

Abstract
Five new chromone derivatives, arthones A–E (1–5), together with eight known biogenetically related cometabolites (6–13), were isolated from a deep-sea-derived fungus Arthrinium sp. UJNMF0008. Their structures were assigned by detailed analyses of spectroscopic data, while the absolute configurations of 1 and 5 were established by electronic circular dichroism (ECD) calculations and that of 2 was determined by modified Mosher ester method. Compounds 3 and 8 exhibited potent antioxidant property with DPPH and ABTS radical scavenging activities, with IC50 values ranging from 16.9 to 18.7 μM. Meanwhile, no compounds indicated obvious bioactivity in our antimicrobial and anti-inflammatory assays at 50.0 μM.
Keywords: Arthrinium sp., chromone, polyketide, antioxidant activity
1. Introduction
The genus Arthrinium has wide geographic distribution and host range as plant pathogens, endophytes, saprobes, etc., while Poaceae and Cyperaceae are the major host plant families [1]. Although some Arthrinium species have been reported as phytopathogens [2,3,4], or even to cause cutaneous infections in humans [5], many others are known to produce diverse bioactive compounds with a variety of pharmacological applications. For instance, cytotoxic cytochalasins, pyridone alkaloids and polyketides, along with naphthalene glycosides with COX-2 inhibitory activity, were obtained from the sponge-derived fungus A. arundinis ZSDS1-F3 [6,7,8,9]; arundifungin with antifungal property was isolated from another A. arundinis species [10]; griseofulvin derivatives showing lethality against the brine shrimp Artemia salina were reported from a gorgonian-derived Arthrinium fungus [11]; antiangiogenic diterpenes were discovered from the marine species A. sacchari [12]; anti-parasitic dihydroisocoumarins were found in an endophytic Arthrinium sp. from Apiospora montagnei [13]; and volatile compounds in endophytic Arthrinium sp. MFLUCC16-0042 from Aquilaria subintegra were also investigated [14].
During the course of our search for new antibiotics from marine resources, an Arthrinium sp. UJNMF0008 from deep-sea sediment gained our interest owing to its strong inhibitory activity against Staphylococcus aureus. Subsequent chemical investigation on this species led to the discovery of a series of pyridone alkaloids with antibacterial and cytotoxic activities [15]. In addition to the pyridones, another class of metabolites with strong UV absorption was also revealed by chemical profiling (HPLC & 1H-NMR). Further study of the remaining fractions have resulted in the isolation and structural characterization of an array of polyketide compounds, including five new chromone arthones A–E (1–5) and eight previously reported analogues, AGI-B4 (6) [16], 1,3,6-trihydroxy-8-methylxanthone (7) [17], 2,3,4,6,8-pentahydroxy-1-methylxanthone (8) [18], sydowinin A (9) [19], sydowinin B (10) [20], conioxanthone A (11) [21], engyodontiumone B (12) [22], and 8-hydroxy-3-hydroxymethyl-9-oxo-9H-xanthene-1-carboxylic acid methyl ester (13) [23]. Herein, we describe the details of the isolation, structure elucidation, and biological evaluations of compounds 1–13.
2. Results and Discussion
2.1. Structure Elucidation
Arthone A (1) was isolated as a pale yellow powder and its molecular formula was established as C16H14O8 by HR-ESIMS analysis (m/z 333.0623 [M − H]−) and NMR data. Detailed analysis of the 1H- and 13C-NMR data (Table 1 and Table 2) showed that 1 possessed the same A and B rings (5-hydroxy-7-(hydroxymethyl)-4H-chromen-4-one, Figure 1) moiety as those in 9–12 [19,20,21,22], which was further confirmed by the HMBC correlations from 10-OH to C-10, H-10 to C-2, C-3 and C-4, H-4 to C-4a, C-9a and C-10, and 1-OH to C-1, C-2 and C-9a (Figure 2). Two groups of olefinic signals (δH/δC 5.72 (J = 5.9 Hz)/95.1; 7.33 (J = 5.9 Hz)/157.1) including one oxygenated (δC 157.1, C–6) and one oxyquaternary sp3 carbon (δC 84.3, C-8) revealed similar features (C ring, Figure 1) to the known compound euparvione [24] with the absence of one methyl (δH/δC 2.06/20.4 in euparvione), along with the appearance of a methyl ester moiety (δH/δC 3.70/52.8, 167.9) and one hydroxymethyl group (δH/δC 3.88, 4.12/63.6). Further HMBC correlations from H-12 to C-8, C-8a and C-11, and OCH3 to C-11 defined the structure of C ring as shown. The planar structure of 1 was thus established with only one chiral center. The absolute configuration of 1 was established as 8R by comparison of its experimental and theoretical ECD spectra (Figure 3) [25].
Table 1.
1H-NMR (600 MHz) data for 1–5 (DMSO-d6).
| Position | 1 | 2 | 3 | 4 | Positon | 5 |
|---|---|---|---|---|---|---|
| 2 | 6.74, brs | 6.70, s | 6.70, brs | 7.27, d (0.9) | 2 | 7.23, brs |
| 4 | 6.96, brs | 6.91, s | 6.94 a, brs | 7.56, brs | 4 | 7.54, brs |
| 5 | 5.72, d (5.9) | 2.79, dt (18.4, 6.4) 2.71, dt (18.4, 6.4) |
6.94 a, s | 6.98, d (8.9) | 10 | 2.84, t (7.9) |
| 6 | 7.33, d (5.9) | 1.87, m 1.80, m |
7.34, d (8.9) | 11 | 2.08, m 1.93, m |
|
| 7 | 3.99, m | 12 | 4.14, m | |||
| 8 | 2.61, dd (16.4, 4.2) 2.32, dd (16.4, 5.8) |
14 | 2.44, brs | |||
| 10 | 4.56, brd (5.6) | 4.54, d (5.8) | 4.56, s | 2.49, s | 13-OCH3 | 3.62, s |
| 12 | 4.12, dd (12.5, 5.8) 3.88, dd (12.5, 7.2) |
15-OCH3 | 3.83, s | |||
| OCH3 | 3.70, s | 3.83, s | 3.89, s | 7-OH | 9.00, brs | |
| 1-OH | 12.37, s | 12.67, s | 12.55, s | 12-OH | 5.63, d (4.3) | |
| 7-OH | 4.92, d (3.7) | 9.45, s | ||||
| 8-OH | 12.05, s | |||||
| 10-OH | 5.50, t (5.6) | 5.47, t (5.8) | ||||
| 12-OH | 5.29, dd (7.2, 5.8) |
a Interchangeable assignments.
Table 2.
13C-NMR (150 MHz) data for 1–5 (DMSO-d6).
| Position | 1 | 2 | 3 | 4 | Position | 5 |
|---|---|---|---|---|---|---|
| 1 | 159.4, C | 159.5, C | 160.5, C | 132.8, C | 1 | 132.7, C |
| 2 | 108.5, CH | 107.6, CH | 107.1, CH | 124.0, CH | 2 | 124.6, CH |
| 3 | 152.0, C | 151.8, C | 152.9, C | 147.7 b, C | 3 | 144.4, C |
| 4 | 104.1, CH | 103.8, CH | 103.8, CH | 119.1, CH | 4 | 119.6, CH |
| 4a | 154.7, C | 155.6, C | 155.3, C | 156.0, C | 5 | 155.0, C |
| 5 | 95.1, CH | 25.0, CH2 | 102.5, CH | 106.3, CH | 6 | 152.7, C |
| 5a | 160.6,C | 165.3, C | 155.2, C | 147.4 b, C | 7 | 138.8, C |
| 6 | 157.1, CH | 28.80 a, CH2 | 151.2, C | 124.6, CH | 8 | 171.1, C |
| 7 | 62.9, CH | 141.4, C | 147.9 b, C | 9 | 117.0, C | |
| 8 | 84.3, C | 28.78 a, CH2 | 117.6, C | 140.5, C | 10 | 24.8, CH2 |
| 8a | 103.8, C | 114.1, C | 108.6, C | 108.7, C | 11 | 31.2, CH2 |
| 9 | 178.2, C | 181.9, C | 179.1, C | 180.9, C | 12 | 69.7, CH |
| 9a | 108.8, C | 107.9, C | 106.3, C | 113.7, C | 13 | 174.6, C |
| 10 | 62.2, CH2 | 62.3, CH2 | 62.4, CH2 | 21.3, CH3 | 14 | 21.4, CH3 |
| 11 | 167.9, C | 166.8, C | 168.8, C | 15 | 169.5, C | |
| 12 | 63.6, CH2 | 13-OCH3 | 52.0, CH3 | |||
| OCH3 | 52.8, CH3 | 52.2, CH3 | 52.7, CH3 | 15-OCH3 | 52.8, CH3 |
a,b Interchangeable assignments.
Figure 1.

Chemical structures of compounds 1–13.
Figure 2.

Key 2D-NMR correlations for 1 and 2.
Figure 3.

Experimental and theoretical ECD spectra for 1.
Arthone B (2) was obtained as a pale yellow powder. The molecular formula was deduced as C14H14O5 based on the HR-ESIMS ion at m/z 263.0916 [M + H]+ (calcd. for C14H15O5, 263.0914), indicating eight degrees of unsaturation. 1D-NMR data analysis (Table 1 and Table 2) revealed that 2 also had a 5-hydroxy-7-(hydroxymethyl)-4H-chromen-4-one fragment but with larger chemical shifts for C-5a (δC 165.3) and C-8a (δC 114.1) compared with those of 1 (δC 160.6 and 103.8 for C-5a and C-8a, respectively). Correlated spectroscopy (COSY) correlations from H-7 (δH 3.99) to H2-6 (δH 1.80/1.87) and H2-8 (δH 2.32/2.61), and H-5 (δH 2.71/2.79) to H-6 (δH 1.80/1.87), revealed the spin-coupling system from H2-5–H2-8 (Figure 2). Key HMBC correlations from H-8 to C-5a, C-8a, and C-9 indicated the connection of C-8 to C-8a, while those from H-5 to C-5a and C-8a identified the connection of C-5 to C-5a (Figure 2). The gross structure of 2 was thus characterized, also bearing only one stereocenter. The absolute configuration of 2 was established by modified mosher ester method [26], where analysis of the 1H-NMR differences between its (R)- and (S)-MTPA esters (Δδ = δS − δR) led to the assignment of 7R-configuration for 2 (Figure 4).
Figure 4.
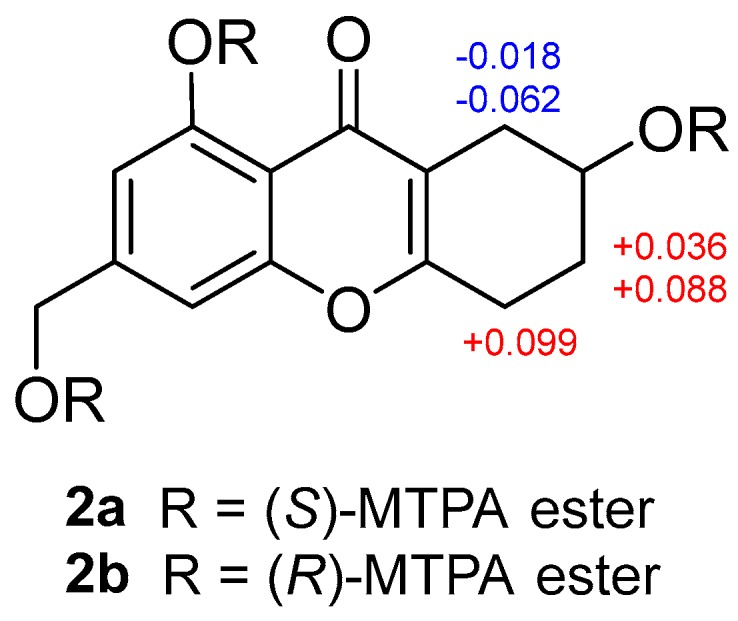
Δδ (δS − δR) values in ppm for MTPA eaters of 2.
Arthone C (3) was yielded as a yellow powder and displayed an HR-ESIMS peak at m/z 333.0607 [M + H]+ (calcd. for C16H13O8, 333.0605) corresponding to the molecular formula C16H12O8, with 16 amu more than that of its cometabolite sydowinin B (10) [20]. A detailed comparison of their 1H- and 13C-NMR data (Table 1 and Table 2) revealed that 3 incorporated the same skeleton as that of sydowinin B (10) [20] with the downfield-shifted chemical shift of C-6 (δC 151.2) and upfield-shifted chemical shift of C-5 (δC 102.5) and C-7 (δC 141.4), implying the presence of a hydroxyl at C-6, and further HMBC correlations from H-5 to C-6, C-7, and C-8a confirmed this moiety. As mentioned above, the structure of compound 3 was thus elucidated, as shown in Figure 1.
Arthone D (4) was isolated as a yellow powder with the molecular formula of C16H12O7 (11 degrees of unsaturation) determined by (+)-HR-ESIMS analysis at m/z 317.0664 ([M + H]+, calcd. for C16H13O7, 317.0656). The 1H- and 13C-NMR spectroscopic data for 4 (Table 1 and Table 2) exhibited a similar skeleton to that of isofusidienol A [27]. However, the methoxycarbonly moiety (δH/δC 3.89/52.7, 168.8) and methyl group (δH/δC 2.49/21.3) were deduced to locate on the same benzene ring as yicathin A [28] based on the HMBC correlations from OCH3 to C-11, H-2 to C-4, C-10, and C-11, H-4 to C-2, C-10, C-4a and C-9a, and H-10 to C-2, C-3 and C-4, and the 1D-NMR data (Figure 5). A proton signal at δH 12.05 in the 1H-NMR accounted for a hydroxyl group at C-8 due to H-bonding with C-9 carbonyl. Hence, two olefinic protons (δH 6.98, d, J = 8.9 Hz and 7.34, d, J = 8.9 Hz) were speculated to be located on C-5 and C-6, respectively. Finally, the hydroxyl resonanced at δH 9.45 could only be assigned to C-7. The seven-membered ring moiety was also supported by the HMBC correlations from H-5 to C-5a and C-8a and H-6 to C-7 and C-5a, as well as long range J4 correlations from H-5 to C-8 and H-6 to C-8a.
Figure 5.

Key 2D-NMR correlations for 4 and 5.
Arthone E (5) was isolated as a pale yellow powder and had a molecular formula of C17H18O8 as inferred from its (+)-HR-ESIMS data at m/z 351.1073 [M + H]+. Its NMR data (Table 1 and Table 2) revealed the same 7-methyl-4-oxo-4H-chromene-5-carboxylate moiety as arthone D (4). COSY correlations from H-11 (δH 1.93/2.08) to H-10 (δH 2.84) and H-12 (δH 4.14), and 12-OH (δH 5.63) to H-12 (δH 4.14), and HMBC correlations from OCH3 to C-13, and H-11/H-12 to C-13, revealed the moiety of C-10–C-13–OCH3 (Figure 5). Finally, the side-chain fragment was proved to be located at C-6, as supported by the HMBC correlations from H2-10 to C-6 and C-7, and a hydroxyl group was suggested at C-7 as indicated by the HMBC correlation from 7-OH to C-6 (Figure 5). The absolute configuration of 5 was established as 12R by comparison of its experimental and theoretical ECD spectra (Supplementary material Figure S31).
All the raw spectroscopic data including 1D/2D NMR and HR-ESIMS spectra for the new compounds 1 (Figures S1–S5), 2 (Figures S6–S11), 3 (Figures S12–S16), 4 (Figures S17–S21) and 5 (Figures S22–S27), and the 1D/2D NMR spectra for MTPA esters of 2 (Figures S28–S30) have been provided in the supplementary material.
2.2. Biological Activity
In order to evaluate the biological properties of 1–13, their antioxidant activity was assayed by DPPH and ABTS radical scavenging methods with curcumin as positive control (IC50 = 24.3 and 9.5 μM, respectively). Only compounds 3 and 8 exhibited significant antioxidant activities, with IC50 values of 16.9 and 22.1 μM for DPPH assay, and 18.7 and 18.0 μM for ABTS assay, respectively, while others showed no significant effect at 50.0 μM. Meanwhile, the antimicrobial activity against Gram-positive bacterial strains Mycobacterium smegmatis ATCC 607 and Staphylococcus aureus ATCC 25923, Gram-negative Escherichia coli ATCC 8739, Pseudomonas aeruginosa ATCC 9027, and fungus Candida albicans ATCC10231, as well as anti-inflammatory activity based on the inhibition effect of NO production in lipopolysaccharide (LPS)-induced mouse macrophages RAW 264.7 cells, were evaluated for 1–13. However, no compounds displayed obvious bioactivity in the two assays up to 50.0 μM.
3. Experimental Section
3.1. General Experimental Procedures
NMR spectra were recorded on a Bruker Avance DRX600 NMR spectrometer (Bruker BioSpin AG, Fällanden, Switzerland), with residual solvent peaks as references (DMSO-d6: δH 2.50, δC 39.52). ESIMS analyses were carried out on an Agilent 1260-6460 Triple Quad LC-MS instrument (Agilent Technologies Inc., Waldbronn, Germany). HR-ESIMS data were acquired on an Agilent 6545 Q-TOF mass spectrometer (Agilent Technologies Inc., Waldbronn, Germany). UV spectra were obtained on a Shimadzu UV-2600 spectrophotometer (Shimadzu, Kyoto, Japan) with 1 cm pathway cell. Optical rotations were measured on a Rudolph VI polarimeter (Rudolph Research Analytical, Hackettstown, NJ, USA) with a 10 cm length cell. ECD spectra were acquired on a Chirascan circular-dichroism spectrometer (Applied Photophysics Ltd., Surrey, UK). IR spectra were recorded on an FT-IR VERTEX 70 (Bruker BioSpin AG, Fremont, CA, USA). All HPLC analyses and separations were carried out on Agilent 1260 series LC instruments (Agilent Technologies Inc., Waldbronn, Germany) and a YMC-Pack ODS-A column (250 × 10 mm, 5 μm) was used for HPLC separations. Column chromatography (CC) was performed on Silica gel (200–300 mesh, Yantai Jiangyou Silica Gel Development Co., Yantai, China) and Sephadex LH-20 gel (GE Healthcare Bio-Sciences AB, Uppsala, Sweden). All solvents used for CC were of analytical grade (Tianjin Fuyu Fine Chemical Co. Ltd., Tianjin, China) and solvents used for HPLC were of HPLC grade (Oceanpak Alexative Chemical Ltd., Goteborg, Sweden).
3.2. Fungal Material
The fungus strain UJNMF0008 was isolated from a marine-sediment sample collected in the South China Sea (17°55′00″ N, 115°55′31″ E; 3858 m depth, Hainan, China). This strain was identified as an Arthrinium sp. based on morphological traits and a molecular biological protocol by DNA amplification and comparison of its ITS region sequence with the GenBank database (100% similarity with Arthrinium sp. zzz1842 (HQ696050.1)). The BLAST sequenced data were deposited at GenBank (No. MG010382). The strain was deposited at the CGMCC center, Institute of Microbiology, Chinese Academy of Sciences (Beijing, China).
3.3. Fermentation and Extraction
Arthrinium sp. UJNMF0008 from a PDA culture plate was inoculated in 500 mL Erlenmeyer flasks containing 150 mL soluble-starch medium (1% glucose, 0.1% soluble starch, 1% MgSO4, 0.1% KH2PO4, 0.1% peptone, and 3% sea salt) at 28 °C on a rotary shaker at 180 rpm for 3 days as seed cultures. Then, each of the seed cultures (20 mL) was transferred into autoclaved 1 L Erlenmeyer flasks with solid-rice medium (each flasks contained 80 g commercially available rice, 0.4 g yeast extract, 0.4 g glucose, and 120 mL water with 3% sea salt). After that, the strain was incubated statically for 30 days at 28 °C.
After fermentation, the total 4.8 kg rice culture was crushed and extracted with 15.0 L 95% EtOH three times. The EtOH extract was evaporated under reduced pressure to afford an aqueous solution and then extracted with 2.0 L ethyl acetate three times to give 80 g crude gum.
3.4. Isolation and Purification
The ethyl acetate extract (80 g) was fractionated by a silica gel column eluting with step gradient CH2Cl2-MeOH (v/v 100:0, 98:2, 95:5, 90:10, 80:20, 70:30, 50:50 and 0:100) to give 10 fractions (Fr.1–Fr.10) based on TLC and HPLC analysis. Fr.6 (30.2 g) was applied to CC over D101-macroporous absorption resin eluted with EtOH-H2O (30%, 50%, 80% and 100%) to afford four subfractions (Fr.6-1–Fr.6-4). Fr.6-3 (11.6 g) was fractionated by the silica gel column with step gradient CH2Cl2-(CH3)2CO (v/v 100:0–0:100) and divided into nine subfractions (Fr.6-3-1–Fr.6-3-9) and a portion of (32.5 mg) Fr.6-3-3 was further purified by HPLC eluting with MeOH-H2O (v/v 55:45, 3.0 mL min−1) to give 13 (tR = 29.3 min, 20.6 mg). Fr.6-3-2 (0.67 g), Fr.6-3-4 (510 mg), Fr.6-3-5 (1.04 g), Fr.6-3-6 (3.9 g), and Fr.6-3-7 (2.2 g) were separated by MPLC with an ODS column eluting with gradient MeOH-H2O (v/v 20:80–100:0) to obtain five, five, six, six and five subfractions, respectively. Fr.6-3-2-4 (37.0 mg) was further purified by HPLC eluting with MeOH-H2O (v/v 55:45, 3.0 mL min−1) to give 9 (tR = 22.2 min, 9.8 mg). Fr.6-3-4-3 (37.0 mg) was further purified by HPLC eluting with MeOH-H2O-CH3CO2H (v/v/v 70:30:10−4, 3.0 mL min−1) to afford 4 (tR = 14.4 min, 1.8 mg), while Fr.6-3-4-5 (50.2 mg) was further purified by HPLC eluting with MeOH-H2O (v/v 84:16, 3.0 mL min−1) to give 7 (tR = 21.9 min, 21.0 mg). Fr.6-3-5-2 (25.0 mg) was further purified by HPLC eluting with MeOH-H2O (v/v 48:52, 3.0 mL min−1) to give 1 (tR = 13.9, 4.5 mg), while Fr.6-3-5-5 (35.2 mg) was further purified by HPLC eluting with MeOH-H2O (v/v 60:40, 3.0 mL min−1) to give 10 (tR = 7.9 min, 4.0 mg) and 11 (tR = 16.4 min, 11.7 mg), and Fr.6-3-5-6 (25.1 mg) was further purified by HPLC eluting with MeOH-H2O (v/v 65:35, 3.0 mL min−1) to give 12 (tR = 20.7 min, 2.4 mg). Fr.6-3-6-2 (55.2 mg) was further purified by HPLC eluting with MeOH-H2O (v/v 34:66, 3.0 mL min−1) to give 6 (tR = 36.9 min, 14.3 mg). Fr.6-3-7-2 (80.1 mg) was further purified by HPLC eluting with MeOH-H2O (v/v 42:58, 3.0 mL min−1) to give 2 (tR = 11.1 min, 2.7 mg). Fr.7 (15.2 g) was chromatographed on a silica gel column with step gradient CH2Cl2-(CH3)2CO (v/v 100:0–0:100) and divided into three subfractions (Fr.7-1–Fr.7-3). Fr.7-1 (3.5 g) was divided into three subfractions (Fr.7-1-1–Fr.7-1-3) by Sephadex LH-20 CC eluting with MeOH-CH2Cl2 (v/v 1:1), and Fr.7-1-2 (1.2 g) was fractionated by MPLC with an ODS column eluting with step gradient MeOH-H2O (v/v 20:80 to 0:100) and further purified by HPLC eluting with MeOH-H2O (v/v 34:66, 3.0 mL min−1) to provide 5 (tR = 46.9 min, 2.6 mg). Fr.9 (7.6 g) was subject to silica gel column with step gradient CH2Cl2-(CH3)2CO (v/v 100:0–0:100) and divided into five subfractions (Fr.9-1–Fr.9-5). Fr.9-2 (1.6 g) was separated by Sephadex LH-20 CC eluting with MeOH-CH2Cl2 (v/v 1:1) to obtain four subfractions (Fr.9-2-1–Fr.9-2-4), and Fr.9-2-2 (42.6 mg) was further purified by HPLC eluting with MeOH-H2O-CH3CO2H (v/v/v 54:46:10−4, 3.0 mL min−1) to give 3 (tR = 17.1 min, 9.3 mg), while Fr.9-2-3 (17.0 mg) was further purified by HPLC eluting with MeOH-H2O (v/v 54:46, 3.0 mL min−1) to give 8 (tR = 26.4 min, 3.7 mg).
3.4.1. Arthone A (1)
Pale yellow powder; −6.3 (c 0.56, MeOH); ECD (0.20 mg mL−1, MeOH) λ (Δε) 322 (2.76), 291 (0.56), 258 (−15.78), 224 (2.50), 212 (−5.55), 204 (1.71) nm; UV (MeOH) λmax (log ε) 238 (4.35), 259 (4.23), 331 (3.64) nm; IR (KBr) νmax 3404, 2957, 1742, 1658, 1594, 1493, 1451, 1299, 1195, 1051, 1022, 821 cm−1; 1H- and 13C-NMR data, Table 1 and Table 2; (−)-ESIMS m/z 332.9 [M − H]−; (−)-HR-ESIMS m/z 333.0623 [M − H]− (calcd. for C16H13O8, 333.0616).
3.4.2. Arthone B (2)
Pale yellow powder; 1.9 (c 0.16, MeOH); ECD (0.20 mg mL−1, MeOH) λ (Δε) 328 (0.42), 286 (0.01), 276 (0.07), 258 (−0.29), 245 (−0.01), 206 (−2.21) nm; UV (MeOH) λmax (log ε) 239 (5.18), 328 (4.35) nm; IR (KBr) νmax 3431, 2935, 1659, 1625, 1597, 1499, 1459, 1292, 1117, 1041 cm−1; 1H- and 13C-NMR data, Table 1 and Table 2; (−)-ESIMS m/z [M − H]− 260.9; (+)-HR-ESIMS m/z 263.0916 [M + H]+ (calcd. for C14H15O5, 263.0914).
3.4.3. Arthone C (3)
Yellow powder; UV (MeOH) λmax (log ε) 233 (4.20), 246 (4.14), 254 (4.13), 296 (3.69), 377 (4.01) nm; IR (KBr) νmax 3307, 1702, 1651, 1607, 1586, 1499, 1436, 1381, 1366, 1286, 1250, 1011, 830 cm−1; 1H- and 13C-NMR data, Table 1 and Table 2; (−)-ESIMS m/z 330.9 [M − H]−; (+)-HR-ESIMS m/z 333.0607 [M + H]+ (calcd. for C16H13O8, 333.0605).
3.4.4. Arthone D (4)
Yellow powder; UV (MeOH) λmax (log ε) 234 (4.27), 267 (4.26), 391 (3.33) nm; IR (KBr) νmax 3427, 1725, 1613, 1501, 1459, 1435, 1297, 1235, 1041 cm−1; 1H- and 13C-NMR data, Table 1 and Table 2; (−)-ESIMS m/z 314.9 [M − H]−; (+)-HR-ESIMS m/z 317.0664 [M + H]+ (calcd. for C16H13O7, 317.0656).
3.4.5. Arthone E (5)
Pale yellow powder; −4.8 (c 0.73, MeOH); UV (MeOH) λmax (log ε) 209 (4.30), 314 (3.64) nm; IR (KBr) νmax 3413, 2957, 1736, 1611, 1438, 1309, 1227, 1176, 1040, 1037, 861 cm−1; 1H- and 13C-NMR data, Table 1 and Table 2; (+)-ESIMS m/z 351.0 [M + H]+; (+)-HR-ESIMS m/z 351.1073 [M + H]+ (calcd. for C17H19O8, 351.1074).
3.5. Antioxidant Assay
The antioxidant activities for compounds 1–13 were determined by DPPH and ABTS methods. DPPH radical scavenging method was conducted as described formerly [29], while ABTS radical scavenging assay was performed according to the method developed by Re et al. [30] with some modifications as below. Briefly, an ABTS radical solution was prepared by mixing equal volumes of aqueous solutions of 7 mM ABTS and 4.9 mM potassium persulfate for 16 h in the dark at room temperature. Then the ABTS radical solution was diluted with EtOH to an absorbance of 0.70 ± 0.02 at 734 nm. 10 μL of the tested compounds in ethanol (final concentrations as 3.13 μM, 6.25 μM, 12.5 μM, 25.0 μM, 50.0 μM, and 100 μM) was mixed with 190 μL of the prepared diluted ABTS radical solution at room temperature, and the absorbance at 734 nm was measured after 6 min in the dark. IC50 values were defined as the concentrations of tested compounds resulting in 50% loss of the ABTS radical. All determinations were carried out in triplicate, and curcumin was applied as positive control.
3.6. Antimicrobial Assays
The antimicrobial activity of compounds 1–13 was assayed against the Gram-positive bacterial strains Mycobacterium smegmatis ATCC 607 and Staphylococcus aureus ATCC 25923, Gram-negative Escherichia coli ATCC 8739, Pseudomonas aeruginosa ATCC 9027, and fungus Candida albicans ATCC10231 by the two-fold serial dilution method in 96-well microplates as described previously [15]. Penicillin was used as positive control in the current assay.
3.7. Anti-Inflammatory Assay
Determination of nitric oxide production. Briefly, RAW 264.7 cells were plated into 96-well plates and pretreated with a series of concentrations of compounds (3.13, 6.25, 12.5, 25.0, 50.0, and 100 μM) for 1 h before treatment with 1 μg mL−1 LPS. After 24 h incubation, detection of accumulated nitric oxide in the cell supernatants was assayed by Griess reagent kit (Beyotime Institute of Biotechnology, Jiangsu, China) according to the manufacturer’s instructions. Equal volumes of culture supernatant and Griess reagent were mixed, and the absorbance at 540 nm was measured using a Microplate Reader (Tecan, Grödig, Austria).
Cell viability assay. RAW 264.7 cells were seeded into 96-well plates at 1 × 104 cells well−1 and allowed to attach for 24 h. The medium was replaced with a 100 μL medium containing the indicated concentrations of compounds and further incubated for 24 h. 10 μL of MTT (5 mg mL−1 in PBS) was added into each well and the plates were incubated for 4 h at 37 °C. Supernatants were aspirated and formed formazan was dissolved in 100 μL of dimethyl sulfoxide (DMSO). The optical density (OD) was measured at an absorbance wavelength of 490 nm using a Microplate Reader (Tecan, Switzerland).
3.8. ECD Calculations
Conformational analysis within an energy window of 3.0 kcal mol−1 was performed using the OPLS3 molecular mechanics force field via the MacroModel [31] panel of Maestro 10.2. The conformers were then further optimized with the software package Gaussian 09 [32] at the CAM-B3LYP/6-311G(d,p) level for 1 and B3LYP/6-311G(d,p) level for 5, respectively, and the harmonic vibrational frequencies were also calculated to confirm their stability. Then, the 30 lowest electronic transitions for the obtained conformers in vacuum were calculated using time-dependent density functional theory (TD-DFT) method at the CAM-B3LYP/6-311G(d,p) level for 1 and B3LYP/6-311G(d,p) level for 5, respectively. ECD spectra of the conformers were simulated using a Gaussian function with a half-bandwidth of 0.25 eV for 1 and 0.35 eV for 5. The overall theoretical ECD spectra were obtained according to the Boltzmann weighting of each conformer.
Supplementary Materials
The following are available online. 1D/2D-NMR and HR-ESIMS spectra of compounds 1–5, the 1D/2D NMR spectra for MTPA esters of 2, along with experimental and theoretical ECD spectra for 5.
Author Contributions
J.B., F.H. and H.Z. (Huijuan Zhai) carried out the microbial fermentation and the isolation of the compounds. Z.-Q.C., C.-S.J., and Y.Z. (Yuying Zhang) performed the biological tests. H.Z. (Hua Zhang), J.B., G.C., and J.-H.Y. analyzed the spectroscopic data and elucidated the structure of the compounds. Y.Z. (Yun Zhang) and X.Z. assisted with the isolation and identification of the fungal material UJNMF0008. H.Z. (Hua Zhang) and J.B. wrote the paper.
Funding
We acknowledge the financial support from the National Natural Science Foundation of China (Nos. 41506148 & 31501104), Natural Science Foundation of Shandong Province (Nos. BS2015HZ005 and JQ201721), the Young Taishan Scholars Program (tsqn20161037), Open Fund of Key Laboratory of Tropical Medicinal Plant Chemistry of Ministry of Education (rdyw2018004), Program for Innovative Research Team in University (IRT-16R19), A Project of Shandong Province Higher Educational Science and Technology Program (J18KA255), and the Shandong Talents Team Cultivation Plan of University Preponderant Discipline (No. 10027).
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Sample Availability: Samples of the compounds 1–13 are available from the authors.
References
- 1.Wang M., Tan X.-M., Liu F., Cai L. Eight new Arthrinium species from China. Mycokeys. 2018;34:1–24. doi: 10.3897/mycokeys.34.24221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li S.-J., Zhu T.-H., Zhu H.-M.-Y., Liang M., Qiao T.-M., Han S., Che G.-N. Purification of protein AP-toxin from Arthrinium phaeospermum causing blight in Bambusa pervariabilis x Dendrocalamopisis grandis and its metabolic effects on four bamboo varieties. Phytopathology. 2013;103:135–145. doi: 10.1094/PHYTO-07-12-0164-R. [DOI] [PubMed] [Google Scholar]
- 3.Li B.J., Liu P.Q., Jiang Y., Weng Q.Y., Chen Q.H. First report of culm rot caused by Arthrinium phaeospennum on Phyllostachys viridis in China. Plant Dis. 2016;100:1013. doi: 10.1094/PDIS-08-15-0901-PDN. [DOI] [Google Scholar]
- 4.Chen K., Wu X.Q., Huang M.X., Han Y.Y. First report of brown culm streak of Phyllostachys praecox caused by Arthrinium arundinis in Nanjing, China. Plant Dis. 2014;98:1274. doi: 10.1094/PDIS-02-14-0165-PDN. [DOI] [PubMed] [Google Scholar]
- 5.Crous P.W., Groenewald J.Z. A phylogenetic re-evaluation of Arthrinium. Ima Fungus. 2013;4:133–154. doi: 10.5598/imafungus.2013.04.01.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang J.F., Wang Z., Ju Z.R., Wan J.T., Liao S.R., Lin X.P., Zhang T.Y., Zhou X.F., Chen H., Tu Z.C., et al. Cytotoxic cytochalasins from marine-derived fungus Arthrinium arundinis. Planta Med. 2015;81:160–166. doi: 10.1055/s-0034-1383403. [DOI] [PubMed] [Google Scholar]
- 7.Wang J.F., Wei X.Y., Qin X.C., Lin X.P., Zhou X.F., Liao S.R., Yang B., Liu J., Tu Z.C., Liu Y.H. Arthpyrones A-C, pyridone alkaloids from a sponge-derived fungus Arthrinium arundinis ZSDS1-F3. Org. Lett. 2015;17:656–659. doi: 10.1021/ol503646c. [DOI] [PubMed] [Google Scholar]
- 8.Li Y.L., Wang J.F., He W.J., Lin X.P., Zhou X.F., Liu Y.H. One strain-many compounds method for production of polyketide metabolites using the sponge-derived fungus Arthrinium arundinis ZSDS1-F3. Chem. Nat. Compd. 2017;53:373–374. doi: 10.1007/s10600-017-1994-3. [DOI] [Google Scholar]
- 9.Wang J.-F., Xu F.-Q., Wang Z., Lu X., Wan J.-T., Yang B., Zhou X.-F., Zhang T.-Y., Tu Z.-C., Liu Y. A new naphthalene glycoside from the sponge-derived fungus Arthrinium sp. ZSDS1-F3. Nat. Prod. Res. 2014;28:1070–1074. doi: 10.1080/14786419.2014.905935. [DOI] [PubMed] [Google Scholar]
- 10.Cabello M.A., Platas G., Collado J., Diez M.T., Martin I., Vicente F., Meinz M., Onishi J.C., Douglas C., Thompson J., et al. Arundifungin, a novel antifungal compound produced by fungi: Biological activity and taxonomy of the producing organisms. Int. Microbiol. 2001;4:93–102. doi: 10.1007/s101230100020. [DOI] [PubMed] [Google Scholar]
- 11.Wei M.-Y., Xu R.-F., Du S.-Y., Wang C.-Y., Xu T.-Y., Shao C.-L. A new griseofulvin derivative from the marine-derived Arthrinium sp. fungus and its biological activity. Chem. Nat. Compd. 2016;52:1011–1014. doi: 10.1007/s10600-016-1849-3. [DOI] [Google Scholar]
- 12.Tsukada M., Fukai M., Miki K., Shiraishi T., Suzuki T., Nishio K., Sugita T., Ishino M., Kinoshita K., Takahashi K., et al. Chemical constituents of a marine fungus, Arthrinium sacchari. J. Nat. Prod. 2011;74:1645–1649. doi: 10.1021/np200108h. [DOI] [PubMed] [Google Scholar]
- 13.Ramos H.P., Simao M.R., de Souza J.M., Magalhaes L.G., Rodrigues V., Ambrosio S.R., Said S. Evaluation of dihydroisocoumarins produced by the endophytic fungus Arthrinium state of Apiospora montagnei against Schistosoma mansoni. Nat. Prod. Res. 2013;27:2240–2243. doi: 10.1080/14786419.2013.811659. [DOI] [PubMed] [Google Scholar]
- 14.Monggoot S., Popluechai S., Gentekaki E., Pripdeevech P. Fungal endophytes: An alternative source for production of volatile compounds from agarwood oil of Aquilaria subintegra. Microb. Ecol. 2017;74:54–61. doi: 10.1007/s00248-016-0908-4. [DOI] [PubMed] [Google Scholar]
- 15.Bao J., Zhai H.J., Zhu K.K., Yu J.-H., Zhang Y.Y., Wang Y.Y., Jiang C.-S., Zhang X.Y., Zhang Y., Zhang H. Bioactive pyridone alkaloids from a deep-sea-derived fungus Arthrinium sp. UJNMF0008. Mar. Drugs. 2018;16 doi: 10.3390/md16050174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim H.S., Park I.Y., Park Y.J., Lee J.H., Hong Y.S., Lee J.J. A novel dihydroxanthenone, AGI-B4 with inhibition of VEGF-induced endothelial cell growth. J. Antibiot. 2002;55:669–672. doi: 10.7164/antibiotics.55.669. [DOI] [PubMed] [Google Scholar]
- 17.Mutanyatta J., Matapa B.G., Shushu D.D., Abegaz B.M. Homoisoflavonoids and xanthones from the tubers of wild and in vitro regenerated Ledebouria graminifolia and cytotoxic activities of some of the homoisoflavonoids. Phytochemistry. 2003;62:797–804. doi: 10.1016/S0031-9422(02)00622-2. [DOI] [PubMed] [Google Scholar]
- 18.Abdel-Lateff A., Klemke C., Konig G.M., Wright A.D. Two new xanthone derivatives from the algicolous marine fungus Wardomyces anomalus. J. Nat. Prod. 2003;66:706–708. doi: 10.1021/np020518b. [DOI] [PubMed] [Google Scholar]
- 19.Hamasaki T., Sato Y., Hatsuda Y. Structure of sydowinin A, sydowinin B, and sydowinol, metabolites from Aspergillus sydowi. Agric. Biol. Chem. 1975;39:2341–2345. doi: 10.1271/bbb1961.39.2341. [DOI] [Google Scholar]
- 20.Little A., Porco J.A., Jr. Total syntheses of graphisin A and sydowinin B. Org. Lett. 2012;14:2862–2865. doi: 10.1021/ol301107m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang Y.C., Zheng Z.H., Liu S.C., Zhang H., Li E.W., Guo L.D., Che Y.S. Oxepinochromenones, furochromenone, and their putative precursors from the endolichenic fungus Coniochaeta sp. J. Nat. Prod. 2010;73:920–924. doi: 10.1021/np100071z. [DOI] [PubMed] [Google Scholar]
- 22.Yao Q.F., Wang J., Zhang X.Y., Nong X.H., Xu X.Y., Qi S.H. Cytotoxic polyketides from the deep-sea-derived fungus Engyodontium album DFFSCS021. Mar. Drugs. 2014;12:5902–5915. doi: 10.3390/md12125902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang H., Umeokoli B.O., Eze P., Heering C., Janiak C., Mueller W.E.G., Orfali R.S., Hartmann R., Dai H.F., Lin W.H., et al. Secondary metabolites of the lichen-associated fungus Apiospora montagnei. Tetrahedron Lett. 2017;58:1702–1705. doi: 10.1016/j.tetlet.2017.03.052. [DOI] [Google Scholar]
- 24.Leon F., Gao J.T., Dale O.R., Wu Y.S., Habib E., Husni A.S., Hill R.A., Cutler S.J. Secondary metabolites from Eupenicillium parvum and their in vitro binding affinity for human opioid and cannabinoid receptors. Planta Med. 2013;79:1756–1761. doi: 10.1055/s-0033-1351099. [DOI] [PubMed] [Google Scholar]
- 25.Berova N., Di B.L., Pescitelli G. Application of electronic circular dichroism in configurational and conformational analysis of organic compounds. Chem. Soc. Rev. 2007;36:914–931. doi: 10.1039/b515476f. [DOI] [PubMed] [Google Scholar]
- 26.Sun Y.-L., Bao J., Liu K.-S., Zhang X.-Y., He F., Wang Y.-F., Nong X.-H., Qi S.-H. Cytotoxic dihydrothiophene-condensed chromones from the marine-derived fungus Penicillium oxalicum. Planta Med. 2013;79:1474–1479. doi: 10.1055/s-0033-1350805. [DOI] [PubMed] [Google Scholar]
- 27.Lösgen S., Magull J., Schulz B., Draeger S., Zeeck A. Isofusidienols: Novel chromone-3-oxepines produced by the endophytic fungus Chalara sp. Eur. J. Org. Chem. 2010;2008:698–703. doi: 10.1002/ejoc.200700839. [DOI] [Google Scholar]
- 28.Sun R.-R., Miao F.-P., Zhang J., Wang G., Yin X.-L., Ji N.-Y. Three new xanthone derivatives from an algicolous isolate of Aspergillus wentii. Magn. Reson. Chem. 2013;51:65–68. doi: 10.1002/mrc.3903. [DOI] [PubMed] [Google Scholar]
- 29.Cao Y.-K., Li H.-J., Song Z.-F., Li Y., Huai Q.-Y. Synthesis and biological evaluation of novel curcuminoid derivatives. Molecules. 2014;19:16349–16372. doi: 10.3390/molecules191016349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Re R., Pellegrini N., Proteggente A., Pannala A., Yang M., Riceevans C. Antioxidant activity applying an improved ABTS radical cation decolorization assay. Free Radic. Biol. Med. 2013;26:1231–1237. doi: 10.1016/S0891-5849(98)00315-3. [DOI] [PubMed] [Google Scholar]
- 31.MacroModel, 9.7.211. Schrödinger; New York, NY, USA: 2009. [Google Scholar]
- 32.Gaussian 09, Revision B.01. Gaussian, Inc.; Wallingford, CT, USA: 2010. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.