

Abstract
Mammalian target of rapamycin complex 2 (mTORC2) has been shown to regulate mTORC1/4E-BP1/eIF4E signaling and collagen I expression in mesenchymal cells (MCs) during fibrotic activation. Here we investigated the regulation of the mTORC2 binding partner mammalian stress-activated protein kinase–interacting protein 1 (mSin1) in MCs derived from human lung allografts and identified a novel role for mSin1 during fibrosis. mSin1 was identified as a common downstream target of key fibrotic pathways, and its expression was increased in MCs in response to pro-fibrotic mediators: lysophosphatidic acid (LPA), transforming growth factor β, and interleukin 13. Fibrotic MCs had higher mSin1 protein levels than nonfibrotic MCs, and siRNA-mediated silencing of mSIN1 inhibited collagen I expression and mTORC1/2 activity in these cells. Autocrine LPA signaling contributed to constitutive up-regulation of mSin1 in fibrotic MCs, and mSin1 was decreased because of LPA receptor 1 siRNA treatment. We identified c-Jun N-terminal kinase (JNK) as a key intermediary in mSin1 up-regulation by the pro-fibrotic mediators, as pharmacological and siRNA-mediated inhibition of JNK prevented the LPA-induced mSin1 increase. Proteasomal inhibition rescued mSin1 levels after JNK inhibition in LPA-treated MCs, and the decrease in mSin1 ubiquitination in response to LPA was counteracted by JNK inhibitors. Constitutive JNK1 overexpression induced mSin1 expression and could drive mTORC2 and mTORC1 activation and collagen I expression in nonfibrotic MCs, effects that were reversed by siRNA-mediated mSIN1 silencing. These results indicate that LPA stabilizes mSin1 protein expression via JNK signaling by blocking its proteasomal degradation and identify the LPA/JNK/mSin1/mTORC/collagen I pathway as critical for fibrotic activation of MCs.
Keywords: mammalian target of rapamycin (mTOR), c-Jun N-terminal kinase (JNK), extracellular matrix protein, siRNA, ubiquitylation (ubiquitination), bronchiolitis obliterans, chronic allograft rejection, lysophosphatidic acid, mesenchymal cells, protein stability
Introduction
Mesenchymal cells (MCs)2 play a key role in fibrotic disease processes, including development of bronchiolitis obliterans syndrome (BOS), the primary cause of chronic graft failure after lung transplantation (1–3). Pro-fibrotic differentiation and collagen synthetic function of MCs can be driven by multiple mediators, including lipids such as lysophosphatidic acid (LPA), cytokines such as interleukin 13 (IL-13), and growth factors such as transforming growth factor β (TGF-β), all of which have been implicated in the pathogenesis of BOS and other fibrotic diseases (4–12). The redundancy of these diverse fibrotic mediators and the inability to halt progression of fibrosis by targeting a specific single pro-fibrotic mediator in human diseases have highlighted the need to identify the common downstream pathways through which these mediators regulate activation of MCs.
mTOR, an evolutionarily conserved serine/threonine protein kinase, functions to control translational machinery and serves as an integration point of large panels of signaling pathways. Structurally, mTOR is a multidomain protein that exists in two distinct complex forms, mTORC1 and mTORC2, with diverse downstream targets. Both mTOR complexes share the catalytic mTOR subunit, but partner proteins (mammalian Sin1 (mSin1), rictor, and Protor1/2) are specific to mTORC2 (13).
The mTORC1-mediated phosphorylation of 4E-BP1 has been identified recently as a key translational mechanism regulating collagen I expression by lung-resident MCs (14). More significantly, these studies demonstrate that the mTORC2 pathway lies upstream of mTORC1 in activated MCs and is critical in sustaining fibrotic functions such as collagen I expression. Inhibition of mTORC2 signaling leads to decreased mTORC1 signaling and collagen I expression in fibrotic but not normal MCs. This dependence of mTORC1 on mTORC2 activity under conditions of cellular activation suggests that mTORC2 intervention could represent a novel strategy to selectively target activated MCs in fibrosis. However, critical for that is a need for increased understanding of the regulatory mechanisms governing mTORC2 activity in MCs.
Of the various mTORC2-specific subunits, mSin1 plays a critical role in the kinase activation of its specific substrates, like AKT, by spatially recruiting it to the mTORC2 complex (15, 16). Furthermore, mSin1 is the mTORC2 component responsible for binding to phosphatidylinositol (3,4,5)-trisphosphate (PIP3) and subsequent activation of mTOR kinase activity downstream of PI3K (17–19). However, little is known about the regulation of mSin1 levels in MCs or its role in fibrotic diseases. In this study, we investigate the regulation of mTORC2 components, specifically mSin1, by fibrotic mediators and elucidate a novel c-Jun N-terminal kinase (JNK)–dependent signaling cascade that regulates mSin1 expression in MCs.
Results
Pro-fibrotic stimulation up-regulates the mTORC2 binding partner mSin1 in lung mesenchymal cells
We have demonstrated previously that the lipid mediator LPA plays a key role in lung MC activation, including induction of mTORC signaling, and is implicated in the pathogenesis of BOS (11, 12, 14). LPA treatment of MCs was shown to induce mTORC2 activation, marked by phosphorylation of AKT at Ser-473 (14). To confirm that mTORC is the primary kinase responsible for LPA-induced phosphorylation of AKT at Ser-473, MCs derived from normal lung allografts (nonfib MCs) were treated with LPA in the presence or absence of a novel ATP-competitive mTORC kinase inhibitor, AZD8055 (20) (Fig. 1A). Treatment with AZD8055 was noted to abrogate the effect of LPA on phosphorylation of AKT Thr-308 and AKT Ser-473. Next, we studied the effect of LPA on the protein expression of mSin1, a critical component of the mTORC2 complex (18, 21). Nonfib MCs treated with LPA demonstrated a significant increase in mSin1 protein expression at 6 h that persisted at 16 h (Fig. 1, B and C). LPA treatment also induced an increase in rictor, another key component of the mTORC2 complex (Fig. 1B). In contrast, the expression level of raptor, a critical and defining partner for mTORC1, was not changed in response to LPA (Fig. 1B). To determine whether mSin1 regulation in lung MCs is exclusive to LPA signaling or a common phenomenon in response to pro-fibrotic mediators, nonfib MCs were treated for 16 h with TGF-β or IL-13, and the level of mSin1 protein expression was assessed by Western blotting (Fig. 1D). mSin1 levels were significantly increased in response to both TGF-β and IL-13, demonstrating a common signaling mechanism by which levels of the mTORC2 component-mSin1 are increased, potentially priming the cell to ramp up mTORC2 assembly and, therefore, total cellular translation.
Figure 1.
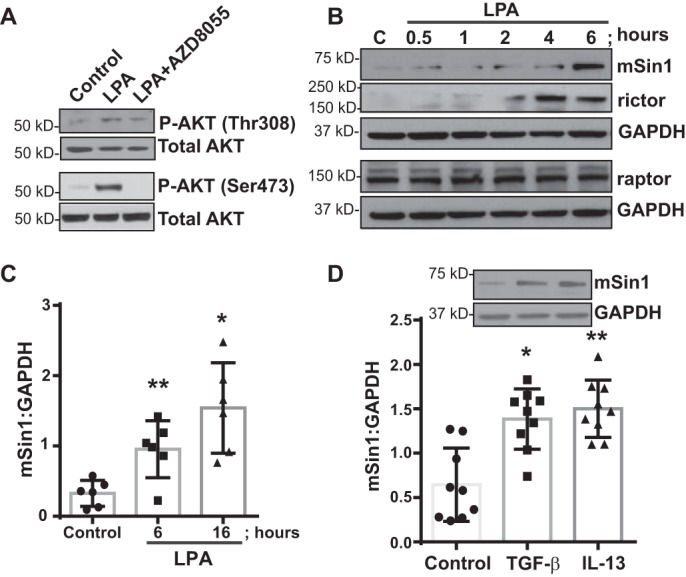
LPA induces mTORC2 signaling-associated mSin1 in mesenchymal cells. A, nonfibrotic MCs were pretreated with the mTORC inhibitor AZD8055 (250 nm for 30 min) and then exposed to LPA (10 μm for 6 h). Protein expression of the total and the phosphorylated forms of AKT at Thr-308 and Ser-473 were measured by immunoblotting. B, cell lysates isolated from normal nonfibrotic MCs were immunoblotted for antibodies against mTORC2-associated mSin1 and rictor, mTORC1-associated raptor, and GAPDH (loading control (C)). C, quantitative densitometry of the mTORC2 binding partner mSin1 was analyzed at 6 and 16 h in response to LPA. Values are means ± S.D., n = 4–6. *, p < 0.05; **, p < 0.01. D, nonfibrotic MCs were exposed to TGF-β (2 ng/ml) or IL-13 (10 ng/ml) for 16 h. Lysates were immunoblotted against mSin1. Quantitative densitometry was normalized using GAPDH (loading control). Values are means ± S.D., n = 9. *, p < 0.05; **, p < 0.01. Statistics: one-way ANOVA; post hoc: Sidak (C and D).
mSin1 is critical in regulating mTORC activation and collagen I expression in mesenchymal cells
To determine the specific role of mSin1 in mTORC activation downstream of LPA, we utilized mSIN1 siRNA–mediated silencing in the presence or absence of LPA. MCs transfected with mSIN1 siRNA demonstrated a 50% decrease in basal mSin1 protein expression and no significant up-regulation with LPA (Fig. 2A). mSIN1 silencing significantly inhibited constitutive and LPA-induced phosphorylation of AKT at Ser-473 (Fig. 2B). Additionally, mSIN1 silencing inhibited LPA-induced downstream phosphorylation of S6K1 and 4E-BP1 (Fig. 2, C and D), consistent with our previous finding that mTORC2 lies upstream of mTORC1 in activated MCs (14).
Figure 2.

mSin1 is required for mTORC regulation and fibrotic functions of mesenchymal cells. A, nonfibrotic MCs (n = 4) were transfected with scrambled or mSIN1-specific siRNA and then treated with LPA (10 μm for 6 h). Densitometry of the replicates was analyzed. Values are means ± S.D. *, p < 0.05; **, p < 0.01; ***, p < 0.001. B–D, cell lysates were subjected to immunoblot analysis for phosphorylated and total forms of AKT (Ser-473, B), p70S6K1 (Thr-389, C), and 4E-BP1 (Thr-37/46, D). Densitometry of the replicates was analyzed. Values are means ± S.D. *, p < 0.05; **, p < 0.01; ***, p < 0.001. E, nonfibrotic MCs and fibrotic MCs (n = 10) were analyzed for mSin1 protein expression by immunoblotting, and the densitometric analysis is presented as ***, p < 0.001. F, mesenchymal cells derived from fibrotic lung allografts (fibrotic MCs, n = 6) were transfected with scrambled or mSIN1-specific siRNA, and the lysates were immunoblotted with antibodies against mSin1, collagen I, phosphorylated and total forms of AKT (Ser-473), TSC2 (Thr-1462), FOXO3a (Ser-253), p70S6K1 (Thr-389), and 4E-BP1 (Thr-37/46) using GAPDH as a loading control. G, fibrotic MCs (n = 10) were transfected with siRNA specific to LPAR1 or the scrambled control, and the lysates were immunoblotted with antibodies against LPA1, collagen I, and mSin1 as well as the phosphorylated and total forms of AKT (Ser-473), p70S6K1 (Thr-389), and 4E-BP1 (Thr-37/46) using GAPDH as a loading control. Representative blots are shown.
We have previously demonstrated elevated mTORC1 and mTORC2 substrate phosphorylation in fibrotic MCs isolated from human lung allografts compared with nonfibrotic allograft MCs (14). To determine whether activated MCs demonstrate higher mSin1 levels, mSin1 protein expression was compared between fibrotic (fib) MCs and nonfibrotic MCs. Fib MCs demonstrated significantly higher expression of mSin1 compared with nonfibrotic MCs (Fig. 2E). siRNA-mediated silencing of mSIN1 in fibrotic MCs significantly blunted collagen I expression and, as expected, phosphorylation of AKT (Ser-473) (Fig. 2F). As expected, mSIN1-silenced fib MCs demonstrated decreased phosphorylation of the direct AKT substrates TSC2 (Thr-1462) and FOXO3a (Ser-253) compared with the scrambled siRNA control (Fig. 2F). Phosphorylation of the mTORC1 substrates 4E-BP1 and S6K1 was also found to be significantly decreased with mSIN1 silencing (Fig. 2F).
Autocrine LPA secretion has been shown previously to contribute to the stable fibrotic activation noted in MCs derived from BOS allografts (11). To determine whether autocrine LPA secretion has a role in the mTORC activation noted in fibrotic MCs, LPA signaling was blocked utilizing LPAR1 siRNA (Fig. 2G). Inhibition of mTORC1 and 2 activities and reduced mSin1 and collagen I protein expression were noted in fibrotic MCs transfected with LPAR1 siRNA (Fig. 2G), further reiterating a potential role of LPA in regulating mSin1 expression.
LPA signals through LPA1/PI3K/JNK to induce mSin1 expression
Next we investigated the downstream receptor-mediated signaling mechanism by which LPA induces mSin1 levels in MCs. LPAR1 is the predominant LPA receptor on MCs (12), and pharmacologic blockade of LPA1 using VPC12249 significantly inhibited LPA-induced AKT phosphorylation at Ser-473 and mSin1 protein expression (Fig. 3A). Additionally, treatment of MCs with LPA in the presence of the Gi-coupled protein inhibitor pertussis toxin (PTX) and the PI3K inhibitor LY294002 significantly inhibited the phosphorylation of AKT at Ser-473 and mSin1 expression, suggesting the LPA1/Gi/PI3K signaling axis (Fig. 3A).
Figure 3.

LPA-mediated induction of the mTORC2 binding partner mSin1 depends on JNK. A, nonfibrotic MCs (n = 8) were pretreated with inhibitors specific to LPA1 (VPC12249), Gi (PTX), or PI3K (LY294002) and then stimulated with LPA (10 μm for 6 h). Cell lysates were immunoblotted with antibodies against phosphorylated and total forms of AKT, mSin1, and GAPDH (loading control). Densitometry analysis of mSin1 was expressed as means ± S.D. **, p < 0.01; n = 9. B, nonfibrotic MCs (n = 9) were pretreated with inhibitors specific to p38MAPK (SB203580), ERK (U0126), and JNK (SP600125) and then stimulated with LPA (10 μm for 6 h). Cell lysates were immunoblotted with antibodies against the phosphorylated and total forms of AKT, mSin1, and GAPDH (loading control). Densitometry analysis of mSin1 was expressed as means ± S.D. **, p < 0.01; *, p < 0.05. C, nonfibrotic MCs (n = 9) were pretreated with a JNK inhibitor (SP600125), followed by stimulation with IL-13 (10 ng/ml) or TGF-β (2 ng/ml) for 16 h. Cell lysates were analyzed for mSin1 by immunoblotting. Densitometry analysis of mSin1 was expressed as means ± S.D. ***, p < 0.001. Statistics: one-way ANOVA; post hoc: Sidak. D, nonfibrotic MCs were transfected with scrambled or JNK1-specific siRNA and then stimulated with LPA (10 μm for 6 h). Cell lysates were immunoblotted with antibodies against mSin1, collagen I, JNK1/2, or phosphorylated and total forms of AKT (Ser-473), p70S6K1 (Thr-389), and 4E-BP1 (Thr-37/46) using GAPDH as a loading control. Representative blots are shown.
PI3K signals downstream via numerous mechanisms to control transcription, translation, and other cellular processes. mSin1, also known as mitogen-activated protein kinase associated protein 1 (MAPKAP1), was first described as a MAPK target in Drosophila and yeast (22). The three MAPK signaling pathways, p38MAPK, JNK, and MEK/ERK, have been reported to cross-talk with PI3K signaling in many cell types during various cellular processes (23). To investigate whether the PI3K/MAPK signaling axis contributes to LPA-induced mSin1 expression, nonfibrotic MCs were treated with LPA in the presence or absence of MAPK inhibitors against p38MAPK (SB203580), MEK/ERK (U0126), or JNK (SP600125). LPA-induced mSin1 expression was not altered in the presence of inhibitors against p38MAPK or MEK/ERK but was significantly blunted in the presence of SP600125 (Fig. 3B). JNK inhibitor II also significantly abrogated TGF-β– and IL-13–induced expression of mSin1 protein in MCs (Fig. 3C). The role of JNK in regulating mSin1 expression in MCs was further reiterated by utilizing siRNA targeted against JNK1. JNK1 silencing in MCs prevented up-regulation of mSin1 in response to LPA. LPA-induced mTORC2 and mTORC1 activity and fibrotic activation were also decreased under these conditions, as signaled by mTORC substrate phosphorylation and collagen I protein expression (Fig. 3D).
JNK1 kinase activity modulates mSin1 expression and mTORC2 stabilization and kinase activity
JNK1 has been implicated in numerous signaling pathways to exert downstream transcription and regulate apoptosis and translational and protein degradation mechanisms (24–26). JNK is regulated by phosphorylation, which occurs via two MAPKs, MKK4 or MKK7. Upon phosphorylation, JNK induces downstream targets such as the transcription factor c-Jun (27). To determine whether JNK activity is sufficient to drive mSin1 expression, we induced the expression of a constitutively active form of JNK1 in nonfibrotic MCs. This tool utilizes the JNK1 protein fused to its MAPK kinase, MKK7, causing JNK1 to be constitutively active. Increased JNK signaling in lentivirus-infected cells was confirmed by immunoblotting for phosphorylation of Jun at Ser-73 (Fig. 4A). A significant increase in mSin1 expression was noted in nonfibrotic MCs with constitutive activation of JNK1 compared with an empty vector control (Fig. 4A). Constitutive activation of JNK1 led to an increase in collagen I expression along with a concomitant increase in phosphorylation of AKT Ser-473 and the mTORC1 substrates 4E-BP1 and S6K1 (Fig. 4A). To determine whether JNK-mediated activation of AKT depended on mTORC2, constitutively active JNK1 was expressed in the presence or absence of the ATP-competitive mTOR inhibitor AZD8055. JNK-mediated AKT phosphorylation at Ser-473 was completely abolished in the presence of AZD8055 (Fig. 4A). Additionally, mTORC1 activity, marked by phosphorylation of 4E-BP1 and S6K1, was significantly blunted, along with down-regulation of collagen I (Fig. 4A).
Figure 4.

JNK1 activation induces mSin1 expression and mTORC2 stabilization. A, nonfibrotic MCs (n = 6) were infected with a lentivirus containing an empty vector or a vector that expresses constitutively active JNK1 tagged with FLAG and then treated with the mTOR inhibitor AZD8055 (250 nm for 24 h). Cell lysates were immunoblotted against mSin1, collagen I, GAPDH (loading control), FLAG, and the phosphorylated and total forms of Jun (Ser-73), AKT (Ser-473), p70S6K1(Thr-389), 4E-BP1 (Thr-37/46), and mTOR (Ser-2481). B, nonfibrotic MCs (n = 6) were infected with a lentivirus containing an empty vector or vector that expresses constitutively active JNK1, followed by transfection with scrambled or mSIN1-specific siRNA. Cell lysates were immunoblotted with antibodies as described in A. C, nonfibrotic MCs (n = 8) were pretreated with 10 μm JNK inhibitor (SP600125) and then stimulated with LPA (10 μm for 6 h). Cell lysates were immunoblotted with antibodies against mSin1, collagen I, and phosphorylated and total forms of JNK1/2 (Thr-183/Tyr-185), AKT (Ser-473), and mTOR (Ser-2481) as well as rictor, raptor, and GAPDH. Cell lysates were immunoprecipitated using antibodies against mSin1, rictor, or mTOR. Each of the immunoprecipitates was analyzed for rictor, mSin1, phosphorylated mTOR (Ser-2481), and total mTOR. Representative blots are shown for each experiment.
To further determine whether the increased mTORC1 and 2 activities seen in the presence of JNK overexpression are mediated via its regulation of mSin1, cells were subjected to siRNA-mediated mSIN1 gene silencing. mSIN1 silencing abrogated the increased phosphorylation of 4E-BP1, S6K1, and AKT473 by constitutively active JNK (Fig. 4B).
Next, to investigate the role of JNK-mediated mSin1 expression on mTORC2 complex kinase activity versus complex formation, we performed co-immunoprecipitation of mSin1, rictor, and mTOR in nonfibrotic MCs treated with LPA in the presence or absence of JNK inhibitor II/SP600125 (Fig. 4C). We observed that increased levels of rictor and mTOR co-immunoprecipitated with mSin1 in response to LPA (Fig. 4C). In addition, an increased amount of mSin1 co-immunoprecipitated with rictor and mTOR in LPA-treated cells, suggesting increased complex formation (Fig. 4C). Co-immunoprecipitated proteins were significantly inhibited in the presence of JNK inhibitor II (Fig. 4C), further demonstrating that JNK is a key intermediary in LPA-induced mSin1 expression and mTORC2 complex formation. In addition, phosphorylation of mTOR at the autocatalytic site, Ser-2481, was significantly inhibited in co-immunoprecipitates treated with JNK inhibitor II (Figs. 4C). This further strengthens the role of JNK signaling in mTORC2 stability, as phosphorylation of mTOR at the autocatalytic site is a key readout for intact mTOR complex assembly and complex activity.
JNK modulates mSin1 expression via posttranslational regulation of protein stability
We next investigated the mechanism by which mSin1 expression is regulated by JNK in MCs. We first assessed mRNA expression of mSIN1 in response to LPA treatment, as canonical JNK signaling involves downstream phosphorylation and activation of the transcription factor c-Jun to increase transcription of target genes (26, 28). Surprisingly, no significant increase in mSIN1 expression was found at the mRNA level in response to LPA treatment (Fig. 5A). Additionally, the transcriptional inhibitor actinomycin D had no effect on LPA-induced mSin1 expression in MCs (Fig. 5B). Similarly, no significant changes in mSIN1 mRNA were observed in response to TGF-β or IL-13 in cells expressing constitutively active JNK expression (data not shown).
Figure 5.

JNK mediates posttranslational regulation of mSin1 induction. A, nonfibrotic MCs (n = 6) were stimulated with LPA (10 μm for 6 h). RNA lysates were analyzed for mSIN1 by qPCR, using β-actin as the endogenous control. B, nonfibrotic MCs (n = 9) were pretreated with actinomycin D (ActD, 5 μg/ml for 30 min), followed by stimulation with LPA. Cell lysates were immunoblotted for antibodies against mSin1 and GAPDH (loading control). **, p < 0.01. Statistics: one-way ANOVA; post hoc: Sidak. C, nonfibrotic MCs (n = 5) were pretreated with CHX (10 μm) for the indicated periods of time. Protein lysates were subjected to a chase assay and analyzed for mSin1. *, p < 0.05; **, p < 0.01; ***, p < 0.001. D, nonfibrotic MCs (n = 12) were treated with the proteasomal inhibitor MG132 (10 μm for 16 h). Protein lysates were immunoblotted with antibodies against mSin1 and GAPDH (loading control). Densitometric values are represented as means ± S.D. ***, p < 0.001. E, nonfibrotic MCs (n = 14) were pretreated with a proteasomal inhibitor (MG132) or JNK inhibitor (SP600125) or both, followed by stimulation with LPA. Cell lysates were analyzed for mSin1 and GAPDH (loading control). Densitometric values are represented as means ± S.D. *, p < 0.05; **, p < 0.01; ***, p < 0.001. Statistics: one-way ANOVA; post hoc: Sidak. F, nonfibrotic MCs (n = 3) were treated with the proteasomal inhibitor MG132 in the presence or absence of JNK inhibitor II (SP600125) with or without LPA stimulation for 16 h. Cell lysates were immunoprecipitated using antibodies against mSin1. mSin1 immunoprecipitates were immunoblotted with antibodies against pan-ubiquitin, mSin1, rictor, and total mTOR. Representative blots are shown.
We further examined the protein levels of mSin1 by utilizing a cycloheximide (CHX) chase assay to assess protein stability in the absence of de novo protein synthesis. The protein levels of mSin1 quickly decreased following CHX treatment, with a half-life of 1 h (Fig. 5C), suggesting a high turnover rate or posttranslational regulation of this protein. Proteasomal inhibition in MCs utilizing MG132 further supported this notion, as mSin1 protein levels were significantly increased following MG132 treatment (Fig. 5D). To confirm the role of posttranslational regulation of mSin1, specifically by the ubiquitin/proteasome pathway, MCs were treated with the proteasome inhibitor MG132 in the presence of LPA and JNK inhibitor II. As shown previously, LPA significantly increased the protein levels of mSin1, which was sensitive to JNK inhibitor II/SP600125 (Fig. 5E). Proteasomal inhibition via MG132 treatment in the presence of LPA and JNK inhibitor II was able to significantly rescue the expression levels of mSin1 protein (Fig. 5E). Ubiquitination of mSin1 was assessed in the presence of LPA and JNK inhibitor II by immunoblotting mSin1 immunoprecipitates for a pan-ubiquitin antibody (Fig. 5F). Mesenchymal mSin1 immunoprecipitates pretreated with MG132 demonstrated a strong baseline level of ubiquitination that was markedly decreased with LPA treatment (Fig. 5F). Finally, we observed an increase in mSin1 ubiquitination in the presence of MG132, LPA, and JNK inhibitor II (Fig. 5F). Collectively, these data suggest that a posttranslational mechanism, specifically the ubiquitin/proteasome pathway, may induce mSin1 expression in response to LPA.
Discussion
Long-term graft survival post-lung transplantation is limited by chronic rejection, which is marked by progressive fibrotic remodeling because of aberrant mesenchymal activation and matrix secretion (4, 29, 30). We have previously identified the role of mTORC2-directed signaling to the mTORC1/4E-BP1 axis as a regulator of translational activation during allograft fibrosis (14). Here we demonstrate that mSin1, a critical mTORC2 component, is a common downstream target of key fibrotic pathways with evidence for up-regulation of mSin1 and subsequent mTORC2 activation by LPA and other fibrotic stimuli, including TGF-β and IL-13. We identify JNK as a key intermediary in mSin1 up-regulation by LPA and provide evidence for a novel fibrotic activation pathway that involves the JNK/mSin1/mTORC/collagen I axis, by which JNK-induced mSin1 expression drives mTORC2 activity and subsequent translational activation.
A key finding in our study is the demonstration of constitutively stable up-regulation of mSin1 protein in fibrotic MCs maintained by autocrine signaling. MCs isolated from fibrotic lung allografts demonstrated significantly higher mSin1 expression compared with MCs isolated from normal lung allografts. Increased expression of mSin1 has been linked to metastatic and activated behavior in cancer cells (31–33). mSin1 was shown to be up-regulated in non-small-cell lung cancer (31) as well as in metastatic carcinoma of the colon (33) and the liver (32), with mSin1 suppression inhibiting proliferation, migration, and invasion rates. MCs derived from fibrotic tissues, similar to cancer cells, demonstrate a stable activated phenotype. However, mSin1 has not been previously investigated in MCs in the context of any organ fibrosis. The finding of high mSin1 is consistent with our previous demonstration of activated mTORC signaling in fibrotic MCs with higher phosphorylation at both mTORC1 (4E-BP1 and S6 kinase) and mTORC2 (AKT) targets (14). That mSin1 is not just a biomarker of mesenchymal activation but plays a key role in sustaining the fibrotic phenotype was established by significant abrogation of collagen expression following mSIN1 silencing in fibrotic MCs. Decreased phosphorylation of 4E-BP1 and S6K1 was noted in cells transfected with mSIN1 siRNA, confirming our previous finding of an essential role for mTORC2 in regulating mTORC1 activity in fibrotic MCs (14). It is noteworthy that our studies utilized MCs derived from fibrotic lung allografts or patients with BOS to demonstrate the constitutively higher expression of mSin1 protein. However, a similar key role of mTORC2 signaling has been demonstrated in TGF-β–stimulated MCs derived from patients with idiopathic pulmonary fibrosis, another chronic fibrotic lung disease (10), and in smooth muscle cells in patients with pulmonary hypertension (34, 35). This increasing awareness of a critical role of mTORC2 activity in regulating translation via mTORC1 in activated diseased cells further underscores the significance of increased constitutive mSin1 expression noted in our study.
We demonstrate for the first time that a pro-fibrotic lipid mediator, LPA, regulates mSin1 expression in lung MCs. Increased mSin1 expression in fibrotic MCs was attenuated by blocking LPA signaling via silencing of LPAR1, a predominant receptor for LPA in MCs (11), suggesting that autocrine LPA contributes to maintaining the sustained higher mSin1 expression in activated MCs. MCs derived from fibrotic lung allografts have been shown to have a higher LPA synthetic capacity, marked by increased secretion of autotaxin, the primary enzyme responsible for generating LPA (11). Furthermore, exogenous LPA was shown to up-regulate mSin1 expression in nonfibrotic MCs. LPA signaling has pleomorphic effects and is known to mediate mitogenic effects via AKT activation in myoblasts (36) by activating p70S6 kinase in airway smooth muscle cells (37) and by activating β-catenin in MCs (11). We have shown previously that LPA induces mTORC1 and mTORC2 activation in MCs. This report shows that pharmacologic blockade of mTORC signaling via AZD8055 treatment prevented AKT phosphorylation at Ser-473, thus establishing mTORC2 as the primary kinase mediating this phosphorylation.
Reports regarding cancer cells also confirm that LPA-induced AKT activation is mediated via mTORC2 activity (38). Although rictor and mSin1 are two key binding partners of mTORC2, our findings demonstrate that mSin1 plays a critical role in the sustained kinase activation of its specific substrates, like AKT. Although studies have demonstrated that the stabilization of rictor (an mTORC2 binding partner) occurs by suppressing its degradation mediated by PI3K/GSK3β signaling (39), little is known regarding the regulation of mSin1 protein. In HeLa cell studies, mSin1 has been implicated in cell survival and stress, and tumor necrosis factor α and H2O2 treatments have been shown to increase mSin1 (40). The demonstration of LPA as a regulator of mSin1 potentially has a significantly wider relevance, as LPA plays a key pathogenic role in malignant and fibrotic disease processes across various organs. In MCs, our pharmacologic inhibition approaches indicate that LPA acts via LPA1/Gi/PI3K signaling to regulate mSin1, suggesting that other extracellular stimuli, such as ligands for growth factor receptors, G protein–coupled receptors, and cytokine receptors, can also potentially regulate mSin1 in MCs. In fact, key pro-fibrotic mediators such as TGF-β and IL-13 were noted to increase mSin1 expression in lung MCs, suggesting that this could be a common downstream mechanism contributing to activation of MCs in fibrosis. Although our studies demonstrate increased expression of mSin1, prior studies have demonstrated the activation of mTORC complexes 1 and 2 in response to TGF-β (41, 42) and IL-13 (43).
JNK-mediated regulation of mTORC2-associated mSin1 has not been shown in any disease pathology to date. Our studies utilized pharmacological and RNAi approaches specific to JNK signaling and demonstrate that mSin1 lies downstream of JNK signaling in response to LPA. mSin1 has been reported to bind to JNK, whereas no interaction with p38- or ERK1/2-family MAPKs was observed in epithelial cells (44). This is consistent with our findings that inhibition of JNK, but not p38MAPK or ERK, is effective in down-regulating LPA-induced mSin1 expression in MCs. Our findings also demonstrate that inhibition of JNK is effective in blunting mSin1 expression in response to IL-13 and TGF-β, suggesting that JNK-mediated mSin1 regulation is a common target for mTORC2 activation in MCs. Importantly, we observed strong suppression of AKT phosphorylation with JNK inhibition. This is consistent with a previous report that shows that TGF-β mediated AKT activation is blocked by JNK inhibition (41). Furthermore, JNK1 overexpression was sufficient to cause mTORC2 and mTORC1 activation as well as fibrotic differentiation of normal MCs (as demonstrated by increased collagen I expression). This depended on mTORC signaling, as the increased mTORC substrate phosphorylation and collagen I expression induced by JNK overexpression were sensitive to the ATP-competitive mTOR inhibitor AZD8055. Our novel findings establish a role of JNK in regulating mSin1 and, therefore, mTORC2 activation, thus linking JNK signaling to AKT phosphorylation, collagen I expression, and fibrotic differentiation.
Our data suggest that LPA, via JNK signaling, stabilizes mSin1 protein expression by blocking its proteasomal degradation. JNK is well-known for its role in regulating cellular functions via modulation of transcription factors such as c-jun (45). However, we noted no change in mesenchymal mSIN1 mRNA expression in the presence of LPA. mSin1 was noted to have a short half-life in the cycloheximide chase assay, and an increase in mSin1 levels was noted in the presence of the proteasomal inhibitor MG132. Proteasomal inhibition rescued mSin1 levels with JNK inhibition in LPA-treated MCs, and LPA treatment decreased the amount of ubiquitinated mSin1 in immunoprecipitates from MCs, further suggesting that the rise in mSin1 levels is likely mediated via its decreased ubiquitination and proteasomal degradation. JNK has been implicated previously in co-translational phosphorylation and stabilization of target proteins at the ribosomal, protein turnover, protein scaffolding, and ubiquitin ligase activation levels (46, 47). With JNK's known role as a kinase, direct phosphorylation of mSin1 could be a potential mechanism mediating its regulation and turnover. Phosphorylation of mSin1 on the Ser-260 residue has been proposed as a mechanism of its stabilization in other cell types where mTOR kinase activity was shown to regulate mSin1 levels (48). In our study, we show that the levels of mSin1 were unchanged in MCs expressing constitutively active JNK in the presence of AZD8055, suggesting that mSin1 phosphorylation at Ser-260 is not in play. Furthermore, phosphorylation at the Ser-260 residue was linked to inhibition of lysosomal rather than proteasomal degradation (49). Additionally, reports suggest that AKT-dependent phosphorylation of mSin1 at Thr-86 and Thr-398 (17, 50) sustains mTORC2 kinase activity and, therefore, maximal activation of AKT at Ser-473. Our results demonstrate that AKT Ser-473 phosphorylation was completely abolished in MCs expressing constitutively active JNK in the presence of AZD8055, suggesting that these phosphorylation events on mSin1 are also not in play. Any phosphorylation changes that occur on mSin1 because of JNK kinase activity and the mechanism by which JNK stabilizes Sin1 expression warrant further investigation.
In conclusion, these data establish, for the first time, the regulation of mSin1 levels by pro-fibrotic mediators such as LPA in MCs and shed new light on regulation of the mTORC2 pathway by JNK via protein stabilization of mSin1 protein. Targeting mSin1 could represent a potential therapeutic approach in fibrotic diseases because mSin1 is a common end point of several upstream stimuli.
Experimental procedures
Isolation of mesenchymal cells from human lung allografts
MCs were isolated from bronchoalveolar lavage fluid obtained from human lung transplant recipients with and without BOS under a protocol on human studies approved by the University of Michigan Institutional Review Board, and the approved studies abide by the Declaration of Helsinki principles. Cells were isolated, cultured, and characterized as described previously (51). MCs grown from each individual bronchoalveolar lavage sample were treated as a separate cell line. Cells at passages three through six were used for all experiments.
Tissue culture, protein isolation, and immunoblotting
MCs were plated and grown to 70% confluence in 60-mm dishes prior to serum deprivation for 24 h. Following serum deprivation, cells were treated in serum-free medium containing the indicated doses of either LPA (10 μm; Cayman Chemical, Ann Arbor, MI), porcine TGF-β (2 ng/ml; R&D Systems, Minneapolis, MN), IL-13 (10 ng/ml; R&D Systems), MG132 (10 μm; Cayman Chemical), or AZD8055 (250 nm; Selleck Chemicals, Houston, TX) for 6 or 24 h. Cells were pretreated for 30 min with MAPK inhibitor SP600125 (10 μm; JNK1/2 inhibitor), SB203580 (10 μm; p38 MAPK inhibitor), or U0126 (10 μm; MEK/ERK1/2 inhibitor) prior to treatment with LPA. MAPK inhibitors were purchased from Selleck Chemicals. VPC12249 (1 μm; LPA1 inhibitor) was purchased from Avanti Polar Lipids (Alabaster, AL). PTX (100 ng/ml) and CHX (10 μg/ml) were purchased from Cayman Chemical. LY294002 (10 μm; PI3K/Akt inhibitor) was purchased from Cell Signaling Technology (Minneapolis, MN). Actinomycin D (5 μg/ml) was purchased from Tocris Biosciences (Minneapolis, MN).
Total protein was collected from treated or untreated cells by cell lysis on ice. Lysates were separated on 4–12% gradient BisTris gels prior to immunoblot analysis for collagen I (Cedarlane Laboratories, ON, Canada). The other antibodies utilized were as follows: phosphorylated p70-S6kinase (Thr-389, 9234), p70-S6Kinase (2708), phosphorylated 4E-BP1 (Thr-37/46, 2855), total 4E-BP1 (9644), phosphorylated AKT (Ser-473, 4058), phosphorylated AKT (Thr-308, 9275), total AKT (9272), phosphorylated TSC2 (Thr-1462, 3611), total TSC2 (3635), total FOXO3a (12829), ubiquitin (3936), rictor (9476), raptor (2280), phosphorylated mTOR (Ser-2481, 2974), total mTOR (2972), JNK2 (9258), and mSin1 (12860) (all from Cell Signaling Technology, Boston, MA) and phosphorylated FOXO3a (Ser-253, ab154786, Abcam). The mouse monoclonal JNK1 antibody was purchased from Santa Cruz Biotechnology (Dallas, TX; sc-1648). Plasmid transfection efficiency was determined by blotting for FLAG M2 (F1804), purchased from Sigma-Aldrich (St. Louis, MO).
siRNA-mediated silencing and plasmid transfection
ON-TARGETplus SMARTpool, siRNA targeting human mSIN1 (79109), JNK1, or scrambled siRNA were obtained from Dharmacon/GE Healthcare. LPAR1 (LPA receptor 1)–specific siRNA (AM51331) was obtained from Ambion/Applied Biosystems (Waltham, MA). Briefly, cells were plated to 50% confluency in 6-well plates, followed by transient transfection utilizing Oligofectamine (Invitrogen) for 24 h. Following transfection, cells were maintained in serum-free medium for 72 h prior to protein harvesting.
pcDNA3-FLAG-MKKB2-JNK1α1 was a kind gift from Dr. Roger Davis (Addgene plasmid 19726). To obtain lentiviral expression plasmids, the fusion sequence was shuttled into pcDNA3.1/hygromycin using HindIII and then inserted into the NheI site of pLentilox-IRES-Puro using NheI and XbaI. The sequence of the insert was confirmed using standard sequencing methods. Briefly, cells were plated to 50% confluence in 6-well plates, followed by infection in serum-free medium utilizing protamine sulfate as a linker. 24 h following infection, fresh serum-containing medium was added for 48 h, followed by protein harvesting.
Statistical analysis
Data are presented as mean values ± S.D. Statistical significance was analyzed using GraphPad Prism 7 software (La Jolla, CA). Significance was assessed with a Student's t test for comparisons of two groups or with one-way analysis of variance (ANOVA) for three or more groups and a post hoc Dunnett's multiple comparisons test unless otherwise specified. Significance was set at p values of less than 0.05.
Author contributions
N. M. W., D. C. F., and V. N. L. conceptualization; N. M. W., R. V., D. C. F., and V. N. L. data curation; N. M. W. software; N. M. W., R. V., and V. N. L. formal analysis; N. M. W., D. C. F., and V. N. L. validation; N. M. W., R. V., and V. N. L. investigation; N. M. W. and D. C. F. methodology; N. M. W., R. V., and V. N. L. writing-original draft; N. M. W., S. M. M., R. V., D. C. F., and V. N. L. writing-review and editing; S. M. M., D. C. F., and V. N. L. visualization; V. N. L. supervision; V. N. L. funding acquisition; V. N. L. project administration.
This work was supported by NHLBI, National Institutes of Health Grants 5R01HL094622-09 and 5R01HL118017-04 (to V. N. L.) and Cystic Fibrosis Foundation Grant LAMA16XX0 (to V. N. L.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- MC
- mesenchymal cell
- BOS
- bronchiolitis obliterans syndrome
- LPA
- lysophosphatidic acid
- IL
- interleukin
- TGF
- transforming growth factor
- PI3K
- phosphatidylinositol 3-kinase
- JNK
- c-Jun N-terminal kinase
- nonfib
- nonfibrotic
- fib
- fibrotic
- PTX
- pertussis toxin
- MAPK
- mitogen-activated protein kinase
- MEK
- mitogen-activated protein kinase/extracellular signal–regulated kinase kinase
- ERK
- extracellular signal–regulated kinase
- CHX
- cycloheximide
- ANOVA
- analysis of variance
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase.
References
- 1. Vock D. M., Durheim M. T., Tsuang W. M., Finlen Copeland C. A., Tsiatis A. A., Davidian M., Neely M. L., Lederer D. J., and Palmer S. M. (2017) Survival benefit of lung transplantation in the modern era of lung allocation. Ann. Am. Thorac. Soc. 14, 172–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Todd J. L., Christie J. D., and Palmer S. M. (2014) Update in lung transplantation 2013. Am. J. Respir. Crit. Care Med. 190, 19–24 10.1164/rccm.201402-0384UP [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Finlen Copeland C. A., Vock D. M., Pieper K., Mark D. B., and Palmer S. M. (2013) Impact of lung transplantation on recipient quality of life: a serial, prospective, multicenter analysis through the first posttransplant year. Chest 143, 744–750 10.1378/chest.12-0971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lama V. N., Harada H., Badri L. N., Flint A., Hogaboam C. M., McKenzie A., Martinez F. J., Toews G. B., Moore B. B., and Pinsky D. J. (2006) Obligatory role for interleukin-13 in obstructive lesion development in airway allografts. Am. J. Pathol. 169, 47–60 10.2353/ajpath.2006.050975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Khalil N., O'Connor R. N., Unruh H. W., Warren P. W., Flanders K. C., Kemp A., Bereznay O. H., and Greenberg A. H. (1991) Increased production and immunohistochemical localization of transforming growth factor-β in idiopathic pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 5, 155–162 10.1165/ajrcmb/5.2.155 [DOI] [PubMed] [Google Scholar]
- 6. Keane M. P., Gomperts B. N., Weigt S., Xue Y. Y., Burdick M. D., Nakamura H., Zisman D. A., Ardehali A., Saggar R., Lynch J. P. 3rd, Hogaboam C., Kunkel S. L., Lukacs N. W., Ross D. J., Grusby M. J., et al. (2007) IL-13 is pivotal in the fibro-obliterative process of bronchiolitis obliterans syndrome. J. Immunol. 178, 511–519 10.4049/jimmunol.178.1.511 [DOI] [PubMed] [Google Scholar]
- 7. Hancock A., Armstrong L., Gama R., and Millar A. (1998) Production of interleukin 13 by alveolar macrophages from normal and fibrotic lung. Am. J. Respir. Cell Mol. Biol. 18, 60–65 10.1165/ajrcmb.18.1.2627 [DOI] [PubMed] [Google Scholar]
- 8. Grande J. P. (1997) Role of transforming growth factor-β in tissue injury and repair. Proc. Soc. Exp. Biol. Med. 214, 27–40 10.3181/00379727-214-44066 [DOI] [PubMed] [Google Scholar]
- 9. Broekelmann T. J., Limper A. H., Colby T. V., and McDonald J. A. (1991) Transforming growth factor β 1 is present at sites of extracellular matrix gene expression in human pulmonary fibrosis. Proc. Natl. Acad. Sci. U.S.A. 88, 6642–6646 10.1073/pnas.88.15.6642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chang W., Wei K., Ho L., Berry G. J., Jacobs S. S., Chang C. H., and Rosen G. D. (2014) A critical role for the mTORC2 pathway in lung fibrosis. PLoS ONE 9, e106155 10.1371/journal.pone.0106155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cao P., Aoki Y., Badri L., Walker N. M., Manning C. M., Lagstein A., Fearon E. R., and Lama V. N. (2017) Autocrine lysophosphatidic acid signaling activates β-catenin and promotes lung allograft fibrosis. J. Clin. Invest. 127, 1517–1530 10.1172/JCI88896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Badri L., and Lama V. N. (2012) Lysophosphatidic acid induces migration of human lung-resident mesenchymal stem cells through the β-catenin pathway. Stem Cells 30, 2010–2019 10.1002/stem.1171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Saxton R. A., and Sabatini D. M. (2017) mTOR signaling in growth, metabolism, and disease. Cell 169, 361–371 10.1016/j.cell.2017.02.004 [DOI] [PubMed] [Google Scholar]
- 14. Walker N. M., Belloli E. A., Stuckey L., Chan K. M., Lin J., Lynch W., Chang A., Mazzoni S. M., Fingar D. C., and Lama V. N. (2016) Mechanistic target of rapamycin complex 1 (mTORC1) and mTORC2 as key signaling intermediates in mesenchymal cell activation. J. Biol. Chem. 291, 6262–6271 10.1074/jbc.M115.672170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yao C. A., Ortiz-Vega S., Sun Y. Y., Chien C. T., Chuang J. H., and Lin Y. (2017) Association of mSin1 with mTORC2 Ras and Akt reveals a crucial domain on mSin1 involved in Akt phosphorylation. Oncotarget 8, 63392–63404 10.18632/oncotarget.18818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cameron A. J., Linch M. D., Saurin A. T., Escribano C., and Parker P. J. (2011) mTORC2 targets AGC kinases through Sin1-dependent recruitment. Biochem. J. 439, 287–297 10.1042/BJ20110678 [DOI] [PubMed] [Google Scholar]
- 17. Yang G., Murashige D. S., Humphrey S. J., and James D. E. (2015) A Positive feedback loop between Akt and mTORC2 via SIN1 phosphorylation. Cell Rep. 12, 937–943 10.1016/j.celrep.2015.07.016 [DOI] [PubMed] [Google Scholar]
- 18. Yang Q., Inoki K., Ikenoue T., and Guan K. L. (2006) Identification of Sin1 as an essential TORC2 component required for complex formation and kinase activity. Genes Dev. 20, 2820–2832 10.1101/gad.1461206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Liu P., Gan W., Chin Y. R., Ogura K., Guo J., Zhang J., Wang B., Blenis J., Cantley L. C., Toker A., Su B., and Wei W. (2015) PtdIns(3,4,5)P3-dependent activation of the mTORC2 kinase complex. Cancer Discov. 5, 1194–1209 10.1158/2159-8290.CD-15-0460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chresta C. M., Davies B. R., Hickson I., Harding T., Cosulich S., Critchlow S. E., Vincent J. P., Ellston R., Jones D., Sini P., James D., Howard Z., Dudley P., Hughes G., Smith L., et al. (2010) AZD8055 is a potent, selective, and orally bioavailable ATP-competitive mammalian target of rapamycin kinase inhibitor with in vitro and in vivo antitumor activity. Cancer Res. 70, 288–298 10.1158/0008-5472.CAN-09-1751 [DOI] [PubMed] [Google Scholar]
- 21. Jacinto E., Facchinetti V., Liu D., Soto N., Wei S., Jung S. Y., Huang Q., Qin J., and Su B. (2006) SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell 127, 125–137 10.1016/j.cell.2006.08.033 [DOI] [PubMed] [Google Scholar]
- 22. Wilkinson M. G., Pino T. S., Tournier S., Buck V., Martin H., Christiansen J., Wilkinson D. G., and Millar J. B. (1999) Sin1: an evolutionarily conserved component of the eukaryotic SAPK pathway. EMBO J. 18, 4210–4221 10.1093/emboj/18.15.4210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Klippel A., Reinhard C., Kavanaugh W. M., Apell G., Escobedo M. A., and Williams L. T. (1996) Membrane localization of phosphatidylinositol 3-kinase is sufficient to activate multiple signal-transducing kinase pathways. Mol. Cell. Biol. 16, 4117–4127 10.1128/MCB.16.8.4117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fuchs S. Y., Fried V. A., and Ronai Z. (1998) Stress-activated kinases regulate protein stability. Oncogene 17, 1483–1490 10.1038/sj.onc.1202184 [DOI] [PubMed] [Google Scholar]
- 25. Patel M. R., Sadiq A. A., Jay-Dixon J., Jirakulaporn T., Jacobson B. A., Farassati F., Bitterman P. B., and Kratzke R. A. (2012) Novel role of c-jun N-terminal kinase in regulating the initiation of cap-dependent translation. Int. J. Oncol. 40, 577–582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Weston C. R., and Davis R. J. (2007) The JNK signal transduction pathway. Curr. Opin. Cell Biol. 19, 142–149 10.1016/j.ceb.2007.02.001 [DOI] [PubMed] [Google Scholar]
- 27. Davis R. J. (2000) Signal transduction by the JNK group of MAP kinases. Cell 103, 239–252 10.1016/S0092-8674(00)00116-1 [DOI] [PubMed] [Google Scholar]
- 28. Wisdom R., Johnson R. S., and Moore C. (1999) c-Jun regulates cell cycle progression and apoptosis by distinct mechanisms. EMBO J. 18, 188–197 10.1093/emboj/18.1.188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lund L. H., Edwards L. B., Kucheryavaya A. Y., Dipchand A. I., Benden C., Christie J. D., Dobbels F., Kirk R., Rahmel A. O., Yusen R. D., Stehlik J., and International Society for Heart and Lung Transplantation (2013) The Registry of the International Society for Heart and Lung Transplantation: Thirtieth Official Adult Heart Transplant Report–2013; focus theme: age. J. Heart Lung Transplant. 32, 951–964 10.1016/j.healun.2013.08.006 [DOI] [PubMed] [Google Scholar]
- 30. Estenne M., Maurer J. R., Boehler A., Egan J. J., Frost A., Hertz M., Mallory G. B., Snell G. I., and Yousem S. (2002) Bronchiolitis obliterans syndrome 2001: an update of the diagnostic criteria. J. Heart Lung Transplant. 21, 297–310 10.1016/S1053-2498(02)00398-4 [DOI] [PubMed] [Google Scholar]
- 31. Hu Z., Wang Y., Wang Y., Zang B., Hui H., You Z., and Wang X. (2017) Epigenetic activation of SIN1 promotes NSCLC cell proliferation and metastasis by affecting the epithelial-mesenchymal transition. Biochem. Biophys. Res. Commun. 483, 645–651 10.1016/j.bbrc.2016.12.089 [DOI] [PubMed] [Google Scholar]
- 32. Xu J., Li X., Yang H., Chang R., Kong C., and Yang L. (2013) SIN1 promotes invasion and metastasis of hepatocellular carcinoma by facilitating epithelial-mesenchymal transition. Cancer 119, 2247–2257 10.1002/cncr.28023 [DOI] [PubMed] [Google Scholar]
- 33. Wang Q., Zhu J., Wang Y. W., Dai Y., Wang Y. L., Wang C., Liu J., Baker A., Colburn N. H., and Yang H. S. (2017) Tumor suppressor Pdcd4 attenuates Sin1 translation to inhibit invasion in colon carcinoma. Oncogene 36, 6225–6234 10.1038/onc.2017.228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Krymskaya V. P., Snow J., Cesarone G., Khavin I., Goncharov D. A., Lim P. N., Veasey S. C., Ihida-Stansbury K., Jones P. L., and Goncharova E. A. (2011) mTOR is required for pulmonary arterial vascular smooth muscle cell proliferation under chronic hypoxia. FASEB J. 25, 1922–1933 10.1096/fj.10-175018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Houssaini A., Abid S., Mouraret N., Wan F., Rideau D., Saker M., Marcos E., Tissot C. M., Dubois-Randé J. L., Amsellem V., and Adnot S. (2013) Rapamycin reverses pulmonary artery smooth muscle cell proliferation in pulmonary hypertension. Am. J. Respir. Cell Mol. Biol. 48, 568–577 10.1165/rcmb.2012-0429OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bernacchioni C., Cencetti F., Ouro A., Bruno M., Gomez-Munoz A., Donati C., and Bruni P. (2018) Lysophosphatidic acid signaling axis mediates ceramide 1-phosphate-induced proliferation of C2C12 myoblasts. Int. J. Mol. Sci. 19, 139–158 10.3390/ijms19010139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ediger T. L., Danforth B. L., and Toews M. L. (2002) Lysophosphatidic acid upregulates the epidermal growth factor receptor in human airway smooth muscle cells. Am. J. Physiol. Lung Cell Mol. Physiol. 282, L91–L98 10.1152/ajplung.2002.282.1.L91 [DOI] [PubMed] [Google Scholar]
- 38. Riaz A., Zeller K. S., and Johansson S. (2012) Receptor-specific mechanisms regulate phosphorylation of AKT at Ser473: role of RICTOR in β1 integrin-mediated cell survival. PLoS ONE 7, e32081 10.1371/journal.pone.0032081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Koo J., Wu X., Mao Z., Khuri F. R., and Sun S. Y. (2015) Rictor undergoes glycogen synthase kinase 3 (GSK3)-dependent, FBXW7-mediated ubiquitination and proteasomal degradation. J. Biol. Chem. 290, 14120–14129 10.1074/jbc.M114.633057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ghosh D., Srivastava G. P., Xu D., Schulz L. C., and Roberts R. M. (2008) A link between SIN1 (MAPKAP1) and poly(rC) binding protein 2 (PCBP2) in counteracting environmental stress. Proc. Natl. Acad. Sci. U.S.A. 105, 11673–11678 10.1073/pnas.0803182105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rahimi R. A., Andrianifahanana M., Wilkes M. C., Edens M., Kottom T. J., Blenis J., and Leof E. B. (2009) Distinct roles for mammalian target of rapamycin complexes in the fibroblast response to transforming growth factor-β. Cancer Res. 69, 84–93 10.1158/0008-5472.CAN-08-2146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ferguson K. T., Torr E. E., Bernau K., Leet J., Sherris D., and Sandbo N. (2017) The novel mTOR complex 1/2 inhibitor P529 inhibits human lung myofibroblast differentiation. J. Cell. Biochem. 118, 2241–2249 10.1002/jcb.25878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Maher P., and Conti B. (2018) Deciphering the pathways that protect from IL-13-mediated potentiation of oxidative stress-induced dopaminergic nerve cell death. Cytokine 103, 114–120 10.1016/j.cyto.2017.09.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Schroder W., Bushell G., and Sculley T. (2005) The human stress-activated protein kinase-interacting 1 gene encodes JNK-binding proteins. Cell Signal. 17, 761–767 10.1016/j.cellsig.2004.10.015 [DOI] [PubMed] [Google Scholar]
- 45. Mesri M., and Altieri D. C. (1999) Leukocyte microparticles stimulate endothelial cell cytokine release and tissue factor induction in a JNK1 signaling pathway. J. Biol. Chem. 274, 23111–23118 10.1074/jbc.274.33.23111 [DOI] [PubMed] [Google Scholar]
- 46. Gao M., Labuda T., Xia Y., Gallagher E., Fang D., Liu Y. C., and Karin M. (2004) Jun turnover is controlled through JNK-dependent phosphorylation of the E3 ligase Itch. Science 306, 271–275 10.1126/science.1099414 [DOI] [PubMed] [Google Scholar]
- 47. Gupta S., Campbell D., Dérijard B., and Davis R. J. (1995) Transcription factor ATF2 regulation by the JNK signal transduction pathway. Science 267, 389–393 10.1126/science.7824938 [DOI] [PubMed] [Google Scholar]
- 48. Chen C. H., and Sarbassov dos D. (2011) The mTOR (mammalian target of rapamycin) kinase maintains integrity of mTOR complex 2. J. Biol. Chem. 286, 40386–40394 10.1074/jbc.M111.282590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chen C. H., Kiyan V., Zhylkibayev A. A., Kazyken D., Bulgakova O., Page K. E., Bersimbaev R. I., Spooner E., and Sarbassov dos D. (2013) Autoregulation of the mechanistic target of rapamycin (mTOR) complex 2 integrity is controlled by an ATP-dependent mechanism. J. Biol. Chem. 288, 27019–27030 10.1074/jbc.M113.498055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Humphrey S. J., Yang G., Yang P., Fazakerley D. J., Stöckli J., Yang J. Y., and James D. E. (2013) Dynamic adipocyte phosphoproteome reveals that Akt directly regulates mTORC2. Cell Metab. 17, 1009–1020 10.1016/j.cmet.2013.04.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Walker N., Badri L., Wettlaufer S., Flint A., Sajjan U., Krebsbach P. H., Keshamouni V. G., Peters-Golden M., and Lama V. N. (2011) Resident tissue-specific mesenchymal progenitor cells contribute to fibrogenesis in human lung allografts. Am. J. Pathol. 178, 2461–2469 10.1016/j.ajpath.2011.01.058 [DOI] [PMC free article] [PubMed] [Google Scholar]