

Abstract
The synthesis of racemic substituted 7-amino-5,7,8,9-tetrahydrobenzocyclohepten-6-one hydrochlorides was optimized to enhance reproducibility and increase the overall yield. In order to investigate their specificity, series of enzyme inhibition assays were carried out against a diversity of proteases, covering representative members of aspartic, cysteine, metallo and serine endopeptidases and including eight members of the monometallic M1 family of aminopeptidases as well as two members of the bimetallic M17 and M28 aminopeptidase families. This aminobenzosuberone scaffold indeed demonstrated selective inhibition of M1 aminopeptidases to the exclusion of other tested protease families; it was particularly potent against mammalian APN and its bacterial/parasitic orthologues EcPepN and PfAM1.
Keywords: M1 aminopeptidases, selective inhibitors, aminobenzosuberone scaffold
1. Introduction
Aminopeptidases (APs) constitute a group of exopeptidases with closely related activities, able to remove amino-acids from unblocked N-termini of peptide and protein substrates. Among them, metallo-aminopeptidases (metallo-APs) are widely distributed in organisms with representative members in animal cells, plant cells, fungi, parasites and bacteria [1,2]. Metallo-APs have been broadly sorted into two important subgroups on the basis of the number of zinc ions in their active site that are involved in catalysis as well as on their protein signatures and folding [1,2]. While some of these enzymes contain only one zinc, others possess a binuclear metal centre, usually referred as a co-catalytic unit [3,4]. Members of the M1 family belong to the subclan MA(E) of metallopeptidases [1], also called Glu-zincins and possess a single essential catalytic zinc ion and a common fold, related to their common ancestry that is related to thermolysin. Present mainly as monomers or homo-dimers (in mammals, mainly), metallo-APs occupy various cell compartments and although some are secreted, the vast majority operates as membrane bound ectoenzymes or cytosolic catalysts [2,5,6,7]. Their evolutionary history indicates that they are related to a complex and ancestral gene family that subsequently yielded series of more divergent sequences [8]. For this reason, their biochemical characterization and respective biological importance has, for a long time, been hindered by considerable confusion, due in part to their often large and sometimes overlapping substrate specificities as well as their very close biochemical properties. For the time being, we have a more precise picture of their highly specialized roles depending on their localization with many related paralogous members in mammals (12 in H. sapiens, 11 in rodents), whereas only a handful are known to be encoded in other species (3 or less; only one in P. falciparum) [1,9].
Human M1 paralogous members [1] have been named Leucyl-cystinyl aminopeptidase (HsIRAP), endoplasmic reticulum aminopeptidase 1 (HsERAP1), endoplasmic reticulum aminopeptidase 2 (HsERAP2), aminopeptidase Q (HsAPQ), puromycin-sensitive aminopeptidase (HsPSA), thyrotropin-releasing hormone-degrading ectoenzyme (HsTRHDE), aminopeptidase A (HsAPA), aminopeptidase N (HsAPN), aminopeptidase O (HsAPO), leukotriene A4 hydrolase (HsLTA4H), arginyl aminopeptidase B (HsAPB) and arginyl aminopeptidase B like 1 (HsRNPL1) and their orthologues in E. coli (EcPepN) and P. falciparum (PfAM1). Their so far recognized biological implications are summarized in Table 1. Broadly, human M1 members play important biological roles in the renin-angiotensin system, the immune system as well as during the inflammatory process, while the bacterial and malarial members are involved in nutrient acquisition.
Table 1.
Current established biological functions for representative members of the M1 aminopeptidases [1,10,11].
| Organism | M1 Family | Biological Roles | Associated Diseases |
|---|---|---|---|
| H. sapiens | APN | Metabolism of regulatory peptides of diverse cell types Processing of peptide hormones (Angiotensin III and IV, neuropeptides and chemokines Regulation of angiogenesis, cell motility, cell-cell adhesion Coronavirus receptor |
Pain sensation, Inflammatory diseases, cancer & upper respiratory tract infections |
| APA | Activation or inactivation of various components of the angiotensin system | Hypertension | |
| LTA4H | Biosynthesis of the proinflammatory mediator LTB-4 | Inflammatory & allergic diseases | |
| TRHDE | Inactivation of Thyrotropin Releasing Hormone (brain) | ||
| PSA | Regulation of neuropeptide activity, digestion of polyQ peptides Antigen processing pathway for MHC class I molecules |
hematologic cancer Impedes the development of neuropathology |
|
| IRAP | Peptide hormone degradation (oxytocin, vasopressin, angiotensin III) Maintain homeostasis during pregnancy Inactivation of neuronal peptide (enkephalin, dynorphin) Generation of antigenic peptides in dendritic cells [12] |
cognitive impairment (Alzheimer’s disease, head trauma, cerebral ischemia) Inflammatory autoimmunity [13] |
|
| APB | Biosynthesis of the proinflammatory mediator LTB-4 | ||
| ERAP1 | Regulation of blood pressure Antigen processing pathway for MHC class I molecules |
Autoimmune diseases (ankylosing spondylitis, psoriasis, type 1 diabetes, Crohn…) Cancer [14,15,16] |
|
| ERAP2 | Antigen processing pathway for MHC class I molecules | ||
| APQ | Placentation: regulation of biological activity of key peptides at the embryo-maternal interface | Pre-eclampsia | |
| APO | Activation or inactivation of various components of the angiotensin system | ||
| P. falciparum | PfAM1 | Catabolism of host haemoglobin in the food vacuole of Plasmodium | Malaria [17] |
| E. coli | PepN | Cytosolic peptide catabolism and adaptation to nutritional downshift and high temperature stress |
As a consequence from the definition of the M1 family, all members (the MEROPS database has so far identified 14718 amino acid sequences belonging to this M1 family) share critical characteristics [18,19]. Sequences alignment of a selection of M1 aminopeptidases highlights the conserved active site residues for substrate recognition and peptide bond hydrolysis that is, the zinc binding motif HExxHx18E, the substrate binding motif, including the GxMEN domain and an extra E or Q distal residue, responsible for the recognition and binding of the key free amino terminus of peptide substrates, as well as the catalytic E residue (part of HExxH motif) and a distal Y residue essential for transition state stabilization in the closed conformations of M1 aminopeptidases (Figure S1) [1].
Although overall amino acid sequence similarity may fall down to a mean value of 20%, a highly conserved 3D structure, reminding that of a sea-horse with 4 domains, has been found for all these M1 APs. Domain I folds as a twisted β-barrel of 200 aa, domain II corresponds to the ancestral thermolysin-like fold, domain III is a beta sandwich built with 2 β-sheets (absent in HsLTA4H) and domain IV is a prominent bowl-like structure composed of α-helices (330 aa) which contains the active site channel entrance (about 40 Å long and 12 Å large) [20].
Dynamic conformational ensembles, involving inter-domains movements, are crucial for catalysis and various conformational states are particularly marked for mammalian enzymes. They include active site opening and closure, particularly important for HsERAP1, HsIRAP and mammalian APN [20,21,22,23,24], suggesting that the peptide-binding channel could accommodate variously exposed N-termini of peptides and/or proteins. The influences of protein dimerization on active site accessibility and specificity as well as on various moonlighting functions have still to be more extensively studied for all M1 family of APs [23,25,26].
Substrate specificities have also been responsible for the confusion in biological roles attributed to these enzymes; as illustrated in Table 2, these substrate specificities are rarely discriminant with overlapping specificities being the rule and specific specificities the rare exceptions.
Table 2.
Substrate specificity: preferred N-terminus amino-acid for a selection of M1 aminopeptidases [1].
| M1 Aminopeptidase | Substrate Specificity: Favoured N-Terminus Amino-Acid |
|---|---|
| APN | Ala, Phe, Tyr, Leu, most of aa including Pro (slow) |
| APA | Glu and to a lesser extent Asp |
| LTA4H | Ala, Arg, Leu, Pro |
| TRHDE | pGlu (pyroglutamyl) |
| PSA | Ala, Leu, Lys, most of aa (except Gly and Pro) |
| IRAP | Cys, Leu, Arg, Ala and most of aa (except Asp, Glu) including cyclic peptides [27] |
| APB | Arg, Lys |
| ERAP1 | Leu and most of aa including Met, Cys, Phe |
| ERAP2 | Arg, Lys |
| RNPL1 | Ala, Lys, Ser, Ile, Met, most of aa |
| APQ | Leu, Arg, Lys, Met, most of aa |
| APO | Arg and to a lesser extent Asn |
| PfAM1 | Ala, Leu, Lys, Arg and most of aa (except Pro, Asp, Glu) |
| PepN | Arg, Ala, most of aa including Pro |
This common substrate specificity, together with a very conserved structure and catalytic mechanism, makes the development of highly selective inhibitors of a given metallo-aminopeptidase obviously challenging [11,28,29,30,31]. Thereby the most notable inhibitors, depicted in Figure 1, consist in tetrahedral intermediate mimics, as for the widely used bestatin [32] and phosphinic [33]/phosphonic [34] acid derivatives, or zinc-chelating group inserted in a peptide-like scaffold, as for hydroxamic acids [35,36,37].
Figure 1.

Representative inhibitors of M1 aminopeptidases (non-exhaustive list) For more detailed structures see references [11,28,29,30,31].
In Table 3, we report inhibition values for several members of the M1 aminopeptidase family and for three selected bi-metallic aminopeptidases, since selectivity towards these other metallo-aminopeptidases remains of major concern and is indeed currently poorly documented.
Table 3.
Ki or * IC50 values in micromoles reported in literature for representative M1 AP’s inhibitors.
| Compound | M1 Aminopeptidases ‘Monometallic’ |
M17 & M28 Aminopeptidases ‘Bimetallic’ |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HsAPA | HsAPB | mAPN | HsERAP1 | HsERAP2 | HsIRAP | HsLTA4H | HsPSA | EcPepN | PfAM1 | AAP | mLAPc | PfAM17 | |
| 1 | NI [38] | 0.014 [39]–6.0 [38] | 0.3 * [37]–89.0 [40] | 11.2 [41] | 0.2 * [37] 0.5 [42] |
0.35 * [37] | 7.3 * [43] | 0.19 [44] | 0.0016 [42] | 0.0006 [38]–0.020 [39] | 0.025 [45] | ||
| 2 | - | - | 0.002 [46] | - | - | - | - | - | - | 0.079 [47] | - | 0.066 [33] | 0.013 [45] |
| 3 [48] | 0.043 * | 0.037 * | 0.002 * | ||||||||||
| 4 [49] | >100 | 1.79 * | >100 | >100 | |||||||||
| 5 [38] | >1000 | >1000 | 0.02 | 3.0 | |||||||||
| 6 [50] | 0.14 | 0.12 | |||||||||||
|
7 [37] 8 [37] |
- | >1 * | 0.22 * | >5 * | - | - | >10 * 0.008 * |
0.15 * 0.85 * |
- | 6 [36] | - | 0.1 * 0.03 * |
0.079 [36] - |
| 9 | >100 [40] | >100 [51] | 0.3 [52]–2.0 * [40] | 2.6 * [51] | |||||||||
| 10 [35,53] | 1.37 * | 0.006 * | |||||||||||
| 11 [54] | 0.92 * | 1.6 * | 0.105 * | ||||||||||
| 12 [55] | >100 | 0.8 * | |||||||||||
| 13 [56] | 0.03 | ||||||||||||
M1 monometallic enzymes: human aminopeptidase A (HsAPA), human arginyl aminopeptidase B (HsAPB), mammalian aminopeptidase N (mAPN), human endoplasmic reticulum aminopeptidase 1 (HsERAP1), human endoplasmic reticulum aminopeptidase 2 (HsERAP2), human Leucyl-cystinyl aminopeptidase (HsIRAP), human leukotriene A4 hydrolase (HsLTA4H) and human puromycine sensitive aminopeptidase (HsPSA); E. coli (EcPepN) and P. falciparum (PfAM1) M1 aminopeptidases; representative members of the bimetallic aminopetidases from A. proteolytica (AAP), mammalian cytosolic leucine aminopeptidase (mLAPc) and P. falciparum M17 aminopeptidase (PfAM17). NI, no inhibition. * IC50.
Inhibitors targeting mammalian APN have been the most extensively developed, since mammalian APN was one of the first metallo-aminopeptidase to be biochemically and enzymatically characterized (although many of its first assigned biological functions were later demonstrated to involve other related M1 paralogous enzymes). Among them, Bestatin 1 has been very often misused as a “specific” inhibitor of this peculiar APN enzyme, since it turns out to be a highly potent inhibitor of bimetallic enzyme families and a quite broad “unspecific” inhibitor of monometallic M1 aminopeptidases. But overall, it appears obvious that the development of M1 aminopeptidase inhibitors remains mainly in its infancy.
Our research group has previously reported the development of potent and selective inhibitors of APN, a human M1 aminopeptidase involved in angiogenesis and tumour metastasis [57]. This work allowed us to identify aminobenzosuberone moiety as a promising scaffold that demonstrates an exceptional selectivity towards mono-metallic aminopeptidases [42,58,59,60,61]. In the present work, we have closely considered the evaluation of these inhibitors on a number of potential interesting targets within the M1 family of aminopeptidases and established their inhibition profiles. We have reported herein an improvement of the aminobenzosuberone synthetic route and the biological evaluation of this family of non-peptidic compounds against 6 human and 2 bacterial/parasitic M1 aminopeptidases, plus members of the M17 and M28 families. We have also verified the observed selectivity on a panel of proteinases, which includes representative members of aspartic, cysteine, metallo and serine endopeptidases.
2. Results
2.1. Chemistry
The aminobenzosuberone’s synthetic route, based on our previously described procedure [42,58,60], was optimized to improve reproducibility and avoid the appearance of side-products (Scheme 1). Indeed, silyl enol ether 15, obtained in four steps from commercially available product 14, was submitted to a Rubottom oxidation to yield the key intermediate silyloxy-ketone 16. This reaction was classically performed by action of m-CPBA but regular and unpredictable failure with this reagent led us to consider alternative oxidizing agents. The best results were obtained with in situ generated dimethyl dioxirane (DMDO) [62,63], which gave only the expected product 16 in a nearly quantitative and reproducible yield (up to 95%). The second optimized step was the conversion of the hydroxy-oxime 17 to its corresponding amino-alcohol 18. Our previously described method for this reduction consisted of a hydrogenation over Raney nickel [42,58]. Although the procedure worked efficiently, it was very dependent on the quality and supply of Raney nickel. Moreover, in the case of brominated compounds it required a careful monitoring to avoid any aromatic debromination. Thus, several conditions were investigated to obtain compound 18 as Mg/ammonium formate [64], SmI2 [65], BH3·THF [66], LiAlH4 [67] or NaBH4/NiCl2·6H2O [68]. Promising results were obtained with this latter but a small amount of debrominated product was still observed. Therefore, addition of NiCl2·6H2O was successfully replaced by addition of CoCl2·6H2O and the expected amino compound was solely obtained with no effect on aromatic bromine, even without strict monitoring of the reaction progress and in acceptable yields (60–80%).
Scheme 1.

Synthetic scheme of racemic substituted 7-amino-5,7,8,9-tetrahydrobenzocyclohepten-6-one hydrochloride salts. Reagents and conditions: (i) NBS, AIBN, CCl4, reflux, 3 h. (ii) Dimethyl-1,3-acetonedicarboxylate, NBu4Br, NaHCO3 1M aq., CH2Cl2, 40 °C, overnight. (iii) H2SO4, CH3CN, reflux, overnight. (iv) TBDMSOTf, NEt3, CH2Cl2, 25 °C, 2 h. (v) Oxone®, NaHCO3, acetone, CH2Cl2, 25 °C, 3 h. (vi) NH2OH. HCl, pyridine, 25 °C, 5 h. (vii) TBAF, THF, 25 °C, 2 h. Potential regioisomers may be separated at this point. (viii) NaBH4, CoCl2·6H2O, MeOH, −30 °C to 25 °C, 2 h. (ix) Boc2O, Na2CO3, MeOH, 25 °C, overnight. (x) Dess-Martin Periodinane, CH2Cl2, 25 °C, 2 h. (xi) PhB(OH)2 or FcB(OH)2, Pd(PPh3)4, K2CO3, DME, H2O, MW 125 °C, 30 min. (xii) 2M HCl in Et2O, 25 °C, overnight or for 21d: TFA, CH2Cl2, 0 °C to RT, 20 min.
2.2. In Vitro Inhibition of A Representative Panel of M1 Aminopeptidases
We have confirmed and extended the outstanding selectivity of aminobenzosuberone derivatives for M1 aminopeptidases over most other classes of proteinases (aspartic, cysteine, metallo and serine endopeptidase) with IC50 value >> 30 µM in all testes cases (Table S1). As expected, the bimetallic aminopeptidases from the M17 (PfAM17) and M28 (AAP) families were not or only very weakly inhibited by the tested compounds (Table 4).
Table 4.
Inhibition of selected aminopeptidases by racemic substituted 7-amino-5,7,8,9-tetrahydrobenzocyclohepten-6-one hydrochloride salts.
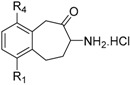
|
R1 | R4 | M1 Aminopeptidases ‘Monometallic’ |
M17 & M28 Aminopeptidases ‘Bimetallic’ |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Papn k [58] | rPfAM1 l | REcPepN m | rHsERAP1 n * | rHsERAP2 o * | rHsPSA p | rHsIRAP q * | rHsLTA4H s [58] | rPfAM17 t | AAP u [58] | |||
| 21a | H | H | 1 | 15 ± 0.07 | 50 ± 3.0 | - | - | - | - | >100 | - | >100 |
| 21b | H | Br | 0.04 | 0.04 ± 0.009 | 1.07 ± 0.11 | 120 ± 11 | 49 ± 3.8 | 1.63 ± 0.08 | 1.0 ± 0.08 | >100 | >100 | >100 |
| 21c | H | Ph | 0.007 | 0.05 ± 0.005 [69] | 0.16 ± 0.012 | 63 ± 3 | 18 ± 1.3 | 0.05 ± 0.004 | 2.4 ± 0.3 | 19 | >100 [69] | 28 |
| 21d | H | Fc | 0.004 [60] | >50 | 0.06 ± 0.008 | - | - | - | - | - | - | - |
| 21e | Br | H | 0.02 | 1 ± 0.04 | 4.59 ± 0.17 | 56 ± 7 | 104 ± 7 | 1.07 ± 0.063 | 2.6 ± 0.2 | >100 | >100 | >100 |
| 21f | Ph | H | 0.25 | 2 ± 0.07 | - | 137 ± 5 | >200 | - | 2.2 ± 0.13 | >100 | >100 | >100 |
| 21g | Benzo [1,2] | 0.04 | 15 ± 1 | - | - | - | - | - | >100 | - | >100 | |
| 21h | Br | Br | 0.006 | 0.005 ± 0.001 | - | 5.5 ± 1.1 | 2.8 ± 0.3 | - | 0.034 ± 0.001 | >100 | - | >100 |
| 21i | Br | Ph | 0.00006 | 0.03 ± 0.005 | 0.05 ± 0.003 | 1.6 ± 0.1 | 0.39 ± 0.013 | 0.021 ± 0.003 | 0.12 ± 0.015 | 68% # | - | 39 |
| 21j | Ph | Br | 0.07 | 4 ± 0.7 | - | - | - | - | - | 54% # | - | >100 |
Ki values (µM) determined from Dixon plots or * IC50 values (µM) of compounds 21a–j with, k—pAPN from porcine kidney, l—P. falciparum rPfAM1. m—E. coli rEcPepN, n—human endoplasmic reticulum aminopeptidase 1 (rHsERAP1), o—human endoplasmic reticulum aminopeptidase 2 (rHsERAP2), p—human puromycine sensitive aminopeptidase (rHsPSA), q—human Leucyl-cystinyl aminopeptidase (rHsIRAP), s—human leukotriene A4 hydrolase (rHsLTA4H), t—P. falciparum rPfAM17 and u—aminopeptidase from A. proteolytica (AAP). r stands for recombinant and Fc for Ferrocenyl. # % of inhibition determined at 100 µM. Inhibition values for pAPN, HsLTA4H and AAP were already reported in literature [58,60].
Among M1 aminopeptidases, the most potent inhibitory values remained on mammalian APN, with a subnanomolar Ki value for compound 21i (Ki = 60 pM). Note that HsLTA4H was not inhibited by this low-molecular weight scaffold, except for 21c (Ki = 19 µM). The SAR determined for mammalian APN was very similar for its M1 orthologues from microorganisms, EcPepN and P. falciparum PfAM1, with an improved affinity when the aminobenzosuberone scaffold was substituted in C-1 or C-4 position with a hydrophobic functional group (for instance 21i exhibited Ki values of 50 nM and 30 nM against EcPepN and PfAM1, respectively). The influence of bromine in C-1 position remained similar for the enzymes of both microorganisms with a 10 fold increase in potency when compared to the unsubstituted derivative 21a. The main differences relied on the lack of synergistic effect (100-fold more potent for APN) when both positions were occupied by these functions (Ph and/or Br). Compounds 21c and 21i showed similar potencies on PfAM1 (Ki values of 50 nM and 30 nM, respectively) and 21i was only 3 times more potent than 21c on EcPepN.
As regard to human enzymes, HsPSA is well-inhibited with Ki values of 55 and 21 nM for 21c and 21i, respectively. Within the oxytocinase subfamily, compounds were less active with inhibitory potencies in the micromolar range, except for 21h on HsIRAP (Ki = 34 nM). Generally, the inhibitors were less potent on HsERAP2 and HsERAP1 even if a similar trend emerged in the SAR studies. Actually, substitution at C-4 position seemed to be more efficient than substitution in the C-1 position, 21c was usually more potent than 21f and 1,4-disubstituted derivatives 21h and 21i displayed the most interesting binding affinity with a precise substitution pattern to achieve an excellent potency.
3. Discussion
3.1. Structure-Based Analysis of the Structure-Activity Relationships
In the present work, we focused on subtle amino acid sequence differences in the active site that may explain the various inhibition values experimentally determined for the selected M1 aminopeptidases. In addition, conformational dynamics have been considered for some members of this family among the structures captured during various crystallization processes [20,21,24,70]. Data remain scarce due to the small number of solved 3D structures and no general statements could be outlined, nevertheless our SAR studies enabled to precise the structural environment required for potent inhibition by the aminobenzosuberone derivatives of a given enzyme within the M1 family. Our discussion will essentially focus on “closed” conformation of different aminopeptidases.
The determined structures from various species indicate that the active site of a M1 aminopeptidase is buried inside the protein and its zinc binding site may be located in a spacious cavity with wide openings, or located in a small and gated compartment as it is the case for bacterial/parasite orthologues [71]: this was in agreement with the active site’s cavity volume of different M1 aminopeptidases analysed by KVFinder (Table S2 and Figure S2) [72]. Local or intradomain/interdomain movements can cause enlargement or contraction of the active site as encountered for EcPepN or mammalian APN [20,25,71,73].
However, the architecture and amino acid composition of the active site are highly conserved within the whole family [1], which permits to predict an identical binding mode of the aminobenzosuberone ligand in the various M1 aminopeptidases similar to the previous model reported for mammalian APN [60] or EcPepN-aminobenzosuberone co-crystallized structures (Figure 2) [74].
Figure 2.

Interactions of aminobenzosuberone derivative 21c with EcPepN. (a) Surface representation of compound 21c in the active site of EcPepN (PDB 5MFS). The binding pocket is shown in light-blue surface representation and the zinc ion is represented by a grey sphere (prepared using PyMOL Molecular Graphics System). (b) Schematic representation of compound 21c binding mode in the active site of EcPepN. Hydrogen bond and metal interactions are shown with dotted purple and red lines, respectively. Green lines represent the hydrophobic residues located in the active site [74].
3.2. S1 Subsite
The S1 subsite is always described as a cylinder formed by hydrophobic residues and capped by two polar amino acids [75,76]. In Table 5, key residues of this constitutive recognition site are summarized. Interestingly, a highly variable residue (highlighted in “red”), both in steric size and polarity is located at the entrance of this pocket and may contribute to the diversity of S1 specificities (Table S3 and Figure S3) [75].
Table 5.
Amino acid composition of the S1 subsite of selected M1 APs.
| M1 AP | PDB | Amino Acid Composition of the S1 Subsite | |||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HsAPN | 4FYT | Q211 | Q213 | N350 | A351 | G352 | A353 | M354 | E355 | H392 | E411 | F472 | Y477 | F896 | |||||||
| 4FYR | Q211 | Q213 | A214 | D216 | N350 | A351 | G352 | A353 | M354 | E355 | H392 | E411 | S469 | F472 | Y477 | S895 | S897 | ||||
| HsERAP1 | 2YD0 | H160 | Q181 | E183 | P184 | A186 | F314 | Q315 | S316 | G317 | A318 | M319 | E320 | H357 | E376 | R430 | F433 | Y438 | E865 | S868 | S869 |
| HsERAP2 | 4JBS | E177 | D198 | E200 | P201 | Q203 | F331 | A332 | P333 | G334 | A335 | M336 | E337 | H374 | E393 | Q447 | F450 | Y455 | Y892 | R895 | |
| HsIRAP | 5MJ6 | Y272 | Q293 | E295 | P296 | F425 | E426 | A427 | G428 | A429 | M430 | E431 | H468 | E487 | E541 | F544 | Y549 | P957 | Y961 | ||
| HsLTA4H | 1HS6 | Q134 | Q136 | A137 | Y267 | G268 | G269 | M270 | E271 | H299 | E318 | D375 | Y378 | Y383 | |||||||
| EcPepN | 5MFS | Q119 | E121 | A122 | M260 | G261 | A262 | M263 | E264 | H301 | E320 | Y376 | Y381 | R825 | |||||||
| 2DQM | Q119 | E121 | A122 | N259 | M260 | G261 | A262 | M263 | E264 | H301 | E320 | N373 | Y376 | Y381 | Q821 | R825 | |||||
| PfAM1 | 3EBH | T305 | Q317 | E319 | A320 | T321 | F457 | N458 | V459 | G460 | A461 | M462 | E463 | H500 | E519 | E572 | Y575 | Y580 | M1034 | ||
In red, the variable residue determined for the corresponding pdb entries. In blue, residue situated at the top of S1 subsite and involved in the capping of the pocket.
The aforementioned variable residue may be flexible upon ligand binding or might be sterically hindered, restricting thus the S1 subsite entrance (Figure 3 and for more details, Figure S3). For instance, in EcPepN it corresponds to Met260 (PDB 2DQM or 5MFS) with a temperature factor value of 28–37 Å2, in agreement with a local significant flexibility of its side chain, which acts as a cushion to accommodate various side chains in P1 position of a substrate/ligand. This local movement of Met residue can cause the contraction of the active site cavity. KVFinder analysis revealed that the cavity volume of EcPepN-bestatin 1 complex (PDB 2DQM) was higher (638.5 Å3), compared to 531.25 Å3 for the EcPepN-aminobenzuberone 21c complex (PDB 5MFS) (Table S2). In this latter complex, this Met260 residue was found at two closed positions and moved slightly inward the S1 subsite, interrupting and shortening its typical cylindrical shape. The distance between the sulphur atom of Met260 and the centroid of the aromatic ring of the aminobenzosuberone scaffold is 5.3–5.7Å, which is consistent with sulphur-carbon van der Waals contacts and the sulphur lone pairs might interact with the edge of the aromatic core, providing additional stability (Figure S4) [77]. On the other hand, Val459 in PfAM1 (PDB 3EBH) and Pro333 in HsERAP2 (PDB 4JBS) might be considered as straight and bulky, shortening the S1 pocket. In HsLTA4H, Tyr267 (PDB 1HS6) in this position induces the formation of a narrow S1 tunnel. Moreover, domain III is missing in this latter enzyme and the presence of Tyr267, Lys565 and particularly Tyr378, which extends into the active site cavity pointing inward toward the Zn ion, reduced substantially the active site cavity volume (Figure S5). As a result of this spatial arrangement, steric clashes with the aromatic core of aminobenzosuberone might explain the very weak inhibitory activities of this scaffold on HsLTA4H.
Figure 3.

Diversity in the S1 subsite of different M1 aminopeptidases (a) Superimposition of the protein backbone of S1 subsite of different M1 APs: HsAPN (cyan, PDB 4FYT), HsERAP1 (orange, PDB 2YD0), HsERAP2 (yellow, PDB 4JBS), HsIRAP (white, PDB 5MJ6), EcPepN (green, PDB 5MFS), PfAM1 (magenta, PDB 3EBH). (b) Surface representation of the S1 subsite of EcPepN (PDB 5MFS). The catalytic zinc ion is shown as grey sphere. Residues involved in S1 plasticity are shown in stick. Images generated with KVFinder plugin in PyMOL.
The aminobenzosuberone derivatives are markedly more potent on M1 aminopeptidases exhibiting a shallow S1 cavity with small to moderate size (EcPepN, HsAPN, PfAM1 and a borderline case for HsIRAP). At the top of the elongated S1 subsite cylinder are located two or three cap residues (presented in bold type in Table 5 and depicted in stick in Figure 3a and Figure S3) from the C-terminal domain of the proteins. These residues are known to influence S1 subsite specificity [78,79]. Indeed, depending on the nature of the residues and through the conformational flexibility of their side chains, they can mitigate any potential unfavourable steric clashes and favour van der Waals, H-bond or electrostatic interactions with the substrates/inhibitors. Moreover, the closing of the S1 subsite induces the expulsion of water molecules from the active site into the bulk (entropically favourable) and may strengthen both van der Waals and electrostatic interactions. The analysis of superimposed structures showed that our aminobenzosuberone inhibitors were not expected to penetrate into the S1 cavity as deeply as other inhibitors (like bestatin) (Figure S4a and Figure S6), indicating they should therefore interact more efficiently with shallow S1 subsite.
However, depending on the nature of the variable amino acid residue, the aminobenzosuberone core might slightly move upward to minimize any potential steric hindrance with this special ‘gatekeeper’ residue. Substituents on position-1 most likely induced steric conflicts with residues at the entrance of S1 cavities. More suitable substitutions in position-9 should offer new scopes for novel series of more potent and selective aminobenzosuberone derivatives (Figure S6).
3.3. S1′ Subsite
In contrast to S1, the S1′ subsite is an open cavity of 9 to 10 residues for all enzymes. For instance, in EcPepN, Gly261, Ala262, Tyr275, Arg293, Val294, His297, Glu298, Val324, Asp327, Tyr381 compose this subsite. The careful analysis of the various 3D structures showed that only 2 to 3 residues (highlighted in blue and red in Table 6) seem to be involved in S1′ plasticity, thereby controlling the cavity width and depth (Figure 4a and Figure S7). Two amino acids, which are Tyr275 (located on the sidewall opposite to the catalytic zinc ion) and Arg293 (partially capping the S1′ cavity) for EcPepN, vary in size and polarity within this enzyme family, as summarized in Table 6 and Figure 4a.
Table 6.
S1′ subsite composition.
| M1 AP | Amino Acid Composition of the S1′ Subsite | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HsAPN | G352 | A353 | R381 | T384 | V385 | H388 | E389 | S415 | E418 | Y477 | |||
| HsERAP1 | G317 | A318 | G346 | M349 | T350 | H353 | E354 | K380 | E383 | Y438 | |||
| HsERAP2 | G334 | A335 | S348 | W363 | R366 | V367 | H370 | E371 | K397 | E400 | Y455 | ||
| HsIRAP | G428 | A429 | T442 | L457 | K460 | I461 | H464 | E465 | T491 | E494 | Y549 | ||
| HsLTA4H | G268 | G269 | N291 | V292 | H295 | E296 | V322 | E325 | Y383 | R563 | K565 | ||
| EcPepN | G261 | A262 | Y275 | R293 | V294 | H297 | E298 | V324 | D327 | Y381 | |||
| PfAM1 | G460 | A461 | R489 | T492 | V493 | H496 | E497 | V523 | E526 | Y580 | |||
Highlighted residues are involved in S1′ site. In blue, residues which reduce the cavity width; in red, residues which modulate the cavity depth and in bold, flexible amino acid side chains pointing either inward or outward the active site.
Figure 4.


Diversity in the S1′ subsite of different M1 aminopeptidases. (a) Superimposition of the protein backbone of S1′ subsite of different M1 APs: HsAPN (cyan, PDB 4FYT), HsERAP1 (orange, PDB 2YD0), HsERAP2 (yellow, PDB 4JBS), HsIRAP (white, PDB 5MJ6), EcPepN (green, PDB 5MFS), PfAM1 (magenta, PDB 3EBH). (b) Surface representation of the S1′ subsite of EcPepN (green, PDB 5MFS). The catalytic zinc ion is shown as grey sphere. Residues involved in S1′ plasticity are shown in stick. Images generated with KVFinder plugin in PyMOL.
For example, PfAM1 Arg489 is found more buried in the cavity than HsAPN Arg381, reducing thus slightly the width of S1′ subsite, whereas the EcPepN Tyr275 and HsAPN Arg381 are positioned at a similar location, modifying thereby the local polarity. Also, the EcPepN Arg293 side chain is located further inside the cavity reducing the cavity depth, whereas the less bulky and polar PfAM1 Thr492 and HsAPN Thr384 occupy a similar position. Finally, local flexibility was observed within captured structures. Indeed, the side chain of residue HsAPN Arg381, HsERAP2 Arg366 and EcPepN Arg293 can adopt different positions upon ligand binding. A similar observation might also be noticed for EcPepN Asp327, this carboxylate side chain is either orientated in- or outside S1′ cavity, with the inside orientation potentially involved in further interactions as exemplified by N. meningitides alanyl aminopeptidase inhibition study [80]. In its close vicinity, a Lys residue is found in HsERAP1 and HsERAP2, Lys380 and Lys397, respectively, with their side chains extending in the cavity (residue in black bold in Figure S6). Developing interactions with this particular residue via para- or meta-substituted phenyl ring at position 4 of the aminobenzosuberone core should improve the selectivity and affinity for both members of this oxytocinase family.
All this information clarifies the inhibition observed for HsAPN and its microbial orthologues, EcPepN and PfAM1. Despite PfAM1 S1′ cavity volume being slightly larger than HsAPN and EcPepN (Table S4), the S1′ subsite cavity width of PfAM1 is shorter, leading to a lack of binding affinity for the bulky ferrocenyl substituent of 21d which cannot fit appropriately the PfAM1 S1′ subsite (Ki > 50 µM). With regard to the SAR experimentally determined, it highlights the importance of the substitution in position-4 of the aminobenzosuberone core, which, according to our model, almost fully occupies this subsite in the evaluated enzymes. Most aminopeptidases can accommodate bromine or a phenyl group with an improved affinity of the inhibitor, when compared to the unsubstituted derivative. Nevertheless, a phenyl group is mostly preferred probably due to a possible π-stacking with EcPepN His297, leading to the low Ki value of 21c when compared to 21a or 21b.
3.4. Intra/Inter-Domain Movements
An important feature to be considered for M1 aminopeptidases molecular recognition is their inter- or intra-domain movements [20,21,22,23,24,25]. Various conformations have been captured during crystallization processes and open, intermediate and closed conformations have been determined for various given enzymes, suggesting that these dynamics are important for ligand binding and peptide hydrolysis [21,22,70,81]. A large inter-domains motion has been markedly observed for HsERAP1, associated with local changes for the highly conserved Tyr residue (Tyr438 for HsERAP1) in the active site (corresponding to the position Tyr381 of EcPepN in Figure S1). In an “open” form of the protein, this residue undergoes a 6 Å shift away from the zinc ion, whereas in a closed form, this Tyr residue is proposed to both assist in the activation of the peptide bond and in the stabilization of the tetrahedral intermediate during the proteolytic catalytic cycle [21,22]. It has been shown, by site directed mutagenesis, that mutation of this Tyr residue into Phe completely abolished the enzyme activity [82,83].
As a result of this dynamic, the open form of the enzyme is likely catalytically inactive and a subsequent allosteric activation upon substrate binding might trigger a catalytically active closed form. This dynamic transition between those different states might help the substrates/products to bind to or to be released from the active site. In solution, a number of conformers might exist, from open to closed states, along with different position of the Tyr residue. We have previously suggested [60] that this Tyr residue probably activates the carbonyl function of the aminobenzosuberone towards a nucleophilic attack of the catalytic water to form a hydrated form of this ketone function and generates in situ a potent chelator of the zinc ion and a perfect mimic of the transition state. So, our low-molecular weight reversible inhibitors would be likely to bind to a closed form of HsERAP1 for optimum interactions. Interestingly, HsIRAP was reported to undergo a similar conformational change with, this time, complete different orientation of the GAMEN motif upon inhibitor binding, leading to a closed conformation of HsIRAP (PDB 5MJ6) and highlighting some significant structural plasticity of the GAMEN loop [24]. Since those enzymes in solution should exist in a conformational equilibrium between different forms (even so small angle X-ray scattering experiments have suggested that the HsERAP1 average structure in solution adopts an open conformation) [70,81], these processes could account for the moderate inhibition activity values observed with the HsERAP sub-family (IC50 = 1.6 to 150 µM) and, to a lesser extent, HsIRAP (IC50 = 0.034 to 2.6 µM), indicating that the aminobenzosuberone scaffolds are not able to induce domain movements to a more closed form of the enzyme.
Intra-domain mobility is also to be considered. For example, besides a more compact folding, a particularity of the human and porcine APN structures determined up to now is the plasticity of the S1 subsite through conformational changes. It looks like a small/shallow hydrophobic pocket, compared to the deeper S1 subsites observed in other crystallized M1 aminopeptidases [20,25]. Indeed, HsAPN Phe896 which serves to cap the S1 pocket belongs to a flexible loop (as indicated by an average temperature factor of 25–35 Å2) of 8 residues (Y891GGGSFSF898) in domain IV and might act as a gatekeeper restricting access and closing the S1 subsite. Upon ligand binding, this S1 pocket might undergo a major conformational change via movement of the flexible 891–898 loop, revealing thus a deeper cylindrical hydrophobic S1 subsite [25]. The cavity volumes of the active site of HsAPN varied from 572.12 Å3 to 811.87 Å3 (Figure S8 and Table S2). Our series of compounds are very potent on this human enzyme, probably through interactions with the “Phe-In” conformation of this loop via the bromine substituent in position-1 of the aminobenzosuberone core and the aromatic ring of Phe896 (8i, Ki = 60 pM) [60,74]. These interactions might induce or stabilize the closure of domain IV, especially that of mammalian APN and hence a closed APN form, which is an important feature to block porcine coronavirus cell entry via its porcine APN receptor [23].
These different conformational dynamic processes seem to be an important aspect of catalytic regulation for some M1 enzymes, although it has yet to be more documented with a larger number of structures. These inter- or intra-domain movements are probably only one remarkable feature of aminopeptidase M1 dynamics, according to their biological functions and their cellular localization.
3.5. ADME-Tox Properties
To gain insight into the physicochemical and ADME-Tox properties of these aminobenzosuberone derivatives, several parameters were experimentally determined or computed (Table S5) [84]. The experimental logD7.4 values of the whole aminobenzosuberone series complied with Lipinski’s “rule of five” [85,86] and the calculated TPSA value is comprised between 44.71 and 68.10 Å2 for the ketone and hydrate forms, respectively. These compounds are predicted to penetrate the blood-brain barrier, although some possible P-glycoprotein (P-gp) efflux issues should be taken into consideration (predicted probability to be P-gp substrate is 0.5). They could be considered as valuable chemical tools to target M1 aminopeptidases implicated in CNS diseases, mostly involved in the brain renin-angiotensin system (HsIRAP, HsAPN, HsAPA) [87,88,89] and in the pain management (HsAPN) [57,90]. However, the aminobenzosuberone scaffold requires careful optimization and increasing selectivity toward targeted M1 aminopeptidase versus HsPSA, which plays major neuroprotective roles [91,92,93,94,95,96]. Analysis of the calculated probabilities of drug-drug interaction with different CYP450 isozymes shows that this series of molecules should inhibit CYP1A2 and to a lesser extent CYP2D6. In addition, the probability of toxicity is high and is mostly associated with cardiotoxicity with the inhibition of hERG channel and also hepatotoxicity. The uncertainties in the predicted outcomes should be taken into account and these potential toxicity liabilities should be investigated further. Thus, considering ADMET properties and inhibitory activities, the most promising compound for subsequent studies is the 1,4-dibromo derivative 21h, which is also the most synthetically feasible molecule. However, according to the predictive method, decreasing the lipholicity (clogP ou logD7.4) by introducing heteroatoms into the phenyl ring of compound 21c might also be a direction of modification since it should improve the ADME properties and have beneficial effects on toxicity.
4. Materials and Methods
4.1. General Information
Reactants were purchased from usual provider. Usual solvents were freshly distilled, dry MeOH distilled over Mg/MgI2, dry DME over Na and benzophenone, dry Et2O was distilled and stored over Na, DCM was distilled over P2O5 and stored over anhydrous K2CO3.
Flash chromatography: silica gel (Merck 60, 230–400 mesh). TLC: Al-roll silica gel (Merck 60, F254). Mp: Kofler hot bench, corrected. 1H- and 13C-NMR (400 MHz and 100.6 MHz resp.) spectra: Bruker Avance 400; δ in ppm and J in Hertz.
All these racemic substituted 7-amino-5,7,8,9-tetrahydrobenzocyclohepten-6-one hydrochloride salts were already fully described in literature [58,60] and were resynthesized to determine their selectivity profiles. Their inhibitory activities against pAPN, hLTA4H and AAP were already reported [58,60].
4.2. General Procedure for Rubottom Oxidation
To an ice-cold mixture of water and acetone (70:35 mL) were added NaHCO3 (6.18 g, 20 eq.) and Oxone® (11.3 g, 5 eq.). The suspension was stirred at 0 °C for 30 min and then a solution of silyl enol ether 15 (1.3 g, 1 eq.) in DCM (70 mL) was dropwise added. The mixture was warmed to r.t. and stirred for 3 h (TLC monitoring). Layers were separated and aqueous layer was extracted with DCM. Combined organic layers were washed with brine, dried on MgSO4, filtered and concentrated to give silyl-oxy ketone 16, which was used without further purification.
4.3. General Procedure for Oxime Reduction
To a solution of hydroxy-oxime 17 (220 mg, 1 eq.) in methanol (13 mL) was added CoCl2·6H2O (388 mg, 2 eq.). The mixture was cooled to −30 °C and NaBH4 (462 mg, 15 eq.) was carefully added. The reaction was slowly warmed to r.t. and stirred for 2 h (TLC monitoring). The mixture was diluted with water and extracted by AcOEt. Organic layers were washed with brine, dried on MgSO4, filtered and concentrated to give amino-alcohol 18, which was N-protected without further purification.
4.4. Production and Purification of Recombinant Aminopeptidases
4.4.1. PfAM1 and PfAM17
Genes encoding residues 192–1085 of native Plasmodium falciparum alanyl aminopeptidase PfAM1 (PlasmoDB PF3D7_1311800) and residues 84–605 of native Plasmodium falciparum leucyl aminopeptidase PfAM17 (PlasmoDB PF3D7_1446200) were synthetized by Genecust (Luxembourg), with the help of algorithms developed for optimizing, DNA sequences to improve proteins expression in Escherichia coli [97]. iSynthetic genes were cloned into the T7 expression pET45b(+) vector (Novagen), which appended a N-terminal hexahistidine tag (KnpI and SalI sites for PfA-M1–BamHI and SalI sites for PfA-M17).
Escherichia coli Rosetta 2 (DE3) bacteria (Novagen) were transformed with these recombinant plasmids, after their validation by Sanger sequencing (Beckman Coulter Genomics). Bacterial cultures were grown in auto-induced LB medium (Merck) supplemented with carbenicillin (50 µg/mL) and chloramphenicol (34 µg/mL), during 24 h at 25 °C, prior to bacterial extract preparations with BugBusterTM (Novagen, Darmstadt, Germany). The clarified lysates were loaded onto Ni2-charged HisTrap column (GE Healthcare) equilibrated in 20 mM imidazole phosphate buffer and washed in the same buffer. Bound recombinant proteins were then eluted in 80 mM imidazole for PfAM1 and 200 mM imidazole for PfAM17, in phosphate buffer. Eluted fractions were finally purified by size exclusion chromatography on a Superdex 200 10 300 (equilibrated with Tris HCl 50 mM, NaCl 200 mM, ZnCl2 10 µM, pH 7.4 for PfAM1 and with Hepes 50 mM, NaCl 300 mM, ZnCl2 10 µM, 5% glycerol, pH 8.5 for PfAM17) using an Äkta purifier chromatography system (GE Healthcare Life Science, Little Chalfont, England).
4.4.2. HsERAP1, HsERAP2 and HsIRAP
The expression and purification of recombinant human endoplasmic reticulum aminopeptidase 1 (HsERAP1), endoplasmic reticulum aminopeptidase 2 (HsERAP2) and insulin-regulated aminopeptidase (HsIRAP) have been described before [79,98,99]. Briefly, recombinant baculovirus containing the gene for each enzyme was produced in sf9 cells according to the manufacturer’s instructions (Bac-to-Bac baculovirus expression system, Invitrogen). Recombinant proteins were expressed in Hi5 cells after infection with the appropriate recombinant baculovirus and purified by NiNTA affinity chromatography as previously described (PMID: 21314638). Proteins were aliquoted and stored at −80 °C in a buffer containing 10 mM Hepes pH 7.0, 100 mM NaCl, 10% glycerol, until needed.
4.4.3. EcPepN and HsPSA
Both the enzymes were purified as reported earlier [73,100].
4.5. Enzymatic Assays and Kinetic Analysis
4.5.1. PfAM1 and PfAM17
Tests were carried out on HP/Agilent UV-Visible diode array spectrophotometer 8453 (HP/Agilent, Santa Clara, CA, USA), at 30 °C in Tris HCl 50 mM pH 7.4 for rPfAM1 and pH 8 for rPfAM17, with a final DMSO concentration of 1%. Enzyme activities of rPfAM1 (10 nM) and rPfAM17 (60 nM) were determined by continuously measuring the release of para-nitroaniline at 405 nm with alanine para-nitroanilide as substrate for rPfAM1 (Km = 1.5 mM) and leucine para-nitroanilide for rPfAM17 (Km = 0.5 mM). After 10 min of incubation, CI50 measurements were conducted during 30 min for rPfAM1 and 3 h for rPfAM17. Initial velocities were measured with increasing concentrations of inhibitors and Ki values were determined by Dixon plots in triplicate.
4.5.2. LTA4H
5 µg of rLTA4H (Sigma) were used to measure the activity of this protein through the release of alanine para-nitroanilide at 405 nm (Km = 2 mM). Experiments were performed on HP/Agilent UV-Visible diode array spectrophotometer 8453 at 30 °C, during 30 min, in Tris HCl 10 mM, KCl 0.1 mM, pH 7.5 to obtain IC50 values.
4.5.3. EcPepN and HsPSA
Enzyme activity assays were carried out in 10 mM Tris-HCl buffer at pH 7.5 with 150 mM NaCl, enzyme, 100 µM substrate (Leu-pNA) in a 100 μL reaction at 37 °C. The absorbance was read at 405 nm. Concentrations ranging from 1 nM to 100 μM of inhibitors (dissolved in DMSO) were incubated with each of the enzymes (75 µM of EcPepN, 120 µM of HsPSA) for 30 min at 37 °C in a 96 well plate. After addition of the substrate, immediate increase in absorption was measured. All inhibition assays were performed in triplicates and initial velocities were measured. Percentage inhibition and log values of inhibitor concentrations were plotted using SigmaPlot to determine IC50 values. Ki values were determined by the Dixon method. The assay consists of reaction buffer, enzyme and varied concentrations of Leu-pNA (25, 50, 100, 200 and 400 µM). Based on the IC50 values, at each substrate concentration, inhibitor concentration was varied in the range of 50 nM to 500 nM or 0.5 µM to 25 µM. Data were plotted as 1/rate versus inhibitor concentration for each substrate concentration and a linear fit was calculated by non-linear regression using SigmaPlot12.5. All the reactions were performed in triplicates, SD values were reported [73].
4.5.4. HsERAP1, HsERAP2 and HsIRAP
The enzymatic activity of HsERAP1, HsERAP2 and HsIRAP was determined by following the time-dependent increase in fluorescence at 460 nm (excitation: 380 nm) of the fluorigenic substrates L-Leucine-7-amido-4-methyl coumarin (L-AMC; Sigma, Darmstatd, Germany) for HsERAP1 and HsIRAP and L-arginyl-7-amido-4-methyl coumarin (R-AMC; Sigma) for HsERAP2. Measurements were performed on a 96-well plate (150 μL total volume in each well) on a TECAN infinite M200 microplate fluorescence reader. 30 nM of HsERAP1, 6 nM of HsERAP2 or 6 nM of HsIRAP was added in each well, along with 50 μM of substrate and varied concentrations of compound (within the range of 1 nM to 200 μM). The reaction was followed for 10 min at room temperature and the remaining enzymatic activity was calculated by measuring the slope of the time course. For calculation of the in vitro IC50 values, experimental data were fit to the following equation using the GraphPad Prism software package:
| Y = Bottom + (Top − Bottom)/(1 + 10^((LogIC50 − X) * HillSlope)) | (1) |
where Y is the enzymatic activity and X the inhibitor concentration.
4.6. Measurement of Octanol/Water Partition Coefficient (logD7.4)
The lipophilicity (logD7.4 values) of the amino-benzosuberones was determined by reversed-phase HPLC on a C-18 column (Eclipse XDB-C18, 5 µm, 4.6 × 150 mm, from Agilent, France) according to the method previously described by Minick and Pomper [101,102]. Measurement of the chromatographic capacity factors (k′) for each compound was done at various concentrations in the range of 90–60% methanol (containing 0.25% octanol) and an aqueous phase consisting of 0.15% n-decylamine in 0.02 M MOPS (3-(N-morpholino)propanesulfonic acid) buffer pH 7.4 (prepared in 1-octanol-satured water). The flow was 0.9 mL/min. The aminobenzosuberone derivatives were dissolved at 0.4 µM in methanol. The standards were also dissolved in methanol. Column void volume was estimated from the retention time of uracil, which was included as a non-retained internal reference with each injection. The capacity factors (k′) are extrapolated to 100% of the aqueous component given the value of k′w. The logD7.4 is then obtained by the formula:
| logD7.4 = 0.13418 + 0.98452k′w | (2) |
Positive control with β-oestradiol, experimental logD7.4 3.043 ± 0.093 (n = 9), literature value 3.261.
4.7. In Silico Prediction of ADMET Properties
The study of ADMET properties was carried out on the website http://admet.scbdd.com [84].
5. Conclusions
M1 family aminopeptidases have broad and overlapping substrate specificity; hence small-molecule inhibitors may not always be specific, especially regarding their selectivity toward bimetallic enzymes. The aminobenzosuberone scaffold demonstrated exclusive selectivity for the monometallic M1 aminopeptidase family with particular potent inhibitory activities against mammalian APN and its microbial orthologues EcPepN and PfAM1 (Ki values in the nanomolar range). It retains interesting potency on HsPSA and also on HsIRAP (Ki values in the nano- to sub-micromolar range) but it is seemingly less active on both oxytocinase enzymes HsERAP1/2 (IC50 values in the micromolar range) and totally inactive against HsLTA4H.
This promising scaffold represents an attractive new class of potent antimalarial compounds targeting PfAM1 [69], a crucial enzyme involved in at least the last committed step of haemoglobin degradation by Plasmodium falciparum [17,31,35]. In addition, growing evidences highlight the crucial role played by mammalian aminopeptidases in a great variety of cancer types, especially HsAPN and HsERAP1/2 via their proteolytic activity or their ability to modulate protein-protein interactions [103]. The aminobenzosuberone core is an attractive starting point to design triple inhibitor of all these three enzymes, acting by interfering with endothelial cell morphogenesis and cell motility [61] and by modulating antigen processing to trigger cancer immunotherapy [14,15,16].
The next challenge is to rationally design selective inhibitors for individual M1 aminopeptidase, to study their biological roles and precise their functions, or to avoid any potential adverse effects following in vivo treatment with aminobenzosuberone derivatives by targeting other members of this diverse family. To achieve this goal, the design process efforts should take into account the plasticity of the active site and the conformational dynamics of these M1 aminopeptidases. Interesting hints suggest deeper interactions into the S1 subsite through a substitution on position-9 of our scaffold should offer new opportunities to improve both activity and selectivity. Another approach for achieving selectivity is to look for the cellular/subcellular localization and/or tissue distribution of the targeted aminopeptidase, to design targeted-prodrug to improve site-specific drug delivery. These different strategies are currently under investigation and will be reported in due course.
Acknowledgments
The support of the Université de Haute-Alsace and the Ecole Nationale Supérieure de Chimie de Mulhouse is gratefully acknowledged.
Supplementary Materials
The following are available online. Figure S1: Conserved active site residues involved in substrate recognition and catalysis, Table S1: Human proteinase selectivity profiles of 21a–c and 21i, Table S2: Cavity volume of the active site of “closed” conformation of M1 APs, Figure S2: Surface representation of the active site of studied M1 Aminopeptidases, Figure S3: Surface representation of the S1 subsite of different M1 aminopeptidases, Table S3: Cavity volume of the S1 subsite of “closed” conformation of M1 APs, Figure S4: Local movement of Met260 in the active site of EcPepN, Figure S5: Superimposition of the protein backbone of the aminobenzosuberone 21c in EcPepN complex (green, PDB 5MFS) with HsLTA4H (slate blue, PDB 1HQ6), Figure S6: Superimposition of the protein backbone of the aminobenzosuberone 21c in EcPepN complex (green, PDB 5MFS) with different M1 aminopeptidases, Figure S7: Surface representation of the S1′ subsite of different M1 aminopeptidases, Table S4: Cavity volume of the S1′ subsite of “closed” conformation of M1 APs, Figure S8: Intra-domain movement in the active site of HsAPN, Table S5: Determination and prediction of various ADME-Tox properties of substituted 7-amino-5,7,8,9-tetrahydrobenzocyclohepten-6-one hydrochloride salts.
Author Contributions
C.T. and S.A. conceived and supervised the study and A.A. and E.S. helped in designing the experiments. E.S. and S.A. performed the synthesis and characterisation of aminobenzosuberone derivatives; M.S., A.K.M., A.S. and C.S. produced proteins and performed enzymatic tests under the guidance of C.T., I.F., A.A. and E.S.; G.R. and S.A. wrote the original draft; C.T. and S.A. wrote the whole article, made discussion and conclusions and S.A. prepared the figures/tables/schemes. All authors reviewed the manuscript.
Funding
This research was funded by Agence Nationale de la Recherche, grant number ANR-12-BS07-0020-01. Funding was also received by the European Union (European Social Fund) and Greek national funds through the Operational Program “Education and Lifelong Learning” of the National Strategic Reference Framework: Research Funding Program of the General Secretariat for Research & Technology and the Harry J. Lloyd Charitable Trust (to E.S.). The use of infrastructure associated with the National Research Infra- structure in Structural Biology, INSTRUCT-EL for recombinant protein production is also acknowledged. A.K.M. thanks Council of Scientific and Industrial Research (CSIR), New Delhi, India, for research fellowship. A.A. thanks Department of Science and Technology (DST), New Delhi, India with the grant number EMR/2015/000461.
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Sample Availability: Samples of the compounds 21a–j are available from the authors.
References
- 1.Rawlings N.D., Barret A.J. Metallopeptidases and their clans. In: Rawlings N.D., Salvesen G.S., editors. Handbook of Proteolytic Enzymes. Volume 1. Academic Press; New York, NY, USA: 2013. pp. 325–370. [Google Scholar]
- 2.Barret A.J., McDonald J.K. Mammalian Proteases: A Glossary and Bibliography, Vol. 2: Exopeptidases. Academic Press; London, UK: 1986. [Google Scholar]
- 3.Lowther W.T., Matthews B.W. Metalloaminopeptidases: Common functional themes in disparate structural surroundings. Chem. Rev. 2002;102:4581–4608. doi: 10.1021/cr0101757. [DOI] [PubMed] [Google Scholar]
- 4.Straeter N., Lipscomb W.N. Two-metal ion mechanism of bovine lens leucine aminopeptidase: Active site solvent structure and binding mode of L-Leucinal, a gem-diolate transition state analog, by X-ray crystallography. Biochemistry. 1995;34:14792–14800. doi: 10.1021/bi00045a021. [DOI] [PubMed] [Google Scholar]
- 5.Hooper N.M. Families of zinc metalloproteases. FEBS Lett. 1994;354:1–6. doi: 10.1016/0014-5793(94)01079-X. [DOI] [PubMed] [Google Scholar]
- 6.Rawlings N.D., Barrett A.J. Methods in Enzymology. Volume 248. Academic Press; San Diego, CA, USA: 1995. Evolutionary families of metallopeptidases; pp. 183–228. [DOI] [PubMed] [Google Scholar]
- 7.Taylor A. Aminopeptidases: Structure and function. FASEB J. 1993;7:290–298. doi: 10.1096/fasebj.7.2.8440407. [DOI] [PubMed] [Google Scholar]
- 8.Maynard K.B., Smith S.A., Davis A.C., Trivette A., Seipelt-Thiemann R.L. Evolutionary analysis of the mammalian M1 aminopeptidases reveals conserved exon structure and gene death. Gene. 2014;552:126–132. doi: 10.1016/j.gene.2014.09.025. [DOI] [PubMed] [Google Scholar]
- 9.Peer W.A. The role of multifunctional M1 metallopeptidases in cell cycle progression. Ann. Bot. 2011;107:1171–1181. doi: 10.1093/aob/mcq265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hooper N.M., Lendeckel U. Aminopeptidases in Biology and Disease. Volume 2. Kluwer Academic/Plenum Publishers; New York, NY, USA: 2004. [Google Scholar]
- 11.Drinkwater N., Lee J., Yang W., Malcolm T.R., McGowan S. M1 aminopeptidases as drug targets: Broad applications or therapeutic niche? FEBS J. 2017;284:1473–1488. doi: 10.1111/febs.14009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Saveanu L., Carroll O., Weimershaus M., Guermonprez P., Firat E., Lindo V., Greer F., Davoust J., Kratzer R., Keller S.R., et al. IRAP identifies an endosomal compartment required for MHC class I cross-presentation. Science. 2009;325:213–217. doi: 10.1126/science.1172845. [DOI] [PubMed] [Google Scholar]
- 13.Cheng H., Li Y., Zuo X.-B., Tang H.-Y., Tang X.-F., Gao J.-P., Sheng Y.-J., Yin X.-Y., Zhou F.-S., Zhang C., et al. Identification of a missense variant in LNPEP that confers psoriasis risk. J. Investig. Dermatol. 2014;134:359–365. doi: 10.1038/jid.2013.317. [DOI] [PubMed] [Google Scholar]
- 14.Stratikos E. Modulating antigen processing for cancer immunotherapy. Oncoimmunology. 2014;3 doi: 10.4161/onci.27568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stratikos E., Stamogiannos A., Zervoudi E., Fruci D. A Role for naturally occurring alleles of endoplasmic reticulum aminopeptidases in tumor immunity and cancer pre-disposition. Front. Oncol. 2014;4 doi: 10.3389/fonc.2014.00363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schmidt K., Keller C., Kühl A.A., Textor A., Seifert U., Blankenstein T., Willimsky G., Kloetzel P.-M. ERAP1-dependent antigen cross-presentation determines efficacy of adoptive T-cell therapy in mice. Cancer Res. 2018;78:3243–3254. doi: 10.1158/0008-5472.CAN-17-1946. [DOI] [PubMed] [Google Scholar]
- 17.Azimzadeh O., Sow C., Gèze M., Nyalwidhe J., Florent I. Plasmodium falciparum PfA-M1 aminopeptidase is trafficked via the parasitophorous vacuole and marginally delivered to the food vacuole. Malar. J. 2010;9:189. doi: 10.1186/1475-2875-9-189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rawlings N.D., Barrett A.J., Thomas P.D., Huang X., Bateman A., Finn R.D. The MEROPS database of proteolytic enzymes, their substrates and inhibitors in 2017 and a comparison with peptidases in the PANTHER database. Nucleic Acids Res. 2018;46:D624–D632. doi: 10.1093/nar/gkx1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.MEROPS: The Peptidase Database. [(accessed on 19 July 2018)]; Available online: https://www.ebi.ac.uk/merops/cgi-bin/famsum?family=M1.
- 20.Chen L., Lin Y.-L., Peng G., Li F. Structural basis for multifunctional roles of mammalian aminopeptidase N. Proc. Natl. Acad. Sci. USA. 2012;109:17966–17971. doi: 10.1073/pnas.1210123109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kochan G., Krojer T., Harvey D., Fischer R., Chen L., Vollmar M., von Delft F., Kavanagh K.L., Brown M.A., Bowness P., et al. Crystal structures of the endoplasmic reticulum aminopeptidase-1 (ERAP1) reveal the molecular basis for N-terminal peptide trimming. Proc. Natl. Acad. Sci. USA. 2011;108:7745–7750. doi: 10.1073/pnas.1101262108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nguyen T.T., Chang S.-C., Evnouchidou I., York I.A., Zikos C., Rock K.L., Goldberg A.L., Stratikos E., Stern L.J. Structural basis for antigenic peptide precursor processing by the endoplasmic reticulum aminopeptidase ERAP1. Nat. Struct. Mol. Biol. 2011;18:604–613. doi: 10.1038/nsmb.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Santiago C., Mudgal G., Reguera J., Recacha R., Albrecht S., Enjuanes L., Casasnovas J.M. Allosteric inhibition of aminopeptidase N functions related to tumour growth and virus infection. Sci. Rep. 2017;7:46045. doi: 10.1038/srep46045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mpakali A., Saridakis E., Harlos K., Zhao Y., Kokkala P., Georgiadis D., Giastas P., Papakyriakou A., Stratikos E. Ligand-induced conformational change of insulin-regulated aminopeptidase: Insights on catalytic mechanism and active site plasticity. J. Med. Chem. 2017 doi: 10.1021/acs.jmedchem.6b01890. [DOI] [PubMed] [Google Scholar]
- 25.Wong A.H.M., Zhou D., Rini J.M. The X-ray crystal structure of human aminopeptidase N reveals a novel dimer and the basis for peptide processing. J. Biol. Chem. 2012;287:36804–36813. doi: 10.1074/jbc.M112.398842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hermans S.J., Ascher D.B., Hancock N.C., Holien J.K., Michell B.J., Chai S.Y., Morton C.J., Parker M.W. Crystal structure of human insulin-regulated aminopeptidase with specificity for cyclic peptides. Protein Sci. 2015;24:190–199. doi: 10.1002/pro.2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wallis M.G., Lankford M.F., Keller S.R. Vasopressin is a physiological substrate for the insulin-regulated aminopeptidase IRAP. Am. J. Physiol.-Endocrinol. Metab. 2007;293:E1092–E1102. doi: 10.1152/ajpendo.00440.2007. [DOI] [PubMed] [Google Scholar]
- 28.Bauvois B., Dauzonne D. Aminopeptidase-N/CD13 (EC 3.4.11.2) inhibitors: Chemistry, biological evaluations and therapeutic prospects. Med. Res. Rev. 2006;26:88–130. doi: 10.1002/med.20044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mucha A., Drag M., Dalton J.P., Kafarski P. Metallo-aminopeptidase inhibitors. Biochimie. 2010;92:1509–1529. doi: 10.1016/j.biochi.2010.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Amin S.A., Adhikari N., Jha T. Design of aminopeptidase N inhibitors as anti-cancer agents. J. Med. Chem. 2018;61:6468–6490. doi: 10.1021/acs.jmedchem.7b00782. [DOI] [PubMed] [Google Scholar]
- 31.Gonzalez-Bacerio J., Fando R., del Monte-Martinez A., Charli J.-L., de los A Chavez M. Plasmodium falciparum M1-aminopeptidase: A promising target for the development of antimalarials. Curr. Drug Targets. 2014;15:1144–1165. doi: 10.2174/1389450115666141024115641. [DOI] [PubMed] [Google Scholar]
- 32.Umezawa H., Aoyagi T., Suda H., Hamada M., Takeuchi T. Bestatin, an inhibitorof aminopeptidase B, produced by actinomycetes. J. Antibiot. (Tokyo) 1976;29:97–99. doi: 10.7164/antibiotics.29.97. [DOI] [PubMed] [Google Scholar]
- 33.Grembecka J., Mucha A., Cierpicki T., Kafarski P. The most potent organophosphorus inhibitors of leucine aminopeptidase. structure-based design, chemistry and activity. J. Med. Chem. 2003;46:2641–2655. doi: 10.1021/jm030795v. [DOI] [PubMed] [Google Scholar]
- 34.Kannan Sivaraman K., Paiardini A., Sieńczyk M., Ruggeri C., Oellig C.A., Dalton J.P., Scammells P.J., Drag M., McGowan S. Synthesis and structure–activity relationships of phosphonic arginine mimetics as inhibitors of the M1 and M17 aminopeptidases from Plasmodium falciparum. J. Med. Chem. 2013;56:5213–5217. doi: 10.1021/jm4005972. [DOI] [PubMed] [Google Scholar]
- 35.Flipo M., Beghyn T., Leroux V., Florent I., Deprez B.P., Deprez-Poulain R.F. Novel selective inhibitors of the zinc plasmodial aminopeptidase PfA-M1 as potential antimalarial agents. J. Med. Chem. 2007;50:1322–1334. doi: 10.1021/jm061169b. [DOI] [PubMed] [Google Scholar]
- 36.Drinkwater N., Bamert R.S., Sivaraman K.K., Paiardini A., McGowan S. X-ray crystal structures of an orally available aminopeptidase inhibitor, Tosedostat, bound to anti-malarial drug targets P f A-M1 and P f A-M17: Structures of Pf A-M1/M17 Bound to Tosedostat. Proteins Struct. Funct. Bioinform. 2015;83:789–795. doi: 10.1002/prot.24771. [DOI] [PubMed] [Google Scholar]
- 37.Krige D., Needham L.A., Bawden L.J., Flores N., Farmer H., Miles L.E.C., Stone E., Callaghan J., Chandler S., Clark V.L., et al. CHR-2797: An antiproliferative aminopeptidase inhibitor that leads to amino acid deprivation in human leukemic cells. Cancer Res. 2008;68:6669–6679. doi: 10.1158/0008-5472.CAN-07-6627. [DOI] [PubMed] [Google Scholar]
- 38.Schalk C., Dorchymont H., Jauch M.F., Tarnus C. 3-Amino-2-tetralone derivatives: Novel potent and selective inhibitors of aminopeptidase-M (EC-3.4.11.2) Arch. Biochem. Biophys. 1994;311:42–46. doi: 10.1006/abbi.1994.1206. [DOI] [PubMed] [Google Scholar]
- 39.Harbeson S.L., Rich D.H. Inhibition of aminopeptidases by peptides containing ketomethylene and hydroxyethylene amide bond replacements. J. Med. Chem. 1989;32:1378–1392. doi: 10.1021/jm00126a039. [DOI] [PubMed] [Google Scholar]
- 40.Tieku S., Hooper N.M. Inhibition of aminopeptidases N, A and W: A re-evaluation of the actions of bestatin and inhibitors of angiotensin converting enzyme. Biochem. Pharmacol. 1992;44:1725–1730. doi: 10.1016/0006-2952(92)90065-Q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee J., Shim J.S., Jung S.-A., Lee S.-T., Kwon H.J. N-hydroxy-2-(naphthalene-2-ylsulfanyl)-acetamide, a novel hydroxamic acid-based inhibitor of aminopeptidase N and its anti-angiogenic activity. Bioorg. Med. Chem. Lett. 2005;15:181–183. doi: 10.1016/j.bmcl.2004.10.010. [DOI] [PubMed] [Google Scholar]
- 42.Albrecht S., Al-Lakkis-Wehbe M., Orsini A., Defoin A., Pale P., Salomon E., Tarnus C., Weibel J.-M. Amino-benzosuberone: A novel warhead for selective inhibition of human aminopeptidase-N/CD13. Bioorg. Med. Chem. 2011;19:1434–1449. doi: 10.1016/j.bmc.2011.01.008. [DOI] [PubMed] [Google Scholar]
- 43.Gumpena R., Kishor C., Ganji R.J., Addlagatta A. Discovery of α,β- and α,γ-diamino acid scaffolds for the inhibition of M1 family aminopeptidases. ChemMedChem. 2011;6:1971–1976. doi: 10.1002/cmdc.201100298. [DOI] [PubMed] [Google Scholar]
- 44.Velmourougane G., Harbut M.B., Dalal S., McGowan S., Oellig C.A., Meinhardt N., Whisstock J.C., Klemba M., Greenbaum D.C. Synthesis of new (−)-bestatin-based inhibitor libraries reveals a novel binding mode in the S1 pocket of the essential malaria M1 metalloaminopeptidase. J. Med. Chem. 2011;54:1655–1666. doi: 10.1021/jm101227t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Skinner-Adams T.S., Lowther J., Teuscher F., Stack C.M., Grembecka J., Mucha A., Kafarski P., Trenholme K.R., Dalton J.P., Gardiner D.L. Identification of Phosphinate Dipeptide Analog Inhibitors Directed against the Plasmodium falciparum M17 Leucine Aminopeptidase as Lead Antimalarial Compounds. J. Med. Chem. 2007;50:6024–6031. doi: 10.1021/jm070733v. [DOI] [PubMed] [Google Scholar]
- 46.Vassiliou S., Węglarz-Tomczak E., Berlicki Ł., Pawełczak M., Nocek B., Mulligan R., Joachimiak A., Mucha A. Structure-guided, single-point modifications in the phosphinic dipeptide structure yield highly potent and selective inhibitors of neutral aminopeptidases. J. Med. Chem. 2014;57:8140–8151. doi: 10.1021/jm501071f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McGowan S., Porter C.J., Lowther J., Stack C.M., Golding S.J., Skinner-Adams T.S., Trenholme K.R., Teuscher F., Donnelly S.M., Grembecka J., et al. Structural basis for the inhibition of the essential Plasmodium falciparum M1 neutral aminopeptidase. Proc. Natl. Acad. Sci. USA. 2009;106:2537–2542. doi: 10.1073/pnas.0807398106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kokkala P., Mpakali A., Mauvais F.-X., Papakyriakou A., Daskalaki I., Petropoulou I., Kavvalou S., Papathanasopoulou M., Agrotis S., Fonsou T.-M., et al. Optimization and structure–activity relationships of phosphinic pseudotripeptide inhibitors of aminopeptidases that generate antigenic peptides. J. Med. Chem. 2016;59:9107–9123. doi: 10.1021/acs.jmedchem.6b01031. [DOI] [PubMed] [Google Scholar]
- 49.Aeluri R., Ganji R.J., Marapaka A.K., Pillalamarri V., Alla M., Addlagatta A. Highly functionalized tetrahydropyridines are cytotoxic and selective inhibitors of human puromycin sensitive aminopeptidase. Eur. J. Med. Chem. 2015;106:26–33. doi: 10.1016/j.ejmech.2015.10.026. [DOI] [PubMed] [Google Scholar]
- 50.Chauvel E.N., Coric P., Llorens-Cortes C., Wilk S., Roques B.P., Fournie-Zaluski M.-C. Investigation of the active site of aminopeptidase A using a series of new thiol-containing inhibitors. J. Med. Chem. 1994;37:1339–1346. doi: 10.1021/jm00035a014. [DOI] [PubMed] [Google Scholar]
- 51.Umezawa H., Aoyagi T., Tanaka T., Suda H., Okuyama A., Naganawa H., Hamada M., Takeuchi T. Production of actinonin, an inhibitor of aminopeptidase M, by actinomycetes. J. Antibiot. (Tokyo) 1985;38:1629–1630. doi: 10.7164/antibiotics.38.1629. [DOI] [PubMed] [Google Scholar]
- 52.Ansorge S., Neubert K., Bank U., Reichstein I., Faust J., Täger M., Fuchs P., Senns B. Novel Dual Peptidase Inhibitors as Prodrugs for the Therapy of Inflammatory and Other Disorders. [(accessed on 5 April 2018)];2007 Google Patent. Available online: https://patents.google.com/patent/WO2007057128A1/en.
- 53.Deprez-Poulain R., Flipo M., Piveteau C., Leroux F., Dassonneville S., Florent I., Maes L., Cos P., Deprez B. Structure–activity relationships and blood distribution of antiplasmodial aminopeptidase-1 inhibitors. J. Med. Chem. 2012;55:10909–10917. doi: 10.1021/jm301506h. [DOI] [PubMed] [Google Scholar]
- 54.Papakyriakou A., Zervoudi E., Tsoukalidou S., Mauvais F.-X., Sfyroera G., Mastellos D.C., van Endert P., Theodorakis E.A., Vourloumis D., Stratikos E. 3,4-Diaminobenzoic acid derivatives as inhibitors of the oxytocinase subfamily of M1 aminopeptidases with immune-regulating properties. J. Med. Chem. 2015 doi: 10.1021/jm501867s. [DOI] [PubMed] [Google Scholar]
- 55.Kakuta H., Tanatani A., Nagasawa K., Hashimoto Y. Specific nonpeptide inhibitors of puromycin-sensitive aminopeptidase with a 2,4(1H,3H)-quinazolinedione skeleton. Chem. Pharm. Bull. (Tokyo) 2003;51:1273–1282. doi: 10.1248/cpb.51.1273. [DOI] [PubMed] [Google Scholar]
- 56.Mountford S.J., Albiston A.L., Charman W.N., Ng L., Holien J.K., Parker M.W., Nicolazzo J.A., Thompson P.E., Chai S.Y. Synthesis, structure–activity relationships and brain uptake of a novel series of benzopyran inhibitors of insulin-regulated aminopeptidase. J. Med. Chem. 2014;57:1368–1377. doi: 10.1021/jm401540f. [DOI] [PubMed] [Google Scholar]
- 57.Mina-Osorio P. The moonlighting enzyme CD13: Old and new functions to target. Trends Mol. Med. 2008;14:361–371. doi: 10.1016/j.molmed.2008.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Maiereanu C., Schmitt C., Schifano-Faux N., Le Nouën D., Defoin A., Tarnus C. A novel amino-benzosuberone derivative is a picomolar inhibitor of mammalian aminopeptidase N/CD13. Bioorg. Med. Chem. 2011;19:5716–5733. doi: 10.1016/j.bmc.2011.06.089. [DOI] [PubMed] [Google Scholar]
- 59.Albrecht S., Salomon E., Defoin A., Tarnus C. Rapid and efficient synthesis of a novel series of substituted aminobenzosuberone derivatives as potent, selective, non-peptidic neutral aminopeptidase inhibitors. Bioorg. Med. Chem. 2012;20:4942–4953. doi: 10.1016/j.bmc.2012.06.041. [DOI] [PubMed] [Google Scholar]
- 60.Revelant G., Al-Lakkis-Wehbe M., Schmitt M., Alavi S., Schmitt C., Roux L., Al-Masri M., Schifano-Faux N., Maiereanu C., Tarnus C., et al. Exploring S1 plasticity and probing S1′ subsite of mammalian aminopeptidase N/CD13 with highly potent and selective aminobenzosuberone inhibitors. Bioorg. Med. Chem. 2015;23:3192–3207. doi: 10.1016/j.bmc.2015.04.066. [DOI] [PubMed] [Google Scholar]
- 61.Schmitt C., Voegelin M., Marin A., Schmitt M., Schegg F., Hénon P., Guenot D., Tarnus C. Selective aminopeptidase-N (CD13) inhibitors with relevance to cancer chemotherapy. Bioorg. Med. Chem. 2013;21:2135–2144. doi: 10.1016/j.bmc.2012.12.038. [DOI] [PubMed] [Google Scholar]
- 62.Adam W., Hadjiarapoglou L., Wang X. Epoxidation of silyl enol ethers, phthalides and enol esters by dimethyldioxirane. Tetrahedron Lett. 1989;30:6497–6500. doi: 10.1016/S0040-4039(01)89004-3. [DOI] [Google Scholar]
- 63.Solladié-Cavallo A., Lupattelli P., Jierry L., Bovicelli P., Angeli F., Antonioletti R., Klein A. Asymmetric oxidation of silyl enol ethers using chiral dioxiranes derived from α-fluoro cyclohexanones. Tetrahedron Lett. 2003;44:6523–6526. doi: 10.1016/S0040-4039(03)01388-1. [DOI] [Google Scholar]
- 64.Abiraj K., Gowda D.C. Magnesium-catalyzed proficient reduction of oximes to amines using ammonium formate. Synth. Commun. 2004;34:599–605. doi: 10.1081/SCC-120027707. [DOI] [Google Scholar]
- 65.Denis J.-N., Jolivalt C.M., Maurin M.M.L., Jeanty M. Novel Bis-Indolic Derivatives, a Process for Preparing the Same and Their Uses as a Drug. [(accessed on 11 October 2013)];2013 Google Patent. Available online: https://patents.google.com/patent/EP2548864A1/un.
- 66.Groneberg R., Zhan J., Askew B., D’Amico D., Han N., Fotsch C., Liu Q., Riahi B., Zhu J., Yang K., et al. Cyclic Amine Derivatives and Methods of Use. [(accessed on 11 October 2013)];2005 Google Patent. Available online: https://patents.google.com/patent/US7199244B2/en.
- 67.Binet J., Guffroy C., Kasai H., Wagatsuma N. 2-Ureido-Benzamide Derivatives. [(accessed on 11 October 2013)];1999 Google Patent. Available online: https://patents.google.com/patent/US5872115A/en.
- 68.Ipaktschi J. Reductive displacement of the acetate group in allyl, propargyl and benzyl acetates by NaBH4/NiCl2·6·H2O. Chem. Ber. 1984;117:3320–3324. doi: 10.1002/cber.19841171116. [DOI] [Google Scholar]
- 69.Bounaadja L., Schmitt M., Albrecht S., Mouray E., Tarnus C., Florent I. Selective inhibition of PfA-M1, over PfA-M17, by an amino-benzosuberone derivative blocks malaria parasites development in vitro and in vivo. Malar. J. 2017;16 doi: 10.1186/s12936-017-2032-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Papakyriakou A., Stratikos E. The Role of conformational dynamics in antigen trimming by intracellular aminopeptidases. Front. Immunol. 2017;8 doi: 10.3389/fimmu.2017.00946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Addlagatta A., Gay L., Matthews B.W. Structure of aminopeptidase N from Escherichia coli suggests a compartmentalized, gated active site. Proc. Natl. Acad. Sci. USA. 2006;103:13339–13344. doi: 10.1073/pnas.0606167103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Oliveira S.H., Ferraz F.A., Honorato R.V., Xavier-Neto J., Sobreira T.J., de Oliveira P.S. KVFinder: Steered identification of protein cavities as a PyMOL plugin. BMC Bioinform. 2014;15:197. doi: 10.1186/1471-2105-15-197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ganji R.J., Reddi R., Gumpena R., Marapaka A.K., Arya T., Sankoju P., Bhukya S., Addlagatta A. Structural basis for the inhibition of M1 family aminopeptidases by the natural product actinonin: Crystal structure in complex with E. coli aminopeptidase N. Protein Sci. 2015;24:823–831. doi: 10.1002/pro.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Peng G., McEwen A.G., Olieric V., Schmitt C., Albrecht S., Cavarelli J., Tarnus C. Insight into the remarkable affinity and selectivity of the aminobenzosuberone scaffold for the M1 aminopeptidases family based on structure analysis. Proteins Struct. Funct. Bioinform. 2017;85:1413–1421. doi: 10.1002/prot.25301. [DOI] [PubMed] [Google Scholar]
- 75.Dalal S., Ragheb D.R.T., Schubot F.D., Klemba M. A Naturally variable residue in the S1 subsite of M1 family aminopeptidases modulates catalytic properties and promotes functional specialization. J. Biol. Chem. 2013;288:26004–26012. doi: 10.1074/jbc.M113.465625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Addlagatta A., Gay L., Matthews B.W. Structural basis for the unusual specificity of Escherichia coli aminopeptidase N. Biochemistry. 2008;47:5303–5311. doi: 10.1021/bi7022333. [DOI] [PubMed] [Google Scholar]
- 77.Beno B.R., Yeung K.-S., Bartberger M.D., Pennington L.D., Meanwell N.A. A Survey of the role of noncovalent sulfur interactions in drug design. J. Med. Chem. 2015;58:4383–4438. doi: 10.1021/jm501853m. [DOI] [PubMed] [Google Scholar]
- 78.Rosati M., Dalal S., Klemba M. Two cap residues in the S1 subsite of a Plasmodium falciparum M1-family aminopeptidase promote broad specificity and enhance catalysis. Mol. Biochem. Parasitol. 2017;217:7–12. doi: 10.1016/j.molbiopara.2017.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zervoudi E., Papakyriakou A., Georgiadou D., Evnouchidou I., Gajda A., Poreba M., Salvesen G.S., Drag M., Hattori A., Swevers L., et al. Probing the S1 specificity pocket of the aminopeptidases that generate antigenic peptides. Biochem. J. 2011;435:411–420. doi: 10.1042/BJ20102049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Węglarz-Tomczak E., Berlicki Ł., Pawełczak M., Nocek B., Joachimiak A., Mucha A. A structural insight into the P1 S1 binding mode of diaminoethylphosphonic and phosphinic acids, selective inhibitors of alanine aminopeptidases. Eur. J. Med. Chem. 2016;117:187–196. doi: 10.1016/j.ejmech.2016.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Stamogiannos A., Maben Z., Papakyriakou A., Mpakali A., Kokkala P., Georgiadis D., Stern L.J., Stratikos E. Critical role of interdomain interactions in the conformational change and catalytic mechanism of endoplasmic reticulum aminopeptidase 1. Biochemistry. 2017;56:1546–1558. doi: 10.1021/acs.biochem.6b01170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cadel S., Darmon C., Pernier J., Hervé G., Foulon T. The M1 family of vertebrate aminopeptidases: Role of evolutionarily conserved tyrosines in the enzymatic mechanism of aminopeptidase B. Biochimie. 2015;109:67–77. doi: 10.1016/j.biochi.2014.12.009. [DOI] [PubMed] [Google Scholar]
- 83.Blomster M., Wetterholm A., Mueller M.J., Haeggström J.Z. Evidence for a catalytic role of tyrosine 383 in the peptidase reaction of leukotriene A4 hydrolase. Eur. J. Biochem. 1995;231:528–534. doi: 10.1111/j.1432-1033.1995.0528d.x. [DOI] [PubMed] [Google Scholar]
- 84.ADMETlab. [(accessed on 19 July 2018)]; Available online: http://admet.scbdd.com/calcpre/index_sys.
- 85.Lipinski C.A., Lombardo F., Dominy B.W., Feeney P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001;46:3–26. doi: 10.1016/S0169-409X(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 86.Lipinski C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004;1:337–341. doi: 10.1016/j.ddtec.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 87.Fournie-Zaluski M.-C., Fassot C., Valentin B., Djordjijevic D., Goazigo A.R.-L., Corvol P., Roques B.P., Llorens-Cortes C. Brain renin-angiotensin system blockade by systemically active aminopeptidase A inhibitors: A potential treatment of salt-dependent hypertension. Proc. Natl. Acad. Sci. USA. 2004;101:7775–7780. doi: 10.1073/pnas.0402312101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wright J.W., Harding J.W. The brain renin–angiotensin system: A diversity of functions and implications for CNS diseases. Pflüg. Arch. Eur. J. Physiol. 2013;465:133–151. doi: 10.1007/s00424-012-1102-2. [DOI] [PubMed] [Google Scholar]
- 89.Farag E., Sessler D.I., Ebrahim Z., Kurz A., Morgan J., Ahuja S., Maheshwari K., John Doyle D. The renin angiotensin system and the brain: New developments. J. Clin. Neurosci. 2017;46:1–8. doi: 10.1016/j.jocn.2017.08.055. [DOI] [PubMed] [Google Scholar]
- 90.Pal Khaket T., Singh J., Attri P., Dhanda S. Enkephalin degrading enzymes: Metalloproteases with high potential for drug development. Curr. Pharm. Des. 2012;18:220–230. doi: 10.2174/138161212799040547. [DOI] [PubMed] [Google Scholar]
- 91.Karsten S.L., Sang T.-K., Gehman L.T., Chatterjee S., Liu J., Lawless G.M., Sengupta S., Berry R.W., Pomakian J., Oh H.S., et al. A genomic screen for modifiers of tauopathy identifies puromycin-sensitive aminopeptidase as an inhibitor of tau-induced neurodegeneration. Neuron. 2006;51:549–560. doi: 10.1016/j.neuron.2006.07.019. [DOI] [PubMed] [Google Scholar]
- 92.Sengupta S., Horowitz P.M., Karsten S.L., Jackson G.R., Geschwind D.H., Fu Y., Berry R.W., Binder L.I. Degradation of tau protein by puromycin-sensitive aminopeptidase in vitro. Biochemistry. 2006;45:15111–15119. doi: 10.1021/bi061830d. [DOI] [PubMed] [Google Scholar]
- 93.Bhutani N., Venkatraman P., Goldberg A.L. Puromycin-sensitive aminopeptidase is the major peptidase responsible for digesting polyglutamine sequences released by proteasomes during protein degradation. EMBO J. 2007;26:1385–1396. doi: 10.1038/sj.emboj.7601592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Menzies F.M., Hourez R., Imarisio S., Raspe M., Sadiq O., Chandraratna D., O’Kane C., Rock K.L., Reits E., Goldberg A.L., et al. Puromycin-sensitive aminopeptidase protects against aggregation-prone proteins via autophagy. Hum. Mol. Genet. 2010;19:4573–4586. doi: 10.1093/hmg/ddq385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kudo L.C., Parfenova L., Ren G., Vi N., Hui M., Ma Z., Lau K., Gray M., Bardag-Gorce F., Wiedau-Pazos M., et al. Puromycin-sensitive aminopeptidase (PSA/NPEPPS) impedes development of neuropathology in hPSA/TAUP301L double-transgenic mice. Hum. Mol. Genet. 2011;20:1820–1833. doi: 10.1093/hmg/ddr065. [DOI] [PubMed] [Google Scholar]
- 96.Kruppa A.J., Ott S., Chandraratna D.S., Irving J.A., Page R.M., Speretta E., Seto T., Camargo L.M., Marciniak S.J., Lomas D.A., et al. Suppression of Aβ toxicity by puromycin-sensitive aminopeptidase is independent of its proteolytic activity. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2013;1832:2115–2126. doi: 10.1016/j.bbadis.2013.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Voorhis W.C.V., Adams J.H., Adelfio R., Ahyong V., Akabas M.H., Alano P., Alday A., Resto Y.A., Alsibaee A., Alzualde A., et al. Open source drug discovery with the malaria box compound collection for neglected diseases and beyond. PLoS Pathog. 2016;12:e1005763. doi: 10.1371/journal.ppat.1005763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Mpakali A., Saridakis E., Harlos K., Zhao Y., Papakyriakou A., Kokkala P., Georgiadis D., Stratikos E. Crystal structure of insulin-regulated aminopeptidase with bound substrate analogue provides insight on antigenic epitope precursor recognition and processing. J. Immunol. 2015;195:2842–2851. doi: 10.4049/jimmunol.1501103. [DOI] [PubMed] [Google Scholar]
- 99.Mpakali A., Giastas P., Mathioudakis N., Mavridis I.M., Saridakis E., Stratikos E. Structural basis for antigenic peptide recognition and processing by endoplasmic reticulum (ER) aminopeptidase 2. J. Biol. Chem. 2015;290:26021–26032. doi: 10.1074/jbc.M115.685909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Golich F.C., Han M., Crowder M.W. Over-expression, purification and characterization of aminopeptidase N from Escherichia coli. Protein Expr. Purif. 2006;47:634–639. doi: 10.1016/j.pep.2005.11.012. [DOI] [PubMed] [Google Scholar]
- 101.Minick D.J., Frenz J.H., Patrick M.A., Brent D.A. A comprehensive method for determining hydrophobicity constants by reversed-phase high-performance liquid chromatography. J. Med. Chem. 1988;31:1923–1933. doi: 10.1021/jm00118a010. [DOI] [PubMed] [Google Scholar]
- 102.Pomper M.G., VanBrocklin H., Thieme A.M., Thomas R.D., Kiesewetter D.O., Carlson K.E., Mathias C.J., Welch M.J., Katzenellenbogen J.A. 11.beta.-Methoxy-, 11.beta.-ethyl and 17.alpha.-ethynyl-substituted 16.alpha.-fluoroestradiols: Receptor-based imaging agents with enhanced uptake efficiency and selectivity. J. Med. Chem. 1990;33:3143–3155. doi: 10.1021/jm00174a009. [DOI] [PubMed] [Google Scholar]
- 103.Hitzerd S.M., Verbrugge S.E., Ossenkoppele G., Jansen G., Peters G.J. Positioning of aminopeptidase inhibitors in next generation cancer therapy. Amino Acids. 2014;46:793–808. doi: 10.1007/s00726-013-1648-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.