

Abstract
Background
Cardiac hypertrophy has been recognized as an important independent risk factor for the development of heart failure and increases the risk of cardiac morbidity and mortality. A disintegrin and metalloprotease 23 (ADAM23), a member of ADAM family, is involved in cancer and neuronal differentiation. Although ADAM23 is expressed in the heart, the role of ADAM23 in the heart and in cardiac diseases remains unknown.
Methods and Results
We observed that ADAM23 expression is decreased in both failing human hearts and hypertrophic mice hearts. Cardiac‐specific conditional ADAM23‐knockout mice significantly exhibited exacerbated cardiac hypertrophy, fibrosis, and dysfunction, whereas transgenic mice overexpressing ADAM23 in the heart exhibited reduced cardiac hypertrophy in response to pressure overload. Consistent results were also observed in angiotensin II‐induced neonatal rat cardiomyocyte hypertrophy. Mechanistically, ADAM23 exerts anti‐hypertrophic effects by specifically targeting the focal adhesion kinase‐protein kinase B (FAK‐AKT) signaling cascade. Focal adhesion kinase inactivation by inhibitor (PF‐562271) greatly reversed the detrimental effects in ADAM23‐knockout mice subjected to aortic banding.
Conclusion
Altogether, we identified ADAM23 as a negative regulator of cardiac hypertrophy through inhibiting focal adhesion kinase‐protein kinase B signaling pathway, which could be a promising therapeutic target for this malady.
Keywords: a disintegrin and metalloprotease23, Akt, cardiac hypertrophy, focal adhesion kinase
Subject Categories: Cardiomyopathy, Heart Failure, Cell Signalling/Signal Transduction
Clinical Perspective
What Is New?
To our knowledge, this study is the first to define the role of ADAM23 in cardiac hypertrophy.
ADAM23 expression is downregulated in human hearts with hypertrophic cardiomyopathy and dilated cardiomyopathy.
ADAM23 is a key suppressor for cardiac hypertrophy and fibrosis by blocking focal adhesion kinase‐protein kinase B (FAK‐AKT) signaling cascade.
What Are the Clinical Implications?
ADAM23 may represent a new therapeutic target for suppressing the onset of cardiac hypertrophy.
Manipulation of focal adhesion kinase‐protein kinase activity may be a promising therapeutic strategy for cardiac hypertrophy.
Pathological cardiac hypertrophy is characterized by an increase in cardiomyocyte size and dysfunctional cardiac contractility.1, 2 Although cardiac hypertrophy initially might be compensatory and adaptive, prolonged pathological hypertrophy eventually leads to functional and histological deterioration of the myocardium, fibrosis, and altered cardiac gene expression and then congestive heart failure, arrhythmia, and sudden death.1, 3 In fact, clinical and epidemiological studies have identified that cardiac hypertrophy is an important independent risk factor for the development of heart failure and increases the risk of cardiac morbidity and mortality.4 Multiple signaling pathways that involved in the development of cardiac hypertrophy have been identified, eg, mitogen‐activated protein kinase (MAPK) signaling3, 5 and protein kinase B (AKT) signaling.3, 6 However, the key components that determine the pathogenesis and progression of cardiac hypertrophy remain incompletely understood.
A disintegrin and metalloprotease (ADAM)23, a member of transmembrane proteins family, mainly contains a disintegrin and a metalloprotease domain. Most members of ADAM families had been recognized to function in a range of biological processes, including myoblast differentiation and growth factor secretion.7, 8 Later on, several studies revealed that ADAM family also participated in cardiovascular development9, 10, 11, 12, 13 or cardiomyopathies.9, 14, 15, 16 Notably, a previous investigation suggested that ADAM10/12/15/17 expressions were increasing in both human dilated cardiomyopathy and (or) hypertrophic obstructive cardiomyopathy.17 Compared with other family members, the biological role of ADAM23 largely depends on its disintegrin domain, while its metalloprotease domain remains inactive.7 ADAM23 acts principally as a mediator of intercellular association by binding to integrin and/or other cell‐surface molecules,18, 19 which could participate in the pathologic process of cardiac hypertrophy. However, the exact role of ADAM23 and its possible mechanisms for regulating hypertrophic cardiomyopathy remains unclear.
In the present study, we observe that ADAM23 expression is downregulated in human hearts with hypertrophic cardiomyopathy (HCM) and dilated cardiomyopathy (DCM), as well as in aortic banding (AB)‐challenged hypertrophic mice hearts. Employing cardiac‐specific ADAM23 knockout (cADAM23‐KO) and transgenic (ADAM23‐TG) mice, we demonstrate that ADAM23 protects against pressure overload‐induced cardiac hypertrophy though focal adhesion kinase‐protein kinase B (FAK‐AKT) signal pathway.
Materials and Methods
The data, analytic methods, and study materials will not be made available to other researchers for purposes of reproducing the results or replicating the procedure.
Human Heart Samples
The failing human heart samples were collected from patients with DCM or HCM. Non‐failing heart tissues were obtained from patients with brain death or those who died from accidents and whose hearts were unsuitable for heart transplantation because of non‐cardiac reasons. Written informed consents were obtained from individual patients or their legal family members. This study was conducted, according to the Declaration of Helsinki and approved by the Ethics Committees of the Central Hospital of Wuhan, Tongji Medical College, Huazhong University of Science and Technology. Clinical Characteristics of human heart samples are shown in Table S1.
Animal Procedures and Models
Generation of cardiac‐specific ADAM23‐TG mice
ADAM23‐TG (C57BL/6J background) mice were established for cardiac‐specific overexpression of ADAM23 using standard protocol. Briefly, the full length of cDNA for mouse ADAM23 was obtained by polymerase chain reaction (PCR) and after being sequenced, the cDNA sequence was inserted into pCAG‐loxP‐CAT‐loxp‐lacZ by replacing the LacZ gene to generate the plasmid of pCAG‐CAT‐mADAM23, which containing the CAG promoter and the LoxP‐flanked CAT gene. The transgene vector was linearized and was microinjected into fertilized murine embryos (C57BL/6J). The genotypes of offspring mice were characterized by PCR on their tail DNA samples to identify potential transgenic founders. The generated CAG‐CAT‐mADAM23 mice were bred with a‐MHC–MerCreMer mice to generate CAG‐CAT‐mADAM23/a‐MHC–MerCreMer double‐transgenic mice. The CAG‐CAT‐mADAM23/a‐MHC–MerCreMer mice at 6 weeks of age were injected intraperitoneally with 25 mg/kg tamoxifen (T‐5648; Sigma‐Aldrich) daily for five consecutive days to generate cardiac‐specific ADAM23‐TG mice. The CAG‐CAT‐mADAM23/a‐MHC–MerCreMer mice without tamoxifen administration (NTG) served as the control group. TG4 with the highest expression was used.
Generation of cADAM23‐KO mice
The cardiac‐specific conditional ADAM23 knockout (cADAM23‐KO) mice were established using the CRISPR/Cas9 system. The exon 3 coding sequence (CDS) region of mouse ADAM23 gene was cloned and flanked by two mLoxP sequences. The recombinant DNA fragment was further cloned into a vector containing 2 homology arms of 1153 and 1414 bp respectively, to generate a circular donor vector, which was used as template for repairing the double‐strand breaks by homologous recombination. Furthermore, two single‐guide RNAs targeting two locations in the CDS region were designed using available online tools (http://crispr.mit.edu/) and synthesized, and their specificity and function were validated in vitro. Subsequently, the Cas9 mRNA, single‐guide RNAs, and donor vector were injected into C57BL/6J mouse zygotes, which were transplanted into surrogate mother mice to generate 2 founder mice with the floxed CDS regions on the same allele. To confirm whether the floxed allele functioned as expected, the genomic DNA was isolated from resistant ES cells and tested for in vitro Cre/loxP‐mediated recombination using two pairs of primers of F1/R1 and F2/R2 to detect the deletion products and the circle product, respectively. The sequences of primer F1/R1 were 5′‐GATTATGCGGTTCGTTTTGG‐3′ (forward), 5′‐AGGGAGAGGTGGTGATTGTCT‐3′ (reverse); F2/R2 were 5′‐TTCCCCACCTCAAATAACCA‐3′ (forward), 5′‐AGCAGAGACACTGTTTTGATGC‐3′ (reverse), respectively.
All PCR products were confirmed by sequencing. The founder mouse 37‐7 was mated with C57BL/6J female mice to obtain ADAM23‐flox/flox mice, which were crossed with MEM‐Cre transgenic mice (a‐MHC–MerCreMer; MEM‐Cre‐Tg [Myh6‐cre/Esr1, 005650]; Jackson Laboratory, Bar Harbor, ME) to produce ADAM23‐flox/flox/MEM‐Cre mice. Furthermore, the ADAM23‐flox/flox/MEM‐Cre mice at 6 weeks of age were injected intraperitoneally with tamoxifen (25 mg/kg) daily for 5 consecutive days to generate cADAM23‐KO mice. ADAM23‐flox/flox/MEM‐Cre mice without tamoxifen treatment (AFMC) were served as control group.
Aortic Banding
The pressure overload induced cardiac hypertrophy mouse model was established via aortic banding (AB) surgery. Adult (8–10 week old) male mice (body weights of 24–27 g) were subjected to AB or a sham operation as previously described.20, 21 Briefly, the mice were anesthetized via an intraperitoneal injection of sodium pentobarbital (50 mg/kg, Sigma‐Aldrich),the left chest of each mouse was opened to identify the thoracic aorta by blunting dissection at the second intercostal space. We then performed AB using 7‐0 silk suture to ligate the ascending aorta against a 26‐ (for BWs of 26–27 g) or 27‐gauge (for BWs of 24–25 g) needle. After aortic constriction, the needle was quickly removed, the thoracic cavity was closed, and mice were extubated and allowed to recover from the anesthesia. A sham‐operated group underwent a similar procedure without aortic constriction. At 1 week of post‐AB, the focal adhesion kinase (FAK) inhibitor (PF‐562271; obtained from Selleck Chemicals) was administered by gavage to cADAM23‐KO and AFMC mice (15 mg/kg) for 3 weeks. This dosage selected here was based on our preliminary study, which confirmed PF‐562271 treatment at 15 mg/kg significantly inhibited FAK activation. The control group (vehicle) was administered the same volume of 0.5% methylcellulose.
At 4 weeks after AB, the internal diameter and wall thickness of the left ventricle were assessed by echocardiography. The mice were euthanitized and their hearts, lungs and tibiae were collected and weighted for calculating the following ratios: heart weight (HW)/body weight (BW) (mg/g), heart weight (HW)/tibia length (TL) (mg/mm), and lung weight (LW)/body weight (BW) (mg/g).
All experiments were approved by the Animal Care and Use Committee of the Central Hospital of Wuhan, Tongji Medical College, Huazhong University of Science and Technology.
Echocardiography Measurements
Echocardiographic assessments were performed to evaluate cardiac function at 4 weeks post‐sham and 4 weeks post‐AB. In brief, echocardiography was performed on anaesthetized (1.5–2% inhaled isoflurane) mice using a MyLab30CV ultrasound system with a 15‐MHz probe. M‐mode tracings derived from the short axis of the left ventricle at the level of the papillary muscles were recorded. The left ventricular end‐diastolic diameter (LVEDd) and left ventricular end‐systolic diameter (LVESd) were measured at the time of the largest and smallest left ventricle areas, respectively. The percentage of left ventricle fractional shortening (FS) was calculated using the formula: FS (%)=(LVEDd−LVESd)/LVEDd×100%.
Histological Analyses
Four weeks after the AB or sham surgery, the mice were euthanized to assess the parameters of hypertrophic growth and cardiac fibrosis. The hearts of the mice were fixed in 10% formalin and embedded in paraffin according to standard histological protocols. The hearts were then sectioned transversely at 5 μm, in the region close to the apex to visualize the left and right ventricles. The sections were stained with hematoxylin and eosin (H&E) and picrosirius red stainAu to evaluate histopathology and collagen deposition, respectively. The cross‐sectional areas of myocytes were detected via fluorescein isothiocyanate (FITC)‐conjugated wheat germ agglutinin (WGA; Invitrogen) staining. The myocyte cross‐sectional and fibrotic areas were measured using Image‐Pro Plus software (version 6.0) with captured images. To detect death cells in the hearts, terminal deoxynucleotidyl transferase‐mediated dUTP nick end‐labeling (TUNEL) assay was performed according to the manufacturer's protocol (S7111, Millipore).
Western Blot Analysis
Total proteins were extracted from the left ventricle tissues and primary cultured cardiomyocytes. Protein concentrations of samples were then measured by a Pierce BCA Protein Assay Kit (Pierce). Fifty micrograms of protein samples were subjected to SDS‐PAGE (Invitrogen), and then were transferred to a polyvinylidene difluoride membrane (Millipore). After blocking with 5% non‐fat milk in Tris‐buffered saline, membranes were hybridized for overnight at 4°C with the following primary antibodies (Cell Signaling Technology) against phospho‐MEK (P‐MEK) (Cat No. 9154), total‐MEK (T‐MEK) (Cat No. 9122), phospho‐ERK (P‐ERK1/2) (Cat No. 4370), total‐ERK (T‐ERK1/2) (Cat No. 4695), P‐JNK1/2 (Cat No. 4668), T‐JNK1/2 (Cat No. 9252), P‐P38 (Cat No. 4511), T‐P38 (Cat No. 9212), P‐AKT (Cat No. 4060), T‐AKT (Cat No. 4691), P‐GSK3β (Cat No. 9322), T‐GSK3β (Cat No. 9315), P‐mTOR (Cat No. 2971), T‐mTOR (Cat No. 2983), GAPDH (Cat No. 2188), P‐FAK (Tyr397) (Cat No. 3283), T‐FAK (Cat No. BS3583, Bioworld), ADAM23 (Cat No. ARP46365‐p050, Aviva Systems Biology), ANP (Cat No. Sc20158, Santa Cruz Biotechnology), and β‐MHC (Cat No. Sc53090, Santa Cruz Biotechnology). The membranes were then incubated with peroxidase‐conjugated secondary antibodies (Jackson Immuno Research Laboratories, at 1:10 000 dilution), and the protein signals were detected with the ChemiDoc TM XRS+ (Bio‐Rad) system (Bio‐Rad, Hercules, CA, USA). Protein expression levels were normalized to corresponding GAPDH levels.
Quantitative Real‐Time PCR
Total mRNA extraction of left ventricles and cultured cells was performed using TRIZol reagent (Invitrogen), and then was used in a reverse transcriptase reaction to synthesize cDNA using a Transcriptor First Strand cDNA Synthesis Kit (Roche) according to the manufacturer's protocol. Quantitative real‐time PCR amplifications of the indicated genes were quantified by using SYBR Green (Roche). Subsequently, the target gene expression was normalized to Gapdh gene expression. The primers for real‐time PCR are shown as follows: Anp‐mouse: 5′‐ACCTGCTAGACCACCTGGAG‐3′ (forward), 5′‐CCTTGGCTG TTATCTTCGGTACCGG‐3′ (reverse). β‐Mhc–mouse: 5′‐CCGAGTCCCAGG TCAACAA‐3′ (forward), 5′‐CTTCACGGGCACCCTTGGA‐3′ (reverse). Bnp‐mouse: 5′‐GAGGTCACTCCTATCCTCTGG‐3′ (forward), 5′‐GCCATTTCCTCCGACTTTTCTC‐3′ (reverse). Ctgf‐mouse: 5′‐TGACCCCTGCGACCCACA‐3′ (forward), 5′‐TACACCGACCCACCGAAGACACAG‐3′ (reverse). CollagenI‐mouse: 5′‐AGGCTTCAGTGGTTTGGATG‐3′ (forward), 5′‐CACCAACAGCACCATCGTTA‐3′ (reverse). Collagen III‐mouse: 5′‐CCCAACCCAGAGATCCCATT‐3′ (forward), 5′‐GAAGCACAGGAGCAGGTGTAGA‐3′ (reverse). ADAM23‐mouse: 5′‐CCTAGCGCCACCAATCTCATA‐3′ (forward), 5′‐GTGGGATCGAATCTCCTCTTCT‐3′ (reverse). Gapdh‐mouse: 5′‐ACTCCACTCACGGCAAATTC‐3′ (forward), 5′‐TCTCCATGGTGGTGAAGACA‐3′ (reverse).
Primary Cardiomyocyte Culture, Recombinant Adenoviral Infection Vectors and Immunofluorescence
Neonatal rat cardiomyocytes (NRCMs) were prepared from 1‐day‐old Sprague–Dawley rat hearts. Whole hearts from newborn rats were excised, cut into pieces, and digested with trypsin. Collected primary cells after passing through a 40‐μm cell strainer were seeded in tissue culture dishes, to remove fibroblasts using a differential attachment technique. The supernatant containing the cardiomyocytes was collected and NRCMs were seeded onto six‐well culture plates in DMEM/F12 medium containing 20% fetal bovine serum, BrdU (to inhibit fibroblast proliferation), and penicillin/streptomycin. To knockdown ADAM23 expression, rat shADAM23 constructs were cloned into adenovirus. Non‐targeting AdshRNA was used as the control. To overexpress ADAM23, the entire coding region of the rat ADAM23 gene, under the control of a cytomegalovirus promoter, was subcloned into replication‐defective adenoviral vectors to construct the adenoviral ADAM23 (AdADAM23). A similar adenoviral vector encoding the green fluorescent protein gene (AdGFP) was used as a control. After 48 hours, the cultured NRCMs were infected with AdshADAM23, AdshRNA, AdADAM23, or AdGFP at a multiplicity of infection of 100 for 24 hours. The medium was then replaced by DMEM/F12 medium containing 1% fetal bovine serum (serum‐free DMEM/F12 medium) for an additional 12 hours to synchronize the cardiomyocytes. Subsequently, these cells were stimulated with angiotensin II (Ang II, 1 μmol/L) or PBS for 48 hours. By immunofluorescence staining to assess the cell surface area, the cardiomyocytes on glass cover slips were fixed with 100% methanol for 20 minutes at room temperature to quench the AdGFP signal; washed 3 times; permeabilized with 0.1% Triton X‐100; and stained with α‐actinin (Sigma‐Aldrich, A7811, 1:100 dilution) under the guidelines of the standard immunofluorescence staining procedure.
Statistical Analysis
The data were showed as mean±SE. All analyses were performed using SPSS Statistics 16.0 software. For data sets with normal distribution, the differences among the groups were analyzed using one‐way ANOVA followed by the least significant difference (equal variances assumed) or Tamhane's T2 (equal variances not assumed) test. For data sets with skewed distribution, non‐parametric statistical analyses were performed using the Kruskal–Wallis test followed by Dunn's test for multiple comparisons.
Results
ADAM23 Expression is Decreased in Hypertrophic Hearts
To study the potential role of ADAM23 in the development of pathological cardiac hypertrophy, at the beginning, we examined the expression level of ADAM23 protein under cardiac pathological conditions. Compared with donor heart controls, the ADAM23 protein levels were significantly reduced in human hearts with hypertrophic cardiomyopathy (HCM) and dilated cardiomyopathy (DCM) (Figure 1A and 1B, Figure S1A), along with increases in the hypertrophic markers atrial natriuretic peptide (ANP) and β‐myosin heavy chain (β‐MHC) (Figure 1A and 1B). In animal experimental hypertrophic models, ADAM23 protein levels were progressively decreased in mouse hearts from 2 to 4 weeks after aortic banding (AB) surgery compared with sham‐operated controls (Figure 1C). And also, the ADAM23 mRNA levels were decreased in the hearts of mice after AB (Figure S2A). In addition, ADAM23 downregulation was confirmed in cultured neonatal rat cardiomyocytes (NRCMs) stimulated with Ang II (1 μmol/L) for 48 hours to induce hypertrophy (Figure 1D, Figure S2B). Collectively, these findings suggest that ADAM23 may be implicated in the pathogenesis of cardiac hypertrophy.
Figure 1.
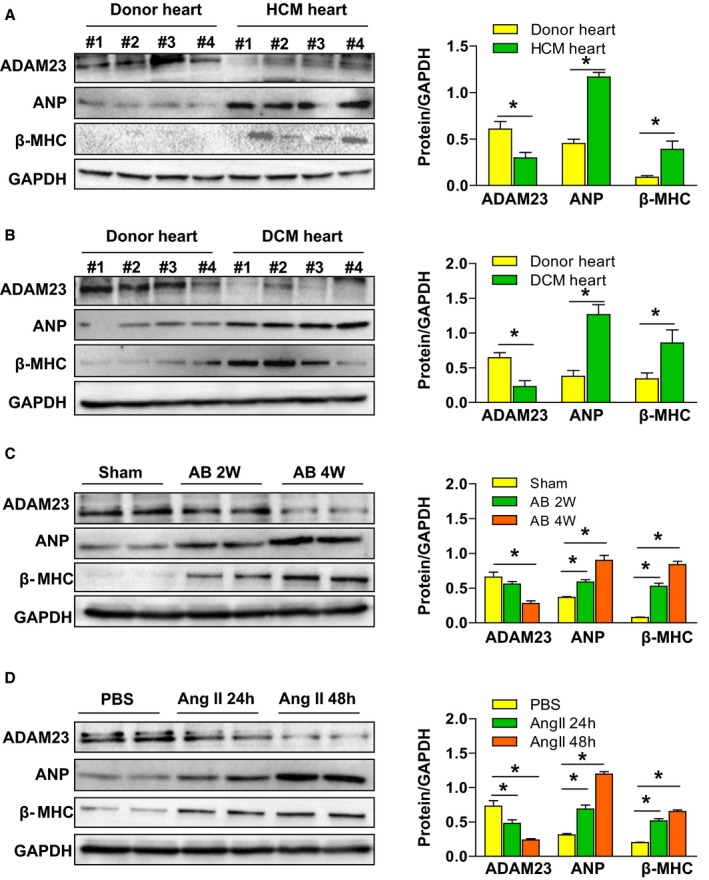
ADAM23 expression is decreased in hypertrophic hearts. A and B, Western blot analysis and quantification of ADAM23, ANP, and β‐MHC protein levels in human heart samples from normal donors and HCM (A) or DCM (B) (n=4 per group, *P<0.05 vs normal hearts). C, Western blot and quantification of ADAM23 and hypertrophic markers (ANP, and β‐MHC) protein levels in the aortic banding (AB)‐induced hypertrophic mouse hearts at the indicated time points (n=4 per group; *P<0.05 vs sham group). D, The expression of ADAM23, ANP, and β‐MHC in neonatal rat cardiomyocytes (NRCMs) treated with PBS, angiotensin II (AngII, 1 μmol/L) for 24 or 48 hours (*P<0.05 vs PBS). AB indicates aortic banding; ANP, atrial natriuretic peptide; β‐MHC, β‐myosin heavy chain; DCM, dilated cardiomyopathy; HCM, hypertrophic cardiomyopathy; NRCM, neonatal rat cardiomyocytes.
Cardiac ADAM23 Deficiency Exacerbates AB‐Induced Hypertrophy
As ADAM23 expression was suppressed in hypertrophic hearts, we explored whether the reduced ADAM23 level affected cardiac hypertrophy and therefore generated cardiac‐specific conditional ADAM23 knockout (cADAM23‐KO) mice (Figure S3A and S3B). Western blot analysis demonstrated a specific ADAM23 deletion in the heart of cADAM23‐KO mice (Figure S3C). Afterwards, cADAM23‐KO and control (AFMC) mice were challenged with AB or sham operation respectively. Then we performed histological analyses at 4 weeks after the operation. Notably, cADAM23‐KO mice did not exhibit marked abnormalities in heart structure or function under basal conditions. However, heart weight/body weight (HW/BW) and HW/tibia length (HW/TL) ratios were dramatically increased in cADAM23‐KO mice compared with the controls after the AB operation for 4 weeks (Figure 2A). Furthermore, AB‐operated cADAM23‐KO mice exhibited remarkably elevated left ventricle end‐diastolic dimension (LVEDd) and decreased fractional shortening (FS) values (Figure 2B). In addition, enlargement of the heart and cardiomyocytes were identified via hematoxylin and eosin (H&E) or wheat germ agglutinin (WGA)‐stained heart sections, and according to the cardiomyocyte cross‐sectional area (CSA) in AB‐operated cADAM23‐KO and control mice (Figure 2C and 2D). The cardiac ADAM23 deficiency leads to a deterioration of fibrosis in both perivascular/interstitial fibrosis area compared with the control group (Figure 2E). Consistently, the mRNA levels of marker genes related to cardiac hypertrophy (Anp, brain natriuretic peptide [Bnp] and β‐Mhc) and fibrosis (collagen I, collagen III and connective tissue growth factor [Ctgf]), were markedly elevated in the hearts of cADAM23‐KO mice compared with the controls (Figure 2F). Collectively, these results indicate that the deficiency of ADAM23 in the heart promotes pressure overload‐induced cardiac hypertrophy.
Figure 2.
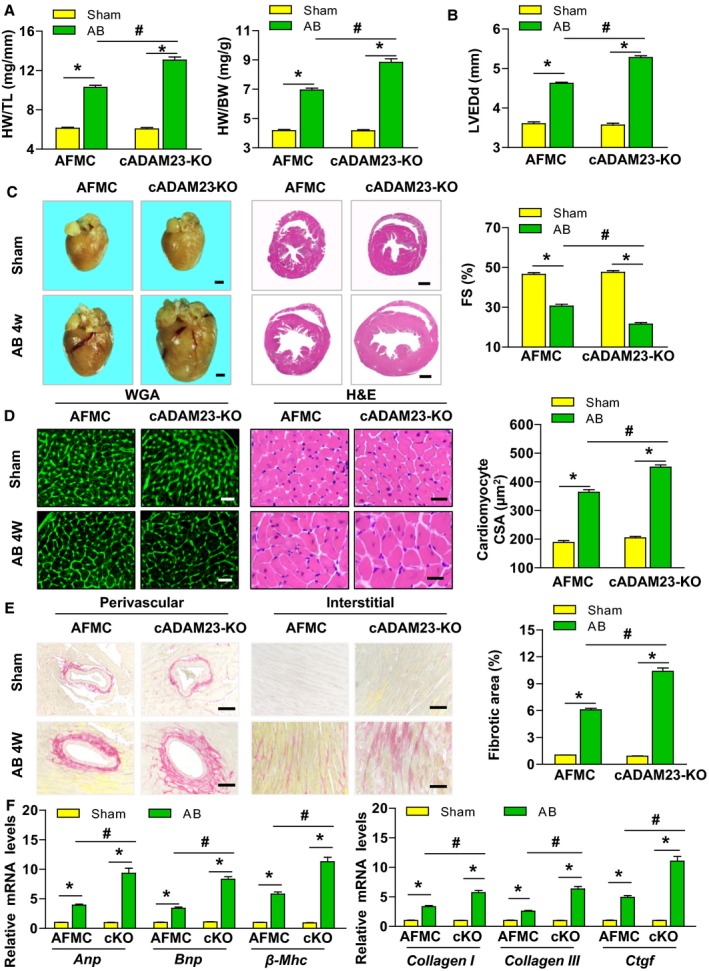
Cardiac ADAM23 deficiency exacerbates aortic banding (AB)‐induced hypertrophy. A, The ratios of heart weight to tibia length (HW/TL) and heart weight to body weight (HW/BW) were determined in the indicated groups 4 weeks after AB surgery (n=13 mice for AFMC sham group, n=12 mice for cADAM23‐KO sham group and AFMC AB group, n=11 mice for cADAM23‐KO AB group). B, Statistical results for the echocardiographic parameters (LVEDd [top] and FS [bottom]) in the indicated groups (n=12 mice for AFMC sham group and AFMC AB group, n=11 mice for cADAM23‐KO sham group and cADAM23‐KO AB group). C, Histological analysis of the whole hearts (left) and heart sections stained with H&E (right) from the indicated groups 4 weeks after sham or AB surgery (n=6 mice per group; scale bar, 1 mm). D, Representative images of heart sections stained with H&E and WGA staining, and statistical results for the cardiomyocyte CSA (n>100 cells per group; scale bar, 20 μm). E, Representative images of histological analysis of cardiac perivascular and interstitial fibrosis, and statistical results for the fibrotic area (n>40 fields per group; scale bar, 20 μm). F, Quantification results for mRNA levels of the hypertrophic marker genes (Anp, Bnp and β‐Mhc) and fibrotic marker genes (collagen I, collagen III, and Ctgf) in the indicated groups (n=4 independent experiments). AB indicates aortic banding; ANP, atrial natriuretic peptide; CSA, cross‐sectional area; DCM, dilated cardiomyopathy; FS, fractional shortening; H&E, hematoxylin and eosin; HCM, hypertrophic cardiomyopathy; HW/BW, heart weight to body weight; HW/TL, heart weight to tibia length; KO, knockout; LVEDd, left ventricular end‐diastolic diameter, WGA, wheat germ agglutinin. *P<0.05 vs sham; # P<0.05 vs AFMC AB group in (A, B, D through F).
Cardiac ADAM23 Overexpression Blunts AB‐Induced Hypertrophy
To further corroborate the role of ADAM23 in cardiac hypertrophy, we generated transgenic mice with cardiac‐specific conditional ADAM23 overexpression (Figure S4A). Four ADAM23‐TG mouse lines were generated, and Western blot analysis revealed ADAM23 overexpression in the heart (Figure S4B). The offspring of the TG4 lines were then selected for further experiments. After 4 weeks of AB, ADAM23‐TG mice showed a notable alleviation of cardiac enlargement and cardiac dysfunction compared with nontransgenic (NTG) mice, as described by the decreased HW/BW and HW/TL ratios (Figure 3A) and ameliorated cardiac dysfunction (Figure 3B). The histological analysis indicated that ADAM23 overexpression attenuated drastically the AB‐induced cardiac hypertrophy and interstitial and perivascular fibrosis (Figure 3C through 3E). Moreover, the transcription levels of hypertrophic and fibrotic markers genes were significantly suppressed in the ADAM23‐TG mice compared with heart samples from the NTG controls (Figure 3F). All together, these data suggest that cardiac ADAM23 overexpression blunts AB‐induced hypertrophy in vivo compared with the controls.
Figure 3.
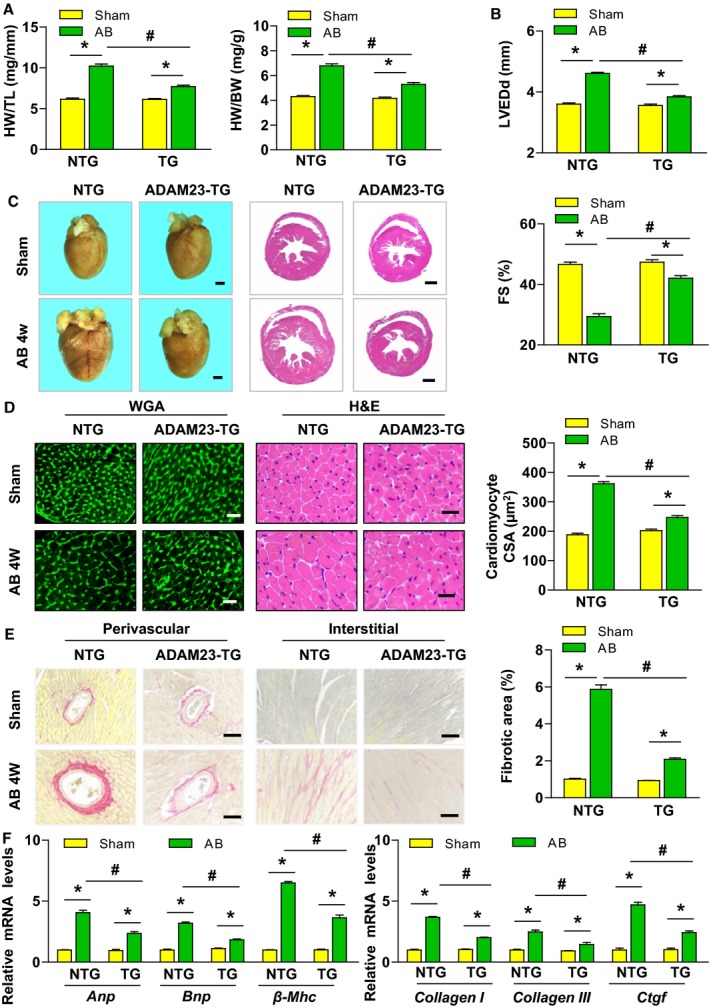
Cardiac ADAM23 overexpression attenuates aortic banding (AB)‐induced hypertrophy. A, Comparison of the HW/TL, and HW/BW ratios in different genotypic mice after sham or AB surgery (n=11 mice for NTG sham group, n=13 mice for TG sham group and NTG AB group, n=12 mice for TG AB group). B, Comparison of the echocardiographic parameters LVEDd and FS in the indicated groups (n=11 mice for NTG sham group, n=13 mice for TG sham group and NTG AB group, n=12 mice for TG AB group). C, Histological analysis of heart sections stained with H&E and WGA staining, and statistical results for the cardiomyocyte CSA) in both NTG and ADAM23‐TG groups (n=6 mice per group; scale bar, 1 mm). D, Representative images of heart sections stained with H&E and WGA staining, and statistical results for the cardiomyocyte CSA (n>100 cells per group; scale bar, 20 μm). E, Representative images of cardiac perivascular and interstitial fibrosis, and statistical results for the fibrotic area (n>40 fields per group; scale bar, 20 μm). F, Quantification results for mRNA levels of the hypertrophic marker genes (Anp, Bnp and, β‐Mhc) and fibrotic marker genes (collagen I, collagen III, and Ctgf) in the indicated groups (n=4 independent experiments). ANP indicates atrial natriuretic peptide; CSA, cross‐sectional area; DCM, dilated cardiomyopathy; FS, fractional shortening; H&E, hematoxylin and eosin; HCM, hypertrophic cardiomyopathy; HW/TL, heart weight to tibia length; HW/BW, heart weight to body weight; LVEDd, left ventricular end‐diastolic diameter; NTG, nontransgenic; TG, transgenic; WGA, wheat germ agglutinin. *P<0.05 vs sham; # P<0.05 vs NTG AB group in (A, B, D through F).
ADAM23 Inhibits AngII‐Induced Cardiomyocyte Hypertrophy In Vitro
We next examined whether the role of ADAM23 in cardiac hypertrophy is directly regulated by the effect of ADAM23 on cardiomyocytes. To test this, we performed loss‐ and gain‐of‐ function experiments in cultured NRCMs. NRCMs were infected with AdshADAM23 to knock down ADAM23 or with AdADAM23 to overexpress ADAM23 (Figure 4A). The cells were subsequently treated with either Ang II (1 μmol/L) or PBS (control) for 48 hours, and then were immunostained with α‐actinin antibody to measure cell size or collected for the analysis of fetal cardiac gene expression. Compared with AdshRNA‐infected controls, ADAM23 depletion cells remarkably increased the cell surface area and the mRNA levels of hypertrophic markers (Anp and β‐Mhc) in response to AngII (Figure 4B and 4C). Conversely, ADAM23 overexpression led to a decrease in these hypertrophic phenotypes (Figure 4D and 4E). These findings confirm that ADAM23 also regulates the development of AngII‐triggered cardiomyocyte hypertrophy in vitro.
Figure 4.
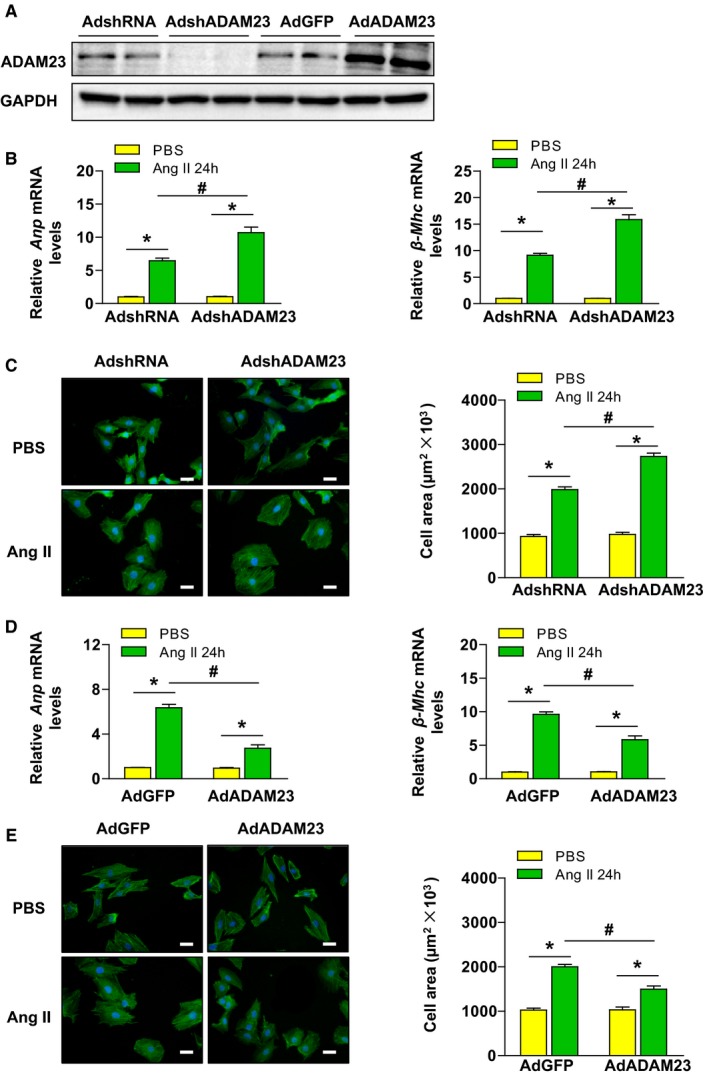
ADAM23 inhibits AngII–induced cardiomyocyte hypertrophy in vitro. A, The protein expression level of ADAM23 in NRCMs after infection with AdshRNA, AdshADAM23, AdGFP or AdADAM23 was determined by Western blotting (n=4 samples per group). B, mRNA levels of the hypertrophic marker genes (ANP and β‐MHC) in NRCMs infected with AdshRNA or AdshADAM23 treated with AngII (1 μmol/L) or PBS for 48 hours (n=4 independent experiments). C, Representative images of a‐actinin (green) and DAPI (blue) stained cardiomyocytes infected with the indicated adenoviruses followed by 48 hours of PBS or AngII treatment, and quantification of the average cell surface area of rat cardiomyocytes (n>50 cells per group; scale bar, 20 μm). D, The relative levels of hypertrophic markers mRNAs in NRCMs infected with AdGFP and AdADAM23 (n=4 independent experiments. E, Microscopic images of NRCMs were infected with the indicated adenovirus followed by treated with PBS or Ang II, and quantification cardiomyocyte size (n>40 cells per group; scale bar, 20 μm). ANP indicates atrial natriuretic peptide; β‐MHC, β‐myosin heavy chain; NRCM, neonatal rat cardiomyocytes. *P<0.05 vs PBS; # P<0.05 vs AdshRNA AngII.
ADAM23 Regulates FAK/AKT Signaling During Cardiac Hypertrophy
Next, we explored the cellular mechanisms by which ADAM23 protects against cardiac hypertrophy. To elucidate these possible mechanisms, we first investigated the expression and activity of mitogen‐activated protein kinase (MAPK) signaling molecules, as the MAPK pathway is known to play a fundamental role in pathological cardiac hypertrophy.3, 5 Compared with the sham operation, pressure overload by AB operation caused an increase in phosphorylation of MAPK signal pathway members. Unexpectedly, MAPK activities were affected by neither ADAM23 deficiency nor overexpression in heart tissue (Figure 5A). According to previous studies, the FAK/AKT signaling cascade is another signaling pathway which plays an important role in the process of cardiac hypertrophy.22, 23 On the contrary, we found that FAK phosphorylation levels and its downstream molecules AKT/GSK3β/mTOR were significantly elevated in the cADAM23‐KO group and blunted in the ADAM23‐TG group in AB‐induced hypertrophy compared with the controls in vivo (Figure 5B). In accordance with the results in vivo, levels of FAK/AKT signaling pathway molecules were increased or decreased after ADAM23 knockdown or overexpression in AngII–treated NRCMs (Figure 5C). Given that FAK/AKT signaling was associated with cardiomyocyte death,24, 25 terminal deoxynucleotidyl transferase‐mediated dUTP nick end‐labeling assay and Western blot analysis for cleaved caspase 3 were performed to assess whether ADAM23 regulates the cell death process in response to pressure overload. However, neither ADAM23 deletion nor ADAM23 overexpression in the heart affects cardiomyocyte death after AB surgery, as compared with their corresponding controls (Figure S5A and S5B).
Figure 5.
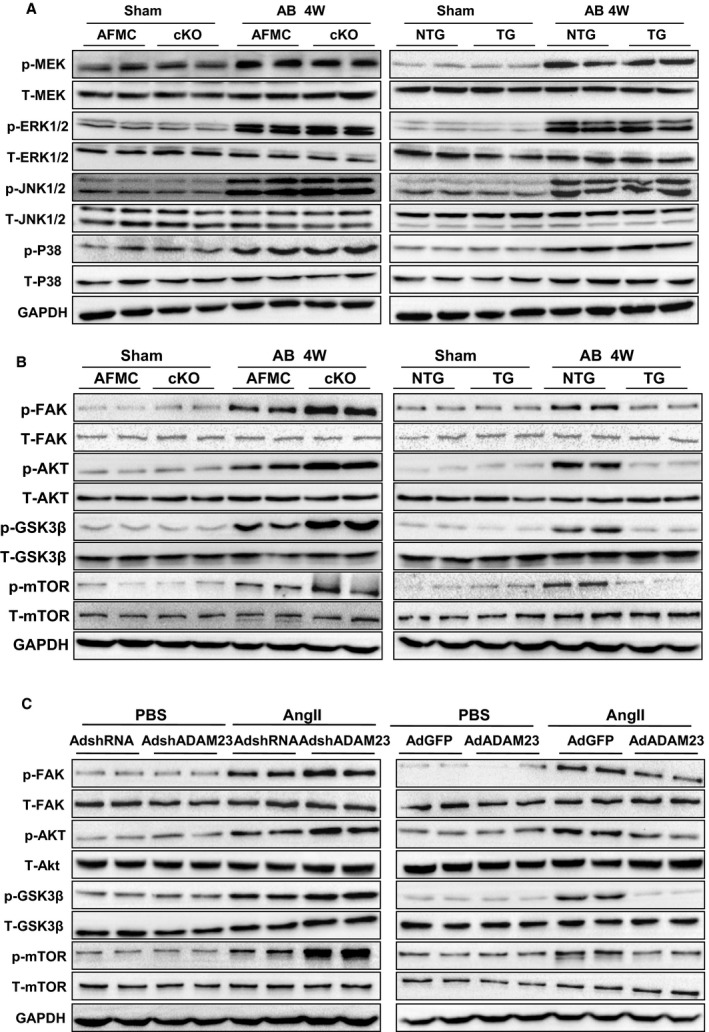
ADAM23 inhibits activation of FAK‐dependent signaling pathways upon hypertrophic stresses. A, Representative Western blot and quantification results of the phosphorylation and total protein levels of MEK, ERK1/2, JNK1/2 and P38 in the hearts of AFMC, cADAM23‐KO, NTG and ADAM23‐TG mice 4 weeks after sham or AB surgery (n=4 mice per group). B, Representative Western blot and quantification results of the phosphorylation and total protein levels of FAK, AKT, GSK3β, m‐TOR in the indicated groups (n=4 mice per group). C, Representative results for Western blot of FAK and its downstream protein levels in NRCMs infected with AdshRNA, AdshADAM23, AdGFP or AdADAM23 treated with AngII or PBS. AB indicates aortic banding; FAK, focal adhesion kinase; NRCM, neonatal rat cardiomyocytes; NTG, nontransgetic.
FAK Blockage Ameliorates Cardiac Hypertrophy in cADAM23‐KO Mice
To further confirm the hypothesis that ADAM23 exerts a cardio‐protective function by inhibiting FAK/AKT signaling pathways, cADAM23‐KO and AFMC mice were treated with FAK inhibitor (PF‐562271). Western blot analysis was taken to confirm the inhibition efficiency of PF‐562271 (Figure 6A). Compared with the vehicle, PF‐562271 almost diminished the phosphorylation of FAK/AKT‐dependent signaling pathways in AB‐operated hearts (Figure 6A). In addition, FAK inactivation significantly attenuated the AB‐induced hypertrophic response characterized by reducing the ratios of HW/TL and HW/BW (Figure 6B); improved cardiac function (Figure 6C); decreased gross size of the heart (Figure 6D), cardiomyocyte cross‐sectional area (Figure 6E and 6F) and cardiac fibrosis (Figure 6G) in cADAM23‐KO mice treated with PF‐562271 compared with vehicle control. In summary, these data robustly implicate the critical involvement of FAK/AKT signaling in ADAM23‐mediated regulation of cardiac hypertrophy.
Figure 6.
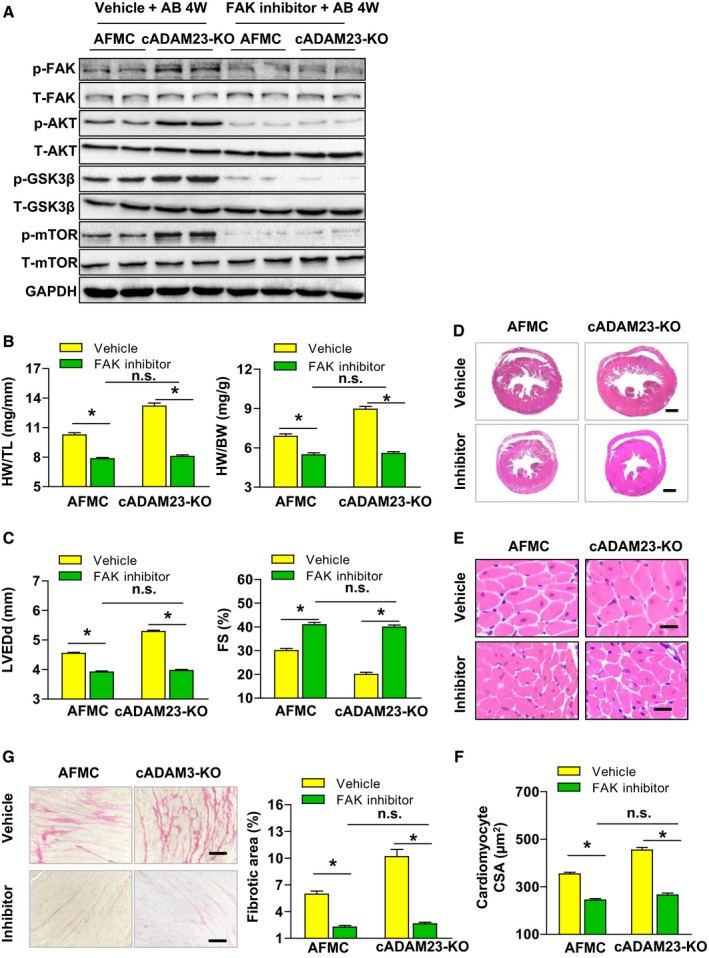
FAK inhibitor blunted aortic banding (AB)‐induced cardiac hypertrophy in cADAM23‐KO mice in vivo. A, AFMC and cADAM23‐KO mice were treated with FAK inhibitor (PF‐562271) or vehicle for control after 4 weeks of AB surgery. Western blot analysis and statistical results of the phosphorylation and total protein levels of FAK, AKT, m‐TOR and GSK3β in heart tissues in the indicated groups (n=4 per group). B, Statistical results for HW/BW, and HW/TL ratios (n=12 mice per group). C, Statistical results for the echocardiographic parameters LVEDd and FS in indicated groups (n=12 mice for AFMC vehicle group, n=11 mice for cADAM23‐KO vehicle group and cADAM23‐KO inhibitor group, n=10 mice for AFMC inhibitor group). D and E, Representative images of the histological analysis of cardiac hypertrophy, as indicated by the whole heart (D) and heart sections (E) stained with H&E in cADAM23‐KO and AFMC mice treated with PF‐562271 or vehicle (for D, scale bars, 1 mm; for E, scale bar, 20 μm). F, Statistical results for the cardiomyocyte CSA in the indicated groups (n>100 cells per group). G, Representative images of cardiac fibrosis and statistical results for the fibrotic area (n>40 fields per group, scale bar, 20 μm). For (B, C, F, and G), CSA indicates cross‐sectional area, FAK, focal adhesion kinase; H&E, hematoxylin and eosin; HW/BW, heart weight to body weight; HW/TL, heart weight to tibia length; LVEDd, left ventricular end‐diastolic diameter; NRCM, neonatal rat cardiomyocytes; n.s., no significance. *P<0.05 vs vehicle.
Discussion
The present study reveals the previously unrecognized biological function of ADAM23 in cardiac hypertrophy. In this study, we identified ADAM23 as an important negative regulator of pathological cardiac hypertrophy. ADAM23 deficiency in cardiomyocyte promoted pressure overload‐induced hypertrophy. In contrast, cardiomyocyte ADAM23 overexpression blunted the AB‐induced hypertrophic response. Consistent with animal studies, in vitro experiments in NRCMs indicated that ADAM23 knockdown significantly enhanced AngII‐induced hypertrophy, whereas ADAM23 overexpression inhibited this hypertrophic response in response to AngII. Furthermore, ADAM23 protected against cardiac hypertrophy in a FAK/AKT‐dependent manner. FAK inactivation by inhibitor (PF‐562271) significantly rescued the adverse effect of AB‐induced cardiac hypertrophy in cADAM23‐KO mice.
ADAM families have been shown to be involved in cardiovascular development or cardiomyopathies.9 A previous investigation suggested that ADAM10 and ADAM15 expressions were increased in human DCM; ADAM12 was elevated in hypertrophic obstructive cardiomyopathy; ADAM17 was upregulated in both DCM and hypertrophic obstructive cardiomyopathy.17 Moreover, inhibition of HB‐EGF shedding by ADAM12 could be a potent therapeutic strategy for cardiac hypertrophy.14 However, Cardiomyocyte‐specific ADAM17 knockdown mice enhanced myocardial hypertrophy, fibrosis, more severe left ventricular dilation, and systolic dysfunction at 5 weeks post transverse aortic constriction.16 We found that ADAM23 expression was downregulated in failing human hearts, AB‐induced hypertrophic mouse hearts, and NRCMs treated with AngII. On the basis of these findings, we hypothesized that ADAM23 plays an important role in cardiac hypertrophy. Our experimental results confirmed that ADAM23 indeed plays a protective role in cardiac hypertrophy.
Other than regulating pathological cardiac hypertrophy, our results show that ADAM23 ameliorated cardiac fibrosis in mice after 4 weeks AB operation. In our opinion, this effect of ADAM23 on cardiac fibrosis is possibly secondary to its alleviation on cardiac hypertrophy. Cardiac hypertrophy and fibrosis would affect each other during heart failure.26 On the other hand, ADAM23 could directly affect the pathogenesis of fibrosis through the same downstream targets FAK, as is in regulating cardiac hypertrophy. Previous studies provided evidence that FAK is involved in cardiac fibrosis in vivo and in vitro, and targeted inhibition of FAK could attenuate cardiac fibrosis and preserve partial cardiac function.23, 27
To investigate the molecular mechanism by which ADAM23 mediates its effects on cardiac hypertrophy, we first examined the MAPK signaling pathway, a pivotal role in the development of cardiac hypertrophy.3 Unexpectedly, we found that MAPK activities were affected by neither ADAM23 deficiency nor overexpression, which indicated that the anti‐hypertrophic effects of ADAM23 were independent of MAPK signaling. In contrast, FAK phosphorylation levels and its downstream AKT‐GSKβ‐mTOR were significantly elevated in the cADAM23‐KO group and blunted in the ADAM23‐TG group in AB‐induced hypertrophy compared with the controls. In addition, we applied FAK inhibitor (PF‐562271) in cADAM23‐KO mice to further confirm the role of FAK/AKT signaling pathway in ADAM23‐mediated regulation of cardiac hypertrophy. Of note, several previous researches supported our findings: FAK deletion attenuates load‐induced hypertrophy,23, 27 while increased FAK activity causes cardiac hypertrophy.22 Importantly, by using the same FAK inhibitor as used in our study, Clemente et al demonstrated that FAK activation causes cardiac concentric hypertrophy by activating the AKT and mTOR pathways.22 However, some studies also reported that deletion of FAK in cardiomyocytes promotes eccentric cardiac hypertrophy in mice.28, 29 Their findings were obtained from cardiomyocyte FAK conditional knockout mice since the embryonic stage, which is different from our studies using FAK inhibitor in 8 weeks‐age mice. The reason for the discrepancies between these studies is unclear, but it might be related to the differences in the mice construction and experiment conditions. Besides, enhancing cardiac FAK activity could attenuate ischemia/reperfusion‐induced myocyte apoptosis,25 however, our results showed that ADAM23 had no effect on cardiomyocyte death during cardiac hypertrophy. These results indicated that modulation FAK activity may lead to different results in response to different situation.
FAK is a non‐receptor tyrosine kinase, which was identified as one of the first integrin signaling molecules.30 As a class of membrane receptors, integrins are major players in transmitting the mechanical force across the plasma membrane and sensing the mechanical load in cardiomyocytes.31 In the integrin initiating signaling pathway, clustering of integrins leads to the recruitment of FAK to the newly formed focal adhesion which results in auto‐phosphorylation and concomitant activation of FAK.30, 32 A recent study supported a role for integrin‐mediated signaling through FAK in the development of cardiac hypertrophy.33 Furthermore, Aikawa and his colleagues demonstrated that the activation of integrin‐FAK might be necessary for stretch‐induced hypertrophic responses.34 Importantly, ADAM23 could directly interact with αvβ3 integrin through its disintegrin domain. By this way, ADAM23 negatively modulates αvβ3 integrin activation and its expression.18 Based on these findings, we postulate that ADAM23 inhibiting FAK/AKT signaling through constraining integrin to regulate cardiac hypertrophy. However, more studies need be further conducted to clearly elucidate the action model of ADAM23‐integrin‐FAK/AKT signaling pathway.
To our knowledge, this study is the first to define the role of ADAM23 in cardiac hypertrophy. Our present work provides in vivo and in vitro evidence to support the concept that ADAM23 protects against pressure overload–induced cardiac hypertrophy through negative regulation of FAK/AKT signaling pathway. These findings suggest that ADAM23 may represent a new therapeutic target for suppressing the onset of cardiac hypertrophy.
Author Contributions
Xiang, Luo, J. Wu and Dr Ren contributed equally to this work. Xiang, Prof M. Chen and Prof Xia participated in the research design. Luo, Ren, Ding, C. Wu, J. Chen, S. Chen, Zhang, Yu, Zou, Xu participated in the performance of the research. Xiang, Luo and J. Wu participated in the writing of the article. Dr Ye, Prof M. Chen and Prof Xia participated in the data analysis.
Sources of Funding
This study was supported by the National Natural Science Foundation of China (No. 81600186).
Disclosures
None.
Supporting information
Table S1. Clinical Characteristics of Human Heart Samples
Figure S1. Western blot analysis of ADAM23 in human heart samples (No. 5 and 6; n=2 per group) from normal donors, hypertrophic cardiomyopathy (HCM) and dilated cardiomyopathy (DCM).
Figure S2. A, The ADAM23 mRNA levels in the hearts of mice at 4 weeks post the sham or aortic banding (AB) surgery (n=4 independent experiments. *P<0.05 vs the sham group). B, The mRNA levels of ADAM23 in cardiomyocytes in response to PBS or Ang II for 24 hours (n=3 independent experiments; *P<0.05 vs PBS group).
Figure S3. Schematic diagram of the construction of cardiac‐specific conditional ADAM23 (cADAM23‐KO) and identification of ADAM23 expression. A, Schematic illustration of cADAM23‐KO generation. B, Amplification of the entire region covering the floxed and homology arm using the F1/R1 primer (left) and the circle excised by Cre using the F2/R2 primer (right). C, Western blot of ADAM23 expression levels in different tissues of WT and cADAM23‐KO mice (n=4 per group).
Figure S4. Generation of cardiac‐specific ADAM23 transgenic (ADAM23‐TG) mice. A, Schematic diagram for the construction of the cardiac‐specific expression of the ADAM23‐transgenic (ADAM23‐TG) mice. B, Western blot analysis of ADAM23 levels in the ADAM23‐TG mice and their NTG littermates (n=4 per group).
Figure S5. ADAM23 had no effect on cardiomyocyte death during cardiac hypertrophy. A, terminal deoxynucleotidyl transferase‐mediated dUTP nick end‐labeling assays in the heart sections of cADAM23‐KO and ADAM23‐TG mice, compared with their respective controls (n=5 mice per group; scale bar, 20 μm). B, Western blot analysis of cleaved caspase3 (C‐Caspase3) in the hearts of cADAM23‐KO and ADAM23‐TG mice, compared with their respective controls (n=4 mice per group). Gapdh served as a loading control.
Acknowledgments
We greatly thank Dr Hongliang Li (Wuhan University) and Keqiong Deng for providing the ADAM23 conditional knockout and transgenic mice and other experimental and technological assistance.
(J Am Heart Assoc. 2018;7:e008604 DOI: 10.1161/JAHA.118.008604.)
Contributor Information
Ping Ye, Email: blue314@163.com.
Manhua Chen, Email: cmh_centre@163.com.
Jiahong Xia, Email: jiahong.xia@hust.edu.cn.
References
- 1. Levy D, Kenchaiah S, Larson MG, Benjamin EJ, Kupka MJ, Ho KK, Murabito JM, Vasan RS. Long‐term trends in the incidence of and survival with heart failure. N Engl J Med. 2002;347:1397–1402. [DOI] [PubMed] [Google Scholar]
- 2. Heineke J, Molkentin JD. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat Rev Mol Cell Biol. 2006;7:589–600. [DOI] [PubMed] [Google Scholar]
- 3. van Berlo JH, Maillet M, Molkentin JD. Signaling effectors underlying pathologic growth and remodeling of the heart. J Clin Invest. 2013;123:37–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tamargo J, Lopez‐Sendon J. Novel therapeutic targets for the treatment of heart failure. Nat Rev Drug Discov. 2011;10:536–555. [DOI] [PubMed] [Google Scholar]
- 5. Li L, Chen W, Zhu Y, Wang X, Jiang DS, Huang F, Wang L, Xiang F, Qin W, Wang Q, Zhang R, Zhu X, Li H, Chen X. Caspase recruitment domain 6 protects against cardiac hypertrophy in response to pressure overload. Hypertension. 2014;64:94–102. [DOI] [PubMed] [Google Scholar]
- 6. Deng KQ, Wang A, Ji YX, Zhang XJ, Fang J, Zhang Y, Zhang P, Jiang X, Gao L, Zhu XY, Zhao Y, Gao L, Yang Q, Zhu XH, Wei X, Pu J, Li H. Suppressor of IKKɛ is an essential negative regulator of pathological cardiac hypertrophy. Nat Commun. 2016;7:11432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Primakoff P, Myles DG. The ADAM gene family: surface proteins with adhesion and protease activity. Trends Genet. 2000;16:83–87. [DOI] [PubMed] [Google Scholar]
- 8. Reiss K, Saftig P. The, “a disintegrin and metalloprotease” (ADAM) family of sheddases: physiological and cellular functions. Semin Cell Dev Biol. 2009;20:126–137. [DOI] [PubMed] [Google Scholar]
- 9. Zhang P, Shen M, Fernandez‐Patron C, Kassiri Z. ADAMs family and relatives in cardiovascular physiology and pathology. J Mol Cell Cardiol. 2016;93:186–199. [DOI] [PubMed] [Google Scholar]
- 10. Vuohelainen V, Raitoharju E, Levula M, Lehtimaki T, Pelto‐Huikko M, Honkanen T, Huovila A, Paavonen T, Tarkka M, Mennander A. Myocardial infarction induces early increased remote ADAM8 expression of rat hearts after cardiac arrest. Scand J Clin Lab Invest. 2011;71:553–562. [DOI] [PubMed] [Google Scholar]
- 11. Al‐Fakhri N, Wilhelm J, Hahn M, Heidt M, Hehrlein FW, Endisch AM, Hupp T, Cherian SM, Bobryshev YV, Lord RS, Katz N. Increased expression of disintegrin‐metalloproteinases ADAM‐15 and ADAM‐9 following upregulation of integrins α5β1 and αvβ3 in atherosclerosis. J Cell Biochem. 2003;89:808–823. [DOI] [PubMed] [Google Scholar]
- 12. Hartmann D, de Strooper B, Serneels L, Craessaerts K, Herreman A, Annaert W, Umans L, Lubke T, Lena Illert A, von Figura K, Saftig P. The disintegrin/metalloprotease ADAM 10 is essential for Notch signalling but not for α‐secretase activity in fibroblasts. Hum Mol Genet. 2002;11:2615–2624. [DOI] [PubMed] [Google Scholar]
- 13. Kurohara K, Komatsu K, Kurisaki T, Masuda A, Irie N, Asano M, Sudo K, Nabeshima Y, Iwakura Y, Sehara‐Fujisawa A. Essential roles of Meltrin β (ADAM19) in heart development. Dev Biol. 2004;267:14–28. [DOI] [PubMed] [Google Scholar]
- 14. Asakura M, Kitakaze M, Takashima S, Liao Y, Ishikura F, Yoshinaka T, Ohmoto H, Node K, Yoshino K, Ishiguro H, Asanuma H, Sanada S, Matsumura Y, Takeda H, Beppu S, Tada M, Hori M, Higashiyama S. Cardiac hypertrophy is inhibited by antagonism of ADAM12 processing of HB‐EGF: metalloproteinase inhibitors as a new therapy. Nat Med. 2002;8:35–40. [DOI] [PubMed] [Google Scholar]
- 15. Wang X, Oka T, Chow F L, Cooper SB, Odenbach J, Lopaschuk GD, Kassiri Z, Fernandez‐Patron C. Tumor necrosis factor‐alpha‐converting enzyme is a key regulator of agonist‐induced cardiac hypertrophy and fibrosis. Hypertension. 2009;54:575–582. [DOI] [PubMed] [Google Scholar]
- 16. Fan D, Takawale A, Shen M, Samokhvalov V, Basu R, Patel V, Wang X, Fernandez‐Patron C, Seubert JM, Oudit GY, Kassiri Z. A disintegrin and metalloprotease‐17 regulates pressure overload‐induced myocardial hypertrophy and dysfunction through proteolytic processing of integrin β1. Hypertension. 2016;68:937–948. [DOI] [PubMed] [Google Scholar]
- 17. Fedak PW, Moravec CS, McCarthy PM, Altamentova SM, Wong AP, Skrtic M, Verma S, Weisel RD, Li RK. Altered expression of disintegrin metalloproteinases and their inhibitor in human dilated cardiomyopathy. Circulation. 2006;113:238–245. [DOI] [PubMed] [Google Scholar]
- 18. Verbisck NV, Costa ET, Costa FF, Cavalher FP, Costa MD, Muras A, Paixao VA, Moura R, Granato MF, Ierardi DF, Machado T, Melo F, Ribeiro KB, Cunha IW, Lima VC, Maciel Mdo S, Carvalho AL, Soares FF, Zanata S, Sogayar MC, Chammas R, Camargo AA. ADAM23 negatively modulates α(v)β(3) integrin activation during metastasis. Cancer Res. 2009;69:5546–5552. [DOI] [PubMed] [Google Scholar]
- 19. Costa MD, Paludo KS, Klassen G, Lopes MH, Mercadante AF, Martins VR, Camargo AA, Nakao LS, Zanata SM. Characterization of a specific interaction between ADAM23 and cellular prion protein. Neurosci Lett. 2009;461:16–20. [DOI] [PubMed] [Google Scholar]
- 20. Ji YX, Zhang P, Zhang XJ, Zhao YC, Deng KQ, Jiang X, Wang PX, Huang Z, Li H. The ubiquitin E3 ligase TRAF6 exacerbates pathological cardiac hypertrophy via TAK1‐dependent signalling. Nat Commun. 2016;7:11267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ren L, Wu C, Yang K, Chen S, Ye P, Wu J, Zhang A, Huang X, Wang K, Deng P, Ding X, Chen M, Xia J. A disintegrin and metalloprotease‐22 attenuates hypertrophic remodeling in mice through inhibition of the protein kinase B signaling pathway. J Am Heart Assoc. 2018;7:e005696 DOI: 10.1161/JAHA.117.005696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Clemente CF, Xavier‐Neto J, Dalla Costa AP, Consonni SR, Antunes JE, Rocco SA, Pereira MB, Judice CC, Strauss B, Joazeiro PP, Matos‐Souza JR, Franchini KG. Focal adhesion kinase governs cardiac concentric hypertrophic growth by activating the AKT and mTOR pathways. J Mol Cell Cardiol. 2012;52:493–501. [DOI] [PubMed] [Google Scholar]
- 23. DiMichele LA, Doherty JT, Rojas M, Beggs HE, Reichardt LF, Mack CP, Taylor JM. Myocyte‐restricted focal adhesion kinase deletion attenuates pressure overload‐induced hypertrophy. Circ Res. 2006;99:636–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bi X, Zhang G, Wang X, Nguyen C, May HI, Li X, Al‐Hashimi AA, Austin RC, Gillette TG, Fu G, Wang ZV, Hill JA. Endoplasmic reticulum chaperone GRP78 protects heart from ischemia/reperfusion injury through Akt activation. Circ Res. 2018;122:1545–1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cheng Z, DiMichele LA, Hakim ZS, Rojas M, Mack CP, Taylor JM. Targeted focal adhesion kinase activation in cardiomyocytes protects the heart from ischemia/reperfusion injury. Arterioscler Thromb Vasc Biol. 2012;32:924–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Manabe I, Shindo T, Nagai R. Gene expression in fibroblasts and fibrosis: involvement in cardiac hypertrophy. Circ Res. 2002;91:1103–1113. [DOI] [PubMed] [Google Scholar]
- 27. Clemente CF, Tornatore TF, Theizen TH, Deckmann AC, Pereira TC, Lopes‐Cendes I, Souza JR, Franchini KG. Targeting focal adhesion kinase with small interfering RNA prevents and reverses load‐induced cardiac hypertrophy in mice. Circ Res. 2007;101:1339–1348. [DOI] [PubMed] [Google Scholar]
- 28. Peng X, Kraus MS, Wei H, Shen TL, Pariaut R, Alcaraz A, Ji G, Cheng L, Yang Q, Kotlikoff MI, Chen J, Chien K, Gu H, Guan JL. Inactivation of focal adhesion kinase in cardiomyocytes promotes eccentric cardiac hypertrophy and fibrosis in mice. J Clin Invest. 2006;116:217–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Peng X, Wu X, Druso JE, Wei H, Park AY, Kraus MS, Alcaraz A, Chen J, Chien S, Cerione RA, Guan JL. Cardiac developmental defects and eccentric right ventricular hypertrophy in cardiomyocyte focal adhesion kinase (FAK) conditional knockout mice. Proc Natl Acad Sci USA. 2008;105:6638–6643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Harburger DS, Calderwood DA. Integrin signalling at a glance. J Cell Sci. 2009;122:159–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Brancaccio M, Hirsch E, Notte A, Selvetella G, Lembo G, Tarone G. Integrin signalling: the tug‐of‐war in heart hypertrophy. Cardiovasc Res. 2006;70:422–433. [DOI] [PubMed] [Google Scholar]
- 32. Mitra SK, Hanson DA, Schlaepfer DD. Focal adhesion kinase: in command and control of cell motility. Nat Rev Mol Cell Biol. 2005;6:56–68. [DOI] [PubMed] [Google Scholar]
- 33. Taylor JM, Rovin JD, Parsons JT. A role for focal adhesion kinase in phenylephrine‐induced hypertrophy of rat ventricular cardiomyocytes. J Biol Chem. 2000;275:19250–19257. [DOI] [PubMed] [Google Scholar]
- 34. Aikawa R, Nagai T, Kudoh S, Zou Y, Tanaka M, Tamura M, Akazawa H, Takano H, Nagai R, Komuro I. Integrins play a critical role in mechanical stress‐induced p38 MAPK activation. Hypertension. 2002;39:233–238. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Clinical Characteristics of Human Heart Samples
Figure S1. Western blot analysis of ADAM23 in human heart samples (No. 5 and 6; n=2 per group) from normal donors, hypertrophic cardiomyopathy (HCM) and dilated cardiomyopathy (DCM).
Figure S2. A, The ADAM23 mRNA levels in the hearts of mice at 4 weeks post the sham or aortic banding (AB) surgery (n=4 independent experiments. *P<0.05 vs the sham group). B, The mRNA levels of ADAM23 in cardiomyocytes in response to PBS or Ang II for 24 hours (n=3 independent experiments; *P<0.05 vs PBS group).
Figure S3. Schematic diagram of the construction of cardiac‐specific conditional ADAM23 (cADAM23‐KO) and identification of ADAM23 expression. A, Schematic illustration of cADAM23‐KO generation. B, Amplification of the entire region covering the floxed and homology arm using the F1/R1 primer (left) and the circle excised by Cre using the F2/R2 primer (right). C, Western blot of ADAM23 expression levels in different tissues of WT and cADAM23‐KO mice (n=4 per group).
Figure S4. Generation of cardiac‐specific ADAM23 transgenic (ADAM23‐TG) mice. A, Schematic diagram for the construction of the cardiac‐specific expression of the ADAM23‐transgenic (ADAM23‐TG) mice. B, Western blot analysis of ADAM23 levels in the ADAM23‐TG mice and their NTG littermates (n=4 per group).
Figure S5. ADAM23 had no effect on cardiomyocyte death during cardiac hypertrophy. A, terminal deoxynucleotidyl transferase‐mediated dUTP nick end‐labeling assays in the heart sections of cADAM23‐KO and ADAM23‐TG mice, compared with their respective controls (n=5 mice per group; scale bar, 20 μm). B, Western blot analysis of cleaved caspase3 (C‐Caspase3) in the hearts of cADAM23‐KO and ADAM23‐TG mice, compared with their respective controls (n=4 mice per group). Gapdh served as a loading control.