunately, we were not able to test the effects of double loop deletion mutants and a complete quadruple loop deletion mutant because these cumulative deletion mutants expressed and refolded poorly and therefore could not be reproducibly reconstituted into proteoliposomes. Nevertheless, the loop deletion results suggest that most likely conformational changes in the barrel region are responsible for the quaternary structure of OprG and are crucial for its function of facilitating transport of small amino acids.
Figure 5.

Amino acid transport of loop deletion mutants of OprG. Liposome-swelling assay with reconstituted loop deletion mutants ΔL1, ΔL2, ΔL3, and ΔL4 of OprG in the presence of amino acids known to be transported by the WT OprG. The data are presented as mean ± S.D. (dashed line) of two to three biological replicates, i.e. repeated purifications and refoldings of loop deletion mutants, each with triplicate permeation measurements. Data for WT OprG (wt) and protein-free liposomes (blank) are shown as positive and negative controls, respectively.
Discussion
In this work, we used three different approaches to show that OprG forms monomers, dimers, and a small population of higher order oligomers in native and reconstituted membranes. Furthermore, we showed that the quaternary structures of WT and the transport-deficient mutant P92A OprG were very similar to each other. Moreover, deletion of each of the four extracellular loops did not inhibit the transport of small amino acids. Cross-linking of double cysteine mutants in native Pseudomonas OMs yielded a strong dimer band and weaker higher order oligomeric bands on SDS gels. It is interesting to note that both cysteines of the most efficient cross-linking double mutant (Cys128–Cys136) are located in extracellular loop 3 suggesting that loop 3 contributes to the oligomeric interaction between OprG subunits (Fig. 1D). However, there must also be additional interfaces in the barrel region that support oligomerization because all individual loop deletion mutants were still active amino acid transporters. For comparison, strands 1–5 and 16 contribute to intra-membranous protein–protein interactions of the trimeric E. coli porin OmpF (26). A computational study using an empirical energy potential function showed that these strands are predicted to be only weakly stable and therefore at a higher energy than the other strands of OmpF. Interactions with strands of a neighboring subunit lowers their energy and thus stabilizes the OmpF trimer through protein–protein interactions within the membrane (26). Therefore, it is possible that weakly stable β-strand regions of different monomers interact with similar regions in neighboring subunits to form functional oligomers of OprG (27).
Monomers and dimers were the predominant species of OprG as assessed by multiple approaches to determine its oligomeric state in membranes. However, trimers and higher order oligomers were also detected by all methods. As mentioned, there is no luminal channel passing through the monomeric OprG structure. Therefore, the detection of dimers and trimers makes them better candidates for the active forms of the OprG transporter even if these species are relatively infrequent. Previously, it has been shown that OprG displays a single channel conductance of 500 pS in 1 m KCl (16). More recently, another group showed that the OprG channel has a conductance of 330 pS (28). These values are large and not consistent with a pore through a small monomeric β-barrel; they are more typical for oligomeric porins. Dimeric porins are rare, but it has been shown previously that dimers of OmpC are stable and have channel activity when reconstituted into membranes (29). Although trimers are the predominant species of OmpF in vivo, OmpF dimers have been observed in vitro and have been proposed to be assembly intermediates toward forming functional trimers of OmpF (30). Thus, the observed OprG dimers could also be incomplete functional intermediates on a pathway to form more active higher order oligomers. It is possible that the actual concentrations of OprG in intact cells could be higher than in our outer membrane vesicles and proteoliposomes. Moreover, the presence of LPS in outer membranes could stabilize certain oligomeric species in native membranes. Therefore, we cannot rule out the possibility that OprG might exist in higher order oligomers in cellular environments and that these may be more populated in cells than in the model membranes. Further experiments will be required to determine the oligomeric state of OprG in cellular environments.
Protein conformations around the highly conserved prolines in the barrel-to-loop transition region are also important for the function of OprG as previously shown for Pro92 (19). Prolines in transmembrane domains and elsewhere have been suggested to be important for substrate transport across membranes in many α-helical transporters (31). For instance, Pro28 has been shown to be important for the function of lac permease (32). In case of the phenylalanine permease PheP, Pro341 has been shown to be critical for substrate transport (33). Therefore and based on the fact that the Pro92 mutation does not appear to change the quaternary structure of OprG, it is likely that Pro92 contributes to local structural features that affect substrate transport by OprG. Although Pro92 provides a good starting point for tracking down the transport mechanism of OprG, further experiments will be needed to find the pathway for substrate transport through the functional oligomer of OprG.
Experimental procedures
Expression plasmids for P. aeruginosa
The oprG gene including the N-terminal leader sequence for periplasmic export was cloned from P. aeruginosa PAO1 into the expression plasmid pHERD30T (a kind gift from J. Goldberg, Emory University) (34) by overlap extension PCR (35) with Q5 high-fidelity DNA polymerase (New England Biololabs), primers 1 and 2 (Table S1) and a boiled suspension of P. aeruginosa PAO1 in water as a source of genomic DNA. Subsequently, a StrepTag II (AS-WSHPQFEK-GS) was inserted at position 22 between the signal sequence and the N terminus of mature OprG by PCR-based insertion using primer pair 3 and 4 (Table S1). Re-circularization with T4 DNA ligase (New England Biololabs) yielded pHERD30T-ST2-oprG.
The double cysteine constructs 65Cys–90Cys, 65 Cys–120Cys, and 44Cys–142Cys were created by multichange isothermal assembly (36) using mixed forward and reverse primer pairs from Table S1 in a PCR and subsequently assembling the resulting two fragments with NEBuilder HiFi DNA Assembly kit (New England Biolabs). The constructs 128Cys–136Cys and 128Cys–137Cys were created by site-directed mutagenesis using primer pairs 11 and 12 and 11 and 13 (Table S1), respectively. Plasmids were propagated in E. coli DH5α in LB medium with 15 μg/ml of gentamicin at 37 °C.
Transformation of P. aeruginosa
P. aeruginosa PAO1 was transformed with pHERD30T-ST2-oprG or the derived double cysteine constructs by electroporation (37). First, electrocompetent cells were prepared from an overnight culture of P. aeruginosa PAO1 in 6 ml of LB by pelleting (5000 × g, 10 min, 4 °C) and resuspending in 1 ml of cold, sterile 300 mm sucrose solution. This procedure was repeated three times and the final pellet was resuspended in only 200 μl of sucrose solution. Electrocompetent cells were kept on ice until immediate use.
For electroporation, 50 μl of electrocompetent cells were mixed with 1 μl of plasmid DNA (approximately 50 ng/μl in H2O) and transferred to an electroporation cuvette with a 2-mm gap (Molecular BioProducts, catalogue no. 5520). Immediately after applying a 5-ms pulse of 2.5 kV (Bio-Rad electroporator; E. coli Pulser), 450 μl of super optimal broth with catabolite repression (SOC) medium were added and the cells were allowed to recover in a shaking incubator at 37 °C for 1 h before plating on LB agar containing 30 μg/ml of gentamicin.
Expression of OprG double cysteine mutants with N-terminal StrepTag II in P. aeruginosa
500 μl of an overnight culture of P. aeruginosa PAO1 transformed with the desired pHERD30T-ST2-oprG variant was used to inoculate 50 ml of LB supplemented with 2 mm MgSO4 and 30 μg/ml of gentamicin. Cells were grown with vigorous shaking at 37 °C. At an OD of 0.8, an additional 2 mm MgSO4 were added, and OprG expression was induced by addition of l-arabinose to 0.4% (w/v). Cells were harvested 2 h after induction by centrifugation (5251 × g, 4 °C, 15 min).
Preparation of outer membrane vesicles
P. aeruginosa outer membrane vesicles were prepared following a procedure modified from Ref. 38. Briefly, harvested cells from a 50 ml of P. aeruginosa culture were resuspended in 4 ml of PBS supplemented with 10 mm EDTA and incubated with end-over-end rotation for 1 h at room temperature with vigorous mixing every 15 min. Cellular debris was removed by low speed centrifugation (5251 × g, 4 °C, 15 min) and the supernatant was subjected to ultracentrifugation (TLA100.3 rotor, 69,000 rpm, 4 °C, 60 min). The pellet containing the membranes was resuspended in 500 μl of PBS by multiple passages through needles with increasingly smaller gauge (19, 25, and 27 gauge). The membrane suspension was flash frozen in liquid nitrogen and stored at −80 °C.
Cross-linking OprG double cysteine mutants in outer membrane vesicles
All cross-linker stocks were freshly prepared immediately before use. 1,11-Bismaleimido-triethyleneglycol (BM(PEG)3) and N,N′-(o-phenylene)dimaleimide (oPDM) were dissolved in anhydrous dimethylformamide at a concentration of 20 mm. 50 mm copper 1,10-phenanthroline sulfate (CuP) was prepared by mixing equal volumes of 100 mm CuSO4 in H2O and 200 mm 1,10-phenanthroline in ethanol. Suspensions of outer membrane vesicles were mixed by end-over-end rotation at room temperature for 1 h while receiving additions of cross-linker every 15 min resulting in a total concentration of 400 μm cross-linker. Reactions were quenched by addition of 1 m EDTA (CuP) or 1 m DTT (BM(PEG)3 and oPDM) to a final concentration of 10 mm. Cross-linked outer membrane vesicles were either analyzed by SDS-PAGE and Western blotting after direct mixing with SDS-PAGE sample buffer or used for the isolation of OprG oligomers.
Isolation of OprG from outer membranes
P. aeruginosa outer membrane suspensions were supplemented with 11 mm MgSO4 and 1% (w/v) Triton X-100 and mixed by end-over-end rotation for 1 h at room temperature to solubilize inner membrane contaminations. The insoluble, LPS-containing outer membrane vesicles were pelleted (TLA120.1 rotor, 80,000 rpm, 4 °C, 1 h) and dissolved in PBS supplemented with 5 mm EDTA and 0.5% (w/v) 3-(N,N-dimethylmyristylammonio) propanesulfonate (SB 3–14) by 30 min incubation at room temperature and 2× 15-min sonications. Insoluble matter was removed by ultracentrifugation (TLA120.1 rotor, 80,000 rpm, 4 °C, 1 h) and the solubilized OprG was purified over Strep-Tactin XT resin (IBA Lifesciences) according to the manufacturer's instructions but with 0.05% (w/v) SB 3–14 in the wash and elution buffers. The eluate was concentrated to less than 250 μl (Amicon Ultracell, 30 kDa MWCO) and subjected to size exclusion chromatography over a Superdex 200 HR 10/30 column in a buffer containing 20 mm Tris-HCl, pH 8.0, 150 mm NaCl, 1 mm EDTA, and 0.05% (w/v) SB 3–14.
Expression of OprG in E. coli and its purification and refolding
The P. aeruginosa strain PA01 OprG construct without its N-terminal signal sequence was cloned into pET30a vector containing the T7 promoter. To prepare a single cysteine mutant, glycine 134 was mutated to cysteine (Stratagene QuikChange Site-directed Mutagenesis Kit 200518). WT, P92A, and G134C OprG were expressed in E. coli strain BL21 (DE3) cells. Cells were grown at 37 °C to an optical density of 0.6. Expression was induced by adding 1 mm isopropyl β-d-thiogalactoside, and cells were further incubated for 4 h at 37 °C. Harvesting, lysis, separation of inclusion bodies from cell fractions, and washing of inclusion bodies was similar to previously published methods (19). Inclusion bodies were solubilized in 10 mm Tris-Cl, pH 8.0, 8 m urea, 0.1 mm EDTA and stirred at room temperature for 1 h. The solubilized inclusion bodies were separated from cell debris by centrifugation (Type 45 Ti rotor, 45,000 rpm, 12 °C, 40 min). The supernatant was filtered (polyvinylidene difluoride, 0.45 μm membrane, Millex) and loaded onto a DEAE-Sepharose column and eluted with a 0–250 mm NaCl gradient in solubilization buffer. Elution fractions were pooled and concentrated using Amicon Ultra-15 centrifugal filter units (10,000 MWCO membrane, Millipore). Refolding of OprG into DHPC micelles was carried out following a published protocol (19). Refolded protein was exchanged into β-octylglucoside (Anatrace) before it was reconstituted into proteoliposomes.
Expression of single loop deletions of OprG and their purification and refolding
Primers 16–23 in Table S1 were used to delete single extracellular loops (ΔL1, ΔL2, ΔL3, ΔL4) from the previously described pET30a-oprG expression plasmid (19) by site-directed mutagenesis (QuikChange Site-directed Mutagenesis Kit 200518). Parent DNA coding for OprG and the forward and reverse primers of each desired mutation were cycled 30 times in a PCR where the annealing step was set to 68 °C. Amplification products were digested for 1 h with DpnI. DNA was transformed into XL1-Blue supercompetent cells, number 200236, from Agilent Technologies. Transformation guidelines were followed as described in the QuikChange mutagenesis kit. Cells were plated and incubated overnight at 37 °C. One colony was selected for a 6-ml overnight culture shaken at 225 rpm and 37 °C. The Qiagen QIAprep Miniprep kit was used to purify DNA for sequence analysis. Successfully mutated DNA was stored at −20 °C. The loop deletion mutants were expressed as inclusion bodies in E. coli, purified, refolded in DHPC, and reconstituted into proteoliposomes for liposome swelling assays as previously described (19).
Reconstitution of refolded WT and P92A OprG into liposomes
1-Palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine (POPE) and 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-(1′-rac-glycerol) (POPG) lipids in chloroform were mixed at a 80:20 ratio and blown dry with N2. Lipid films were further dried overnight in a desiccator using house vacuum. The dried lipid films were suspended in 10 mm Tris-Cl, pH 7.6, 150 mm KCl and incubated at room temperature for 1 h. Unilamellar liposomes were prepared by extrusion through polycarbonate membranes (∼100 nm pore diameter, Avestin) 30 times. To prepare proteoliposomes, the liposomes were saturated using a detergent (β-octylglucoside)-to-lipid (∼1 mm) ratio of 1.3 (39). After saturating for 3 h, OprG in β-octylglucoside was added at ∼1 μm and incubated for 1 h at room temperature. To remove the detergent, proteoliposomes were dialyzed (10,000 MWCO, Slide-A-Lyzer Dialysis Cassette) overnight at 4 °C against 10 mm Tris-Cl, pH 7.6, 150 mm KCl buffer. The proteoliposomes were isolated by centrifugation (TLS-55 rotor, 259,000 × g, 4 °C, 40 min). The pellet was suspended in 10 mm Tris-Cl, pH 7.6, 150 mm KCl buffer, and the suspension was stored at −80 °C. More detailed methods for expression, purification, refolding, and reconstitution of OprG and for the liposome swelling assay to measure the amino acid transport activity of WT and loop deletion mutants of OprG can be found elsewhere (19).
Atomic force microscopy imaging and data analysis
Proteoliposomes were diluted to ∼100 nm OprG, 100 μm lipid in imaging buffer (10 mm Tris, pH 7.6, 150 mm KCl), deposited on freshly cleaved highest grade V1 mica (TedPella), and incubated for 30 min. During this incubation time proteoliposomes were allowed to form supported lipid bilayers via vesicle fusion. The sample was then rinsed three times with ∼100 μl of imaging buffer to remove loosely attached vesicles. AFM images were acquired in imaging buffer in force-distance mode using a Bruker Nanoscope Multimode 8 (Bruker, Santa Barbara, CA) equipped with a 10 μm scanner (E-scanner). Biolever mini-cantilevers (BL-AC40TS, Olympus) with measured spring constants ∼0.06 newton/m were used to collect images. Images were collected with an applied maximum force <100 piconewton, an oscillation frequency of 2 kHz, and an oscillation amplitude of 25 nm. Imaging conditions were similar for all data sets reported in this work. Individual molecules of OprG do not laterally diffuse in these membranes that are directly supported on mica. However, all our images are those of samples that are equilibrated in proteoliposomes before they were deposited and therefore should represent snapshots of free standing membranes in equilibrium.
Individual OprG molecules were automatically identified and extracted from within large-scale AFM images via the Hessian blob algorithm (40) in Igor Pro 7 (WaveMetrics, Inc.), allowing for precise determination of particle boundaries. Local background levels within individual particles were obtained via Laplace interpolation with boundary conditions provided by neighboring background pixels. Particle heights were measured as the maximum heights above the background level attained within the particle. Particle volumes were measured from the sums over all pixels within the particles. The sums of background-subtracted pixel heights were scaled by the pixel areas to determine the volumes of the space occupied by each particle.
Distributions of particle heights and volumes were generated via kernel density estimation, which is a smoother and more stable alternative to standard histograms. The optimal Epanechnikov kernel (41) was used to generate distributions using custom software in Igor Pro (WaveMetrics, Inc.), with a bandwidth of 2 Å for height distributions and 0.35 × 105 Å3 for volume distributions. Two-dimensional heat maps, displaying the distribution of heights and volumes simultaneously, were analogously generated using a bivariate kernel density estimation. A deconvolution of the volume distributions into individual Gaussian populations was performed via the least-squares fit of a sum of independent Gaussian peaks to the distributions. The number of Gaussian populations in each fit was determined to produce the best fits with the smallest number of peaks. The curve fitting functionality of Igor Pro was used to compute the fits.
Alexa 647 labeling of OprG G134C and its reconstitution into liposomes
Following the manufacturer's protocol, the maleimide derivative of Alexa Fluor® 647 (catalogue number A20347, ThermoFisher Scientific) was conjugated to the thiol group in purified OprG G134C before it was refolded. Labeled OprG was separated from the free dye by Sephadex G-25 gel filtration over a PD-10 column (GE Healthcare Life Sciences). The labeling efficiency was ∼100% as assessed by UV-visible spectroscopy. Labeled proteins were refolded into DHPC micelles as described above and exchanged into β-octylglucoside detergent (Fig. S6) before reconstituting them into liposomes composed of POPE:POPG (80:20) at a protein-to-lipid ratio of 1:107 following the above described reconstitution protocol, except that the ultracentrifugation step was omitted.
Single molecule photobleaching and analysis
Proteoliposomes were allowed to form supported lipid bilayers on piranha solution (mixture of concentrated sulfuric acid with hydrogen peroxide, in a ratio of 3:1) cleaned quartz slides. After 30 to 60 min incubation, the slides were rinsed with imaging buffer (10 mm Tris-Cl, pH 7.6, 150 mm KCl) to remove loosely attached vesicles. Single molecule photobleaching experiments were carried out on a Zeiss Axiovert 200 fluorescence microscope (Carl Zeiss, Thornwood, NY), equipped with a ×63 water immersion objective (Zeiss; numerical aperture 0.95) and a prism-based total internal reflection fluorescence illumination system. A laser beam from a diode laser (Cube 640, Coherent) emitting light at 640 nm was directed into a prism above the quartz slide at an angle of 72° from the normal to illuminate the sample by total internal reflection at the quartz/water interface, creating an evanescent wave with a characteristic penetration depth ∼130 nm. Fluorescence signals were recorded by an electron-multiplying charge-coupled device camera (iXon DV887ESC-BV, Andor Technologies). The laser intensity, light-blocking shutters, and cameras were controlled by a homemade program written in LabVIEW (National Instruments). Images of 512 × 512 pixel2 (corresponding to a sample area of 128 × 128 μm2) were acquired with an exposure time of 100 ms and a frame rate of 10 Hz. Particles were identified using an adapted and previously described single particle tracking algorithm (42, 43). The mean fluorescence intensities of a 5 × 5 pixel2 region of interest around each identified particle was plotted as a function of time. The numbers and intensities of bleaching steps were recorded manually for each trace. We used the Cheng-Kennedy filtering technique (44) to accurately identify drops in signal intensities that directly correlate with bleaching steps in each trace. In our experiments, it took roughly 10 s to focus on the sample before data acquisition was started. To accurately quantify the oligomeric state, bleaching during the focusing on the sample was measured on a sample with a high protein-to-lipid ratio (1:1000). Approximately 30% of the fluorophores were bleached during the first 10 s of image acquisition. Binomial distributions for varying stoichiometries of OprG were calculated using the probability that 70% of all Alexa 647 molecules were fluorescent during the recording time.
Author contributions
R. R. S. G., P. S., and L. K. T. conceptualization; R. R. S. G. data curation; R. R. S. G., P. S., and B. M. formal analysis; R. R. S. G. and P. S. investigation; R. R. S. G. and P. S. visualization; R. R. S. G., P. S., V. K., and C. E. C. methodology; R. R. S. G. writing-original draft; B. M. and V. K. software; C. E. C. and L. K. T. supervision; L. K. T. funding acquisition; L. K. T. writing-review and editing.
Supplementary Material
Acknowledgment
We thank Dr. Iga Kucharska for preparing the loop deletion plasmids of OprG.
This work was supported by National Institutes of Health Grant R01 GM051329. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Table S1 and Figs. S1–S6.
- OM
- outer membrane
- LPS
- lipopolysaccharide
- AFM
- atomic force microscopy
- BM(PEG)3
- 1,11-bismaleimido-triethyleneglycol
- CuP
- copper 1,10-phenanthroline sulfate
- oPDM
- N,N′-(o-phenylene)dimaleimide)
- SB 3–14
- 3-(N,N-dimethylmyristylammonio)propanesulfonate
- DHPC
- 1,2-diheptanoyl-sn-glycero-3-phosphocholine
- POPE
- 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine
- POPG
- 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-(1′-rac-glycerol)
- PDB
- Protein Data Bank.
References
- 1. Strateva T., and Yordanov D. (2009) Pseudomonas aeruginosa: a phenomenon of bacterial resistance. J. Med. Microbiol. 58, 1133–1148 10.1099/jmm.0.009142-0 [DOI] [PubMed] [Google Scholar]
- 2. Kang C. I., Kim S. H., Kim H. B., Park S. W., Choe Y. J., Oh M. D., Kim E. C., and Choe K. W. (2003) Pseudomonas aeruginosa bacteremia: risk factors for mortality and influence of delayed receipt of effective antimicrobial therapy on clinical outcome. Clin. Infect. Dis. 37, 745–751 10.1086/377200 [DOI] [PubMed] [Google Scholar]
- 3. Hancock R. E., and Brinkman F. S. (2002) Function of pseudomonas porins in uptake and efflux. Annu. Rev. Microbiol. 56, 17–38 10.1146/annurev.micro.56.012302.160310 [DOI] [PubMed] [Google Scholar]
- 4. Davies D. (2003) Understanding biofilm resistance to antibacterial agents. Nat. Rev. Drug Discov. 2, 114–122 10.1038/nrd1008 [DOI] [PubMed] [Google Scholar]
- 5. Hall-Stoodley L., Costerton J. W., and Stoodley P. (2004) Bacterial biofilms: from the natural environment to infectious diseases. Nat. Rev. Microbiol. 2, 95–108 10.1038/nrmicro821 [DOI] [PubMed] [Google Scholar]
- 6. Delcour A. H. (2009) Outer membrane permeability and antibiotic resistance. Biochim. Biophys. Acta 1794, 808–816 10.1016/j.bbapap.2008.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Masuda N., Sakagawa E., and Ohya S. (1995) Outer membrane proteins responsible for multiple drug resistance in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 39, 645–649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Silhavy T. J., Kahne D., and Walker S. (2010) The bacterial cell envelope. Cold Spring Harb. Perspect. Biol. 2, a000414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ruiz N., Kahne D., and Silhavy T. J. (2006) Advances in understanding bacterial outer-membrane biogenesis. Nat. Rev. Microbiol. 4, 57–66 10.1038/nrmicro1322 [DOI] [PubMed] [Google Scholar]
- 10. Clifton L. A., Holt S. A., Hughes A. V., Daulton E. L., Arunmanee W., Heinrich F., Khalid S., Jefferies D., Charlton T. R., Webster J. R., Kinane C. J., and Lakey J. H. (2015) An accurate in vitro model of the E. coli envelope. Angew. Chem. Int. Ed. Engl. 54, 11952–11955 10.1002/anie.201504287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chevalier S., Bouffartigues E., Bodilis J., Maillot O., Lesouhaitier O., Feuilloley M. G., Orange N., Dufour A., and Cornelis P. (2017) Structure, function and regulation of Pseudomonas aeruginosa porins. FEMS Microbiol. Rev. 41, 698–722 10.1093/femsre/fux020 [DOI] [PubMed] [Google Scholar]
- 12. Pagès J., James C. E., and Winterhalter M. (2008) The porin and the permeating antibiotic: a selective diffusion barrier in Gram-negative bacteria. Nat. Rev. Microbiol. 6, 893–903 10.1038/nrmicro1994 [DOI] [PubMed] [Google Scholar]
- 13. Kim K. H., Aulakh S., and Paetzel M. (2012) The bacterial outer membrane β-barrel assembly machinery. Protein Sci. 21, 751–768 10.1002/pro.2069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gensberg K., Smith A. W., Brinkman F. S., and Hancock R. E. (1999) Identification of oprG, a gene encoding a major outer membrane protein of Pseudomonas aeruginosa. J. Antimicrob. Chemother. 43, 607–608 10.1093/jac/43.4.607 [DOI] [PubMed] [Google Scholar]
- 15. Touw D. S., Patel D. R., and van den Berg B. (2010) The crystal structure of OprG from Pseudomonas aeruginosa, a potential channel for transport of hydrophobic molecules across the outer membrane. PloS One 5, e15016 10.1371/journal.pone.0015016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. McPhee J. B., Tamber S., Bains M., Maier E., Gellatly S., Lo A., Benz R., and Hancock R. E. (2009) The major outer membrane protein OprG of Pseudomonas aeruginosa contributes to cytotoxicity and forms an anaerobically regulated, cation-selective channel. FEMS Microbiol. Lett. 296, 241–247 10.1111/j.1574-6968.2009.01651.x [DOI] [PubMed] [Google Scholar]
- 17. Yates J. M., Morris G., and Brown M. R. (1989) Effect of iron concentration and growth rate on the expression of protein G in Pseudomonas aeruginosa. FEMS Microbiol. Lett. 58, 259–262 10.1111/j.1574-6968.1989.tb03055.x [DOI] [PubMed] [Google Scholar]
- 18. Catel-Ferreira M., Marti S., Guillon L., Jara L., Coadou G., Molle V., Bouffartigues E., Bou G., Shalk I., Jouenne T., Vila-Farrés X., and Dé E. (2016) The outer membrane porin OmpW of Acinetobacter baumannii is involved in iron uptake and colistin binding. FEBS Lett. 590, 224–231 10.1002/1873-3468.12050 [DOI] [PubMed] [Google Scholar]
- 19. Kucharska I., Seelheim P., Edrington T., Liang B., and Tamm L. K. (2015) OprG harnesses the dynamics of its extracellular loops to transport small amino acids across the outer membrane of Pseudomonas aeruginosa. Structure 23, 2234–2245 10.1016/j.str.2015.10.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nitao L. K., and Reisler E. (1998) Probing the conformational states of the SH1-SH2 helix in myosin: a cross-linking approach. Biochemistry 37, 16704–16710 10.1021/bi9817212 [DOI] [PubMed] [Google Scholar]
- 21. Hong H., Patel D. R., Tamm L. K., and van den Berg B. (2006) The outer membrane protein OmpW forms an eight-stranded beta-barrel with a hydrophobic channel. J. Biol. Chem. 281, 7568–7577, March 17 [DOI] [PubMed] [Google Scholar]
- 22. Müller D. J., and Dufrêne Y. F. (2008) Atomic force microscopy as a multifunctional molecular toolbox in nanobiotechnology. Nat. Nanotechnol. 3, 261–269 10.1038/nnano.2008.100 [DOI] [PubMed] [Google Scholar]
- 23. Sanganna Gari R. R., Frey N. C., Mao C., Randall L. L., and King G. M. (2013) Dynamic structure of the translocon SecYEG in membrane: direct single molecule observations. J. Biol. Chem. 288, 16848–16854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Subburaj Y., Cosentino K., Axmann M., Pedrueza-Villalmanzo E., Hermann E., Bleicken S., Spatz J., and García-Sáez A. J. (2015) Bax monomers form dimer units in the membrane that further self-assemble into multiple oligomeric species. Nat. Commun. 6, 8042 10.1038/ncomms9042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Blackburn M. R., Hubbard C., Kiessling V., Bi Y., Kloss B., Tamm L. K., and Zimmer J. (2018) Distinct reaction mechanisms for hyaluronan biosynthesis in different kingdoms of life. Glycobiology 28, 108–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Naveed H., Jackups R. Jr., and Liang J. (2009) Predicting weakly stable regions, oligomerization state, and protein-protein interfaces in transmembrane domains of outer membrane proteins. Proc. Natl. Acad. Sci. U.S.A. 106, 12735–12740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Naveed H., and Liang J. (2014) Weakly stable regions and protein-protein interactions in β-barrel membrane proteins. Curr. Pharm. Des. 20, 1268–1273 10.2174/13816128113199990071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Schwarzer T. S., Hermann M., Krishnan S., Simmel F. C., and Castiglione K. (2017) Preparative refolding of small monomeric outer membrane proteins. Protein Expr. Purif. 132, 171–181 10.1016/j.pep.2017.01.012 [DOI] [PubMed] [Google Scholar]
- 29. Rocque W. J., and McGroarty E. J. (1989) Isolation and preliminary characterization of wild-type OmpC porin dimers from Escherichia coli K-12. Biochemistry 28, 3738–3743 10.1021/bi00435a017 [DOI] [PubMed] [Google Scholar]
- 30. Visudtiphole V., Thomas M. B., Chalton D. A., and Lakey J. H. (2005) Refolding of Escherichia coli outer membrane protein F in detergent creates LPS-free trimers and asymmetric dimers. Biochem. J. 392, 375–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Brandl C. J., and Deber C. M. (1986) Hypothesis about the function of membrane-buried proline residues in transport proteins. Proc. Natl. Acad. Sci. U.S.A. 83, 917–921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Consler T. G., Tsolas O., and Kaback H. R. (1991) Role of proline residues in the structure and function of a membrane transport protein. Biochemistry 30, 1291–1298 10.1021/bi00219a019 [DOI] [PubMed] [Google Scholar]
- 33. Pi J., Dogovski C., and Pittard A. J. (1998) Functional consequences of changing proline residues in the phenylalanine-specific permease of Escherichia coli. J. Bacteriol. 180, 5515–5519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Qiu D., Damron F. H., Mima T., Schweizer H. P., and Yu H. D. (2008) PBAD-based shuttle vectors for functional analysis of toxic and highly regulated genes in Pseudomonas and Burkholderia spp., and other bacteria. Appl. Environ. Microbiol. December; 74, 7422–7426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. van den Ent F., and Löwe J. (2006) RF cloning: a restriction-free method for inserting target genes into plasmids. J. Biochem. Biophys. Methods 67, 67–74 10.1016/j.jbbm.2005.12.008 [DOI] [PubMed] [Google Scholar]
- 36. Mitchell L. A., Cai Y., Taylor M., Noronha A. M., Chuang J., Dai L., and Boeke J. D. (2013) Multichange isothermal mutagenesis: a new strategy for multiple site-directed mutations in plasmid DNA. ACS Synth. Biol. 2, 473–477 10.1021/sb300131w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Smith A. W., and Iglewski B. H. (1989) Transformation of Pseudomonas aeruginosa by electroporation. Nucleic Acids Res. 17, 10509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Benz R., Jones M. D., Younas F., Maier E., Modi N., Mentele R, Lottspeich F, Kleinekathöfer U., and Smit J. (2015) OmpW of Caulobacter crescentus functions as an outer membrane channel for cations. PloS One 10, e0143557 10.1371/journal.pone.0143557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rigaud J. L., and Lévy D. (2003) Reconstitution of membrane proteins into liposomes. Methods Enzymol. 372, 65–86 10.1016/S0076-6879(03)72004-7 [DOI] [PubMed] [Google Scholar]
- 40. Marsh B. P., Chada N., Sanganna Gari R. R., Sigdel K. P., and King G. M. (2018) The Hessian blob algorithm: precise particle detection in atomic force microscopy imagery. Sci. Rep. 8, 978 10.1038/s41598-018-19379-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Epanechnikov V. A. (1969) Non-parametric estimation of a multivariate probability density. Theory Prob. Appl. 14, 153–158 10.1137/1114019 [DOI] [Google Scholar]
- 42. Domanska M. K., Kiessling V., Stein A., Fasshauer D, and Tamm L. K. (2009) Single vesicle millisecond fusion kinetics reveals number of SNARE complexes optimal for fast SNARE-mediated membrane fusion. J. Biol. Chem. 284, 32158–32166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kiessling V., Crane J. M., and Tamm L. K. (2006) Transbilayer effects of raft-like lipid domains in asymmetric planar bilayers measured by single molecule tracking. Biophys. J. 91, 3313–3326 10.1529/biophysj.106.091421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chung S. H., and Kennedy R. A. (1991) Forward-backward non-linear filtering technique for extracting small biological signals from noise. J. Neurosci. Methods 40, 71–86 10.1016/0165-0270(91)90118-J [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.