

Abstract
Mitochondrial reactive oxygen species (ROS) production is a tightly regulated redox signal that transmits information from the organelle to the cell. Other mitochondrial signals, such as ATP, are sensed by enzymes, including the key metabolic sensor and regulator, AMP-activated protein kinase (AMPK). AMPK responds to the cellular ATP/AMP and ATP/ADP ratios by matching mitochondrial ATP production to demand. Previous reports proposed that AMPK activity also responds to ROS, by ROS acting on redox-sensitive cysteine residues (Cys-299/Cys-304) on the AMPK α subunit. This suggests an appealing model in which mitochondria fine-tune AMPK activity by both adenine nucleotide–dependent mechanisms and by redox signals. Here we assessed whether physiological levels of ROS directly alter AMPK activity. To this end we added exogenous hydrogen peroxide (H2O2) to cells and utilized the mitochondria-targeted redox cycler MitoParaquat to generate ROS within mitochondria without disrupting oxidative phosphorylation. Mitochondrial and cytosolic thiol oxidation was assessed by measuring peroxiredoxin dimerization and by redox-sensitive fluorescent proteins. Replacing the putative redox-active cysteine residues on AMPK α1 with alanines did not alter the response of AMPK to H2O2. In parallel with measurements of AMPK activity, we measured the cell ATP/ADP ratio. This allowed us to separate the effects on AMPK activity due to ROS production from those caused by changes in this ratio. We conclude that AMPK activity in response to redox changes is not due to direct action on AMPK itself, but is a secondary consequence of redox effects on other processes, such as mitochondrial ATP production.
Keywords: AMP-activated kinase (AMPK), reactive oxygen species (ROS), mitochondria, hydrogen peroxide, redox signaling
Introduction
AMP-activated protein kinase (AMPK)2 is a key sensor and regulator of cellular energy metabolism, which helps match mitochondrial ATP production to the energy demands of the cell (1–5). A decrease in ATP production, or an increase in its demand, elevates AMP and ADP levels relative to ATP. This results in increased binding of AMP (and ADP) to the AMPK γ subunit, which enhances AMPK activity (hereafter AMP-dependent regulation) and thus phosphorylation of a range of downstream target proteins (1–5).
AMPK can also be activated by atypical mechanisms, independently of changes in the cell's ATP/ADP or ATP/AMP ratio (hereafter AMP-independent regulation) (6–11) (Fig. 1A). The small molecule AMPK activators A-769662, 991, and salicylate (a metabolite of aspirin), bind at the interface of the phosphorylated β subunit carbohydrate-binding module and the N-lobe of the α subunit kinase domain, known as the Allosteric Drug and Metabolite (ADaM)-binding site (6–9). Binding of compounds at this site increases AMPK activity independently of adenine nucleotide-binding to the γ subunit by inducing a conformational change that puts AMPK into the active conformation. More recent is the finding that AMPK can also be activated in an AMP-independent manner by glucose deprivation, specifically due to a decrease in the intracellular level of fructose 1,6-bisphosphate, which promotes formation of a multienzyme complex necessary for AMPK activation (10).
Figure 1.

Overview of AMPK regulation and experimental rationale. A, established and hypothesized regulators of AMPK activity. B, summary of hypothesis showing that ROS could affect AMPK activity directly, or indirectly by altering adenine nucleotide ratios or other pathways.
It has also been proposed that reactive oxygen species (ROS) can directly regulate AMPK activity independently of changes in adenine nucleotides (12–18). Redox regulation typically occurs by the reversible oxidation of cysteine thiols, thereby altering the activity, cellular localization, or binding interactions of a protein (19–22). Mitochondria are an important site of ROS production. ROS generation by complex I through reverse electron transport (RET) is particularly attractive as a potential mitochondrial redox signal because its magnitude responds sensitively to redox status and occurs under physiological conditions (23–26). Thus, a scenario in which mitochondrial functional status and ROS production can alter AMPK activity by two parallel but independent pathways: through changes in adenine nucleotides and by redox signaling from mitochondria, has considerable appeal (Fig. 1B). In support of AMPK redox regulation, its activity was reported to be increased by exogenous hydrogen peroxide (H2O2), independently of changes in adenine nucleotides and this alteration required redox changes to key cysteine residues, Cys-299 and Cys-304, in the AMPK α1 catalytic subunit (13). However, other reports have challenged whether the AMPK activation by H2O2 in cells is independent of changes in the ATP/ADP or ATP/AMP ratio (17, 18) and furthermore, another laboratory has shown that exogenous H2O2 can inhibit AMPK (12). These discrepancies could arise from methodological or biological differences, such as alternative ways of measuring adenine nucleotides or different levels of ROS scavenging systems in the cells types used (12, 18). In addition, as AMPK is localized to the cytosol, it is important to assess whether changes in AMPK activity in response to ROS are associated with reversible cytosolic protein thiol oxidation, e.g. peroxiredoxin (Prx) dimerization (27), which would be consistent with direct ROS signaling and/or redox relay signaling (28, 29). Furthermore, mitochondrial redox signaling could also affect AMPK activity independently of either direct effects on the enzyme, or on the ATP/ADP ratio (Fig. 1B).
We set out to determine the possibility of AMP-independent regulation of AMPK activity in response to physiological levels of ROS generated from mitochondria in cells. To do this we altered ROS levels by addition of hydrogen peroxide. In addition, we utilized the mitochondria-targeted redox cycler MitoParaquat (MitoPQ), which can generate physiological ROS levels within mitochondria at complex I without disrupting oxidative phosphorylation (30). MitoPQ also mimics production of mitochondrial ROS at complex I by RET, a physiologically plausible mechanism of mitochondrial redox signaling (24–26, 31). To determine where protein thiol redox changes were occurring, we analyzed redox-dependent protein thiol oxidation in the mitochondria and cytosol. We further investigated the regulatory effects on AMPK activity by replacing the key redox-sensitive residues Cys-299 and Cys-304 in the AMPK α subunit (13). In parallel, we measured the cell ATP/ADP ratio to determine whether the effects on AMPK activity could be accounted for by secondary changes. These methods allowed us to separate the effects of mitochondrial ROS production on AMPK activity from those of a changing ATP/ADP ratio. We found that the effects of mitochondrial and cytosolic redox changes on AMPK activity were indirect and could be largely, but not entirely, accounted for by the effect of ROS on the ATP/ADP ratio. We conclude that ROS affect AMPK activity indirectly.
Results
Characterizing AMPK activity in C2C12 mouse myotubes
To characterize the putative regulation of AMPK activity by redox signals we used differentiated C2C12 mouse myotubes, a model of skeletal muscle fibers in which AMPK is a key regulator of mitochondrial and cellular function (11, 32, 33) (Fig. S1A). We first established how AMPK activity responded to the ATP/ADP ratio, which determines cellular AMP and thus accounts for AMPK regulation by adenine nucleotides (3, 4). AMPK activity was assessed by the phosphorylation of Thr-172 of AMPK's catalytic α subunit; by phosphorylation of Ser-79 on the AMPK target acetyl-CoA carboxylase (ACC); and by determining AMPK-specific activity using the SAMS kinase assay. Decreasing mitochondrial ATP production with the complex I inhibitors rotenone or phenformin or with the F0F1-ATP synthase inhibitor oligomycin all increased AMPK activity (Fig. 2, A and B). The ATP/ADP ratio in control myotubes (16.1 ± 2.1) (Fig. S1B) decreased on inhibition of oxidative phosphorylation to between 7.4 ± 1.1 (rotenone) and 8.3 ± 2.1 (phenformin), ∼46 and ∼52% of control cells, respectively (Fig. 2C). Thus, the cell ATP/ADP ratio correlates inversely with AMPK activity.
Figure 2.

AMPK activity markers and ATP/ADP ratios in C2C12 myotubes in response to AMP-dependent and AMP-independent activators. A–C, C2C12 myotubes were treated with mitochondrial inhibitors: rotenone (2 μg/ml), phenformin (5 mm), or oligomycin (100 ng/ml) for 30 min. A, Western blotting of AMPKα-phospho-Thr-172, ACC-phospho-Ser-79, and AMPKβ (loading control). B, AMPK-specific activity was assessed by the SAMS kinase assay, measured as picomole of [γ-32P]ATP/μg of cell protein/min and expressed as % untreated control. C, ATP/ADP ratios in C2C12 myotubes were measured by a luciferase/luciferin bioluminescence assay and expressed as % untreated control. D and E, C2C12 myotubes were treated with 991 (1 or 10 μm) for 30 min. D, Western blotting was performed as previously described. E, AMPK-specific activity was measured as previously described. F, ATP/ADP ratios were measured as previously described. All Western blots are representative of 3 or 4 biological replicates. Bar graphs indicate mean ± S.D. of 3 biological replicates. Statistical analysis was performed by one-way ANOVA with Dunnett's multiple comparison post-test comparing all treatments to untreated controls; *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001.
If redox signals activate AMPK independently of adenine nucleotides they will mimic pharmacological AMPK activators such as 991, A-769662, and salicylate, which bind directly to the regulatory ADaM-binding site (6–8). Treatment with 991 increased AMPK activity dose-dependently (Fig. 2, D and E) without decreasing the ATP/ADP ratio (Fig. 2F).
Combining the data where AMPK was activated indirectly (Fig. 2, A–C) or with the selective activator 991 (Fig. 2, D–F) showed a clear divergence in the relationship between the ATP/ADP ratio and AMPK activity for all three measurements of activity individually (Fig. S1, B–G). In Fig. S1, H and I changes in AMPK activation by alterations in the ATP/ADP ratio (AMP-dependent activation) are shown upon the red background, whereas AMP-independent activation (green background) induces AMPK activation without decreasing ATP/ADP ratios. In fact AMP-independent activation occurred with a slight increase in the ATP/ADP ratio. Combining all these data shows clearly the different trajectories of dependence of AMPK activity on ATP/ADP ratio for these two modes of regulation (Fig. 3). Next, we set out to use this approach to assess the mechanism by which redox signals affect AMPK activity.
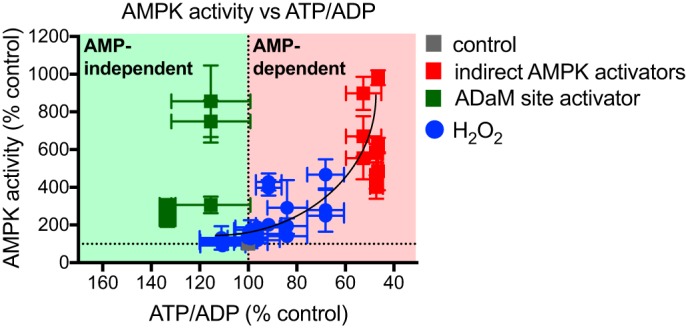
Figure 3.

Reference plot showing AMP-dependent (red background) and AMP-independent (green background) AMPK activation in C2C12 myotubes. Data points are measures of AMPKα-phospho-Thr-172 (Fig. S1H), ACC-phospho-Ser-79 (Fig. S1I), and AMPK-specific activity (Fig. S1J) plotted against corresponding cell ATP/ADP ratios. Values are % untreated control and presented as mean ± S.E. of ≥3 biological replicates.
Exogenous H2O2 leads to dose-dependent, reversible protein thiol oxidation in the cytosol and mitochondrial matrix of myotubes
We first assessed the effects of exogenous H2O2 on thiol redox status in C2C12 myotubes. Myotubes were treated with a range of H2O2 concentrations administered as single boluses. Cytosolic Prx 2 dimerization, measured 10 min after treatment, peaked upon treatment with the 75 μm H2O2 bolus (Fig. 4A). Extended oxidation induces thiol modifications (sulfinic acid (–SO2H) and sulfonic acid (–SO3H)), which block the formation of Prx dimers (36), hence the decrease in Prx 2 dimerization and increase in Prx-SO2/3 formation (Fig. 4A) observed in response to higher levels of H2O2. Exogenous H2O2 (≥75 μm) also oxidized the mitochondrial matrix Prx 3 pool (Fig. S2A). Dimers of Prx 3 were visible as double bands, likely due to the different electrophoretic mobility of dimers linked by one or two disulfide bridges (37–39).
Figure 4.

Effects of H2O2 boluses on subcellular redox state, AMPK activity, and ATP/ADP ratios in C2C12 myotubes. A–H, C2C12 myotubes were treated with serially diluted boluses of H2O2 (7.5, 75, 375, or 750 μm) in serum-free media for 10 or 30 min. A, Prx 2 dimerization and Prx-SO2/3 formation were assessed by Western blotting. B–D, Western blots and quantification of AMPKα-phospho-Thr-172 and ACC-phospho-Ser-79 levels in H2O2-treated cells. Graphed values are mean ± S.D. of 3 biological replicates. Results at each time point were assessed by one-way ANOVA with Dunnett's multiple comparison post-test comparing all H2O2 treatments to untreated controls; *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001. E, AMPK-specific activity was measured as previously described. Graphed data are mean ± S.D. of 3 or mean ± range of 2 biological replicates, expressed as % untreated control. Statistical analysis was performed as described above. F, ATP/ADP ratios were measured as previously described. Graphed data are mean ± S.D. of 4 biological replicates, expressed as % untreated control. Statistical analysis was performed as described above. G and H, graphed data shows the relationship between Prx-SO2/3 formation (Fig. 4A) and (E) AMPK-specific activity or (F) ATP/ADP ratios in response to H2O2 in cells. Values are mean ± S.E. of ≥3 or mean ± range of 2 biological replicates, expressed as % untreated control.
We conclude that within the range of H2O2 concentrations tested, a period of reversible thiol oxidation occurred in both the cytosol and mitochondrial matrix, which is an environment conducive to thiol redox signaling (28, 29). The rapidly changing Prx redox states in response to single additions of H2O2 indicated rapid H2O2 metabolism. At higher H2O2 concentrations, thiol hyperoxidation occurred (Prx-SO2/3 formation), which is evidence of oxidative stress. We thus used this Prx redox mapping to assess the response of AMPK to protein thiol redox changes by H2O2.
Exogenous H2O2 activates AMPK indirectly in myotubes by decreasing the ATP/ADP ratio
Addition of exogenous H2O2 that led to protein thiol oxidation in the cytosol (Fig. 4A) induced a dose-dependent increase in AMPK activity measured 10 min after addition of H2O2 (Fig. 4, B–E). H2O2 also had a dose-dependent effect on cellular ATP/ADP ratios at 10 min (Fig. 4F).
The dose-dependent increases in AMPK-specific activity at 10 min did not correlate with reversible Prx 2 dimerization (Fig. S2B), but did correlate with formation of the thiol hyperoxidation marker Prx-SO2/3 (Fig. 4G), implicating oxidative damage rather than redox signaling in the mechanism of AMPK activation. Furthermore, decreased ATP/ADP ratios at 10 min also correlated with increasing Prx-SO2/3 formation (Fig. 4H). Although H2O2 boluses lower than 750 μm were sufficient to oxidize the cytosol without affecting the cell ATP/ADP ratio, AMPK activity did not change significantly under these conditions, indicating that AMPK activity only responds to the ATP/ADP ratio and not directly to H2O2.
Of further interest, we note that 30 min after H2O2 treatment, cell ATP/ADP ratios approached recovery (Fig. 4F) and accordingly AMPK-specific activity decreased from that measured at 10 min (Fig. 4E). However, AMPK and ACC phosphorylation was sustained at 30 min (Fig. 4, B–D) as was Prx-SO2/3 formation (Fig. S2C).
All ways of measuring the changes in AMPK activity showed that addition of exogenous H2O2 increased AMPK activity (Fig. 4, B–E). Graphing these data confirmed that increased AMPK activity occurred only when ATP/ADP ratios were decreased (Fig. S2, D–F). Most significantly, the effect of exogenous H2O2 on AMPK activity was described by the same relationship as when the ATP/ADP ratio was decreased by the use of mitochondrial inhibitors (Fig. 5). We conclude that AMPK activation in C2C12 myotubes by added H2O2 occurs indirectly as a consequence of altering the cell's ATP/ADP ratio.
Figure 5.
Reference plot showing AMP-dependent and -independent AMPK activation in C2C12 myotubes. H2O2 data points are measures of AMPKα-Thr-172 phosphorylation (Fig. S2D), ACC-Ser-79 phosphorylation (Fig. S2E), and AMPK-specific activity (Fig. S2E) measured at 10 and 30 min, plotted against corresponding cell ATP/ADP ratios. Values are % untreated control and presented as mean ± S.E. of ≥3 biological replicates.
Selective generation of ROS within mitochondria does not induces AMPK activation
The effect of exogenous H2O2 on AMPK activity in C2C12 myotubes occurred indirectly, via changes in the ATP/ADP ratio (Fig. 5). To further analyze any effects of mitochondrial redox signals on AMPK activity, which will come from mitochondria within the cell rather than from outside the cell, we next assessed the effect of mitochondria-generated ROS on AMPK activity. Mitochondrial ROS signaling is likely to arise from enhanced superoxide generation and subsequent H2O2 production within the organelle that leads to the propagation of a signal from the mitochondria to the rest of the cell (24–26, 31). To mimic this scenario, we assessed the effects on AMPK activity of the mitochondria-targeted redox cycler, MitoPQ. MitoPQ generates superoxide selectively and continuously within mitochondria at complex I, which is then rapidly dismutated to H2O2 (30) (Fig. 6A). To confirm that MitoPQ generated ROS in the mitochondrial matrix and to assess potential flux of H2O2 to the cytosol, we measured the extent of redox-dependent dimerization of Prx 3 (matrix isoform) and Prx 2 (cytosolic isoform) in response to MitoPQ (Fig. 6B and Fig. S3, A and B). MitoPQ led to a dose- and time-dependent increase in Prx 3 dimerization from 6 to 24 h, but had no effect on Prx 2 dimerization (Fig. 6B), consistent with persistent ROS production in the matrix. These data indicated that MitoPQ generated ROS locally in the mitochondria, but that these ROS did not diffuse to the cytosol. Redox compartmentalization was confirmed using cytosol-specific (CytoORP) and mitochondria-specific (MitoORP) fluorescent protein probes for H2O2 (34, 35) (Fig. 6, C and D). This analysis showed enhanced H2O2-dependent fluorescence in the mitochondria in response to MitoPQ (Fig. 6C), but not in the cytosol (Fig. 6D).
Figure 6.

Effects of MitoPQ on subcellular redox state, AMPK activity, and ATP/ADP ratios in C2C12 myotubes. A, MitoPQ mechanism of action. B, C2C12 myotubes were treated with 5 μm MitoPQ or vehicle (EtOH) for 0.5, 6, or 24 h. Prx 2 and 3 dimerization was measured from Western blotting target bands (Fig. S3, A and B). Graphed data show mean ± S.D. of ≥3 biological replicates. Results at each time point were analyzed by unpaired, one-tailed t tests compared with EtOH controls; **, p < 0.01. C and D, C2C12 myoblast cells were transfected with redox-sensitive roGFP coupled to ORP1, targeted to the mitochondria (MitoORP) or cytosol (CytoORP) and treated with MitoPQ (1 or 5 μm), MitoPQ control (5 μm), or H2O2 (50 μm). roGFP-ORP1 alters excitation from 488 to 405 when oxidized by H2O2 (shown here as increasing levels of blue). C, treatment with MitoPQ induced dose-dependent oxidation of MitoORP, peaking at 5 μm. Treatment with the MitoPQ control did not oxidize MitoORP (baseline value of 1). Graphed data are mean ± S.D. of ≥5. Statistical analysis was performed by one-way ANOVA with Dunnett's multiple comparison post-test comparing all treatments to MitoPQ control; ***, p < 0.001. D, MitoPQ specifically oxidized MitoORP, with no changes observed in CytoORP. CytoORP was oxidized by addition of a single H2O2 bolus to cell media. Graphed data are mean ± S.D. of ≥4 except the H2O2 treatment, which is mean ± range of 2. Statistical analysis was performed by one-way ANOVA with Dunnett's multiple comparison post-test comparing all treatments; **, p < 0.01; ***, p < 0.001. E, ATP/ADP ratios in cells treated with 5 μm or EtOH control. Data are presented as mean ± S.D. of 3 biological replicates. Results at each time point were analyzed by unpaired, one-tailed t tests compared with EtOH controls; *, p < 0.05. F, reference plot showing AMP-dependent and -independent AMPK activation in C2C12 myotubes. MitoPQ data points are measures of AMPKα-Thr-172 phosphorylation (Fig. S3D) and ACC-Ser-79 phosphorylation (Fig. S3E) (at 6 and 24 h) plotted against corresponding cell ATP/ADP ratios. Values are % untreated control or % EtOH control (MitoPQ) and presented as mean ± S.E. of ≥3 biological replicates.
There was negligible alteration to AMPK activity in response to short-term incubation with MitoPQ (Fig. S3, C–E). However, there were suggestions of an increase in AMPK activity upon sustained incubation with MitoPQ (Fig. S3, C–E). This most likely reflects the decrease in the ATP/ADP ratio upon long-term incubation with MitoPQ (Fig. 6E). However, the effects of MitoPQ on AMPK activity were negligible compared with either direct or indirect AMPK activators (Fig. 6F). That MitoPQ could significantly alter the mitochondrial thiol redox state without considerably affecting AMPK activity suggests that mitochondrial redox signaling is unlikely to regulate AMPK, either directly or indirectly.
Mutation of the putative redox active cysteines in AMPK does not affect its activation by ROS
Previous studies proposed direct redox regulation of the key cysteine residues Cys-299 and Cys-304 in the α catalytic subunit of AMPK (13). To assess whether these residues could facilitate a direct effect of ROS on AMPK activation we expanded our study to an AMPK KO HEK 293T cell line and compared WT AMPK α1/β1/γ1 versus mutant AMPK α1(C299A/C304A)/β1/γ1 (Fig. 7A and Fig. S4A). In WT HEK 293T cells addition of exogenous H2O2 increased AMPK activity (Fig. 7, B–D, and Fig. S4, B and C). Importantly, the H2O2 led to identical activation of mutant AMPK α1(C299A/C304A)/β1/γ1 (Fig. 7, B–D, and Fig. S4, B and C). Therefore, AMPK activation by H2O2 is independent of any redox changes on the putative regulatory cysteine residues on the AMPK α subunit. The lowest concentration of H2O2 used had no effect on the ATP/ADP ratio in the HEK 293T cells (Fig. 7E), even though this concentration of H2O2 lead to activation of AMPK (Fig. 7, B–D). Higher concentrations of H2O2 affected both the ATP/ADP ratio and AMPK activity (Fig. S4, B–D). Therefore, these experiments show clearly that these cysteine residues on AMPK are not involved in regulating its activity. However, in contrast to C2C12 myotubes, in HEK 293T cells exogenous H2O2 can increase AMPK activity by an indirect mechanism that is distinct from changes in the ATP/ADP ratio.
Figure 7.

Effects of H2O2 boluses on WT AMPK α1/β1/γ1 or AMPK α1(C299A/C304A)/β1/γ1 activity and ATP/ADP ratios in HEK 293T cells. A, location of Cys-299 and Cys-304 in the AMPK α subunit. AID, autoinhibitory domain; ST loop, Ser/Thr-rich loop. B–E, AMPK KO HEK 293T cells overexpressing WT AMPK α1/β1/γ1 or AMPK α1(C299A/C304A)/β1/γ1 were treated with serially diluted boluses of H2O2 (75, 375, or 750 μm) in serum-free media for 10 min. B–D, Western blots and quantification of AMPKα-phospho-Thr-172 and ACC-phospho-Ser-79 levels in H2O2 (75 μm)-treated cells. E, ATP/ADP ratios in cells treated with 75 μm H2O2. Graphed values are mean ± S.D. of 3 biological replicates.
Conclusions
The appealing possibility that redox signaling from mitochondria can modulate the activity of the key energy regulator, AMPK, independently of the effects of adenine nucleotides offers possibilities of new modes of regulation of mitochondrial function. Thus, reports of the direct redox regulation of AMPK attracted considerable attention (13). However, other labs have not observed redox regulation of AMPK in response to exogenous oxidants (18), or have instead found inhibition (12). Therefore, here we set out to assess the possibility of redox regulation of AMPK in differentiated C2C12 mouse skeletal myotubes. To do this we used exogenous H2O2, or the mitochondria-targeted redox cycler MitoPQ. The rationale for using MitoPQ was that this closely mimics mitochondrial redox signaling by RET at complex I, and is thus a good model for physiological redox signaling. However, even though MitoPQ led to the selective oxidation of protein thiols within the mitochondria, suggestive of redox signaling, this did not considerably impact AMPK activity in the cytosol. Furthermore, whereas we found that exogenous H2O2 activated AMPK, this activation was not by direct redox signaling to AMPK, but was secondary to changes in cellular ATP/ADP. This was extended to HEK 293T cells where AMPK activity was also altered by exogenous H2O2. This change in AMPK activity was unaffected by replacing the putative redox-sensitive cysteine residues by redox-insensitive alanine residues. In contrast to C2C12 myotubes, in HEK 293T cells exogenous H2O2 activated AMPK independently of the ATP/ADP ratio, suggesting a further indirect response of AMPK to ROS exposure. Possible mechanisms of this response, such as changes in the level of fructose 1,6-bisphosphate (10), will be explored in future work.
Our results complement and expand previous studies into the role of ROS in AMPK activity (17, 18). In addition, some interesting new insights into AMPK regulation emerge. We observed that AMPK activity in C2C12 myotubes was increased at 10 min after addition of H2O2, which correlated with a decreased cell ATP/ADP ratio. The specific activity of AMPK later decreased at 30 min as ATP/ADP ratios recovered. As Prx-SO2/3 formation was increased at 30 min compared with 10 min, one possibility is that the increased Prx hyperoxidation served as a protection mechanism. A previous study has shown that under conditions of oxidative stress dimerized Prxs can compete with other cellular proteins for cysteine thiol reduction catalyzed by thioredoxins (40). Thus, Prx hyperoxidation may promote cell survival, and allow ATP/ADP ratio recovery, under conditions of oxidative stress. In agreement with a previous study (18), our data suggest that H2O2 may inhibit AMPK and ACC dephosphorylation, as AMPK and ACC phosphorylation were maintained and increased at 30 min after H2O2 addition, even as cell ATP/ADP ratios recovered.
Our work also provides insights into the redox regulation of cellular processes. Monitoring the redox states of cytosolic and mitochondrial Prx isoforms enabled us to assess the effects of redox signals on different cell compartments simultaneously and also monitor ROS flux between cell compartments. Extracellular H2O2 was capable of oxidizing mitochondrial matrix Prx 3, but only at high concentrations (≥75 μm). MitoPQ generated superoxide within the mitochondrial matrix, inducing significant Prx 3 dimerization, but there was no increase in cytosolic Prx 2 dimerization. Together these data suggest that whereas H2O2 can cross the mitochondrial inner membrane, it will more usually be degraded in the matrix by the action of Prx 3.
In conclusion, we found that the effects of redox changes on AMPK activity were indirect and could be largely, but not entirely, accounted for by the effect of ROS on the ATP/ADP ratio. We conclude that ROS affect AMPK activity indirectly.
Experimental procedures
Antibodies
All commercial antibodies were purchased from Cell Signaling Technology, unless otherwise stated. Primary antibodies were rabbit ACC1/2-phospho-Ser79 (number 3661); rabbit ACC1/2 (number 3662); rabbit AMPKα1/2-phospho-Thr-172 mAb 40H9 (number 2535); rabbit AMPKα1/2 (number 2532); rabbit-AMPKβ1/2 mAb 57C12 (number 4150); mouse α-tubulin clone B-5–1-2 (Sigma: number T5168); mouse FLAG M2 (Sigma, number F1804); mouse Myc (Merck, number 05724); mouse Prx 2 (Abcam: number ab50862); rabbit-Prx 3 (Abcam: number ab73349); rabbit Prx-SO2/3 (Cambridge Research Biochemicals: number crb2005004e); rabbit myogenin (Abcam, number ab124800); and rabbit anti-pan-AMPKβ antibody (42). Secondary antibodies were IRDye® 800 goat anti-rabbit IgG F(c) (Rockland Antibodies and Assays: number 611-132-003) and IRDye® 680RD goat anti-mouse IgG (H+L) (LI-COR Biosciences: number 926–68070).
Plasmids
cDNA encoding WT human AMPKα1, or harboring mutation of cysteine residues 299 and 304 to alanine, were cloned into pLPC, and engineered to encode an N-terminal FLAG epitope tag. cDNAs encoding rat AMPKβ1 and rat AMPKγ1 (harboring an N-terminal Myc epitope tag) were cloned into pCDNA3. All constructs were confirmed by DNA sequencing.
Cell culture
HEK 293T AMPK knockout (KO) cells were generated by using CRISPR/Cas9 to remove expression of both AMPKβ1 and -β2, as previously described (41). Transient transfections were performed using Lipofectamine® 2000 (according to the manufacturer's instructions), 24 h prior to cell treatments. C2C12 mouse myoblast cells and HEK 293T cells were cultured in standard Dulbecco's modified Eagle's medium/GlutaMAX medium (glucose (4.5 g/liter), sodium pyruvate (100 mg/ml)) supplemented with 10% (v/v) fetal bovine serum (FBS), penicillin (100 units/ml), and streptomycin (100 μg/ml). Cells were incubated in a humidified atmosphere (5% CO2, 95% air) at 37 °C. C2C12s were differentiated to myotubes by serum depletion to 1% (v/v) FBS for 7 days, with media changes every 2 days. Treatments were performed on day 7 or 8 of serum depletion. H2O2 treatments were performed in serum-free medium. After treatments, cells were rapidly washed in ice-cold PBS before rapid lysis on an ice-cold aluminum block. For AMPK assays, cells were lysed in 250 μl of ice-cold Hepes lysis buffer: Hepes (50 mm, pH (7.4)), EDTA (1 mm), glycerol (10% (v/v)), sodium fluoride (50 mm), sodium pyrophosphate (5 mm), Triton X-100 (1% (v/v)). This was supplemented just before use with dithiothreitol (DTT) (1 mm) and protease inhibitors: soybean trypsin-chymotrypsin inhibitor (4 μg/ml), phenylmethanesulfonyl fluoride (0.1 mm), and 1 mm benzamidine (1 mm). Cells for Prx oxidation assays were lysed in 250 μl of RIPA buffer: Tris (50 mm, pH 8.0, at 4 °C), sodium chloride (150 mm), Triton X-100 (1.0% (v/v)), sodium deoxycholate (0.5% (w/v)) and SDS (0.1% (w/v)). This was supplemented just before use with protease inhibitors and methyl methanethiosulfonate (80 mm). Prior to lysis, cells were incubated with methyl methanethiosulfonate (80 mm) for 5 min at room temperature. Cells for ATP/ADP assays were lysed in 500 μl of ice-cold perchloric acid extractant: HClO4 (3% (v/v)), Na2EDTA (2 mm), and Triton X-100 (0.5% (v/v)). Lysed cell supernatants were stored on ice for immediate use or aliquoted, snap-frozen on dry ice, and stored at −20 or −80 °C (long-term).
SDS-PAGE
Cell lysate was heated in Laemmli sample buffer with freshly added DTT (25 mm) for 5 min at 95 °C (DTT was excluded for Prx Western blots). Samples (∼25 μg of protein) were loaded on Mini-PROTEAN® TGXTM Precast 4–20% gradient gels (Bio-Rad). Proteins were electrophoresed at 100 volts in the presence of Tris (25 mm, pH (8.3)), glycine (192 mm), and SDS (0.1% (w/v)).
Western blotting
Electrophoresed proteins were transferred to Immobilon®-FL PVDF membranes by wet transfer performed at 100 volts for 1 h at 4 °C in the presence of Tris (25 mm), glycine (192 mm), and methanol (20% (v/v), pH 8.4) at 4 °C. Where appropriate, membranes were sectioned according to molecular weight before incubation with primary antibodies, overnight at 4 °C. Signal intensities of target bands were measured as fluorescence emission at 800 or 680 nm with the Odyssey CLx IR Imaging System and quantified with LI-COR Biosciences Image Studio Lite software. In C2C12 lysates, phospho-AMPKα1/2(Thr-172) and phospho-ACC1/2(Ser-79) signal intensities (SI) were normalized to AMPKβ2. In HEK 293T cells, overexpressed phospho-AMPKα1(Thr-172) and endogenous phospho-ACC1/2(Ser-79) SIs were normalized to overexpressed AMPKα1-FLAG. Prx dimerization was calculated as % dimer (SI dimer/(SI dimer + SI monomer) × 100). Prx-SO2/3 formation was normalized to α-tubulin.
AMPK SAMS kinase assay
The specific kinase activity of AMPK was determined by the radiometric SAMS peptide assay (43). Kinase assays were performed on AMPK immunoprecipitated from 100 to 200 μg of whole cell lysate using a rabbit-pan β antibody conjugated to protein-A (from Staphylococcus aureus)-Sepharose beads. Kinase assays were performed in duplicate and normalized to SAMS-blank controls.
ATP/ADP assays
ATP and ADP levels were measured by luciferase/luciferin bioluminescence using an AutoLumat LB-953-Plus multi-tube luminometer (Berthold), fitted with an autoinjector, and quantified against standard curves of purified ATP and ADP. For ATP measurements, 100 μl of lysate was added to 400 μl of Tris acetate buffer (Tris (100 mm), Na2EDTA (2 mm), MgCl2 (50 mm), pH 7.75) with glacial acetic acid) in luminometer tubes. Luciferase/luciferin solution (DTT (7.5 mm), BSA (0.4 mg/ml), firefly luciferase (1.92 μg of protein/ml), and d-luciferin (120 μm) was made just before use and 100 μl was delivered to each sample tube via autoinjection. Reactions were performed at 30 °C and light emission (relative light units) was recorded 30 s post-injection. ADP was measured by first degrading endogenous ATP with 2× ATP sulfurylase solution followed by incubation at 30 °C for 30 min with agitation (500 rpm) and then heat inactivation. 200 μl of each ATP sulfurylase-treated sample was added to 400 μl of Tris acetate buffer in luminometer tubes (in duplicate). To convert endogenous ADP to ATP, 10 μl of pyruvate kinase/phospho(enoyl)pyruvate solution (type II protein kinase from rabbit muscle (5 units), phosphoenolpyruvate (100 mm)) was added to one of the duplicate tubes (and to all ADP standards) and incubated at 30 °C for 30 min prior to ATP measurement. The protein kinase/phosphoenolpyruvate-blank tubes served as blanks for quantification. Quantification was performed with Excel or GraphPad Prism 7.0. ATP and ADP values were expressed as ATP/ADP ratios.
Microscopy
C2C12 mouse myoblast cells were seeded at a density of 20,000 cells on pre-coated glass coverslips in 6-well plates (Nunclon Delta Surface, Thermo Scientific). 24 h after plating, cells were transfected with Lipofectamine® 2000 and 1 μg of a plasmid encoding either the cytoplasmic targeted roGFP2 ORP1, or the mitochondria-targeted roGFP2 ORP1 (34) (Addgene numbers 64993 and 64992). 48 h after transfection, C2C12 cells were imaged using a Zeiss LSM microscope every 30 s for 1 h in Dulbecco's modified Eagle's medium + 10% FBS. Compounds were added immediately prior to imaging and remained present throughout. The roGFP was sequentially excited at 405 and 488 nm with emission collected at 500–550 nm for both excitation wavelengths. Images were analyzed in ImageJ where the channels were split dependent upon excitation, regions of interest were drawn around the cells, and fluorescence intensity was measured across the full hour. A ratio of 405/488 fluorescence was calculated before the initial intensity was normalized to 1, and subsequent time points were calculated as a derivative. Data are mean ± S.D.
Statistical analysis
Data analysis was performed with GraphPad Prism 7.0 (unless otherwise stated). Data (n = 2) were expressed as mean ± range. Data (n ≥ 3) were analyzed by unpaired, one-tailed t tests (2 groups) or one-way analysis of variance (ANOVA) (>2 groups) unless otherwise stated. Data were expressed as mean ± S.D. (bar graphs) or mean ± S.E. (xy graphs). p values < 0.05 were considered statistically significant.
Author contributions
E. C. H. and M. P. M. conceptualization; E. C. H. data curation; E. C. H. formal analysis; E. C. H. and M. P. M. supervision; E. C. H., A. V. G., N. N., A. R. H., G. B., T. P. B., and T. K. investigation; E. C. H., A. V. G., R. W., N. N., A. R. H., G. B., T. P. B., T. K., and D. C. methodology; E. C. H. writing-original draft; E. C. H., and M. P. M. project administration; E. C. H., D. C., and M. P. M. writing-review and editing; R. W., N. N., and D. C. resources; M. P. M. funding acquisition.
Supplementary Material
Acknowledgments
We thank Dr. Stuart T. Caldwell and Professor Richard C. Hartley, University of Glasgow, for synthesizing MitoPQ.
This work was supported by Medical Research Council UK Grants MC_UU_00015/3 (to M. P. M.) and MC_US_A654_5QB10 (to D. C.) and Wellcome Trust Investigator Award 110159/Z/15/Z (to M. P. M.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S4.
- AMPK
- AMP-activated protein kinase
- ROS
- reactive oxygen species
- ADaM
- allosteric drug and metabolite
- RET
- reverse electron transport
- Prx
- peroxiredoxin
- ACC
- acetyl-CoA carboxylase
- FBS
- fetal bovine serum
- SI
- signal intensities
- ANOVA
- analysis of variance'
- roGFP
- reduction-oxidation sensitive green fluorescent protein.
References
- 1. Carling D., Mayer F. V., Sanders M. J., and Gamblin S. J. (2011) AMP-activated protein kinase: nature's energy sensor. Nat. Chem. Biol. 7, 512–518 10.1038/nchembio.610 [DOI] [PubMed] [Google Scholar]
- 2. Hardie D. G., Ross F. A., and Hawley S. A. (2012) AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 13, 251–262 10.1038/nrm3311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hardie D. G., Carling D., and Gamblin S. J. (2011) AMP-activated protein kinase: also regulated by ADP? Trends Biochem. Sci. 36, 470–477 10.1016/j.tibs.2011.06.004 [DOI] [PubMed] [Google Scholar]
- 4. Oakhill J. S., Steel R., Chen Z. P., Scott J. W., Ling N., Tam S., and Kemp B. E. (2011) AMPK is a direct adenylate charge-regulated protein kinase. Science 332, 1433–1435 10.1126/science.1200094 [DOI] [PubMed] [Google Scholar]
- 5. Garcia D., and Shaw R. J. (2017) AMPK: mechanisms of cellular energy sensing and restoration of metabolic balance. Mol. Cell 66, 789–800 10.1016/j.molcel.2017.05.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Xiao B., Sanders M. J., Carmena D., Bright N. J., Haire L. F., Underwood E., Patel B. R., Heath R. B., Walker P. A., Hallen S., Giordanetto F., Martin S. R., Carling D., and Gamblin S. J. (2013) Structural basis of AMPK regulation by small molecule activators. Nat. Commun. 4, 3017 10.1038/ncomms4017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hawley S. A., Fullerton M. D., Ross F. A., Schertzer J. D., Chevtzoff C., Walker K. J., Peggie M. W., Zibrova D., Green K. A., Mustard K. J., Kemp B. E., Sakamoto K., Steinberg G. R., and Hardie D. G. (2012) The ancient drug salicylate directly activates AMP-activated protein kinase. Science 336, 918–922 10.1126/science.1215327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Langendorf C. G., and Kemp B. E. (2015) Choreography of AMPK activation. Cell Res. 25, 5–6 10.1038/cr.2014.163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hardie D. G. (2016) Regulation of AMP-activated protein kinase by natural and synthetic activators. Acta Pharmaceut. Sin. B 6, 1–19 10.1016/j.apsb.2015.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhang C. S., Hawley S. A., Zong Y., Li M., Wang Z., Gray A., Ma T., Cui J., Feng J. W., Zhu M., Wu Y. Q., Li T. Y., Ye Z., Lin S. Y., Yin H., Piao H. L., Hardie D. G., and Lin S. C. (2017) Fructose-1,6-bisphosphate and aldolase mediate glucose sensing by AMPK. Nature 548, 112–116 10.1038/nature23275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hardie D. G. (2017) Targeting an energy sensor to treat diabetes. Science 357, 455–456 10.1126/science.aao1913 [DOI] [PubMed] [Google Scholar]
- 12. Shao D., Oka S., Liu T., Zhai P., Ago T., Sciarretta S., Li H., and Sadoshima J. (2014) A redox-dependent mechanism for regulation of AMPK activation by thioredoxin1 during energy starvation. Cell Metab. 19, 232–245 10.1016/j.cmet.2013.12.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zmijewski J. W., Banerjee S., Bae H., Friggeri A., Lazarowski E. R., and Abraham E. (2010) Exposure to hydrogen peroxide induces oxidation and activation of AMP-activated protein kinase. J. Biol. Chem. 285, 33154–33164 10.1074/jbc.M110.143685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Emerling B. M., Weinberg F., Snyder C., Burgess Z., Mutlu G. M., Viollet B., Budinger G. R., and Chandel N. S. (2009) Hypoxic activation of AMPK is dependent on mitochondrial ROS but independent of an increase in AMP/ATP ratio. Free Radic. Biol. Med. 46, 1386–1391 10.1016/j.freeradbiomed.2009.02.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hart P. C., Mao M., de Abreu A. L., Ansenberger-Fricano K., Ekoue D. N., Ganini D., Kajdacsy-Balla A., Diamond A. M., Minshall R. D., Consolaro M. E., Santos J. H., and Bonini M. G. (2015) MnSOD upregulation sustains the Warburg effect via mitochondrial ROS and AMPK-dependent signalling in cancer. Nat. Commun. 6, 6053 10.1038/ncomms7053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mungai P. T., Waypa G. B., Jairaman A., Prakriya M., Dokic D., Ball M. K., and Schumacker P. T. (2011) Hypoxia triggers AMPK activation through reactive oxygen species-mediated activation of calcium release-activated calcium channels. Mol. Cell. Biol. 31, 3531–3545 10.1128/MCB.05124-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hawley S. A., Ross F. A., Chevtzoff C., Green K. A., Evans A., Fogarty S., Towler M. C., Brown L. J., Ogunbayo O. A., Evans A. M., and Hardie D. G. (2010) Use of cells expressing γ subunit variants to identify diverse mechanisms of AMPK activation. Cell Metab. 11, 554–565 10.1016/j.cmet.2010.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Auciello F. R., Ross F. A., Ikematsu N., and Hardie D. G. (2014) Oxidative stress activates AMPK in cultured cells primarily by increasing cellular AMP and/or ADP. FEBS Lett. 588, 3361–3366 10.1016/j.febslet.2014.07.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. van Leeuwen L. A. G., Hinchy E. C., Murphy M. P., Robb E. L., and Cocheme H. M. (2017) Click-PEGylation: a mobility shift approach to assess the redox state of cysteines in candidate proteins. Free Radic. Biol. Med. 108, 374–382 10.1016/j.freeradbiomed.2017.03.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Vissers M. C. M., Hampton M., and Kettle A. J. (2018) Hydrogen peroxide metabolism in health and disease, Taylor & Francis/CRC Press, Boca Raton, FL [Google Scholar]
- 21. Holmström K. M., and Finkel T. (2014) Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat. Rev. Mol. Cell Biol. 15, 411–421 10.1038/nrm3801 [DOI] [PubMed] [Google Scholar]
- 22. Collins Y., Chouchani E. T., James A. M., Menger K. E., Cochemé H. M., and Murphy M. P. (2012) Mitochondrial redox signalling at a glance. J. Cell Sci. 125, 801–806 10.1242/jcs.098475 [DOI] [PubMed] [Google Scholar]
- 23. Chouchani E. T., Pell V. R., Gaude E., Aksentijević D., Sundier S. Y., Robb E. L., Logan A., Nadtochiy S. M., Ord E. N., Smith A. C., Eyassu F., Shirley R., Hu C. H., Dare A. J., James A. M., et al. (2014) Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 515, 431–435 10.1038/nature13909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chouchani E. T., Pell V. R., James A. M., Work L. M., Saeb-Parsy K., Frezza C., Krieg T., and Murphy M. P. (2016) A unifying mechanism for mitochondrial superoxide production during ischemia-reperfusion injury. Cell Metab. 23, 254–263 10.1016/j.cmet.2015.12.009 [DOI] [PubMed] [Google Scholar]
- 25. Mills E. L., Kelly B., Logan A., Costa A. S., Varma M., Bryant C. E., Tourlomousis P., Däbritz J. H., Gottlieb E., Latorre I., Corr S. C., McManus G., Ryan D., Jacobs H. T., Szibor M., et al. (2016) Succinate dehydrogenase supports metabolic repurposing of mitochondria to drive inflammatory macrophages. Cell 167, 457–470.e413 10.1016/j.cell.2016.08.064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Scialo F., Sriram A., Fernández-Ayala D., Gubina N., Lõhmus M., Nelson G., Logan A., Cooper H. M., Navas P., Enriquez J. A., Murphy M. P., and Sanz A. (2016) Mitochondrial ROS produced via reverse electron transport extend animal lifespan. Cell Metab. 23, 725–734 10.1016/j.cmet.2016.03.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sobotta M. C., Barata A. G., Schmidt U., Mueller S., Millonig G., and Dick T. P. (2013) Exposing cells to H2O2: a quantitative comparison between continuous low-dose and one-time high-dose treatments. Free Radic. Biol. Med. 60, 325–335 10.1016/j.freeradbiomed.2013.02.017 [DOI] [PubMed] [Google Scholar]
- 28. Murphy M. P. (2012) Mitochondrial thiols in antioxidant protection and redox signaling: distinct roles for glutathionylation and other thiol modifications. Antioxid. Redox Signal. 16, 476–495 [DOI] [PubMed] [Google Scholar]
- 29. Sobotta M. C., Liou W., Stöcker S., Talwar D., Oehler M., Ruppert T., Scharf A. N., and Dick T. P. (2015) Peroxiredoxin-2 and STAT3 form a redox relay for H2O2 signaling. Nat. Chem. Biol. 11, 64–70 10.1038/nchembio.1695 [DOI] [PubMed] [Google Scholar]
- 30. Robb E. L., Gawel J. M., Aksentijevic D., Cocheme H. M., Stewart T. S., Shchepinova M. M., Qiang H., Prime T. A., Bright T. P., James A. M., Shattock M. J., Senn H. M., Hartley R. C., and Murphy M. P. (2015) Selective superoxide generation within mitochondria by the targeted redox cycler MitoParaquat. Free Radic. Biol. Med. 89, 883–894 10.1016/j.freeradbiomed.2015.08.021 [DOI] [PubMed] [Google Scholar]
- 31. Buskiewicz I. A., Montgomery T., Yasewicz E. C., Huber S. A., Murphy M. P., Hartley R. C., Kelly R., Crow M. K., Perl A., Budd R. C., and Koenig A. (2016) Reactive oxygen species induce virus-independent MAVS oligomerization in systemic lupus erythematosus. Sci. Signal. 9, ra115 10.1126/scisignal.aaf1933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. O'Neill H. M., Maarbjerg S. J., Crane J. D., Jeppesen J., Jørgensen S. B., Schertzer J. D., Shyroka O., Kiens B., van Denderen B. J., Tarnopolsky M. A., Kemp B. E., Richter E. A., and Steinberg G. R. (2011) AMP-activated protein kinase (AMPK) β1β2 muscle null mice reveal an essential role for AMPK in maintaining mitochondrial content and glucose uptake during exercise. Proc. Natl. Acad. Sci. U.S.A. 108, 16092–16097 10.1073/pnas.1105062108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jäger S., Handschin C., St-Pierre J., and Spiegelman B. M. (2007) AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1α. Proc. Natl. Acad. Sci. U.S.A. 104, 12017–12022 10.1073/pnas.0705070104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gutscher M., Sobotta M. C., Wabnitz G. H., Ballikaya S., Meyer A. J., Samstag Y., and Dick T. P. (2009) Proximity-based protein thiol oxidation by H2O2-scavenging peroxidases. J. Biol. Chem. 284, 31532–31540 10.1074/jbc.M109.059246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Albrecht S. C., Barata A. G., Grosshans J., Teleman A. A., and Dick T. P. (2011) In vivo mapping of hydrogen peroxide and oxidized glutathione reveals chemical and regional specificity of redox homeostasis. Cell Metab. 14, 819–829 10.1016/j.cmet.2011.10.010 [DOI] [PubMed] [Google Scholar]
- 36. Riquier S., Breton J., Abbas K., Cornu D., Bouton C., and Drapier J. C. (2014) Peroxiredoxin post-translational modifications by redox messengers. Redox Biol. 2, 777–785 10.1016/j.redox.2014.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dietz K. J., Horling F., König J., and Baier M. (2002) The function of the chloroplast 2-cysteine peroxiredoxin in peroxide detoxification and its regulation. J. Exp. Bot. 53, 1321–1329 10.1093/jexbot/53.372.1321,10.1093/jxb/53.372.1321 [DOI] [PubMed] [Google Scholar]
- 38. Hall A., Karplus P. A., and Poole L. B. (2009) Typical 2-Cys peroxiredoxins: structures, mechanisms and functions. FEBS J. 276, 2469–2477 10.1111/j.1742-4658.2009.06985.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Peskin A. V., Cox A. G., Nagy P., Morgan P. E., Hampton M. B., Davies M. J., and Winterbourn C. C. (2010) Removal of amino acid, peptide and protein hydroperoxides by reaction with peroxiredoxins 2 and 3. Biochem. J. 432, 313–321 10.1042/BJ20101156 [DOI] [PubMed] [Google Scholar]
- 40. Day A. M., Brown J. D., Taylor S. R., Rand J. D., Morgan B. A., and Veal E. A. (2012) Inactivation of a peroxiredoxin by hydrogen peroxide is critical for thioredoxin-mediated repair of oxidized proteins and cell survival. Mol. Cell 45, 398–408 10.1016/j.molcel.2011.11.027 [DOI] [PubMed] [Google Scholar]
- 41. Woods A., Cheung P. C., Smith F. C., Davidson M. D., Scott J., Beri R. K., and Carling D. (1996) Characterization of AMP-activated protein kinase β and γ subunits: Assembly of the heterotrimeric complex in vitro. J. Biol. Chem. 271, 10282–10290 10.1074/jbc.271.17.10282 [DOI] [PubMed] [Google Scholar]
- 42. Willows R., Navaratnam N., Lima A., Read J., and Carling D. (2017) Effect of different γ-subunit isoforms on the regulation of AMPK. Biochem. J. 474, 1741–1754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hardie D. G., Salt I. P., and Davies S. P. (2000) Analysis of the role of the AMP-activated protein kinase in the response to cellular stress. Methods Mol. Biol. 99, 63–74 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.