

Abstract
Colorectal cancer (CRC) is one of the leading cancers throughout the world. It represents the third most common cancer and the fourth in mortality. Most of CRC are sporadic, arise with no known high-penetrant genetic variation and with no previous family history. The etiology of sporadic CRC is considered to be multifactorial and arises from the interaction of genetic variants of low-penetrant genes and environmental risk factors. The most common well-studied genetic variation is single nucleotide polymorphisms (SNPs). SNP arises as a point mutation. If the frequency of the sequence variation reaches 1% or more in the population, it is referred to as polymorphism, but if it is lower than 1%, the allele is typically considered as a mutation. Lots of SNPs have been associated with CRC development and progression, for example, genes of TGF-β1 and CHI3L1 pathways. TGF-β1 is a pleiotropic cytokine with a dual role in cancer development and progression. TGF-β1 mediates its actions through canonical and noncanonical pathways. The most important negative regulatory protein for TGF-β1 activity is termed SMAD7. The production of TGF-β can be controlled by another protein called YKL-40. YKL-40 is a glycoprotein with an important role in cancer initiation and metastasis. YKL-40 is encoded by the CHI3L1 gene. The aim of the present review is to give a brief introduction of CRC, SNP, and examples of some SNPs that have been documented to be associated with CRC. We also discuss two important signaling pathways TGF-β1 and CHI3L1 that influence the incidence and progression of CRC.
1. Colorectal Cancer
Colorectal cancer (CRC) has attracted significant attention as it represents the third most common cancer and fourth cancer in mortality in the world after lung, stomach, and liver cancers [1]. Colorectal cancer accounts for approximately 10% of all new cancer cases, affecting one million people every year throughout the world [2]. The highest incidence rates are mainly found in developed countries, whereas the lowest rates are found in developing countries (Figure 1) [3]. From the genetic standpoint, CRC can be divided into three types: sporadic, familial, and hereditary CRC [4] as shown in Table 1.
Figure 1.

Age-standardized CRC incidence rates by sex and world area, GLOBOCAN 2012.
Table 1.
Genetic classification of CRC.
| Sporadic CRC | Familial CRC | Hereditary CRC |
|---|---|---|
| Occurs entirely by chance throughout life without any previous family history | Occurs when there are two or more family members with a history of CRC | When people inherit a high penetrant gene mutation from either of their parents |
| No specific inherited gene mutation has been identified to explain the cancer yet. | ||
|
| ||
| ~60%–80% | ~15%–30% | ~5% |
The etiology of sporadic CRC is considered to be multifactorial and arises from the interaction between allelic variants in low-penetrant genes and environmental risk factors [5, 6]. Penetrance is the frequency with which the characteristics transmitted by a gene appear in individuals possessing it. A highly penetrant gene almost always expresses its phenotypes regardless of other environmental influence, while low-penetrant genes express its phenotype in the presence of other genetic and/or environmental influence [7]. The genetic contribution of high- and low-penetrant genes to CRC is shown in Figure 2. Risk factors for CRC may be nonmodifiable or modifiable [8] as shown in Table 2.
Figure 2.

Genetic contribution to CRC.
Table 2.
Risk factors of CRC.
| Nonmodifiable |
| (i) Age: the incidence of CRC diagnosis increases after the age of 40 and rises sharply after age 50, but there is an increase in the young-onset rate due to the adoption of a Westernized lifestyle and diet [9] (ii) Family history of CRC (especially a first-degree relative diagnosed at age 49 or younger) [10] (iii) Hereditary predisposition (a) Hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) (b) Familial adenomatous polyposis (FAP) [4, 9] (iv) Inflammatory bowel disease (IBD): chronic inflammation is assumed to underlie the cause of colitis-associated cancer, which is associated with oxidative stress-induced DNA damage resulting in the activation of procarcinogenic genes and silencing of tumor-suppressor pathways [11] (v) Adenomatous polyp: polyps are abnormal growths of the large intestine lining that protrude into the intestinal lumen. Polyps greater than one centimeter in diameter are associated with a greater risk of cancer [12] |
|
|
| Modifiable |
| (i) Diets: Western diet rich in red meat, refined grains, desserts, and low in fiber was reported to be associated with increased CRC risk [10, 13, 14] (ii) Cigarette smoking: carcinogens as aromatic amines, nitrosamines, and polycyclic aromatic hydrocarbons in tobacco smoke produce metabolites that can react with DNA or other macromolecules to form DNA adducts inducing genetic mutations [15] (iii) Obesity: obese women have higher risk of CRC than obese men due to higher abdominal visceral adipose tissue volume [16, 17] (iv) High alcohol consumption (>2 glasses per day): ethanol increases the activation of various procarcinogens present in tobacco smoke, diets, and industrial chemicals to carcinogens through the induction of CYP2E1 [18] |
Vogelstein model, also known as the adenoma-carcinoma sequence, is a multistep model [19] that describes the progression of CRC carcinogenesis from a benign adenoma to a malignant carcinoma through a series of well-defined histological stages (Figure 3). The main features of the model include a mutational activation of oncogenes and/or the inactivation of tumor suppressor genes. At least four or five genetic alterations must take place for the formation of malignant tumors. The characteristics of the tumor are dependent upon the accumulation of multiple genetic mutations rather than a certain sequence of mutations of these genes.
Figure 3.

The colorectal adenoma-carcinoma sequence (Vogelstein model). Progression from normal epithelium through adenoma to CRC is characterized by accumulated abnormalities of multiple genes.
Dukes' colorectal cancer staging and Tumors/Nodes/Metastases (TNM) are the two classification system that are used for the staging of CRC (Table 3). There has been a gradual move from Dukes' to the TNM classification system as TNM was reported to give a more accurate independent description of the primary tumors and its spread [20].
Table 3.
Staging and survival of CRC.
| Dukes' staging | TNM staging | Description | Survival (%) |
|---|---|---|---|
| Stage 0 | Carcinoma in situ | ||
| A | Stage I | No nodal involvement, no metastasis, tumor invades submucosa (T1, N0, M0), tumor invades muscularis (T2, N0, M0) | 90–100% |
| B | Stage II | No nodal involvement, no metastasis, tumor invades subserosa (T3, N0, M0), invade other organ (T4, N0, M0) | 75–85% |
| C | Stage III | Regional lymph nodes involved (any T, N1, M0) | 30–40% |
| D | Stage IV | Distant metastasis (any T, any N, M1) | <5% |
2. Prevention of Colorectal Cancer
Several approaches have been developed to reduce CRC incidence and mortality. Prevention includes primary and secondary strategies. Primary strategy includes dietary changes, increasing physical activity, and the use of nonsteroidal anti-inflammatory drugs (NSAIDs), while the secondary strategy is based on screening tests (Table 4).
Table 4.
Primary and secondary prevention strategies of CRC.
| Primary |
| (i) Diet. A diet high in vegetables, fruits, dairy products, olive oil, fish, and whole grains and low in red and processed meats has been shown to lower CRC risk [21–23]. |
| (ii) Physical Activity. Physically active individuals have 24% lower risk of CRC development than those who have a sedentary lifestyle. Physical activity promotes the production of interleukin-6 (IL-6) and decreases the expression of inducible nitric oxide synthase (iNOS) and tumor necrosis factor-alpha (TNF-α) in plasma and colon, leading to enhanced immunity [24, 25]. |
| (iii) NSAIDs. They reduce the risk of CRC by blocking cyclooxygenase (COX) enzymes, so inhibit prostaglandin production, which are known to promote tumor angiogenesis and cell proliferation [26]. |
|
|
| Secondary |
| (i) Fecal Tests. Fecal occult blood test (FOBT) and fecal immunochemical test (FIT) detect hidden blood in the stool, while fecal DNA test detects DNA in the stool [27–29]. |
| (ii) Flexible Sigmoidoscopy. It is performed using an endoscope that allows the examination of the surface up to 60 cm from the anal verge (rectum, sigmoid colon, and part of the descending colon). It is done after colon lavage using enema or administering laxatives without the need of sedation [30]. |
| (iii) Colonoscopy. It is performed using an endoscope, which allows an examination of the entire colon surface. It must be done under intravenous sedation and requires being on a low-residue diet, colon lavage using laxatives, and drinking plenty of water the day before the test [31]. |
Interestingly, dietary factors are responsible for 70% to 90% of CRC. The relatively low CRC rates in the Mediterranean area compared with most Western countries are mostly because the traditional Mediterranean diet is characterized by high consumption of foods of plant origin, relatively low consumption of red meat, and high consumption of olive oil [32]. Therefore, diet modification could potentially help to reduce the incidence of CRC [33, 34]. Examples of some dietary components that lower CRC risk are shown in Table 5.
Table 5.
Examples of some dietary components that decrease risk of CRC.
| Fiber | (i) A high-fiber diet has a protective effect from CRC as it decreases transit time through the gastrointestinal tract, dilutes colonic contents, and enhances bacterial fermentation. This can increase the production of short-chain fatty acids that interfere with numerous regulators of the cell cycle, proliferation, and apoptosis such as β-catenin, p53, and caspase 3 genes [35, 36] (ii) Corn, beans, avocado, brown rice, lentils, pear, artichoke, carrots, oatmeal, broccoli, and apples are examples of diet rich in fiber [37] |
|
| |
| Fish oil | (i) Fish oil rich in omega-3 fatty acids may inhibit the promotion and progression of cancer through suppression of arachidonic acid-derived eicosanoid biosynthesis, which results in altered immune response to cancer and modulation of inflammation, cell proliferation, apoptosis, metastasis, and angiogenesis [38] (ii) It also influences transcription factor activity, gene expression, and signal transduction, which leads to changes in metabolism, cell growth, and differentiation [38–40] |
|
| |
| Olive oil | (i) Olive oil reduces deoxycholic acid in the human colon and rectum (ii) Deoxycholic acid was found to reduce diamine oxidase, a main enzyme for the metabolism of ingested histamine and control of mucosal proliferation in the ileal and the colonic mucosa [41] |
|
| |
| Folate | (i) Folate acts as donors of methyl groups in the biosynthesis of nucleotide precursors used for DNA synthesis and methylation of DNA, RNA, and protein and participates in the maintenance of genomic stability [42, 43] (ii) Spinach, broccoli, strawberries, raspberries, beans, peas, lettuce, lentils, and celery are examples of diet rich in folate [37] |
|
| |
| Calcium | (i) Calcium can suppress epithelial cell proliferation in the colon by binding to bile acids and ionized fatty acids [44] (ii) Calcium can act directly by reducing proliferation, stimulating differentiation, and inducing apoptosis via upregulation of p21 and Bcl-2 in the colonic mucosa [44–47] |
Early diagnosis of CRC is important to improve outcomes. Fecal occult blood testing (FOBT) or fecal immunochemical test (FIT) is routinely used prior to colonoscopy, and only patients with a positive test result are referred to a specialist. Although these assays are useful screening tools, patient compliance with these stool-based assays tends to be low. Serum-based assays for the early detection of CRC are highly attractive, as they could be integrated into any regular health checkup without the need for additional stool sampling, thereby increasing acceptance among patients [29].
3. Gene Polymorphism
Polymorphism is the occurrence of two or more clearly different morphs or forms of a species in the population. Poly means many; morph means form [48]. The colored flowers of mustard, butterflies, and human ABO blood group system are obvious examples of polymorphisms [49, 50].
Genetic polymorphisms are different forms of the DNA sequence, which may or may not affect biological function depending on its exact nature. Polymorphism arises as a result of mutation. If the frequency of a specific sequence variant reaches 1% or more in the population, it is referred to as polymorphism, and if it is lower than 1%, the allele is typically regarded as mutation [51]. Molecular polymorphism, first demonstrated in Drosophila pseudoobscura, stimulated molecular studies of many other organisms and led to vigorous theoretical debate about the significance of the observed polymorphisms [52, 53].
Single nucleotide polymorphism (SNP) is a variation in a single nucleotide that occurs at a specific position in the genome. Single nucleotide polymorphisms are the most abundant type of genetic variation in the human genome, accounting for more than 90% of all differences between individuals [54]. Single nucleotide may be changed (substitution), removed (deletion), or added (insertion) to a polynucleotide sequence [54].
Single nucleotide polymorphisms are also thought to be the keys in realizing the concept of personalized medicine as it can affect how humans develop diseases and respond to pathogens, chemicals, drugs, vaccines, and other agents. Single nucleotide polymorphisms underlie the differences in the susceptibility to a wide range of human diseases, for example, a single base mutation in the apolipoprotein E gene is associated with a higher risk for Alzheimer's disease. The severity of illness and the way the body responds to treatments are also manifestations of genetic variations [55, 56].
According to their location in the genome, SNPs are classified into cSNP in the coding region (exons), rSNP in the regulatory region, and iSNP located in the intronic region [54].
Polymorphisms in the coding region are either synonymous or nonsynonymous (Figure 4). Synonymous polymorphisms do not result in a change of amino acid in the protein but still can affect its function in other ways. Silent mutation in the multidrug resistance gene 1, which codes for a cellular membrane pump that expels drugs from the cell, is an example of synonymous polymorphism. It can slow down translation and allow unusual folding of the peptide chain, causing the mutant pump to be less functional [57, 58].
Figure 4.

Genetic polymorphism in the coding region (http://academic.pgcc.edu/).
Nonsynonymous polymorphisms, on the other hand, can change the amino acid sequence of the protein and subclassified into missense and nonsense. Missense polymorphism results in different amino acids such as single base change G > T in LMNA gene that results in the replacement of the arginine by the leucine at the protein level, which manifests progeria syndrome [59]. Nonsense polymorphism results in a premature stop codon and usually nonfunctional protein product such as that manifested in cystic fibrosis caused by mutation in the cystic fibrosis transmembrane conductance regulator gene [60].
Promoter polymorphism can cause variations in gene expression as it affects the DNA binding site and alters the affinity of the regulatory protein while intronic region polymorphism may affect gene splicing and messenger RNA degradation [61, 62].
Genotyping technologies typically involve the generation of allele-specific products for SNPs of interest followed by their detection for genotype determination. All current genotyping technologies with only a few exceptions require the polymerase chain reaction (PCR) amplification step. In most technologies, PCR amplification of a desired SNP-containing region is performed initially to introduce specificity and increase the number of molecules for detection following allelic discrimination [63]. Enzymatic cleavage, primer extension, hybridization, and ligation are four popular methods used for allelic discrimination (Table 6).
Table 6.
Methods of allelic discrimination used in SNP genotyping [63].
| Enzymatic cleavage | Enzymatic cleavage is based on the ability of certain classes of enzymes to cleave DNA by recognition of specific sequences and structures. Such enzymes can be used for discrimination between alleles when SNP sites are located in an enzyme recognition sequence and allelic differences affect recognition. For example, restriction fragment length polymorphism (RFLP) is based on genotyping a SNP located in a restriction enzyme site using PCR product containing the SNP that is incubated with corresponding restriction enzyme. The reaction product is run on a gel, and SNP genotype is easily determined from the product sizes [64]. |
|
| |
| Primer extension | In a typical primer extension reaction, a primer is designed to anneal with its 3\ end adjacent to a SNP site and extended with nucleotides by polymerase enzyme. The identity of the extended base is determined either by fluorescence or mass to reveal SNP genotype, for example, the PinPoint assay, MassEXTEND tm, SPC-SBE, and GOODassay primer extension-based methods, where SNP-specific primers are simultaneously extended with various nucleotides using PCR products as a template [65]. |
|
| |
| Hybridization | Hybridization approaches use differences in the thermal stability of double-stranded DNA to distinguish between perfectly matched and mismatched target-probe. For example, the TaqMan® genotyping assay combines hybridization and 5\ nuclease activity of polymerase coupled with fluorescence detection. The allele-specific probes carry a fluorescent dye at one end (reporter) and a nonfluorescent dye at the other end (quencher). The intact probes show no fluorescence owing to the close proximity between the reporter and quencher dyes. During PCR primer extension, the enzyme only cleaves the hybridized probe that is perfectly matched, freeing the reporter dye from the quencher. The reporter dye generates a fluorescent signal, whereas the mismatched probe remains intact and shows no fluorescence [66]. |
|
| |
| Ligation | Ligation approach employs specificity of ligase enzymes. When two oligonucleotides hybridize to single-stranded template DNA with perfect complementarity, adjacent to each other, ligase enzymes join them to form a single oligonucleotide. Three oligonucleotide probes are used in traditional ligation assays, 2 of which are allele-specific and bind to the template at the SNP site. The third probe is common and binds to the template adjacent to the SNP immediately next to the allele-specific probe. For example, combinatorial fluorescence energy transfer tags are composed of fluorescent dyes that can transfer energy when they are in close proximity. Tags with different fluorescence signatures can be created using a limited number of dyes by varying the number of dyes used and spacing between the dyes [67]. |
4. Genome-Wide Association Study and Colorectal Cancer
Genome-wide association study (GWAS), also known as whole genome association study, is defined as an examination of many common SNPs in different individuals to see if any SNP is associated with a disease. Genome-wide association study compares the DNA of participants having a disease with similar people without the disease. The ultimate goal is to determine genetic risk factors that can be used to make predictions about who is at risk for a disease and to identify their role in disease development for developing new prevention and treatment strategies [68].
The availability of chip-based microarray technology that assay hundreds and thousands of SNPs made genome-wide association studies easy to be performed (Table 7). Genome-wide association study identifies a specific location, not complete genes. Many SNPs identified in GWAS are near a protein-coding gene or are within genes that were not previously believed to associate with the disease. So, researchers use data from this type of study to pinpoint genes that may contribute to a person's risk of developing a certain disease [69].
Table 7.
Some of the published GWASs on CRC (100).
| Reference SNP (rs) | Gene or region | Population | Sample size for stage | Sample size for subsequent stages | Genotyping platform (Nb. of SNPs) | Study reference |
|---|---|---|---|---|---|---|
| rs4939827 | 18q21 SMAD7 | First stage: UK | 940 cases/965 controls | 7473 cases/5984 controls | Affymetrix (550,163) | (101) |
| Second stage: UK | ||||||
| rs6983267 | 8q24 | First stage: UK | 930 cases/960 controls | 7334 cases/5246 controls | Illumina (547,647) | (102) |
| Second stage: UK | ||||||
| rs10505477 | 8q24 | First stage: Canada | 1257 cases/1336 controls | 4024 cases/4042 controls | Illumina and Affymetrix (99,632) | (103) |
| rs719725 | 9p24 | Other stages: Canada, US, and Scotland | ||||
| rs4779584 | 15q13 CRAC1 | First stage: UK | 730 cases/960 controls | 4500 cases/3860 controls | Illumina (547,647) | (104) |
| Second stage: UK | ||||||
| rs4939827 | 18q21 SMAD7 | First stage: Scotland | 98 cases/1002 controls | 16476 cases/15351 controls | Illumina (541,628) | (105) |
| rs7014346 | 8q24 | Second stage and replication: Canada, UK, Israel, Japan, and EU | ||||
| rs3802842 | 11q23 | |||||
| rs4444235 | 14q22.2 BMP4 | First stage: UK | 6780 cases/6843 controls | 13406 cases/14012 controls | Multiple (38,710) | (106) |
| rs9929218 | 16q22.1 CDH1 | Replication: EU, Canada | ||||
| rs10411210 | 19q13 RHPN2 | |||||
| rs961253 | 20p12.3 |
Genome-wide association study is built on the expanding knowledge of the relationships among SNPs generated by the international HapMap project. The HapMap project is an international scientific effort to identify common SNPs among people from different ethnic populations. When several SNPs cluster together on a chromosome, they are inherited as a block known as a haplotype. The HapMap describes haplotypes, including their locations in the genome, and how common they are present in different populations throughout the world [70].
Genome-wide association study is an important tool for discovering genetic variants influencing a disease, but it has important limitations, including their potential for false-positive and false-negative results and for biases related to selection of study participants and genotyping errors [71]. The gold standard for validation of any GWAS is replication in an additional independent sample. Replication studies are performed in an independent set of data drawn from the same population as the GWAS, in an attempt to confirm the effect in the GWAS target population. Once an effect is confirmed in the target population, other populations may be sampled to determine if the SNP has an ethnic-specific effect [72].
It has been recognized that SNPs play an important role in conferring risk of CRC. Genome-wide association studies have reported multiple risk loci associated with risk CRC, some of which are involved in the transforming growth factor-β (TGF-β) signaling pathway [73]. For example, SMAD7 rs4939827 was found to be associated with CRC in two GWASs [74, 75]. The association of SMAD7 rs4939827 with CRC was confirmed by other replication studies [76, 77]. A summary of other SNPs studied as risk factors for CRC is shown in Table 8.
Table 8.
Gene polymorphisms associated with CRC.
| Gene | Reference SNP (rs) | Effect on CRC | Reference |
|---|---|---|---|
| Matrix metalloproteinases-9 (MMP 9) | rs34016235 | A promoter polymorphism due to a C to T substitution results in the loss of the binding site of a nuclear protein to this region of the MMP 9 gene promoter. The polymorphism is associated with lymph node metastasis of CRC. | [78] |
| COX-2 | rs20417 | The C allele has lower promoter activity than the G allele, and GG genotype in smokers is associated with a significant increase in the risk of CRC compared to nonsmokers. | [79] |
| Vitamin D receptor | rs1544410 | Polymorphism of the vitamin D receptor gene to be associated with an increased risk of colon cancer. | [80] |
| Bone morphogenetic protein 4 (BMP 4) | rs4444235 | The rs4444235 increases risk of CRC development through its cis-acting regulatory influence on BMP4 expression. | [81] |
| Phospholipase A2 | rs9657930 | Polymorphisms in the phospholipase A2 gene is associated with the risk of the rectal cancer. | [82] |
| Colorectal adenoma and carcinoma 1 | rs4779584 | The rs4779584 polymorphism is associated with increased risk of CRC among Caucasian not Asian populations. | [83] |
| Eukaryotic translation initiation factor 3 | rs16892766 | The rs16892766 polymorphism is associated with increased risk of CRC but not adenoma among Caucasian. | [84] |
| Cadherin-1 | rs9929218 | The minor allele of rs9929218 has reduced E-cadherin expression and resulted in worsening the survival of CRC patients. | [85] |
| FAS | rs2234767 | The rs2234767 contributes to an increased risk of CRC by altering recruitment of SP1/STAT1 complex to the FAS promoter for transcriptional activation. | [86] |
| Maternally expressed gene 3 | rs7158663 | The rs7158663 changes the folding structures of maternally expressed gene 3; therefore, it contributes to genetic susceptibility of CRC. | [87] |
| Fc-g receptor gene | rs1801274 | The rs1801274 changes the amino acid from histidine (H) to arginine. CRC patients with Fc-g receptor H/H genotype have better survival. | [88] |
| SPSB2 gene | rs11064437 | The rs11064437 contributes to an increased risk of CRC by disrupting the splicing and introduction of a transcriptional isoform with a shortened untranslated region of SPSB2 gene. | [89] |
| TPP1 | rs149418249 | Prevents TPP1-TIN2 interaction, shortening the telomere length, and as a consequence, enhances cell proliferation | [90] |
| SLC22A5 | rs27437 | The G allele decreases the expression of SLC22A5 via influencing the TF-binding upstream of the gene, leading to higher CRC risk. | [91] |
| KBTBD11 | rs11777210 | C allele allows binding of MYC, a potent oncogene, preventing the expression of KBTBD11, a potent tumor suppressor. | [92] |
| miR-17-92 cluster | rs9588884 | The G allele lowers the CRC risk by decreasing transcriptional activity and consequently lowering levels of miR-20a. | [93] |
5. Transforming Growth Factor-β Signaling and Its Regulatory Smad7
Mothers against decapentaplegic homolog 7 (Smad7) is a key inhibitor of TGF-β [94, 95]. Smad7 was named after mothers against decapentaplegic (mad), an intermediate of the decapentaplegic signaling pathway in Drosophila melanogaster and sma-gene in Caenorhabditis elegans that has mutant phenotype similar to that observed for the TGF-β-like receptor gene [96]. Regulation of TGF-β by Smad7 is crucial to maintain gastrointestinal homeostasis [97]. Smad7 overexpression is commonly found in patients with chronic inflammatory conditions of the colon [98] and may be associated with prognosis in patients with CRC [99]. Loss of Smad/TGF-β signaling interrupts the principal role of TGF-β as a growth inhibitor, allowing unchecked cellular proliferation [100].
In the early 1980s, Roberts and his colleagues isolated two fractions that could induce growth of normal fibroblasts from murine sarcoma cell extracts and were named TGFα and TGF-β [101, 102]. Transforming growth factor-β is a prototype of a large family of cytokines that includes the TGF-βs, activins, inhibins, and bone morphogenetic proteins (BMPs) [103].
In mammals, TGF-β has 3 isoforms (TGF-β1, TGF-β2, and TGF-β3), with similar biological properties. The TGF-β isoforms are encoded from genes located on different chromosomes. The TGF-β1 gene is located in chromosome 19q13.1, while TGF-β2 and TGF-β3 genes are located in chromosomes 1q4.1 and 14q24.3, respectively [104].
The isoforms of TGF-β1, TGF-β2, and TGF-β3 are encoded as large precursor, which undergo proteolytic digestion by the endopeptidase furin, yielding two products that assemble into dimers. One is latency-associated peptide (LAP), a dimer from the N-terminal region. The other is mature TGF-β, a dimer from the C-terminal portion. A common feature of TGF-β is that its N-terminal portion (LAP) remains noncovalently associated with the mature TGF-β forming a small latent complex [105, 106]. The small latent complex is associated with a large protein termed latent TGF-β binding protein (LTBP) via disulfide bonds forming large latent complex for targeted export to the extracellular matrix (ECM) [107, 108]. For TGF-β to bind its receptors, the latent complex must be removed so that the receptor-binding site in TGF-β is not masked by LAP. Latent TGF-β is cleaved by several factors, including proteases, thrombospondin, reactive oxygen species (ROS), and integrins (Figure 5) [109, 110].
Figure 5.

The sequential steps in the synthesis and secretion of active TGF-β.
Transforming growth factor-β is a pleiotropic cytokine that has a dual function in cancer development, where it acts as a tumor suppressor in the early stages and a tumor promoter in the late stages [111]. The main actions of TGF-β are summarized in Table 9.
Table 9.
The role of TGF-β in various cell processes.
| Cytostasis | (i) TGF-β can activate cytostatic gene responses at any point in the cell cycle phases G1, S, or G2 [112] (ii) TGF-β induces activation of the cyclin-dependent kinase (CDK) inhibitors [113–115] and repression of the growth-promoting transcription factors c-MYC and inhibitors of differentiation (ID1, ID2, and ID3) [116]. |
|
| |
| Apoptosis | TGF-β induces apoptosis through (i) upregulation of SH2-domain-containing inositol-5-phosphatase expression, which inhibits signaling via the survival protein kinase AKT [117] (ii) induction of TGF-β-inducible early-response gene, which induces the generation of ROS and the loss of the mitochondrial membrane potential preceding the apoptotic death [118, 119] (iii) induction of death-associated protein kinase [117] |
|
| |
| Immunity | For immune suppression, TGF-β plays a critical role through (i) blocking antigen-presenting cells such as dendritic cells, which acquire the ability to effectively stimulate T cells during an immune response [120] (ii) decreasing the activity of natural killer cells and neutrophils [121] |
|
| |
| Angiogenesis | (i) TGF-β induces the expression of matrix metalloproteinases (MMPs) on both endothelial cells and tumor cells, allowing the release of the endothelial cells from the basement membrane [122] (ii) TGF-β can also induce the expression of angiogenic factors such as vascular endothelial growth factor (VEGF) and connective-tissue growth factor (CTGF) in epithelial cells and fibroblasts [123, 124] |
|
| |
| Epithelial-mesenchymal transition (EMT) | The migratory ability of epithelial cells relies on loss of cell–cell contacts, a process that is commonly referred to as the EMT. It is marked by the loss of E-cadherin and the expression of mesenchymal proteins such as vimentin and N-cadherin [125]. (i) TGF-β was reported to destabilize the E-cadherin adhesion complex resulting in its loss in pancreatic cancer [126]. Alternatively, in epithelial cell lines, TGF-β can deacetylate the E-cadherin promoter, thus repressing its transcription [127] (ii) TGF-β was found to upregulate vimentin in prostate cancer [128] (iii) TGF-β upregulates MMPs to promote invasion through proteolytic degradation and remodeling of the ECM [129] |
The active TGF-β binds to transforming growth factor-β receptor 2 (TGF-βR2), a serine/threonine kinase receptor, leading to the recruitment and phosphorylation of the TGF-βR1 (Figure 6). The activated TGF-βR1 interacts with and phosphorylates a number of proteins, thereby activating multiple downstream signaling pathways in either a Smad-dependent (canonical) or Smad-independent (noncanonical) signaling pathway (Figure 6) [96].
Figure 6.

Canonical and noncanonical pathways of TGF-β.
In the canonical pathway, TGF-βR1 propagates the signal through a family of intracellular signal mediators known as Smads. To date, eight mammalian Smad proteins have been characterized and are grouped into three functional classes: receptor-activated Smads (R-Smads) including Smad1, Smad2, Smad3, Smad5, and Smad8, common mediator Smad (Smad4), and inhibitory Smads (I-Smads) including Smad6 and Smad7. Receptor-activated Smads are retained in the cytoplasm by binding to SARA (Smad anchor for receptor activation). Receptor-activated Smads are released from SARA when they are phosphorylated by the activated TGF-βR1 [130, 131].
Once R-Smads (Smad2/3) are activated through phosphorylation by TGF-βR1, they form an oligomeric complex with Smad4 and translocate into the nucleus, where it modulates the transcription of specific genes. Ability of Smads to target a particular gene and the decision to activate or repress gene transcription are determined by many cofactors that affect the Smad complex [130].
In the noncanonical pathway, TGF-β activates other non-Smad signaling pathways (Table 10). Some of these pathways can regulate Smad activation, but others might induce responses unrelated to Smad [132].
Table 10.
TGF-β-induced non-Smad signaling pathways.
| c-Jun N-terminal kinases (JNK)/p38 activation | (i) TGF-β can rapidly activate JNK and p38 through MAPK kinases (MKK4, MKK 3/6) in various cell lines [133, 134]. Activation of JNK/P38 plays a role in TGF-β-induced apoptosis and in TGF-β-induced EMT [135]. |
| Extracellular signal-regulatedkinase (ERK) activation | (i) TGF-β was found to activate the mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase (ERK) pathway which are important for TGF-β mediated EMT [125, 136]. |
| Phosphoinositide 3-kinase(PI3-K)/AKT activation | (i) TGF-β was reported to rapidly activate phosphoinositide 3-kinase (PI3-K) as indicated by the phosphorylation of its downstream effector Akt [137] (ii) Although the PI3-K/Akt pathway is a non-Smad pathway contributing to TGF-β-induced EMT, it can antagonize Smad-induced apoptosis and growth inhibition [138] |
| Rho-like GTPases | (i) The Rho-like GTPases, such as Ras homolog gene family, member A (RhoA) plays an important role in controlling dynamic cytoskeletal organization, cell motility, and gene expression and is a key player in TGF-β-induced EMT [139] (ii) TGF-β regulates RhoA activity in two different modes as it induces a rapid activation of RhoA during the early phase of stimulation and then downregulates the level of RhoA protein at later stages, both of these modes of regulation appear to be essential for TGF-β-induced EMT [140] |
Transforming growth factor-β is strongly implicated in cancer as genetic alterations of some common components of TGF-β pathway (Table 11) that have been identified in human tumors [141].
Table 11.
Alterations of some components of TGF-β pathway in human tumors.
| TGF-βR2 | (i) The TGF-βR2 gene has been mapped to chromosome 3p, a chromosome in which mutation was observed in small cell lung carcinoma (SCLC), non-small-cell lung carcinoma (NSCLC), CRCs, and ovarian and breast cancers [142–144] (ii) Besides mutations in the coding region of TGF-βR2, loss of expression of TGF-βR2 in NSCLCs, bladder cancer, and breast cancer were reported [145–147] |
|
| |
| TGF-βR1 | (i) The TGF-βR1 gene has been mapped to chromosome 9q (ii) Mutation in TGF-β gene was reported in ovarian cancer, head and neck squamous cell carcinomas (HNSCC), and breast cancer [148–150] (iii) Homozygous deletion of TGF-βR1 was also identified in pancreatic and biliary adenocarcinomas [151] |
|
| |
| SMAD3 | (i) The gene for SMAD3 is located in chromosome 15q21-q22 (ii) The rate of mutation in the SMAD3 gene is rare, and there are only few examples of such defects in Smad3 expression that was found in some gastric cancer and leukemia [152, 153] |
|
| |
| SMAD2/SMAD4 and SMAD7 | (i) Chromosome 18q has genes encodes for SMAD2, SMAD4, and SMAD7 (ii) Mutation in chromosome 18q was found in about 30% of neuroblastoma, breast, prostate, and cervical cancers and even more frequently in HNSCC (40%), NSCLC (56%), colon cancer (60%), gastric cancer (61%), and 90% of pancreatic tumors [154, 155] |
6. Inhibitory Smad (I-Smad, Smad7)
Mothers against decapentaplegic homolog 7 (Smad7) belongs to the third type of Smads, the I-Smads that also include Smad6. The structure of the Smads is characterized by two conserved regions known as the amino terminal (N-terminal) Mad homology domain-1 (MH1) and C-terminal Mad homology domain-2 (MH2), which are joined by a short poorly conserved linker region. The MH1 domain is highly conserved among the R-Smads and the Co-Smad, whereas the I-Smads lack a MH1. The MH2 domain is conserved among all of the Smad proteins but I-Smads lack SXSS motif, which is needed for phosphorylation following TGF-βR1 activation (Figure 7). Thus, I-Smads are not phosphorylated upon binding of TGF-β to its receptors. The L3 loop in the MH2 domain of the R-Smads is a specific binding site for the TGF-βR1 [95, 156].
Figure 7.
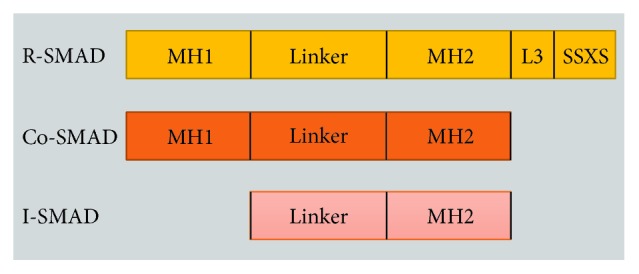
Gene constructions of SMADs.
Smad7 antagonizes TGF-β signaling through multiple mechanisms, both in the cytoplasm and the nucleus (Figure 8). Smad7 antagonizes TGF-β in the cytoplasm through the formation of a stable complex with TGF-βR1, leading to inhibition of R-Smad phosphorylation. Smad7 can recruit E3 ubiquitin ligases that induce the degradation of activated TGF-βR1 complexes [156, 157]. Also, Smad7 forms a heteromeric complex with R-Smads through the MH2 domain and hence interferes with R-Smad (Smad2/3)-Smad4 oligomerization in a competitive manner. Additionally, Smad7 can bind to DNA disrupting the formation of functional Smad-DNA complexes [158, 159].
Figure 8.

Smad7 antagonizes TGF-β signaling in the cytoplasm and the nucleus, respectively [160].
Inhibitory Smads can mediate the cross talking of TGF-β with other signaling pathways. Various extracellular stimuli such as interferon-γ (IFN-γ) can induce Smad7 expression to exert opposite effects on diverse cellular functions modulated by TGF-β [161]. In addition, Smad7 was found to be a key regulator of Wnt/β-catenin pathway that is responsible for the TGF-β-induced apoptosis and survival in various cell types [162].
There is a controversy regarding the role of Smad7 in tumor development depending on the type of the tumor. High Smad7 expression was reported to be correlated with the clinical prognosis of patients with colorectal, pancreatic, liver, and prostate cancer. In contrast, a protective role of high Smad7 expression was reported in other tumors [163]. Boulay et al. [164] found that CRC patients with deletion of Smad7 had a favorable clinical outcome compared with patients with Smad7 expression. Additionally, Smad7 was found to act as a scaffold protein to facilitate TGF-β-induced activation of p38 and subsequent apoptosis in prostate cancer cells [162].
Even in the same tumor, the function of Smad7 can switch from tumor suppressive to tumor promoting depending on the tumor stage (i.e., early versus advanced). These apparently contradictory functions are in harmony with the opposite roles of TGF-β signaling pathway in the early versus advanced tumor stages and the interaction of Smad7 with a vast array of functionally heterogeneous molecules that may be differently expressed during the carcinogenic process [160].
The overexpression of Smad7 in CRC cell was reported to enhance cell growth and inhibit apoptosis through a mechanism dependent on suppression of TGF-β signaling [100]. In addition, Smad7-deficient CRC cells were reported to enhance the accumulation of CRC cells in S phase of cell cycle and cell death through a pathway independent on TGF-β [165]. Genetic variants in SMAD7 gene have been extensively studied in CRC patients (Table 12).
Table 12.
Association studies of SNPs in SMAD7 gene and CRC.
| Population | Reference SNP (rs) | Location | Association | Reference |
|---|---|---|---|---|
| African American and Caucasian | rs4939827 | Intron 3 | In women: yes | [166] |
| rs4464148 | Intron 3 | Yes | ||
| Caucasian | rs12953717 | Intron 3 | Yes | [167] |
| rs4939827 | Intron 3 | Yes | ||
| rs4464148 | Intron 3 | No | ||
| Swedish | rs4939827 | Intron 3 | Yes | [168] |
| European | rs4464148 | Intron 3 | Yes | [169] |
| rs4939827 | Intron 3 | No | ||
| Chinese | rs4939827 | Intron 3 | No | [170] |
| rs12953717 | Intron 3 | Yes | ||
| rs4464148 | Intron 3 | No | ||
| African American | rs4939827 | Intron 3 | Yes | [171] |
| Chinese | rs4939827 | Intron 3 | Yes | [76] |
| Romanian | rs4939827 | Intron 3 | CRC vs control: no |
[172] |
| Rectal vs colon cancer: yes | ||||
| Caucasian | rs4939827 | Intron 3 | Yes | [173] |
| Croatian | rs4939827 | Intron 3 | Yes | [77] |
| Italian | rs4939827 | Intron 3 | Yes | [174] |
| Korean | rs4939827 | Intron 3 | Yes | [175] |
| Spanish | rs4939827 | Intron 3 | Yes | [176] |
| French | rs4939827 | Intron 3 | Yes | [177] |
| rs58920878 | Intron 3 | Yes |
7. Chitinase 3 Like 1/YKL-40
YKL-40 is a mammalian member of the chitinase protein family. YKL-40 is a 40 kDa heparin- and chitin-binding glycoprotein. The human protein was named YKL-40 based on its three N-terminal amino acids tyrosine (Y), lysine (K), and leucine (L) and its 40 kDa molecular mass [178]. This protein has several names, YKL-40 [178], human cartilage glycoprotein-39 (HC-gp39) [179], 38 kDa heparin-binding glycoprotein (Gp38k) [180], chondrex [181], and 40 kDa mammary gland protein (MGP-40) [182].
In a search of new bone proteins, the glycoprotein YKL-40 was identified in 1989 to be secreted in vitro by the human osteosarcoma cell line MG63. The protein was later found to be secreted by differentiated smooth muscle cells, macrophages, human synovial cells, and nonlactating mammary gland [178, 181, 182]. In 1997, the chitinase 3 like 1 (CHI3L1) gene encoding for YKL-40 was isolated. It is assigned to chromosome 1q31-q32 and consists of 10 exons and spans about 8 kilobases of genomic DNA [178, 183].
Based on amino acid sequence, it was found that YKL-40 belongs to the glycosyl hydrolase family 18 that hydrolyses the glycosidic bond between two or more carbohydrates or between a carbohydrate and a noncarbohydrate moiety. Based on sequence similarity, there are more than 100 different families of glycosyl hydrolases [184–186].
Chitin, a polymer of N-acetyl glucosamine, is the second most abundant polysaccharide in nature, following cellulose. It is found in the walls of fungi, the exoskeleton of crabs, shrimp and insects, and the micro filarial sheath of parasitic nematodes [187]. Chitin accumulation is regulated by the balance of chitin synthase-mediated biosynthesis and degradation by chitinases. Although YKL-40 contains highly conserved chitin-binding domains, it functionally lacks chitinase activity due to the mutation of catalytic glutamic acid into leucine [183].
Several types of solid tumors can express YKL-40 such as osteosarcoma [178], CRC [188], thyroid carcinoma [189], breast [190], ovarian [191], lung [192], pancreatic cancer [193], glioblastoma [194–196], and cholangiocarcinoma [197].
There are several synergistic and antagonistic factors that modulate the regulatory functions of YKL-40 (Figure 9) in both normal and pathological conditions [198].
Figure 9.
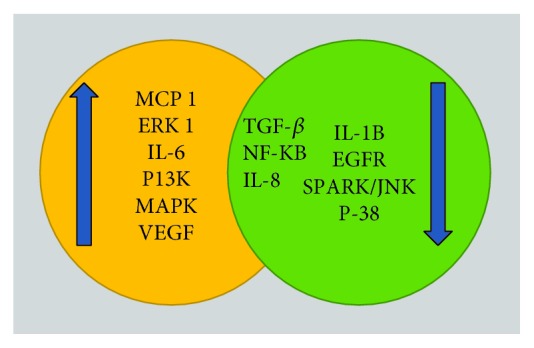
Several synergistic and antagonistic factors modulate the regulatory functions of YKL-40. EGFR: epidermal growth factor receptor; SAPK: stress-activated protein kinases; MCP-1: monocyte chemoattractant protein-1.
8. CHI3L1/YKL-40 Targets and Actions
Although the biological function of YKL-40 is not fully understood, the pattern of its expression suggests function in remodeling or degradation of ECM. The diverse roles of YKL-40 in cell proliferation, differentiation, survival, inflammation, and tissue remodeling have been suggested [199]. Aberrant expression of YKL-40 is associated with the pathogenesis of an array of human diseases (Figure 10).
Figure 10.

YKL-40 regulates the pathogenesis of cancer and inflammatory disorders [198].
Elevated serum YKL-40 levels were reported to be associated with a wide range of inflammatory diseases (Table 13). More than 75% of patients with streptococcus pneumoniae bacteremia had elevated serum levels of YKL-40 compared with age-matched healthy subjects. Treatment of these patients with antibiotics resulted in reaching serum YKL-40 normal level within few days in most patients before the serum C-reactive protein (CRP) reach the normal level [200].
Table 13.
Serum YKL-40 levels (ng/ml) in patients with inflammation, tissue remodeling, or fibrosis [201].
| Disease | Median serum YKL-40 (ng/l) | Reference |
|---|---|---|
| Viral hepatitis | 83 | [202] |
| Noncirrhotic fibrosis | 158 | |
| Posthepatitis cirrhosis | 204 | |
| Rheumatoid arthritis | 110 | [203] |
| Streptococcus pneumoniae bacteremia | 342 | [200] |
| Osteoarthritis | 112 | [204] |
| UC, severe | 59 | [205] |
| CD, severe | 59 | |
| Pulmonary sarcoidosis | 201 | [206] |
| Asthma | 92 | [207] |
Biologically, YKL-40 was found to activate a wide range of inflammatory responses. An inflammatory stimulus can trigger the secretion of a variety of cytokines that in turn may regulate YKL-40 (Figure 11). Increased YKL-40 was reported to regulate chronic inflammatory responses like asthma, chronic obstructive pulmonary disease (COPD), cardiovascular disease (CVD), and arthritis. Inhibition of YKL-40 by utilizing anti-CHI3L1 antibody may be a useful therapeutic strategy to control/reduce the effect of inflammatory diseases [198].
Figure 11.
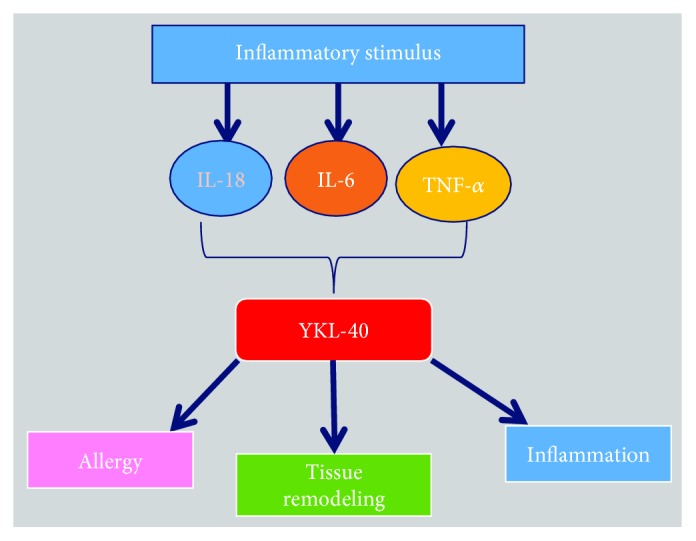
Role of inflammatory cytokines in YKL-40-mediated allergy and inflammation.
Over the past three decades, a considerable attention has been focused on the potential role of YKL-40 in the development of a variety of human cancers. Serum levels of YKL-40 (Table 14) were independent of serum carcinoembryonic antigen (CEA) in CRC [188], serum cancer antigen 125 (CA-125) in ovarian cancer [191], serum human epidermal growth factor receptor 2 (HER-2) in metastatic breast cancer [190], serum lactate dehydrogenase (LDH) in small cell lung cancer [192], and serum prostate-specific antigen (PSA) in metastatic prostate cancer [208]. Therefore, it may be of value to include serum YKL-40 as a biomarker for screening of cancer together with a panel of other tumor markers as it can reflect other aspects of tumor growth and metastasis than the routine tumor markers [201].
Table 14.
Serum YKL-40 levels (ng/ml) in patients with localized or advanced cancer [201].
| Disease | Median serum YKL-40 (ng/l) | Reference |
|---|---|---|
| Metastatic breast cancer | 80 | [209] |
| CRC | 160 | [210] |
| Glioblastoma multiforme | 130 | [195] |
| Lower grade gliomas | 101 | |
| Primary breast cancer | 57 | [211] |
| Small cell lung cancer | 82 | [192] |
| Local disease | 71 | |
| Extensive disease | 101 | |
| Metastatic prostate cancer | 112 | [208] |
| Ovarian cancer, all stages | 94 | [212] |
| Ovarian cancer, stage III | 168 | |
| Ovarian cancer, relapse | 94 |
Macrophages and neutrophils in tumor microenvironment or tumor cells were found to secrete YKL-40 into extracellular space, which can enhance tumor initiation, proliferation, angiogenesis, and metastasis (Figure 12).
Figure 12.

YKL-40 supports tumor progression.
The ability of YKL-40 to induce cytokine secretion, proliferation, and migration of target cells suggests the existence of their receptors on the cell surface. However, receptors interacting with YKL-40 are incompletely characterized, and only limited information is available about YKL-40-induced signaling pathways. There are evidences to strengthen a hypothesis that a cross talk between adjacent membrane-anchored receptors plays a key role in transmitting “outside-in” signaling to the cells, leading to a diverse array of intracellular signaling [213, 214].
YKL-40 possesses heparin-binding affinity, which enables it to specifically bind heparan sulfate (HS) fragments [215]. Syndecans are transmembrane molecules with cytoplasmic domains that can interact with a number of regulators [216]. Syndecan-1 is the major source of cell surface HS. There is compelling evidence demonstrating that syndecan-1 can act as a matrix coreceptor with adjacent membrane-bound receptors such as integrins to mediate cell adhesion and/or spreading [217]. It was found that YKL-40 could induce the coupling of syndecan-1 and αvβ3 integrin (Figure 13), resulting in phosphorylation of focal adhesion kinase (FAK) and activation of downstream ERK1/2 signaling pathway, which enhance vascular endothelial growth factor (VEGF) expression in tumor cells, angiogenesis, and tumor growth [214]. Additionally, ERK1/2 and JNK signaling pathways were reported to upregulate proinflammatory mediators such as C-chemokine ligand 2 (CCL2), chemokine CX motif ligand 2 (CXCL2), and MMP-9; all of which contribute to tumor growth and metastasis [218].
Figure 13.
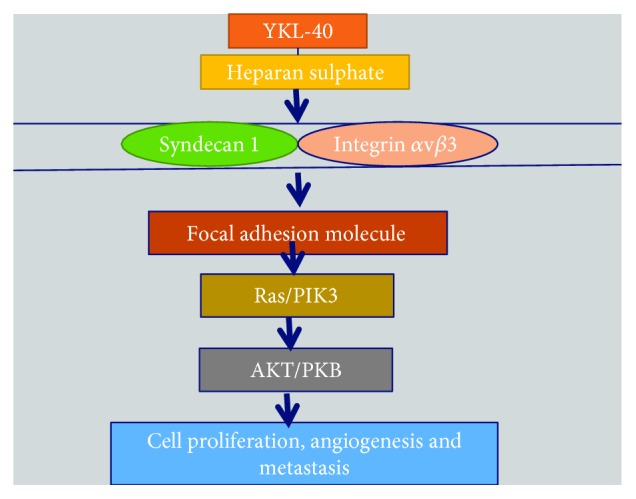
Involvement of YKL-40 in pathways pertaining to cell proliferation, survival, differentiation, and tumorigenesis.
Another VEGF-independent pathway was reported to mediate angiogenic activity of YKL-40, as an anti-VEGF neutralizing antibody failed to impede YKL-40-induced migration [219]. Therefore, targeting both YKL-40 and VEGF could be an efficient course of therapy along with radiotherapy for eventual eradication of deadly diseases.
Furthermore, YKL-40 was demonstrated to stimulate TGF-β1 production in malignant cells via interleukin-13 receptor α2- (IL-13Rα2-) dependent mechanism (Figure 14). The binding of YKL-40 to IL-13Rα2 results in the activation of MAPK, AKT, and Wnt/β-catenin which play an important role in inhibiting apoptosis and interleukin-1β (IL-1β) production thereby acting as a potential cancer promoter [220].
Figure 14.

YKL-40 function through IL-13Rα2-dependent mechanism.
Recently, Low et al. [221] showed that YKL-40 can also bind surface receptor for advanced glycation end product (RAGE), which is involved in tumor cell proliferation, migration, and survival through β-catenin- and nuclear factor kappa-B- (NF-κB-) associated signaling pathways [221, 222].
Most of the ongoing researches have been carried out on SNP rs4950928 in the promoter region of CHI3L1 gene as it was found to be associated with the serum/plasma YKL-40 levels [223, 224] and diseases such as asthma, bronchial hyperresponsiveness [207], and the severity of hepatitis C virus-induced liver fibrosis [225]. Some of the association studies of CHI3L1 SNPs with different diseases are shown in Table 15.
Table 15.
Association of some CHI3L1 SNPs with diseases.
| Disease | Population | Reference SNP (rs) | Location | Association | Reference |
|---|---|---|---|---|---|
| Sarcoidosis | Caucasian | rs10399931 | Promoter | No | [226] |
| Schizophrenia | Caucasian | rs10399805 | Promoter | Yes | [163] |
| Liver fibrosis | Caucasian | rs4950928 | Promoter | Yes | [225] |
| Glioblastoma | Caucasian | rs4950928 | Promoter | No | [227] |
| Asthma and atopy | Danish | rs4950928 | Promoter | Yes | [228] |
| Rheumatoid arthritis | Danish | rs4950928 | Promoter | No | [229] |
| rs6691378 | Promoter | No | |||
| rs10399931 | Promoter | No | |||
| rs880633 | Exon 5 | No | |||
| Coronary heart disease | Chinese | rs10399931 | Promoter | No | [230] |
| Schizophrenia | Japanese | rs4950928 | Promoter | Yes | [231] |
| Atrial fibrillation | Danish | rs4950928 | Promoter | No | [232] |
| Asthma | African Americans | rs4950928 | Promoter | Yes | [233] |
| Cervical cancer | Taiwanese | rs10399805 | Promoter | Yes | [234] |
| rs4950928 | Promoter | No | |||
| Asthma | Taiwanese | rs10399931 | Promoter | Yes | [235] |
| rs1538372 | Intron2/exon3 | Yes | |||
| Atherosclerosis | Taiwanese | rs10399931 | Promoter | No | [236] |
| Asthma | Indian | rs4950928 | Promoter | No | [237] |
| Non-Hodgkin's lymphoma | Danish | rs4950928 | Promoter | Yes | [238] |
| Asthma | Swedish | rs4950928 | Promoter | No | [239] |
| Venous thromboembolism | Danish | rs4950928 | Promoter | No | [240] |
| Coronary artery disease | Taiwanese | rs4950928 | Promoter | Yes | [241] |
Conflicts of Interest
The authors declare that they have no conflict of interest.
References
- 1.Jemal A., Bray F., Center M. M., Ferlay J., Ward E., Forman D. Global cancer statistics. CA: A Cancer Journal for Clinicians. 2011;61(2):69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2.Tenesa A., Dunlop M. G. New insights into the aetiology of colorectal cancer from genome-wide association studies. Nature Reviews Genetics. 2009;10(6):353–358. doi: 10.1038/nrg2574. [DOI] [PubMed] [Google Scholar]
- 3.Torre L. A., Bray F., Siegel R. L., Ferlay J., Lortet-tieulent J., Jemal A. Global cancer statistics, 2012. CA: A Cancer Journal of Clinicians. 2015;65(2):87–108. doi: 10.3322/caac.21262. [DOI] [PubMed] [Google Scholar]
- 4.Bogaert J., Prenen H. Molecular genetics of colorectal cancer. Annals of Gastroenterology. 2014;27(1):9–14. [PMC free article] [PubMed] [Google Scholar]
- 5.Hutter C. M., Chang-Claude J., Slattery M. L., et al. Characterization of gene-environment interactions for colorectal cancer susceptibility loci. Cancer Research. 2012;72(8):2036–2044. doi: 10.1158/0008-5472.CAN-11-4067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Johnson C. M., Wei C., Ensor J. E., et al. Meta-analyses of colorectal cancer risk factors. Cancer causes & control. 2013;24(6):1207–1222. doi: 10.1007/s10552-013-0201-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de la Chapelle A. Genetic predisposition to colorectal cancer. Nature Reviews Cancer. 2004;4(10):769–780. doi: 10.1038/nrc1453. [DOI] [PubMed] [Google Scholar]
- 8.Tárraga López P. J., Albero J. S., Rodríguez-Montes J. A. Primary and secondary prevention of colorectal cancer. Clinical Medicine Insights: Gastroenterology. 2014;7 doi: 10.4137/CGast.S14039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grady W. M. Genetic testing for high-risk colon cancer patients. Gastroenterology. 2003;124(6):1574–1594. doi: 10.1016/S0016-5085(03)00376-7. [DOI] [PubMed] [Google Scholar]
- 10.Slattery M. L., Potter J. D., Ma K. N., Caan B. J., Leppert M., Samowitz W. Western diet, family history of colorectal cancer, NAT2, GSTM-1 and risk of colon cancer. Cancer causes & control. 2000;11(1):1–8. doi: 10.1023/A:1008913619957. [DOI] [PubMed] [Google Scholar]
- 11.Terzić J., Grivennikov S., Karin E., Karin M. Inflammation and colon cancer. Gastroenterology. 2010;138(6):2101–2114.e5. doi: 10.1053/j.gastro.2010.01.058. [DOI] [PubMed] [Google Scholar]
- 12.Koenig M., Schofield J. B. The pathology of colorectal polyps and cancers. Surgery. 2011;29(1):11–14. doi: 10.1016/j.mpsur.2010.10.003. [DOI] [Google Scholar]
- 13.Chavez J. A., Summers S. a. Lipid oversupply, selective insulin resistance, and lipotoxicity: molecular mechanisms. Biochimica et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids. 2010;1801(3):252–265. doi: 10.1016/j.bbalip.2009.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huxley R. R., Ansary-Moghaddam A., Clifton P., Czernichow S., Parr C. L., Woodward M. The impact of dietary and lifestyle risk factors on risk of colorectal cancer: a quantitative overview of the epidemiological evidence. International Journal of Cancer. 2009;125(1):171–180. doi: 10.1002/ijc.24343. [DOI] [PubMed] [Google Scholar]
- 15.Hecht S. S. Tobacco smoke carcinogens and lung cancer. Journal of National Cancer Institute. 1999;91(14):1194–1210. doi: 10.1093/jnci/91.14.1194. [DOI] [PubMed] [Google Scholar]
- 16.Gutierrez D. A., Puglisi M. J., Hasty A. H. Impact of increased adipose tissue mass on inflammation, insulin resistance, and dyslipidemia. Current Diabetes Reports. 2009;9(1):26–32. doi: 10.1007/s11892-009-0006-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kumor A., Daniel P., Pietruczuk M., Małecka-Panas E. Serum leptin, adiponectin, and resistin concentration in colorectal adenoma and carcinoma (CC) patients. International Journal of Colorectal Disease. 2009;24(3):275–281. doi: 10.1007/s00384-008-0605-y. [DOI] [PubMed] [Google Scholar]
- 18.Seitz H. K., Stickel F. Molecular mechanisms of alcohol-mediated carcinogenesis. Nature Reviews Cancer. 2007;7(8):599–612. doi: 10.1038/nrc2191. [DOI] [PubMed] [Google Scholar]
- 19.Vogelstein B., Fearon E. R., Hamilton S. R., et al. Genetic alternations during colorectal tumor development. The New England Journal of Medicine. 1988;319(9):525–532. doi: 10.1056/nejm198809013190901. [DOI] [PubMed] [Google Scholar]
- 20.Sameer A. S. Colorectal cancer: molecular mutations and polymorphisms. Frontiers in Oncology. 2013;3:p. 114. doi: 10.3389/fonc.2013.00114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chan A., Giovannucci E. Primary prevention of colorectal cancer. Gastroenterology. 2010;138(6):2029–2043.e10. doi: 10.1053/j.gastro.2010.01.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vargas P. A., Alberts D. S. Primary prevention of colorectal cancer through dietary modification. Cancer. 1991;70(5):1229–1235. doi: 10.1002/1097-0142(19920901)70:3+<1229::aid-cncr2820701507>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 23.Chan D. S. M., Aune D., Norat T. Red meat intake and colorectal cancer risk: a summary of epidemiological studies. Current Nutrition Reports. 2012;2(1):56–62. doi: 10.1007/s13668-012-0035-x. [DOI] [Google Scholar]
- 24.Wolin K. Y., Yan Y., Colditz G. A., Lee I.-M. Physical activity and colon cancer prevention: a meta-analysis. British Journal of Cancer. 2009;100(4):611–616. doi: 10.1038/sj.bjc.6604917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim S.-Y., Jun T.-W., Lee Y.-S., Na H.-K., Surh Y.-J., Song W. Effects of exercise on cyclooxygenase-2 expression and nuclear factor-κB DNA binding in human peripheral blood mononuclear cells. Annals of the New York Academy of Sciences. 2009;1171(1):464–471. doi: 10.1111/j.1749-6632.2009.04915.x. [DOI] [PubMed] [Google Scholar]
- 26.Wang D., DuBois R. N. The role of COX-2 in intestinal inflammation and colorectal cancer. Oncogene. 2010;29(6):781–788. doi: 10.1038/onc.2009.421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Allison J. E., Sakoda L. C., Levin T. R., et al. Screening for colorectal neoplasms with new fecal occult blood tests: update on performance characteristics. Journal of the National Cancer Institute. 2007;99(19):1462–1470. doi: 10.1093/jnci/djm150. [DOI] [PubMed] [Google Scholar]
- 28.Beg M., Singh M., Saraswat M., Rewar B. Occult gastro-intestinal bleeding: detection, interpretation, and evaluation. Journal, Indian Academy of Clinical Medicine. 2002;3(2):153–158. [Google Scholar]
- 29.Stracci F., Zorzi M., Grazzini G. Colorectal cancer screening: tests, strategies, and perspectives. Frontiers in Public Health. 2014;2:p. 210. doi: 10.3389/fpubh.2014.00210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Atkin W. S., Edwards R., Kralj-Hans I., et al. Once-only flexible sigmoidoscopy screening in prevention of colorectal cancer: a multicentre randomised controlled trial. The Lancet. 2010;375(9726):1624–1633. doi: 10.1016/S0140-6736(10)60551-X. [DOI] [PubMed] [Google Scholar]
- 31.Brenner H., Hoffmeister M., Arndt V., Stegmaier C., Altenhofen L., Haug U. Protection from right- and left-sided colorectal neoplasms after colonoscopy: population-based study. Journal of the National Cancer Institute. 2010;102(2):89–95. doi: 10.1093/jnci/djp436. [DOI] [PubMed] [Google Scholar]
- 32.Trichopoulou A., Lagiou P., Kuper H., Trichopoulos D. Cancer and Mediterranean dietary traditions. Cancer Epidemiology Biomarkers & Prevention. 2000;9(9):869–873. [PubMed] [Google Scholar]
- 33.Shannon J., White E., Shattuck A. L., Potter J. D. Relationship of food groups and water intake to colon cancer risk. Cancer Epidemiology, Biomarkers & Prevention. 1996;5(7):495–502. [PubMed] [Google Scholar]
- 34.Ahmed F. E. Effect of diet, life style, and other environmental/chemopreventive factors on colorectal cancer development, and assessment of the risks. Journal of Environmental Science and Health. 2004;22(2):91–148. doi: 10.1081/LESC-200038263. [DOI] [PubMed] [Google Scholar]
- 35.Scharlau D., Borowicki A., Habermann N., et al. Mechanisms of primary cancer prevention by butyrate and other products formed during gut flora-mediated fermentation of dietary fibre. Mutation Research/Reviews in Mutation Research. 2009;682(1):39–53. doi: 10.1016/j.mrrev.2009.04.001. [DOI] [PubMed] [Google Scholar]
- 36.Feregrino-Pérez A. A., Berumen L. C., García-Alcocer G., et al. Composition and chemopreventive effect of polysaccharides from common beans (Phaseolus vulgaris L.) on azoxymethane-induced colon cancer. Journal of Agricultural and Food Chemistry. 2008;56(18):8737–8744. doi: 10.1021/jf8007162. [DOI] [PubMed] [Google Scholar]
- 37.Pennington J. A. T., Fisher R. A. Classification of fruits and vegetables. Journal of Food Composition and Analysis. 2009;22:S23–S31. doi: 10.1016/j.jfca.2008.11.012. [DOI] [Google Scholar]
- 38.Larsson S. C., Kumlin M., Ingelman-sundberg M., Wolk A. Dietary long-chain n−3 fatty acids for the prevention of cancer: a review of potential mechanisms. The American Journal of Clinical Nutrition. 2004;79(6):935–945. doi: 10.1093/ajcn/79.6.935. [DOI] [PubMed] [Google Scholar]
- 39.Sanders L. M., Henderson C. E., Hong M. Y., et al. An increase in reactive oxygen species by dietary fish oil coupled with the attenuation of antioxidant defenses by dietary pectin enhances rat colonocyte apoptosis. The Journal of Nutrition. 2004;134(12):3233–3238. doi: 10.1093/jn/134.12.3233. [DOI] [PubMed] [Google Scholar]
- 40.Vanamala J., Glagolenko A., Yang P., et al. Dietary fish oil and pectin enhance colonocyte apoptosis in part through suppression of PPARδ/PGE 2 and elevation of PGE 3. Carcinogenesis. 2008;29(4):790–796. doi: 10.1093/carcin/bgm256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Maintz L., Novak N. Histamine and histamine intolerance. American journal of clinical nutrition. 2007;85(5):1185–1196. doi: 10.1093/ajcn/85.5.1185. [DOI] [PubMed] [Google Scholar]
- 42.Duthie S. J. Folate and cancer: how DNA damage, repair and methylation impact on colon carcinogenesis. Journal of Inherited Metabolic Disease. 2011;34(1):101–109. doi: 10.1007/s10545-010-9128-0. [DOI] [PubMed] [Google Scholar]
- 43.Powers H. J. Interaction among folate, riboflavin, genotype, and cancer, with reference to colorectal and cervical cancer. The Journal of Nutrition. 2005;135(12):2960S–2966S. doi: 10.1093/jn/135.12.2960s. [DOI] [PubMed] [Google Scholar]
- 44.Buset M., Lipkin M., Winawer S., Swaroop S., Friedman E. Inhibition of human colonic epithelial cell proliferation in vivo and in vitro by calcium. Cancer Research. 1986;46(10):5426–5430. [PubMed] [Google Scholar]
- 45.Wu K., Willett W. C., Fuchs C. S., Colditz G. A., Giovannucci E. L. Calcium intake and risk of colon cancer in women and men. Journal of the National Cancer Institute. 2002;94(6):437–446. doi: 10.1093/jnci/94.6.437. [DOI] [PubMed] [Google Scholar]
- 46.Fedirko V., Bostick R. M., Flanders W. D., et al. Effects of vitamin D and calcium supplementation on markers of apoptosis in normal colon mucosa: a randomized, double-blind, placebo-controlled clinical trial. Cancer Prevention Research. 2009;2(3):213–223. doi: 10.1158/1940-6207.CAPR-08-0157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fedirko V., Bostick R. M., Flanders W. D., et al. Effects of vitamin D and calcium on proliferation and differentiation in normal colon mucosa: a randomized clinical trial. Cancer Epidemiology Biomarkers & Prevention. 2009;18(11):2933–2941. doi: 10.1158/1055-9965.EPI-09-0239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Leimar O. Environmental and genetic cues in the evolution of phenotypic polymorphism. Evolutionary Ecology. 2009;23(1):125–135. doi: 10.1007/s10682-007-9194-4. [DOI] [Google Scholar]
- 49.Chester M. A., Olsson M. L. The ABO blood group gene: a locus of considerable genetic diversity. Transfusion Medicine Reviews. 2001;15(3):177–200. doi: 10.1053/tmrv.2001.24591. [DOI] [PubMed] [Google Scholar]
- 50.Dick C. A., Buenrostro J., Butler T., Carlson M. L., Kliebenstein D. J., Whittall J. B. Arctic mustard flower color polymorphism controlled by petal-specific downregulation at the threshold of the anthocyanin biosynthetic pathway. PLoS One. 2011;6(4, article e18230) doi: 10.1371/journal.pone.0018230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Harris H., Hopkinson D. A. Average heterozygosity per locus in man: an estimate based on the incidence of enzyme polymorphisms. Annals of Human Genetics. 1972;36(1):9–20. doi: 10.1111/j.1469-1809.1972.tb00578.x. [DOI] [PubMed] [Google Scholar]
- 52.Lewontin R. C. Twenty-five years ago in genetics: electrophoresis in the development of evolutionary genetics: milestone or millstone? Genetics. 1991;128(4):657–662. doi: 10.1093/genetics/128.4.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hubby J. L., Lewontin R. C. A molecular approach to the study of genic heterozygosity in natural populations. I. The number of alleles at different loci in Drosophila pseudoobscura. Genetics. 1966;54(2):577–594. doi: 10.1093/genetics/54.2.577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Collins F. S., Brooks L. D., Chakravarti A. A DNA polymorphism discovery resource for research on human genetic variation. Genome Research. 1998;8(12):1229–1231. doi: 10.1101/gr.8.12.1229. [DOI] [PubMed] [Google Scholar]
- 55.Hamosh A., King T. M., Rosenstein B. J., et al. Cystic fibrosis patients bearing both the common missense mutation, Gly→Asp at codon 551 and the ΔF508 mutation are clinically indistinguishable from ΔF508 homozygotes, except for decreased risk of meconium ileus. American Journal of Human Genetics. 1992;51(2):245–250. [PMC free article] [PubMed] [Google Scholar]
- 56.Wolf A. B., Caselli R. J., Reiman E. M., Valla J. APOE and neuroenergetics: an emerging paradigm in Alzheimer’s disease. Neurobiology of Aging. 2013;34(4):1007–1017. doi: 10.1016/j.neurobiolaging.2012.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sauna Z. E., Kimchi-Sarfaty C., Ambudkar S. V., Gottesman M. M. Silent polymorphisms speak: how they affect pharmacogenomics and the treatment of cancer. Cancer Research. 2007;67(20):9609–9612. doi: 10.1158/0008-5472.CAN-07-2377. [DOI] [PubMed] [Google Scholar]
- 58.Kimchi-Sarfaty C., Oh J. M., Kim I.-W., et al. A “silent” polymorphism in the MDR1 gene changes substrate specificity. Science. 2007;315(5811):525–528. doi: 10.1126/science.1135308. [DOI] [PubMed] [Google Scholar]
- 59.Al-Haggar M., Madej-Pilarczyk A., Kozlowski L., et al. A novel homozygous p.Arg527Leu LMNA mutation in two unrelated Egyptian families causes overlapping mandibuloacral dysplasia and progeria syndrome. European journal of human genetics. 2012;20(11):1134–1140. doi: 10.1038/ejhg.2012.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cordovado S. K., Hendrix M., Greene C. N., et al. CFTR mutation analysis and haplotype associations in CF patients. Molecular Genetics and Metabolism. 2012;105(2):249–254. doi: 10.1016/j.ymgme.2011.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Modrek B., Lee C. A genomic view of alternative splicing. Nature Genetics. 2002;30(1):13–19. doi: 10.1038/ng0102-13. [DOI] [PubMed] [Google Scholar]
- 62.Fareed M., Afzal M. Single nucleotide polymorphism in genome-wide association of human population: a tool for broad spectrum service. Egyptian Journal of Medical Human Genetics. 2012;14(2):123–134. doi: 10.1016/j.ejmhg.2012.08.001. [DOI] [Google Scholar]
- 63.Kim S., Misra A. SNP genotyping: technologies and biomedical applications. Annual Review of Biomedical Engineering. 2007;9(1):289–320. doi: 10.1146/annurev.bioeng.9.060906.152037. [DOI] [PubMed] [Google Scholar]
- 64.Botstein D., White R. L., Skolnick M., Davis R. W. Construction of a genetic linkage map in man using restriction fragment length polymorphisms. American Journal of Human Genetics. 1980;32(3):314–331. [PMC free article] [PubMed] [Google Scholar]
- 65.Ross P., Hall L., Smirnov I., Haff L. High level multiplex genotyping by MALDI-TOF mass spectrometry. Nature Biotechnology. 1998;16(13):1347–1351. doi: 10.1038/4328. [DOI] [PubMed] [Google Scholar]
- 66.Livak K. J. Allelic discrimination using fluorogenic probes and the 5′ nuclease assay. Genetic Analysis - Biomolecular Engineering. 1999;14(5-6):143–149. doi: 10.1016/S1050-3862(98)00019-9. [DOI] [PubMed] [Google Scholar]
- 67.Tong A. K., Li Z., Jones G. S., Russo J. J., Ju J. Combinatorial fluorescence energy transfer tags for multiplex biological assays. Nature Biotechnology. 2001;19(8):756–759. doi: 10.1038/90810. [DOI] [PubMed] [Google Scholar]
- 68.Vineis P., Brennan P., Canzian F., et al. Expectations and challenges stemming from genome-wide association studies. Mutagenesis. 2008;23(6):439–444. doi: 10.1093/mutage/gen042. [DOI] [PubMed] [Google Scholar]
- 69.Ku C. S., Loy E. Y., Pawitan Y., Chia K. S. The pursuit of genome-wide association studies: where are we now? Journal of Human Genetics. 2010;55(4):195–206. doi: 10.1038/jhg.2010.19. [DOI] [PubMed] [Google Scholar]
- 70.Frazer K. A., Ballinger D. G., Cox D. R., et al. A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449(7164):851–861. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hirschhorn J. N., Daly M. J. Genome-wide association studies for common diseases and complex traits. Nature Reviews Genetics. 2005;6(2):95–108. doi: 10.1038/nrg1521. [DOI] [PubMed] [Google Scholar]
- 72.Pearson T. A., Manolio T. A. How to interpret a genome-wide association study. JAMA. 2008;299(11):1335–1344. doi: 10.1001/jama.299.11.1335. [DOI] [PubMed] [Google Scholar]
- 73.Peters U., Hutter C. M., Hsu L., et al. Meta-analysis of new genome-wide association studies of colorectal cancer risk. Human Genetics. 2012;131(2):217–234. doi: 10.1007/s00439-011-1055-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Broderick P., Carvajal-Carmona L., Pittman A. M., et al. A genome-wide association study shows that common alleles of SMAD7 influence colorectal cancer risk. Nature Genetics. 2007;39(11):1315–1317. doi: 10.1038/ng.2007.18. [DOI] [PubMed] [Google Scholar]
- 75.Tenesa A., Farrington S. M., Prendergast J. G. D., et al. Genome-wide association scan identifies a colorectal cancer susceptibility locus on 11q23 and replicates risk loci at 8q24 and 18q21. Nature Genetics. 2008;40(5):631–637. doi: 10.1038/ng.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Song Q., Zhu B., Hu W., et al. A common SMAD7 variant is associated with risk of colorectal cancer: evidence from a case-control study and a meta-analysis. PLoS One. 2012;7(3, article e33318) doi: 10.1371/journal.pone.0033318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kirac I., Matošević P., Augustin G., et al. SMAD7 variant rs4939827 is associated with colorectal cancer risk in Croatian population. PLoS One. 2013;8(9, article e74042) doi: 10.1371/journal.pone.0074042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Xing L., Wang Z., Jiang L., et al. Matrix metalloproteinase-9-1562C>T polymorphism may increase the risk of lymphatic metastasis of colorectal cancer. Journal of Gastroenterology. 2007;13(34):4626–4629. doi: 10.3748/wjg.v13.i34.4626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Xing L. L., Wang Z. N., Jiang L., et al. Cyclooxygenase 2 polymorphism and colorectal cancer: -765G>C variant modifies risk associated with smoking and body mass index. World Journal of Gastroenterology. 2008;14(11):1785–1789. doi: 10.3748/wjg.14.1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bai Y.-H., Lu H., Hong D., Lin C.-C., Yu Z., Chen B.-C. Vitamin D receptor gene polymorphisms and colorectal cancer risk: a systematic meta-analysis. World journal of gastroenterology. 2012;18(14):1672–1679. doi: 10.3748/wjg.v18.i14.1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lubbe S. J., Pittman A. M., Olver B., et al. The 14q22.2 colorectal cancer variant rs4444235 shows cis-acting regulation of BMP4. Oncogene. 2012;31(33):3777–3784. doi: 10.1038/onc.2011.564. [DOI] [PubMed] [Google Scholar]
- 82.Abbenhardt C., Poole E. M., Kulmacz R. J., et al. Phospholipase A2G1B polymorphisms and risk of colorectal neoplasia. International journal of molecular epidemiology and genetics. 2013;4(3):140–149. [PMC free article] [PubMed] [Google Scholar]
- 83.Yang H., Gao Y., Feng T., Jin T. B., Kang L. L., Chen C. Meta-analysis of the rs4779584 polymorphism and colorectal cancer risk. PLoS One. 2014;9(2, article e89736) doi: 10.1371/journal.pone.0089736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Li M., Gu Y. Quantitative assessment of the influence of common variation rs16892766 at 8q23.3 with colorectal adenoma and cancer susceptibility. Molecular Genetics and Genomics. 2015;290(2):461–469. doi: 10.1007/s00438-014-0928-z. [DOI] [PubMed] [Google Scholar]
- 85.Smith C. G., Fisher D., Harris R., et al. Analyses of 7,635 patients with colorectal cancer using independent training and validation cohorts show that rs9929218 in CDH1 is a prognostic marker of survival. Clinical Cancer Research. 2015;21(15):3453–3461. doi: 10.1158/1078-0432.CCR-14-3136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wang S., Wu S., Meng Q., et al. FAS rs2234767 and rs1800682 polymorphisms jointly contributed to risk of colorectal cancer by affecting SP1/STAT1 complex recruitment to chromatin. Scientific Reports. 2016;6, article 19229 doi: 10.1038/srep19229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Cao X., Zhuang S., Hu Y., Xi L., Deng L. Associations between polymorphisms of long non-coding RNA MEG3 and risk of colorectal cancer in Chinese. Oncotarget. 2016;7(14):19054–19059. doi: 10.18632/oncotarget.7764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Liu G., Tu D., Lewis M., et al. Fc-γ receptor polymorphisms, cetuximab therapy, and survival in the NCIC CTG CO.17 trial of colorectal cancer. Clinical Cancer Research. 2016;22(10):2435–2444. doi: 10.1158/1078-0432.CCR-15-0414. [DOI] [PubMed] [Google Scholar]
- 89.Zeng C., Matsuda K., Jia W.-H., et al. Identification of susceptibility loci and genes for colorectal cancer risk. Gastroenterology. 2016;150(7):1633–1645. doi: 10.1053/j.gastro.2016.02.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Li J., Chang J., Tian J., et al. A rare variant P507L in TPP1 interrupts TPP1–TIN2 interaction, influences telomere length, and confers colorectal cancer risk in Chinese population. Cancer Epidemiology Biomarkers & Prevention. 2018;27(9):1029–1035. doi: 10.1158/1055-9965.EPI-18-0099. [DOI] [PubMed] [Google Scholar]
- 91.Zou D., Lou J., Ke J., et al. Integrative expression quantitative trait locus–based analysis of colorectal cancer identified a functional polymorphism regulating SLC22A5 expression. European Journal of Cancer. 2018;93(4):1–9. doi: 10.1016/j.ejca.2018.01.065. [DOI] [PubMed] [Google Scholar]
- 92.Shimizu T., Nakai T., Deguchi K., Yamada K., Yue B., Ye J. A polymorphic MYC response element in KBTBD11 influences colorectal cancer risk, especially in interaction with an MYC-regulated SNP rs6983267. Annals Oncology. 2018;29(3):632–639. doi: 10.1093/annonc/mdx789. [DOI] [PubMed] [Google Scholar]
- 93.Sun R., Liang Y., Yuan F., et al. Functional polymorphisms in the promoter region of miR-17-92 cluster are associated with a decreased risk of colorectal cancer. Oncotarget. 2017;8(47):82531–82540. doi: 10.18632/oncotarget.19753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sporn M. B., Roberts A. B. Transforming growth factor-beta: recent progress and new challenges. The Journal of Cell Biology. 1992;119(5):1017–1021. doi: 10.1083/jcb.119.5.1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Nakao A., Afrakhte M., More A., Itoh S., Kawabata M., Heldin N. Identification of Smad7, a TGFβ-inducible antagonist of TGF-β signalling. Nature. 1997;389(6651):631–635. doi: 10.1038/39369. [DOI] [PubMed] [Google Scholar]
- 96.Chaudhury A., Howe P. H. The tale of transforming growth factor-beta (TGFbeta) signaling: a soigné enigma. IUBMB Life. 2009;61(10):929–939. doi: 10.1002/iub.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hong S., Lee H. J., Kim S. J., Hahm K. B. Connection between inflammation and carcinogenesis in gastrointestinal tract: focus on TGF-β signaling. World Journal of Gastroenterology. 2010;16(17):2080–2093. doi: 10.3748/wjg.v16.i17.2080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Monteleone G., Pallone F., MacDonald T. T. Smad7 in TGF-β-mediated negative regulation of gut inflammation. Trends in Immunology. 2004;25(10):513–517. doi: 10.1016/j.it.2004.07.008. [DOI] [PubMed] [Google Scholar]
- 99.Fukushima T., Mashiko M., Takita K., et al. Mutational analysis of TGF-beta type II receptor, Smad2, Smad3, Smad4, Smad6 and Smad7 genes in colorectal cancer. Journal of experimental & clinical cancer research. 2003;22(2):315–320. [PubMed] [Google Scholar]
- 100.Halder S. K., Beauchamp R. D., Datta P. K. Smad7 induces tumorigenicity by blocking TGF-β-induced growth inhibition and apoptosis. Experimental Cell Research. 2005;307(1):231–246. doi: 10.1016/j.yexcr.2005.03.009. [DOI] [PubMed] [Google Scholar]
- 101.Roberts A. B., Anzano M. A., Lamb L. C., Smith J. M., Sporn M. B. New class of transforming growth factors potentiated by epidermal growth factor: isolation from non-neoplastic tissues. Proceedings of the National Academy of Sciences of the United States of America. 1981;78(9):5339–5343. doi: 10.1073/pnas.78.9.5339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Roberts A., Lamb L. Transforming growth factors: isolation of polypeptides from virally and chemically transformed cells by acid/ethanol extraction. Proceedings of the National Academy of Sciences of the United States of America. 1980;77(6):3494–3498. doi: 10.1073/pnas.77.6.3494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Tsushima H., Kawata S., Tamura S., et al. High levels of transforming growth factor beta 1 in patients with colorectal cancer: association with disease progression. Gastroenterology. 1996;110(2):375–382. doi: 10.1053/gast.1996.v110.pm8566583. [DOI] [PubMed] [Google Scholar]
- 104.Roberts A. B., Thompson N. L., Heine U., Flanders C., Sporn M. B. Transforming growth factor-β: possible roles in carcinogenesis. British Journal of Cancer. 1988;57:594–600. doi: 10.1038/bjc.1988.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Blanchette F., Day R., Dong W., Laprise M. H., Dubois C. M. TGFbeta1 regulates gene expression of its own converting enzyme furin. The Journal of Clinical Investigation. 1997;99(8):1974–1983. doi: 10.1172/JCI119365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Schlunegger M. P., Grütter M. G. An unusual feature revealed by the crystal structure at 2.2 Å resolution of human transforming growth factor-β2. Nature. 1992;358(6385):430–434. doi: 10.1038/358430a0. [DOI] [PubMed] [Google Scholar]
- 107.Taipale J., Miyazono K., Heldin C. H., Keski-Oja J. Latent transforming growth factor-beta 1 associates to fibroblast extracellular matrix via latent TGF-beta binding protein. Journal of Cell Biology. 1994;124(1):171–181. doi: 10.1083/jcb.124.1.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Hayashi H., Sakai T. Biological significance of local TGF-β activation in liver diseases. Frontiers in Physiology. 2012;3:p. 12. doi: 10.3389/fphys.2012.00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Isogai Z., Ono R. N., Ushiro S., et al. Latent transforming growth factor β-binding protein 1 interacts with fibrillin and is a microfibril-associated protein. Journal of Biological Chemistry. 2003;278(4):2750–2757. doi: 10.1074/jbc.M209256200. [DOI] [PubMed] [Google Scholar]
- 110.Wipff P., Hinz B. Integrins and the activation of latent transforming growth factor β1 – an intimate relationship. European Journal of Cell Biology. 2008;87(8-9):601–615. doi: 10.1016/j.ejcb.2008.01.012. [DOI] [PubMed] [Google Scholar]
- 111.Gulubova M., Manolova I., Ananiev J., Julianov A., Yovchev Y., Peeva K. Role of TGF-β1, its receptor TGFβRII, and Smad proteins in the progression of colorectal cancer. International Journal of Colorectal Disease. 2010;25(5):591–599. doi: 10.1007/s00384-010-0906-9. [DOI] [PubMed] [Google Scholar]
- 112.Laiho M., DeCaprio J. A., Ludlow J. W., Livingston D. M., Massagué J. Growth inhibition by TGF-β linked to suppression of retinoblastoma protein phosphorylation. Cell. 1990;62(1):175–185. doi: 10.1016/0092-8674(90)90251-9. [DOI] [PubMed] [Google Scholar]
- 113.Hannon G. J., Beach D. p15INK4B is a potential effector of TGF-beta-induced cell cycle arrest. Nature. 1994;371(6494):257–261. doi: 10.1038/371257a0. [DOI] [PubMed] [Google Scholar]
- 114.Reynisdottir I., Polyak K., Iavarone A., Massague J. Kip/Cip and Ink4 Cdk4 inhibitors cooperate to induce cell cycle arrest in response to TGF-beta. Genes and Development. 1995;9(15):1831–1845. doi: 10.1101/gad.9.15.1831. [DOI] [PubMed] [Google Scholar]
- 115.Datto M. B., Li Y., Panus J. F., Howe D. J., Xiong Y., Wang X. F. Transforming growth factor beta induces the cyclin-dependent kinase inhibitor p21 through a p53-independent mechanism. Proceedings of the National Academy of Sciences of the United States of America. 1995;92(12):5545–5549. doi: 10.1073/pnas.92.12.5545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Chen C. R., Kang Y., Siegel P. M., Massagué J. E2F4/5 and p107 as Smad cofactors linking the TGFβ receptor to c-myc repression. Cell. 2002;110(1):19–32. doi: 10.1016/S0092-8674(02)00801-2. [DOI] [PubMed] [Google Scholar]
- 117.Jang C.-W., Chen C.-H., Chen C.-C., Chen J., Su Y.-H., Chen R.-H. TGF-β induces apoptosis through Smad-mediated expression of DAP-kinase. Nature Cell Biology. 2002;4(1):51–58. doi: 10.1038/ncb731. [DOI] [PubMed] [Google Scholar]
- 118.Ribeiro A., Bronk S. F., Roberts P. J., Urrutia R., Gores G. J. The transforming growth factor β1 – inducible transcription factor, TIEG1, mediates apoptosis through oxidative stress. Hepatology. 1999;30(6):1490–1497. doi: 10.1002/hep.510300620. [DOI] [PubMed] [Google Scholar]
- 119.Tachibana I., Imoto M., Adjei P. N., et al. Overexpression of the TGFbeta-regulated zinc finger encoding gene, TIEG, induces apoptosis in pancreatic epithelial cells. The Journal of Clinical Investigation. 1997;99(10):2365–2374. doi: 10.1172/JCI119418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Torre-Amione G., Beauchamp R. D., Koeppen H., et al. A highly immunogenic tumor transfected with a murine transforming growth factor type beta 1 cDNA escapes immune surveillance. Proceedings of the National Academy of Sciences of the United States of America. 1990;87(4):1486–1490. doi: 10.1073/pnas.87.4.1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Arteaga C. L., Hurd S. D., Winnier A. R., Johnson M. D., Fendly B. M., Forbes J. T. Anti-transforming growth factor (TGF)-beta antibodies inhibit breast cancer cell tumorigenicity and increase mouse spleen natural killer cell activity. Implications for a possible role of tumor cell/host TGF-beta interactions in human breast cancer progression. Journal of Clinical Investigation. 1993;92(6):2569–2576. doi: 10.1172/JCI116871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Derynck R., Akhurst R. J., Balmain A. TGF-β signaling in tumor suppression and cancer progression. Nature Genetics. 2001;29(2):117–129. doi: 10.1038/ng1001-117. [DOI] [PubMed] [Google Scholar]
- 123.Kang Y., Siegel P. M., Shu W., et al. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell. 2003;3(6):537–549. doi: 10.1016/S1535-6108(03)00132-6. [DOI] [PubMed] [Google Scholar]
- 124.Pertovaara L., Kaipainen A., Mustonen T., et al. Vascular endothelial growth factor is induced in response to transforming growth factor-beta in fibroblastic and epithelial cells. Journal of biological chemistry. 1994;269(9):6271–6274. [PubMed] [Google Scholar]
- 125.Lee J. M. The epithelial–mesenchymal transition: new insights in signaling, development, and disease. Journal of Cell Biology. 2006;172(7):973–981. doi: 10.1083/jcb.200601018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Vogelmann R., Nguyen-Tat M.-D., Giehl K., Adler G., Wedlich D., Menke A. TGFβ-induced downregulation of E-cadherin-based cell-cell adhesion depends on PI3-kinase and PTEN. Journal of Cell Science. 2005;118(20):4901–4912. doi: 10.1242/jcs.02594. [DOI] [PubMed] [Google Scholar]
- 127.Peinado H., Ballestar E., Esteller M., Cano A. Snail mediates E-cadherin repression by the recruitment of the Sin3A/histone deacetylase 1 (HDAC1)/HDAC2 complex. Molecular and Cellular Biology. 2003;24(1):306–319. doi: 10.1128/mcb.24.1.306-319.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Zhang Q., Helfand B. T., Jang T. L., et al. Nuclear factor-κB-mediated transforming growth factor-β-induced expression of vimentin is an independent predictor of biochemical recurrence after radical prostatectomy. Clinical Cancer Research. 2009;15(10):3557–3567. doi: 10.1158/1078-0432.CCR-08-1656. [DOI] [PubMed] [Google Scholar]
- 129.Radisky E. S., Radisky D. C. Matrix metalloproteinase-induced epithelial-mesenchymal transition in breast cancer. Journal of Mammary Gland Biology and Neoplasia. 2010;15(2):201–212. doi: 10.1007/s10911-010-9177-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Siegel P. M., Massagué J. Cytostatic and apoptotic actions of TGF-beta in homeostasis and cancer. Nature Reviews. Cancer. 2003;3(11):807–820. doi: 10.1038/nrc1208. [DOI] [PubMed] [Google Scholar]
- 131.Singh P., Wig J. D., Srinivasan R. The Smad family and its role in pancreatic cancer. Indian Journal of Cancer. 2011;48(3):351–360. doi: 10.4103/0019-509X.84939. [DOI] [PubMed] [Google Scholar]
- 132.Derynck R., Zhang Y. E. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. 2003;425(6958):577–584. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- 133.Yu L., Hébert M. C., Zhang Y. E. TGF‐β receptor‐activated p38 MAP kinase mediates Smad‐independent TGF‐β responses. EMBO Journal. 2002;21(14):3749–3759. doi: 10.1093/emboj/cdf366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Bhowmick N. A., Zent R., Ghiassi M., McDonnell M., Moses H. L. Integrin β1 signaling is necessary for transforming growth factor-β activation of p38MAPK and epithelial plasticity. Journal of Biological Chemistry. 2001;276(50):46707–46713. doi: 10.1074/jbc.M106176200. [DOI] [PubMed] [Google Scholar]
- 135.Yamashita M., Fatyol K., Jin C., Wang X., Liu Z., Zhang Y. E. TRAF6 mediates Smad-independent activation of JNK and p38 by TGF-β. Molecular Cell. 2008;31(6):918–924. doi: 10.1016/j.molcel.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Thiery J. P. Epithelial–mesenchymal transitions in development and pathologies. Current Opinion in Cell Biology. 2003;15(6):740–746. doi: 10.1016/j.ceb.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 137.Wilkes M. C., Mitchell H., Penheiter S. G., et al. Transforming growth factor-β activation of phosphatidylinositol 3-kinase is independent of Smad2 and Smad3 and regulates fibroblast responses via p21-activated kinase-2. Cancer Research. 2005;65(22):10431–10440. doi: 10.1158/0008-5472.CAN-05-1522. [DOI] [PubMed] [Google Scholar]
- 138.Shin I., Bakin A. V., Rodeck U., Brunet A., Arteaga C. L. Transforming growth factor β enhances epithelial cell survival via Akt-dependent regulation of FKHRL1. Molecular Biology of the Cell. 2001;12(11):3328–3339. doi: 10.1091/mbc.12.11.3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Jaffe A. B., Hall A. RHO GTPASES: biochemistry and biology. Annual Review of Cell and Developmental Biology. 2005;21(1):247–269. doi: 10.1146/annurev.cellbio.21.020604.150721. [DOI] [PubMed] [Google Scholar]
- 140.Zhang Y. E. Non-Smad pathways in TGF-beta signaling. Cell Research. 2009;19(1):128–139. doi: 10.1038/cr.2008.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Levy L., Hill C. S. Alterations in components of the TGF-β superfamily signaling pathways in human cancer. Cytokine & Growth Factor Reviews. 2006;17(1-2):41–58. doi: 10.1016/j.cytogfr.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 142.Grady W. M., Myeroff L. L., Swinler S. E., et al. Mutational inactivation of transforming growth factor β receptor type II in microsatellite stable colon cancers. Cancer Research. 1999;59(2):320–324. [PubMed] [Google Scholar]
- 143.Lynch M. A., Nakashima R., Song H., et al. Mutational analysis of the transforming growth factor β receptor type II gene in human ovarian carcinoma. Cancer Research. 1998;58(19):4227–4232. [PubMed] [Google Scholar]
- 144.Lucke C. D., Philpott A., Metcalfe J. C., et al. Inhibiting mutations in the transforming growth factor β type 2 receptor in recurrent human breast cancer. Cancer Research. 2001;61(2):482–485. [PubMed] [Google Scholar]
- 145.Kim I. Y., Ahn H. J., Lang S., et al. Loss of expression of transforming growth factor-beta receptors is associated with poor prognosis in prostate cancer patients. Clinical Cancer Research. 1998;4(7):1625–1630. [PubMed] [Google Scholar]
- 146.Zhang H. T., Chen X. F., Wang M. H., et al. Defective expression of transforming growth factor β receptor type II is associated with CpG methylated promoter in primary non-small cell lung cancer. Clinical Cancer Research. 2004;10(7):2359–2367. doi: 10.1158/1078-0432.ccr-0959-3. [DOI] [PubMed] [Google Scholar]
- 147.Gobbi H., Arteaga C. L., Jensen R. A., et al. Loss of expression of transforming growth factor beta type II receptor correlates with high tumour grade in human breast in-situ and invasive carcinomas. Histopathology. 2000;36(2):168–177. doi: 10.1046/j.1365-2559.2000.00841.x. [DOI] [PubMed] [Google Scholar]
- 148.Chen T., Triplett J., Dehner B., et al. Transforming growth factor-β receptor type I gene is frequently mutated in ovarian carcinomas. Cancer Research. 2001;61(12):4679–4682. [PubMed] [Google Scholar]
- 149.Wang D., Kanuma T., Mizunuma H., et al. Analysis of specific gene mutations in the transforming growth factor-β signal transduction pathway in human ovarian cancer. Journal of controlled release. 2000;60(16):4507–4512. [PubMed] [Google Scholar]
- 150.Chen T., Yan W., Wells R. G., et al. Novel inactivating mutations of transforming growth factor-β type I receptor gene in head-and-neck cancer metastases. International Journal of Cancer. 2001;93(5):653–661. doi: 10.1002/ijc.1381. [DOI] [PubMed] [Google Scholar]
- 151.Goggins M., Shekher M., Turnacioglu K., Yeo C. J., Hruban R. H., Kern S. E. Genetic alterations of the transforming growth factor β receptor genes in pancreatic and biliary adenocarcinomas. Cancer research. 1998;58(23):5329–5332. [PubMed] [Google Scholar]
- 152.Wolfraim L. A., Fernandez T. M., Mamura M., et al. Loss of Smad3 in acute T-cell lymphoblastic leukemia. The New England Journal of Medicine. 2004;351(6):552–559. doi: 10.1056/NEJMoa031197. [DOI] [PubMed] [Google Scholar]
- 153.Fleming N. I., Jorissen R. N., Mouradov D., et al. SMAD2, SMAD3 and SMAD4 mutations in colorectal cancer. Cancer Research. 2013;73(2):725–735. doi: 10.1158/0008-5472.CAN-12-2706. [DOI] [PubMed] [Google Scholar]
- 154.Thiagalingam S., Lengauer C., Leach F., et al. Evaluation of candidate tumour suppressor genes on chromosome 18 in colorectal cancers. Nature Genetics. 1996;13(3):343–346. doi: 10.1038/ng0796-343. [DOI] [PubMed] [Google Scholar]
- 155.Boulay J. L., Mild G., Reuter J., et al. Combined copy status of 18q21 genes in colorectal cancer shows frequent retention of SMAD7. Genes, Chromosomes & Cancer. 2001;31(3):240–247. doi: 10.1002/gcc.1140. [DOI] [PubMed] [Google Scholar]
- 156.Hayashi H., Abdollah S., Qiu Y., et al. The MAD-related protein Smad7 associates with the TGFβ receptor and functions as an antagonist of TGFβ signaling. Cell. 1997;89(7):1165–1173. doi: 10.1016/S0092-8674(00)80303-7. [DOI] [PubMed] [Google Scholar]
- 157.Kavsak P., Rasmussen R. K., Causing C. G., et al. Smad7 binds to Smurf2 to form an E3 ubiquitin ligase that targets the TGFβ receptor for degradation. Molecular cell. 2000;6(6):1365–1375. doi: 10.1016/s1097-2765(00)00134-9. [DOI] [PubMed] [Google Scholar]
- 158.Yan X., Liao H., Cheng M., et al. Smad7 protein interacts with receptor-regulated Smads (R-Smads) to inhibit transforming growth factor-β (TGF-β)/Smad signaling. Journal of Biological Chemistry. 2016;291(1):382–392. doi: 10.1074/jbc.M115.694281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Zhang S., Fei T., Zhang L., et al. Smad7 antagonizes transforming growth factor β signaling in the nucleus by interfering with functional Smad-DNA complex formation. Molecular and cellular biology. 2007;27(12):4488–4499. doi: 10.1128/MCB.01636-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Stolfi C., Marafini I., De Simone V., Pallone F., Monteleone G. The dual role of Smad7 in the control of cancer growth and metastasis. International Journal of Molecular Sciences. 2013;14(12):23774–23790. doi: 10.3390/ijms141223774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Ulloa L., Doody J., Massagué J. Inhibition of transforming growth factor-β/SMAD signalling by the interferon-γ/STAT pathway. Nature. 1999;397(6721):710–713. doi: 10.1038/17826. [DOI] [PubMed] [Google Scholar]
- 162.Edlund S., Lee S. Y., Grimsby S., et al. Interaction between Smad7 and β-Catenin: importance for transforming growth factor β-induced apoptosis. Molecular and cellular biochemistry. 2005;25(4):1475–1488. doi: 10.1128/mcb.25.4.1475-1488.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Yang M. S., Morris D. W., Donohoe G., et al. Chitinase-3-like 1 (CHI3L1) gene and schizophrenia: genetic association and a potential functional mechanism. Biological Psychiatry. 2008;64(2):98–103. doi: 10.1016/j.biopsych.2007.12.012. [DOI] [PubMed] [Google Scholar]
- 164.Boulay J.-L., Mild G., Lowy A., et al. SMAD7 is a prognostic marker in patients with colorectal cancer. International journal of cancer. 2003;104(4):446–449. doi: 10.1002/ijc.10908. [DOI] [PubMed] [Google Scholar]
- 165.Stolfi C., De Simone V., Colantoni A., et al. A functional role for Smad7 in sustaining colon cancer cell growth and survival. Cell death & disease. 2014;5(2, article e1073) doi: 10.1038/cddis.2014.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Thompson C. L., Plummer S. J., Acheson L. S., Tucker T. C., Casey G., Li L. Association of common genetic variants in SMAD7 and risk of colon cancer. Carcinogenesis. 2009;30(6):982–986. doi: 10.1093/carcin/bgp086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Slattery M. L., Herrick J., Curtin K., et al. Increased risk of colon cancer associated with a genetic polymorphism of SMAD7. Cancer Research. 2010;70(4):1479–1485. doi: 10.1158/0008-5472.CAN-08-1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.von Holst S., Picelli S., Edler D., et al. Association studies on 11 published colorectal cancer risk loci. British journal of cancer. 2010;103(4):575–580. doi: 10.1038/sj.bjc.6605774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Loh Y. H., Mitrou P. N., Wood A., et al. SMAD7 and MGMT genotype variants and cancer incidence in the European Prospective Investigation into Cancer and Nutrition (EPIC)-Norfolk Study. Cancer epidemiology. 2011;35(4):369–374. doi: 10.1016/j.canep.2010.09.011. [DOI] [PubMed] [Google Scholar]
- 170.Li X., Yang X.-X., Hu N.-Y., Sun J.-Z., Li F.-X., Li M. A risk-associated single nucleotide polymorphism of SMAD7 is common to colorectal, gastric, and lung cancers in a Han Chinese population. Molecular biology reports. 2011;38(8):5093–5097. doi: 10.1007/s11033-010-0656-3. [DOI] [PubMed] [Google Scholar]
- 171.Passarelli M. N., Coghill A. E., Hutter C. M., et al. Common colorectal cancer risk variants in SMAD7 are associated with survival among prediagnostic nonsteroidal anti-inflammatory drug users: a population-based study of postmenopausal women. Genes, Chromosomes & Cancer. 2011;50(11):875–886. doi: 10.1002/gcc.20913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Mates I. N., Jinga V., Csiki I. E., et al. Single nucleotide polymorphisms in colorectal cancer: associations with tumor site and TNM stage. Journal of Gastrointestinal and Liver Diseases. 2012;21(1):45–52. [PubMed] [Google Scholar]
- 173.Garcia-Albeniz X., Nan H., Valeri L., et al. Phenotypic and tumor molecular characterization of colorectal cancer in relation to a susceptibility SMAD7 variant associated with survival. Carcinogenesis. 2013;34(2):292–298. doi: 10.1093/carcin/bgs335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Noci S., Dugo M., Bertola F., et al. A subset of genetic susceptibility variants for colorectal cancer also has prognostic value. The Pharmacogenomics Journal. 2015;16(2):173–179. doi: 10.1038/tpj.2015.35. [DOI] [PubMed] [Google Scholar]
- 175.Jung K. J., Won D., Jeon C., et al. A colorectal cancer prediction model using traditional and genetic risk scores in Koreans. BMC genetics. 2015;16(1):1–7. doi: 10.1186/s12863-015-0207-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Abulí A., Castells A., Bujanda L., et al. Genetic variants associated with colorectal adenoma susceptibility. PLoS One. 2016;11(4, article e0153084) doi: 10.1371/journal.pone.0153084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Baert-Desurmont S., Charbonnier F., Houivet E., et al. Clinical relevance of 8q23, 15q13 and 18q21 SNP genotyping to evaluate colorectal cancer risk. European Journal of Human Genetics. 2015;24(1):99–105. doi: 10.1038/ejhg.2015.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Johansen J. S., Williamson M. K., Rice J. S., Price P. A. Identification of proteins secreted by human osteoblastic cells in culture. Journal of Bone and Mineral Research. 1992;7(5):501–512. doi: 10.1002/jbmr.5650070506. [DOI] [PubMed] [Google Scholar]
- 179.Hakala B. E., White C., Recklies A. D. Human cartilage gp-39, a major secretory product of articular chondrocytes and synovial cells, is a mammalian member of a chitinase protein family. Journal of Biological Chemistry. 1993;268(34):25803–25810. [PubMed] [Google Scholar]
- 180.Shackelton L. M., Mann D. M., Millis A. J. T. Identification of a 38-kDa heparin-binding glycoprotein (gp38k) in differentiating vascular smooth muscle cells as a member of a group of proteins associated with tissue remodeling. Journal of Biological Chemistry. 1995;270(22):13076–13083. doi: 10.1074/jbc.270.22.13076. [DOI] [PubMed] [Google Scholar]
- 181.Harvey S., Weisman M., O’Dell J., et al. Chondrex: new marker of joint disease. Clinical chemistry. 1998;44(3):509–516. [PubMed] [Google Scholar]
- 182.Mohanty A. K., Singh G., Paramasivam M., et al. Crystal structure of a novel regulatory 40-kDa mammary gland protein (MGP-40) secreted during involution. Journal of Biological Chemistry. 2003;278(16):14451–14460. doi: 10.1074/jbc.M208967200. [DOI] [PubMed] [Google Scholar]
- 183.Rehli M., Krause S. W., Andreesen R. Molecular characterization of the gene for human cartilage gp-39 (CHI3L1), a member of the chitinase protein family and marker for late stages of macrophage differentiation. Genomics. 1997;43(2):221–225. doi: 10.1006/geno.1997.4778. [DOI] [PubMed] [Google Scholar]
- 184.Buhi W. C. Characterization and biological roles of oviduct-specific, oestrogen-dependent glycoprotein. Reproduction. 2002;123(3):355–362. doi: 10.1530/reprod/123.3.355. [DOI] [PubMed] [Google Scholar]
- 185.Henrissat B., Bairoch A. New families in the classification of glycosyl hydrolases based on amino acid sequence similarities. The Biochemical Journal. 1993;293(3):781–788. doi: 10.1042/bj2930781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Renkema G. H., Boot R. G., Au F. L., et al. Chitotriosidase, a chitinase, and the 39-kDa human cartilage glycoprotein, a chitin-binding lectin, are homologues of family 18 glycosyl hydrolases secreted by human macrophages. European Journal of Biochemistry. 1998;251(1-2):504–509. doi: 10.1046/j.1432-1327.1998.2510504.x. [DOI] [PubMed] [Google Scholar]
- 187.Kramer K. J., Koga D. Insect chitin: physical state, synthesis, degradation and metabolic regulation. Insect Biochemistry. 1986;16(6):851–877. doi: 10.1016/0020-1790(86)90059-4. [DOI] [Google Scholar]
- 188.Cintin C., Johansen J. S., Christensen I. J., Price P. A., Sørensen S., Nielsen H. J. Serum YKL-40 and colorectal cancer. British journal of cancer. 1999;79(9-10):1494–1499. doi: 10.1038/sj.bjc.6690238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Huang Y., Prasad M., Lemon W. J., et al. Gene expression in papillary thyroid carcinoma reveals highly consistent profiles. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(26):15044–15049. doi: 10.1073/pnas.251547398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Jensen B. V., Johansen J. S., Price P. A. High levels of serum HER-2/neu and YKL-40 independently reflect aggressiveness of metastatic breast cancer. 2003;9(12):4423–4434. [PubMed] [Google Scholar]
- 191.Dehn H., Høgdall E. V. S., Johansen J. S., et al. Plasma YKL-40, as a prognostic tumor marker in recurrent ovarian cancer. Acta Obstetricia et Gynecologica Scandinavica. 2003;82(3):287–293. doi: 10.1034/j.1600-0412.2003.00010.x. [DOI] [PubMed] [Google Scholar]
- 192.Johansen J. S., Drivsholm L., Price P. a., Christensen I. J. High serum YKL-40 level in patients with small cell lung cancer is related to early death. Lung cancer. 2004;46(3):333–340. doi: 10.1016/j.lungcan.2004.05.010. [DOI] [PubMed] [Google Scholar]
- 193.Fukushima N., Koopmann J., Sato N., et al. Gene expression alterations in the non-neoplastic parenchyma adjacent to infiltrating pancreatic ductal adenocarcinoma. Modern pathology. 2005;18(6):779–787. doi: 10.1038/modpathol.3800337. [DOI] [PubMed] [Google Scholar]
- 194.Nigro J. M., Misra A., Zhang L., et al. Integrated array-comparative genomic hybridization and expression array profiles identify clinically relevant molecular subtypes of glioblastoma. Cancer Research. 2005;65(5):1678–1686. doi: 10.1158/0008-5472.CAN-04-2921. [DOI] [PubMed] [Google Scholar]
- 195.Tanwar M. K., Gilbert M. R., Holland E. C. Gene expression microarray analysis reveals YKL-40 to be a potential serum marker for malignant character in human glioma. Cancer Research. 2002;62(15):4364–4368. [PubMed] [Google Scholar]
- 196.Lal A., Lash A. E., Altschul S. F., et al. A public database for gene expression in human cancers. Cancer Research. 1999;59(21):5403–5407. [PubMed] [Google Scholar]
- 197.Thongsom S., Chaocharoen W., Silsirivanit A., et al. YKL-40/chitinase-3-like protein 1 is associated with poor prognosis and promotes cell growth and migration of cholangiocarcinoma. Tumour biology. 2016;37(7):9451–9463. doi: 10.1007/s13277-016-4838-z. [DOI] [PubMed] [Google Scholar]
- 198.Prakash M., Bodas M., Prakash D., et al. Diverse pathological implications of YKL-40: answers may lie in ‘outside-in’ signaling. Cellular signalling. 2013;25(7):1567–1573. doi: 10.1016/j.cellsig.2013.03.016. [DOI] [PubMed] [Google Scholar]
- 199.Lee C. G., Hartl D., Lee G. R., et al. Role of breast regression protein 39 (BRP-39)/chitinase 3-like-1 in Th2 and IL-13–induced tissue responses and apoptosis. Journal of Experimental Medicine. 2009;206(5):1149–1166. doi: 10.1084/jem.20081271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Kronborg G., Østergaard C., Weis N., et al. Serum level of YKL-40 is elevated in patients with streptococcus pneumoniae bacteremia and is associated with the outcome of the disease. Scandinavian Journal of Infectious Diseases. 2002;34(5):323–326. doi: 10.1080/00365540110080233. [DOI] [PubMed] [Google Scholar]
- 201.Johansen J. S. Studies on serum YKL-40 as a biomarker in diseases with inflammation, tissue remodelling, fibroses and cancer. Danish Medical Bulletin. 2006;53(2):172–209. [PubMed] [Google Scholar]
- 202.Johansen J. S., Christoffersen P., Møller S., et al. Serum YKL-40 is increased in patients with hepatic fibrosis. Journal of hepatology. 2000;32(6):911–920. doi: 10.1016/s0168-8278(00)80095-1. [DOI] [PubMed] [Google Scholar]
- 203.Combe B., Dougados M., Goupille P., et al. Prognostic factors for radiographic damage in early rheumatoid arthritis: a multiparameter prospective study. Arthritis & Rheumatism. 2001;44(8):1736–1743. doi: 10.1002/1529-0131(200108)44:8<1736::aid-art308>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 204.Abe M., Takahashi M., Naitou K., Ohmura K., Nagano A. Investigation of generalized osteoarthritis by combining X-ray grading of the knee, spine and hand using biochemical markers for arthritis in patients with knee osteoarthritis. Clinical Rheumatology. 2003;22(6):425–431. doi: 10.1007/s10067-003-0802-6. [DOI] [PubMed] [Google Scholar]
- 205.Vind I., Johansen J. S., Price P. a., Munkholm P. Serum YKL-40, a potential new marker of disease activity in patients with inflammatory bowel disease. Scandinavian Journal of Gastroenterology. 2003;38(6):599–605. doi: 10.1080/00365520310000537. [DOI] [PubMed] [Google Scholar]
- 206.Johansen J. S., Milman N., Hansen M., Garbarsch C., Price P. A., Graudal N. Increased serum YKL-40 in patients with pulmonary sarcoidosis—a potential marker of disease activity? Respiratory Medicine. 2005;99(4):396–402. doi: 10.1016/j.rmed.2004.09.016. [DOI] [PubMed] [Google Scholar]
- 207.Ober C., Tan Z., Sun Y., et al. Effect of variation in CHI3L1 on serum YKL-40 level, risk of asthma, and lung function. The New England Journal of Medicine. 2008;358(16):1682–1691. doi: 10.1056/nejmoa0708801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Brasso K., Christensen I. J., Johansen J. S., et al. Prognostic value of PINP, bone alkaline phosphatase, CTX-I, and YKL-40 in patients with metastatic prostate carcinoma. The Prostate. 2006;66(5):503–513. doi: 10.1002/pros.20311. [DOI] [PubMed] [Google Scholar]
- 209.Johansen J. S., Cintin C., Jørgensen M., Kamby C., Price P. A. Serum YKL-40: a new potential marker of prognosis and location of métastases of patients with recurrent breast cancer. European Journal of Cancer. 1995;31(9):1437–1442. doi: 10.1016/0959-8049(95)00196-p. [DOI] [PubMed] [Google Scholar]
- 210.Cintin C., Johansen J. S., Christensen I. J., Price P. A., Sørensen S., Nielsen H. J. High serum YKL-40 level after surgery for colorectal carcinoma is related to short survival. Cancer. 2002;95(2):267–274. doi: 10.1002/cncr.10644. [DOI] [PubMed] [Google Scholar]
- 211.Johansen J. S., Christensen I. J., Riisbro R., et al. High serum YKL-40 levels in patients with primary breast cancer is related to short recurrence free survival. Breast Cancer Research and Treatment. 2003;80(1):15–21. doi: 10.1023/a:1024431000710. [DOI] [PubMed] [Google Scholar]
- 212.Høgdall E. V. S., Ringsholt M., Høgdall C. K., et al. YKL-40 tissue expression and plasma levels in patients with ovarian cancer. BMC Cancer. 2009;9(1):p. 8. doi: 10.1186/1471-2407-9-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Kzhyshkowska J., Yin S., Liu T., Riabov V., Mitrofanova I. Role of chitinase-like proteins in cancer. Biological Chemistry. 2016;397(3):231–247. doi: 10.1515/hsz-2015-0269. [DOI] [PubMed] [Google Scholar]
- 214.Shao R., Hamel K., Petersen L., et al. YKL-40, a secreted glycoprotein, promotes tumor angiogenesis. Oncogene. 2009;28(50):4456–4468. doi: 10.1038/onc.2009.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Fusetti F., Pijning T., Kalk K. H., Bos E., Dijkstra B. W. Crystal structure and carbohydrate-binding properties of the human cartilage glycoprotein-39. Journal of Biological Chemistry. 2003;278(39):37753–37760. doi: 10.1074/jbc.M303137200. [DOI] [PubMed] [Google Scholar]
- 216.Maeda T., Desouky J., Friedl A. Syndecan-1 expression by stromal fibroblasts promotes breast carcinoma growth in vivo and stimulates tumor angiogenesis. Oncogene. 2006;25(9):1408–1412. doi: 10.1038/sj.onc.1209168. [DOI] [PubMed] [Google Scholar]
- 217.McQuade K. J., Beauvais D. M., Burbach B. J., Rapraeger A. C. Syndecan-1 regulates αvβ5 integrin activity in B82L fibroblasts. Journal of Cell Science. 2006;119(12):2445–2456. doi: 10.1242/jcs.02970. [DOI] [PubMed] [Google Scholar]
- 218.Libreros S., Garcia-Areas R., Iragavarapu-Charyulu V. CHI3L1 plays a role in cancer through enhanced production of pro-inflammatory/pro-tumorigenic and angiogenic factors. Immunologic research. 2013;57(1–3):99–105. doi: 10.1007/s12026-013-8459-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Francescone R. A., Scully S., Faibish M., et al. Role of YKL-40 in the angiogenesis, radioresistance, and progression of glioblastoma. Journal of Biological Chemistry. 2011;286(17):15332–15343. doi: 10.1074/jbc.M110.212514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.He C. H., Lee C. G., Dela Cruz C. S., et al. Chitinase 3-like 1 regulates cellular and tissue responses via IL-13 receptor α2. Cell Reports. 2013;4(4):830–841. doi: 10.1016/j.celrep.2013.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Low D., Subramaniam R., Lin L., et al. Chitinase 3-like 1 induces survival and proliferation of intestinal epithelial cells during chronic inflammation and colitis-associated cancer by regulating S100A9. Oncotarget. 2015;6(34):36535–36550. doi: 10.18632/oncotarget.5440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Malik P., Chaudhry N., Mittal R., Mukherjee T. K. Role of receptor for advanced glycation end products in the complication and progression of various types of cancers. Biochimica et Biophysica Acta (BBA) - General Subjects. 2015;1850(9):1898–1904. doi: 10.1016/j.bbagen.2015.05.020. [DOI] [PubMed] [Google Scholar]
- 223.Kjaergaard A. D., Johansen J. S., Nordestgaard B. G., Bojesen S. E. Genetic variants in CHI3L1 influencing YKL-40 levels: resequencing 900 individuals and genotyping 9000 individuals from the general population. Journal of Medical Genetics. 2013;50(12):831–837. doi: 10.1136/jmedgenet-2013-101908. [DOI] [PubMed] [Google Scholar]
- 224.Zhao X., Tang R., Gao B., et al. Functional variants in the promoter region of chitinase 3–like 1 (CHI3L1) and susceptibility to schizophrenia. AJHG. 2007;80(1):12–18. doi: 10.1086/510438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Berres M.-L., Papen S., Pauels K., et al. A functional variation in CHI3L1 is associated with severity of liver fibrosis and YKL-40 serum levels in chronic hepatitis C infection. Journal of Hepatology. 2009;50(2):370–376. doi: 10.1016/j.jhep.2008.09.016. [DOI] [PubMed] [Google Scholar]
- 226.Kruit A., Grutters J. C., Ruven H. J. T., van Moorsel C. C. M., van den Bosch J. M. M. A CHI3L1 gene polymorphism is associated with serum levels of YKL-40, a novel sarcoidosis marker. Respiratory Medicine. 2007;101(7):1563–1571. doi: 10.1016/j.rmed.2006.12.006. [DOI] [PubMed] [Google Scholar]
- 227.Boisselier B., Marie Y., El Hallani S., et al. No association of (−131C→G) variant of CHI3L1 gene with risk of glioblastoma and prognosis. Journal of Neuro-Oncology. 2009;94(2):169–172. doi: 10.1007/s11060-009-9817-4. [DOI] [PubMed] [Google Scholar]
- 228.Rathcke C. N., Holmkvist J., Husmoen L. L. N., et al. Association of polymorphisms of the CHI3L1 gene with asthma and atopy: a populations-based study of 6514 Danish adults. PLoS One. 2009;4(7, article e6106) doi: 10.1371/journal.pone.0006106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Nielsen K. R., Steffensen R., Boegsted M., et al. Promoter polymorphisms in the chitinase 3-like 1 gene influence the serum concentration of YKL-40 in Danish patients with rheumatoid arthritis and in healthy subjects. Arthritis Research & Therapy. 2011;13(3, article R109) doi: 10.1186/ar3391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Xie F., Qian Q., Chen Z., Ma G., Feng Y. Chitinase 3-like 1 gene-329G/A polymorphism, plasma concentration and risk of coronary heart disease in a Chinese population. Gene. 2012;499(1):135–138. doi: 10.1016/j.gene.2012.03.036. [DOI] [PubMed] [Google Scholar]
- 231.Yamamori H., Hashimoto R., Ohi K., et al. A promoter variant in the chitinase 3-like 1 gene is associated with serum YKL-40 level and personality trait. Neuroscience letters. 2012;513(2):204–208. doi: 10.1016/j.neulet.2012.02.039. [DOI] [PubMed] [Google Scholar]
- 232.Henningsen K. M. A., Olesen M. S., Sajadieh G., Haunsoe S., Svendsen J. H. A polymorphism associated with increased levels of YKL-40 and the risk of early onset of lone atrial fibrillation. Journal of Negative Results in Biomedicine. 2013;12(1):p. 1. doi: 10.1186/1477-5751-12-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Ortega H., Prazma C., Suruki R. Y., Li H., Anderson W. H. Association of CHI3L1 in African-Americans with prior history of asthma exacerbations and stress. Journal of asthma. 2013;50(1):7–13. doi: 10.3109/02770903.2012.733991. [DOI] [PubMed] [Google Scholar]
- 234.Lin Y.-S., Liu Y.-F., Chou Y.-E., et al. Correlation of chitinase 3-like 1 single nucleotide polymorphisms and haplotypes with uterine cervical cancer in Taiwanese women. PLoS One. 2014;9, article e104038(9) doi: 10.1371/journal.pone.0104038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Tsai Y., Ko Y., Huang M., et al. CHI3L1 polymorphisms associate with asthma in a Taiwanese population. BMC Medical Genetics. 2014;15(1):p. 86. doi: 10.1186/1471-2350-15-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Wu S., Hsu L.-A., Cheng S.-T., et al. Circulating YKL-40 level, but not CHI3L1 gene variants, is associated with atherosclerosis-related quantitative traits and the risk of peripheral artery disease. International Journal of Molecular Sciences. 2014;15(12):22421–22437. doi: 10.3390/ijms151222421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Naglot S., Dalal K. Association of CG genotype at rs4950928 promoter in CHI3L1 gene with YKL-40 levels and asthma susceptibility in North Indian asthma patients. Indian Journal of Clinical Biochemistry. 2015;30(4):403–411. doi: 10.1007/s12291-015-0478-0. [DOI] [Google Scholar]
- 238.Nielsen K. R., Steffensen R., Bendtsen M. D., et al. Inherited inflammatory response genes are associated with B-cell non-Hodgkin’s lymphoma risk and survival. PLoS One. 2015;10(10):e0139329–e0139318. doi: 10.1371/journal.pone.0139329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.James A. J., Reinius L. E., Verhoek M., et al. Increased YKL-40 and chitotriosidase in asthma and chronic obstructive pulmonary disease. American Journal of Respiratory and Critical Care Medicine. 2015;193(2):131–142. doi: 10.1164/rccm.201504-0760OC. [DOI] [PubMed] [Google Scholar]
- 240.Kjaergaard A. D., Johansen J. S., Bojesen S. E., Nordestgaard B. G. Observationally and genetically high YKL-40 and risk of venous thromboembolism in the general population: cohort and Mendelian randomization studies. Arteriosclerosis, Thrombosis, and Vascular Biology. 2016;36(5):1030–1036. doi: 10.1161/ATVBAHA.116.307251. [DOI] [PubMed] [Google Scholar]
- 241.Ting K., Ueng K., Yang S., Wang P. The association of YKL-40 genetic polymorphisms with coronary artery disease in Taiwan population. International Journal of Clinical and Experimental Medicineis. 2016;9(2):4211–4221. [Google Scholar]