

Abstract
Background
We have reported that cytochrome P450 1B1 (CYP1B1), expressed in cardiovascular tissues, contributes to angiotensin II–induced vascular smooth muscle cell (VSMC) migration and proliferation and development of hypertension in various experimental animal models via generation of reactive oxygen species. This study was conducted to determine the contribution of CYP1B1 to platelet‐derived growth factor‐BB–induced VSMC migration and proliferation in vitro and to neointimal growth in vivo.
Methods and Results
VSMCs isolated from aortas of male Cyp1b1 +/+ and Cyp1b1 −/− mice were used for in vitro experiments. Moreover, carotid arteries of Cyp1b1 +/+ and Cyp1b1 −/− mice were injured with a metal wire to assess neointimal growth after 14 days. Platelet‐derived growth factor‐BB–induced migration and proliferation and H2O2 production were found to be attenuated in VSMCs from Cyp1b1 −/− mice and in VSMCs of Cyp1b1 +/+ mice treated with 4‐hydroxy‐2,2,6,6‐tetramethylpiperidin‐1‐oxyl, a superoxide dismutase and catalase mimetic. In addition, wire injury resulted in neointimal growth, as indicated by increased intimal area, intima/media ratio, and percentage area of restenosis, as well as elastin disorganization and adventitial collagen deposition in carotid arteries of Cyp1b1 +/+ mice, which were minimized in Cyp1b1 −/− mice. Wire injury also increased infiltration of inflammatory and immune cells, as indicated by expression of CD68+ macrophages and CD3+ T cells, respectively, in the injured arteries of Cyp1b1 +/+ mice, but not Cyp1b1 −/− mice. Administration of 4‐hydroxy‐2,2,6,6‐tetramethylpiperidin‐1‐oxyl attenuated neointimal growth in wire‐injured carotid arteries of Cyp1b1 +/+ mice.
Conclusions
These data suggest that CYP1B1‐dependent oxidative stress contributes to the neointimal growth caused by wire injury of carotid arteries of male mice.
Keywords: cytochrome P450 enzymes, neointima, platelet‐derived growth factor, reactive oxygen species, vascular smooth muscle cell
Subject Categories: Growth Factors/Cytokines, Inflammation, Smooth Muscle Proliferation and Differentiation, Remodeling, Restenosis
Clinical Perspective
What Is New?
This study demonstrated that platelet‐derived growth factor‐BB, which is known to be involved in neointimal growth, stimulates vascular smooth muscle cell migration and proliferation via cytochrome P450 1B1–dependent reactive oxygen species production.
Cyp1b1 gene disruption minimized neointimal growth and associated pathophysiological changes, including superoxide production, in wire‐injured carotid artery of male mice.
This study demonstrated that neointimal growth and associated superoxide produced by vascular injury were attenuated by 4‐hydroxy‐2,2,6,6‐tetramethylpiperidin‐1‐oxyl (Tempol), a superoxide dismutase mimetic.
What Are the Clinical Implications?
Development of selective cytochrome P450 1B1 inhibitors could be useful to treat restenosis after balloon angioplasty in men.
Vascular endothelial cell injury caused by angioplasty results in inflammation and activation of multiple signaling pathways. This leads to migration of vascular smooth muscle cells (VSMCs) from the media into the intimal layer, where they proliferate and synthesize extracellular matrix (ECM) components, resulting in vascular remodeling.1, 2 The neointimal growth associated with vascular injury is responsible for arterial restenosis after balloon angioplasty and formation of atherosclerotic lesions.3 The development of drug‐eluting stents to inhibit VSMC hyperplasia has reduced the incidence of restenosis after angioplasty, but their usefulness is still limited because of stent‐initiated thrombosis requiring antiplatelet therapy.2, 4 Therefore, elucidation of the molecular mechanisms involved in neointimal growth caused by vascular injury would allow the development of novel therapeutic agents to treat vascular pathogenesis and restenosis associated with angioplasty.
We have previously reported that cytochrome P450 1B1 (CYP1B1), a heme‐thiolate monooxygenase expressed in cardiovascular tissues, contributes to the development of hypertension, and its associated pathogenesis.5 CYP1B1 also mediates angiotensin II (Ang II)–induced migration, proliferation, and growth of rat VSMCs, via generation of reactive oxygen species (ROS).6 The present study was conducted to determine if CYP1B1 also contributes, via ROS production, to the action of platelet‐derived growth factor‐BB (PDGF‐BB) on VSMC migration and proliferation. Because VSMC migration and proliferation, initiated by vascular injury, are hallmarks of neointima formation, and PDGF and ROS have been implicated in this process,7, 8 these data led us to the hypothesis that CYP1B1‐generated oxidative stress contributes to the pathogenesis of vascular remodeling and restenosis after vascular injury. To test this hypothesis, we investigated the effects of Cyp1b1 gene disruption and 4‐hydroxy‐2,2,6,6‐tetramethylpiperidin‐1‐oxyl (Tempol), a superoxide dismutase and catalase mimetic that scavenges ROS, including superoxide, and facilitates H2O2 metabolism,9 on vascular remodeling and associated pathogenesis in wire‐injured carotid artery in male mice. The results of our study show that CYP1B1 contributes to PDGF‐BB–induced VSMC migration and proliferation via ROS production, and that Cyp1b1 gene disruption attenuates neointimal growth in these mice, most likely by reducing oxidative stress.
Methods
Specific data, analytic methods, and study materials, outlined below, will be made available on reasonable request to the corresponding author.
Materials
The following reagents were used for the study: Amplex Red Hydrogen Peroxide/Peroxidase Assay Kit, dihydroethidium, DMEM, M199 media, fetal bovine serum, and trypsin from Invitrogen (Carlsbad, CA); von Willebrand factor, CD3, CD68, Alexa Fluor 488 goat anti‐rabbit, and Dylight 549 goat anti‐rat antibodies from Abcam (Cambridge, MA); PDGF‐B and CYP1B1 antibodies from Santa Cruz Biotechnology (Dallas, TX); [3H]‐thymidine from Perkin‐Elmer (Boston, MA); Wizard SV Genomic DNA Purification System from Promega (Madison, WI); and 2,3′,4,5′‐tetramethoxystilbene from Cayman Chemical (Ann Arbor, MI). All other chemicals, kits, and antibodies were purchased from Sigma (St Louis, MO).
Animals
All experiments were performed according to protocols approved by the Institutional Animal Care and Use Committee of University of Tennessee in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Cyp1b1 −/− mice were generated at the National Cancer Institute, transferred to the University of Tennessee, backcrossed 10 generations to a C57BL/6J background, and then brother‐sister mated to generate a homozygous line. Male C57BL/6J (Cyp1b1 +/+) mice were purchased from Jackson Laboratory (Bar Harbor, ME). All animals weighed 20 to 30 g and were ≈8 weeks of age at the beginning of the experiment. VSMCs were isolated from Cyp1b1 +/+ and Cyp1b1 −/− mice and cultured, as described.10 Wire injury was performed on the carotid artery of Cyp1b1 +/+ and Cyp1b1 −/− mice, as described.11
Genotyping of Mice
The genotype of Cyp1b1 +/+ and Cyp1b1 −/− mice was routinely assessed by polymerase chain reaction, as described.12 For polymerase chain reaction analysis, genomic DNA was obtained from tail snips using the Wizard SV Genomic DNA Purification System, according to the manufacturer's instructions. Animals of both genotypes were randomized for control and experimental groups.
Wire Injury and Measurement of Neointimal Growth
Wire injury was performed on the carotid artery of Cyp1b1 +/+ and Cyp1b1 −/− mice, as described.11 Briefly, mice were anesthetized by IP injection of ketamine/xylazine (87 mg/kg per 13 mg/kg). The right common carotid artery of the anesthetized mouse was exposed through a midline cervical incision, and blood flow to the site was temporarily interrupted by ligating with microclamps and silk suture. A small incision was made, and a metal guide wire (0.381 mm in diameter) (Cook Incorporated, Bloomington, IN) was introduced into the artery and passed up and down 5 times with rotation through the clamped section to denude the endothelium. Blood flow was reestablished by removing the microclamps. The mice were euthanized (1) 1 day, (2) 5 days, and (3) 14 days after injury. Both injured and uninjured carotid arteries were harvested and frozen for subsequent histological analysis. The tissues were sectioned, as described below, and stained with hematoxylin and eosin for analysis of neointimal growth. The surface areas of the intima and media were calculated using National Institutes of Health ImageJ 1.48v software, and the intima/media ratio of randomly selected sections of carotid arteries was analyzed.
In a separate series of experiments, Cyp1b1 +/+ mice were administered with the following: (1) Tempol, a superoxide dismutase mimetic known to scavenge ROS,13 in drinking water (2 mmol/L), 7 days before wire injury of carotid artery; and (2) 2,3′,4,5′‐tetramethoxystilbene, a selective inhibitor of CYP1B1, or its vehicle dimethyl sulfoxide, injected IP 300 μg/kg every third day, subjected to wire injury of carotid artery and continued for 14 days before euthanasia.
Immunohistochemical Analysis
At the completion of the experiments, mice were anesthetized, as described above, and the animals were perfused with saline (3 minutes). The carotid arteries (both injured and uninjured) were dissected and placed in optimal cutting temperature compound (Sakura Finetek USA Inc, Torrance, CA) and frozen at −80°C. Carotid artery sections (5 μm thick) were cut using a cryostat (Leica Microsystems, Bannockburn, IL; model CM1850) and stained for histological and immunohistochemical analysis. Briefly, sections were air dried, fixed in 10% formalin or cold acetone for 10 minutes, washed with PBS 3 times (3 minutes each), incubated with 0.3% H2O2 for 10 minutes to quench endogenous peroxides, washed with PBS for 3 minutes, and blocked with serum appropriate for the species of the secondary antibody for 1 hour. Sections were then incubated with primary antibodies against von Willebrand factor (for endothelial cells), α‐actin (for smooth muscle cells), CD68 (for macrophages), and CD3 (for T cells), overnight at 4°C. The sections were then washed with PBS 3 times (3 minutes each) and incubated with corresponding horseradish peroxidase– or fluorophore‐tagged secondary antibodies for 1 hour, for chromogenic detection of horseradish peroxidase activity with 3,3′‐diaminobenzidine substrate or immunofluorescent detection, respectively. In addition, sections were stained with hematoxylin and eosin, Masson's trichrome for collagen deposition, and Verhoeff–Van Gieson for elastin fibers, according to the manufacturer's instructions (Sigma). Sections were viewed in a double‐blinded manner using an Olympus inverted system microscope (Olympus America, Inc, Melville, NY; model BX41), photographed using a SPOT Insight digital camera (Diagnostic Instruments, Inc, Sterling Heights, MI; model Insight 2MP Firewire), and analyzed using ImageJ.
Measurement of ROS Production
To measure ROS production, carotid artery sections were exposed to dihydroethidium, as described.14 Fresh, unfixed tissue samples were placed in optimal cutting temperature compound and frozen at −80°C. Tissue segments were cut into 5‐μm sections using a cryostat, sections were incubated in PBS for 30 minutes at 37°C, and then further incubated in dihydroethidium, 2 μmol/L, at 37°C in a light‐protected humidified chamber for 45 minutes. Sections were then rinsed in PBS, and fluorescence was detected using a 585‐nm filter using an Olympus inverted system microscope (Olympus America Inc; model IX50). Images were photographed with an Olympus digital camera (Olympus America Inc; model DP71) and analyzed using ImageJ.
VSMC Culture
Eight‐week‐old male Cyp1b1 +/+ and Cyp1b1 −/− mice (n=10 per genotype) were anesthetized with ketamine/xylazine (as described above), and the thoracic aorta was excised and rapidly removed. VSMCs were isolated and cultured as described.10 Cultured cells were maintained under 5% CO2 in DMEM with fetal bovine serum (10%) and penicillin/streptomycin (1%), at 37°C. Before each experiment, cells were quiesced for 24 to 48 hours in M199 medium without serum or antibiotics.
Migration Assay
To determine VSMC migration, we measured wound closure at different time points by using the wound healing approach.15 VSMCs from Cyp1b1 +/+ and Cyp1b1 −/− mice were treated with PDGF‐BB (10 ng/mL) or its vehicle in the presence or absence of Tempol (2 mmol/L) in 6‐well plates. The wells were scratched with a sterile pipette tip across the diameter to produce 1‐ to 1.5‐mm‐wide wounds. Images of the wounds were collected at ×10 magnification with an Olympus CKX41 inverted microscope equipped with an Olympus Altra20 camera using Microsoft Analytical Suite software. Using a motorized stage system (Prior OptiScan II, Rockland, MA) along with marking x, y, and z coordinates of each wound location allowed us to acquire images of the same wound locations at different time points. The outline of the wound in the field was measured, and the wound area was calculated using ImageJ at the initial time of wounding (0) and after 24 hours. Data are shown as a percentage of the 0‐hour wound area closure to normalize variability in wounding from well to well and experiment to experiment. Wound widths measured from 5 locations in the same well from 3 different wells were averaged. Experiments were repeated at least 3 times for confirmation.
Proliferation Assay
DNA synthesis for proliferation was determined by measuring the incorporation of [3H]‐thymidine in VSMCs, as described.16 Subconfluent VSMCs from Cyp1b1 +/+ and Cyp1b1 −/− mice were treated with PDGF‐BB (20 ng/mL) or its vehicle in the presence or absence of Tempol (2 mmol/L) in 24‐well plates along with addition of [3H]‐thymidine (0.5 μCi/well), and radioactivity in the cells was measured after 24 hours. To determine radioactivity, after washing the cells with ice‐cold PBS, trichloroacetic acid (10%), and ethanol/ether solution (2:1), [3H]‐labeled cells were incubated with 0.1% SDS and 0.1 mmol/L NaOH for 3 hours; then, the radioactivity was measured as disintegrations per minutes per well by using liquid scintillation spectrometry. The data are shown as an average fold change from vehicle from at least 6 different wells of each treatment condition. Experiments were repeated at least 3 times for confirmation.
H2O2 Production Assay
H2O2 released from VSMCs was measured using an Amplex Red Hydrogen Peroxide/Peroxidase Assay Kit following manufacturer's instructions. Briefly, subconfluent VSMCs from Cyp1b1 +/+ and Cyp1b1 −/− mice were quiesced for 24 hours, and then pretreated with Tempol (2 mmol/L) or its vehicle in 24‐well plates for 1 hour. The cells were then stimulated with PDGF‐BB (20 ng/mL) or its vehicle in the presence or absence of Tempol for 30 minutes in a prewarmed reaction mixture (37°C; 10 minutes) containing 50 μmol/L Amplex Red reagent and 0.1 U/mL horseradish peroxidase in Krebs‐Ringer phosphate buffer (145 mmol/L NaCl, 5.7 mmol/L NaH2PO4, 4.86 mmol/L KCl, 0.54 mmol/L CaCl2, 1.22 mmol/L MgSO4, and 5.5 mmol/L glucose, pH 7.35). The cell supernatant‐reaction mixture (100 µL) was then transferred to a 96‐well plate, and fluorescence was measured (excitation, 530 nm; emission, 590 nm) using a Synergy H1 multiplate reader (BioTek, Winooski, VT).
Western Blot Analysis
Western blot analysis was performed, as described previously.17 Briefly, VSMCs were scraped from the culture plates and lysed, and protein was isolated. An equal amount of protein from each lysate was subjected to SDS‐PAGE and transferred onto a nitrocellulose membrane. The blots were then probed with anti‐CYP1B1, anti–PDGF‐B, and anti–β‐actin primary antibodies and corresponding secondary antibodies (anti‐rabbit and anti‐mouse), and intensity of the bands was measured using ImageJ.
CYP1B1 Activity Assay
To determine CYP1B1 activity in VSMCs, we used a P450 Glo assay kit, according to the manufacturer's instructions (Promega, Madison, WI). Briefly, cells were incubated with PDGF‐BB (20 ng/mL) or its vehicle for 24 hours. Cells were washed with PBS. KPO4 solution (0.1 mol/L) was added to the plate, and cells were scraped and sonicated. Protein concentrations of the samples were quantitated by Bradford assay, and luciferin‐6′ chloroethyl ether (100 μmol/L), a substrate for CYP1B1, along with nicotinamide‐adenine dinucleotide phosphate, reduced form (NADPH; 100 μmol/L), were added to the samples in a white 96‐well plate. After 45 minutes, an equal volume of luciferin detection reagent was added to each well, mixed by gently shaking, and equilibrated at room temperature for 20 minutes. Luminescence was measured from each well using a Synergy H1 spectrophotometer (BioTek), and relative luminescent units were measured.
Statistical Analysis
Data were analyzed by 1‐way ANOVA, followed by Dunnett's post hoc test; 2‐way ANOVA, followed by Tukey's multiple comparison test; or Student t test with Welch's correction. The values of a minimum of 3 different experiments are expressed as the mean±SEM. P<0.05 was considered statistically significant and expressed as *P<0.05, **P<0.01, ***P<0.001, and ****P<0.0001.
Results
Cyp1b1 Gene Disruption Prevents PDGF‐BB–Induced Migration and Proliferation of VSMCs via Production of ROS
To determine if CYP1B1 contributes to PDGF‐BB–induced VSMC migration and proliferation through ROS generation, we examined the effect of PDGF‐BB on VSMCs isolated from Cyp1b1 +/+ and Cyp1b1 −/− mice. Treatment with PDGF‐BB stimulated migration of VSMCs from Cyp1b1 +/+ mice, following a scratch wound, as indicated by the percentage of 24‐hour wound closure, whereas PDGF‐BB–induced wound healing was attenuated in VSMCs from Cyp1b1 −/− mice (Figure 1A and Figure S1A). PDGF‐BB also increased the proliferation of VSMCs from Cyp1b1 +/+ mice, as determined by [3H]‐thymidine uptake, which was attenuated in cells from Cyp1b1 −/− mice (Figure 1B). PDGF‐BB did not alter expression of CYP1B1 protein levels in VSMCs from Cyp1b1 +/+ mice, and as expected, CYP1B1 expression was absent in VSMCs from Cyp1b1 −/− mice (Figure S2A and S2B). PDGF‐B expression was also not affected in VSMCs from Cyp1b1 +/+ and Cyp1b1 −/− mice (Figure S2C and S2D). PDGF‐BB also did not alter the activity of CYP1B1 in VSMCs (Figure S3).
Figure 1.
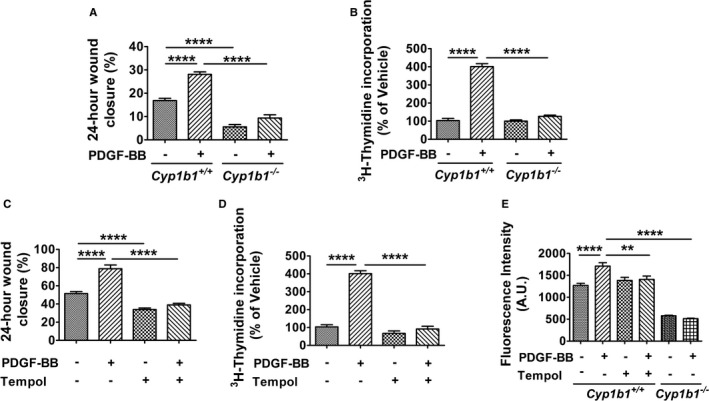
Cytochrome P450 1B1 (Cyp1b1) gene disruption prevents platelet‐derived growth factor‐BB (PDGF‐BB)–induced migration and proliferation of vascular smooth muscle cells (VSMCs) via production of reactive oxygen species. Quantitative analysis of migration of VSMCs expressed as percentage of wound closure after 24 hours of scratch wound healing assay in VSMCs from Cyp1b1 +/+ and Cyp1b1 −/− mice treated with vehicle or PDGF‐BB (10 ng/mL; A) and VSMCs from Cyp1b1 +/+ mice treated with vehicle or PDGF‐BB (10 ng/mL; C) in presence or absence of 4‐hydroxy‐2,2,6,6‐tetramethylpiperidin‐1‐oxyl (Tempol; 2 mmol/L). n=15 (5 scratch wound areas per well for 3 wells) for each treatment condition, for each experiment, with experiments repeated at least 3 times. Quantitative analysis of proliferation (as measured by [3H]‐thymidine uptake) expressed as average fold change from vehicle in VSMCs from Cyp1b1 +/+ and Cyp1b1 −/− mice treated with vehicle or PDGF‐BB (20 ng/mL; B) and VSMCs from Cyp1b1 +/+ mice treated with vehicle or PDGF‐BB (20 ng/mL; D) in presence or absence of Tempol (2 mmol/L). n=6 (6 different wells) for each treatment condition, for each experiment, with experiments repeated at least 3 times. E, Quantitative analysis of H2O2 production measured by Amplex Red assay kit and expressed as arbitrary units (A.U.s) of fluorescence intensity in VSMCs from Cyp1b1 +/+ mice treated with vehicle or PDGF‐BB (20 ng/mL) in presence or absence of Tempol (2 mmol/L). n=5 for Cyp1b1 +/+ group (5 different wells), and n=6 for Cyp1b1 −/− group (6 different wells). Data are expressed as mean±SEM. **P<0.01, ****P<0.0001.
To determine if ROS contributes to PDGF‐BB–induced VSMC migration and proliferation, we examined the effect of Tempol, a superoxide dismutase and catalase mimetic,9 on VSMC migration and proliferation. Tempol attenuated PDGF‐BB–induced migration (Figure 1C and Figure S1B) and proliferation of VSMCs from Cyp1b1 +/+ mice (Figure 1D). PDGF‐BB also increased ROS production, measured as H2O2 generated from metabolism of superoxide by superoxide dismutase, in VSMCs from Cyp1b1 +/+ mice; this increase was reduced by Cyp1b1 gene disruption as well as by Tempol (Figure 1E).
Cyp1b1 Gene Disruption Attenuates Neointimal Growth and Associated Pathophysiological Changes in Wire‐Injured Carotid Artery of Male Mice
To determine the contribution of CYP1B1 to neointimal growth, the effect of Cyp1b1 gene disruption on neointima formation in carotid arteries of male Cyp1b1 +/+ and Cyp1b1 −/− mice, injured with a metal guide wire, was analyzed for neointimal growth 14 days after injury. Denudation of the endothelial layer by wire injury was confirmed by absence of staining with antibody against von Willebrand factor, a marker of endothelial cells, after 1 day of injury in Cyp1b1 +/+ and Cyp1b1 −/− mice (Figure S4A and S4B). Extensive neointimal growth was observed in sections of injured but not uninjured contralateral carotid arteries of Cyp1b1 +/+ mice (Figure 2A). Neointimal growth in response to injury was markedly lower in Cyp1b1 −/− mouse arteries examined after 14 days (Figure 2B). Quantification of the hematoxylin and eosin staining revealed a significant reduction in total intimal area (Figure 2C), intima/media area ratio (Figure 2D), and percentage of total area of carotid artery restenosis in the injured carotid arteries of Cyp1b1 −/− mice compared with Cyp1b1 +/+ mice (Figure 2E).
Figure 2.
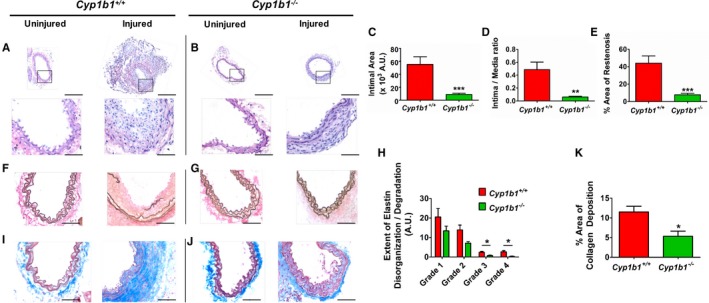
Cyptochrome P450 1B1 (Cyp1b1) gene disruption attenuates neointimal growth and associated pathophysiological changes in wire‐injured carotid artery of male mice. Representative images of hematoxylin and eosin staining in uninjured and injured carotid arteries of Cyp1b1 +/+ mice (A) and Cyp1b1 −/− mice (B) after 14 days of injury. Bars: 200 μm (top panels); 50 μm (bottom panels). Quantitative analysis of neointimal growth in injured carotid arteries of Cyp1b1 +/+ and Cyp1b1 −/− mice, as represented by total intimal area (C), intima/media ratio (D), and percentage area of restenosis (E) (n=20 for Cyp1b1 +/+ group, and n=14 for Cyp1b1 −/− group). Representative images of Verhoeff–Van Gieson staining of elastin fibers in uninjured and injured carotid arteries of Cyp1b1 +/+ mice (F) and Cyp1b1 −/− mice (G). Bar=50 μm. H, Quantitative analysis of extent of elastin disorganization/degradation in injured carotid arteries of Cyp1b1 +/+ and Cyp1b1 −/− mice (average number [arbitrary units {A.U.s}] of assigned scores of images from 3 different sections of each mouse; n=12 for Cyp1b1 +/+ group, and n=7 for Cyp1b1 −/− group). Representative images of Masson's trichrome staining of collagen deposition in uninjured and injured carotid arteries of Cyp1b1 +/+ mice (I) and Cyp1b1 −/− mice (J) (collagen: blue; smooth muscle cell: red). Bar=50 μm. K, Quantitative analysis of percentage area of collagen deposition in injured carotid arteries of Cyp1b1 +/+ and Cyp1b1 −/− mice (average areas of images from 3 different sections of each mouse; n=4 for Cyp1b1 +/+ group, and n=5 for Cyp1b1 −/− group). Data are expressed as mean±SEM. *P<0.05, **P<0.01, ***P<0.001.
After 14 days of injury, carotid arteries demonstrated extensive elastin disorganization, degradation, and disruption in the Cyp1b1 +/+ mice, whereas the uninjured carotid arteries exhibited the usual wavy appearance of the elastin fibers (Figure 2F). In contrast, elastin disorganization and fragmentation were attenuated in the injured carotid arteries of Cyp1b1 −/− mice (Figure 2G). The extent of elastin damage in the injured carotid arteries was quantified by scoring them into different grades of damage: grade 1 (straightening of elastin fibers), grade 2 (lighter elastin/degradation of elastin), grade 3 (short breaks in elastin fibers), and grade 4 (long breaks in elastin fibers). All 4 grades of elastin disorganization were minimized, with a significant reduction in grades 3 and 4 of elastin fragmentation, in the injured carotid arteries of Cyp1b1 −/− mice, compared with those in Cyp1b1 +/+ mice (Figure 2H).
Injured carotid arteries of Cyp1b1 +/+ mice displayed extensive deposition of collagen, a marker of ECM formation, in the adventitial layers, which was attenuated in injured arteries of Cyp1b1 −/− mice (Figure 2I through 2K). Uninjured carotid arteries of both Cyp1b1 +/+ and Cyp1b1 −/− mice had much lower collagen deposition in the adventitia (Figure 2I and 2J). In addition, the medial layers and neointimal area in injured carotid arteries of Cyp1b1 +/+ mice stained positively for presence of α‐smooth muscle actin, indicating that most cells in the neointimal lesion were VSMCs (Figure S5A and S5B). Because no differences were observed in the structural and pathological features of uninjured carotid arteries between Cyp1b1 +/+ and Cyp1b1 −/− mice, we used parameters of only injured carotid arteries of Cyp1b1 +/+ and Cyp1b1 −/− mice for quantitation.
Cyp1b1 Gene Disruption Prevents Macrophage and T‐Cell Infiltration in Wire‐Injured Carotid Artery of Mice
Carotid arteries of Cyp1b1 +/+ mice displayed infiltration of CD68+ macrophages after 5 days of wire injury, which was diminished in the injured carotid arteries of Cyp1b1 −/− mice (Figure 3A and 3B). Also, after 14 days of wire injury, extensive accumulations of CD3+ T cells were observed in carotid arteries of Cyp1b1 +/+ mice, which were attenuated in the Cyp1b1 −/− mice (Figure 3C and 3D).
Figure 3.

Cytochrome P450 1B1 (Cyp1b1) gene disruption prevents macrophage and T‐cell infiltration in wire‐injured carotid artery of mice. A, Representative images of infiltration of CD68+ macrophages (red) and nuclear staining with 4′,6‐diamidino‐2‐phenylindole (DAPI) (blue). B, Quantitative analysis of total number of CD68+ macrophages per high‐power field (hpf) in injured carotid arteries of Cyp1b1 +/+ and Cyp1b1 −/− mice after 5 days of injury. n=5 for each group (A and B). C, Representative image of infiltration of CD3+ T cells (green) and nuclear staining with DAPI. D, Quantitative analysis of total number of CD3+ T cells per hpf in injured carotid arteries of Cyp1b1 +/+ and Cyp1b1 −/− mice after 14 days of injury. n=5 for each group (C and D). Scale bars: 200 μm (left panels); 50 μm (right panels). Data are expressed as mean±SEM. **P<0.01.
Tempol Administration Prevents Neointimal Growth and Associated Pathological Changes in Wire‐Injured Carotid Artery of Cyp1b1 +/+ Mice
To determine if CYP1B1‐generated ROS contributes to the neointimal growth after vascular injury, we exposed the arteries to dihydroethidium and analyzed the fluorescence of 2‐hydroxyethidium as a marker of ROS generation in injured and uninjured carotid arteries of Cyp1b1 +/+ and Cyp1b1 −/− mice. ROS production was increased after wire injury in carotid arteries of Cyp1b1 +/+ but not Cyp1b1 −/− mice (Figure S6). Hence, to determine if inhibition of ROS production attenuates neointimal growth, we administered Tempol to Cyp1b1 +/+ mice in drinking water, 7 days before wire injury of the carotid artery, and continued until euthanizing the animals after 14 days of injury. Administration of Tempol attenuated neointimal growth, as evaluated by total intimal area, intima/media ratio, and percentage area of restenosis in wire‐injured carotid arteries of Cyp1b1 +/+ mice (Figure 4A through 4D). Tempol also minimized elastin disorganization and fragmentation (Figure 4E) and adventitial ECM formation (Figure 4G). Treatment with Tempol decreased all 4 grades of elastin disorganization/fragmentation (Figure 4F), as well as the percentage area of collagen deposition (Figure 4H), in wire‐injured carotid arteries of Cyp1b1 +/+ mice. Also, Tempol reduced ROS generation, as measured by 2‐hydroxyethidium fluorescence, in wire‐injured carotid arteries of Cyp1b1 +/+ mice (Figure 4I and 4J).
Figure 4.
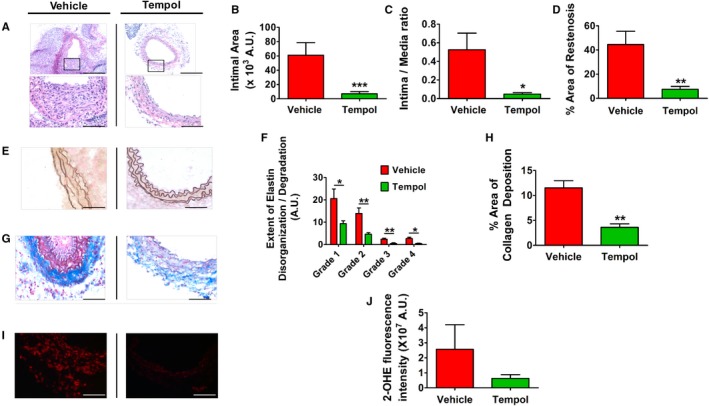
4‐Hydroxy‐2,2,6,6‐tetramethylpiperidin‐1‐oxyl (Tempol) administration prevents neointimal growth and associated pathophysiological features in wire‐injured carotid artery of cytochrome P450 B1 (Cyp1b1) +/+ mice. A, Representative images of hematoxylin and eosin staining in injured carotid arteries of Cyp1b1 +/+ mice treated with vehicle or Tempol in drinking water (2 mmol/L) (started 7 days before injury) after 14 days of injury. Bars: 200 μm (top panels); 50 μm (bottom panels). Quantitative analysis of neointimal growth in injured carotid arteries of Cyp1b1 +/+ mice treated with vehicle or Tempol, as represented by total intimal area (B), intima/media ratio (C), and percentage area of restenosis (D) (n=11 for vehicle group, and n=10 for Tempol group). Representative images of Verhoeff–Van Gieson staining of elastin fibers (E) and quantitative analysis of extent of elastin disorganization/degradation (F) in injured carotid arteries of Cyp1b1 +/+ mice treated with vehicle or Tempol. Bar=50 μm. (Average number [arbitrary units {A.U.s}] of assigned scores of images from 3 different sections of each mouse; n=12 for vehicle group, and n=9 for Tempol group). Representative images of Masson's trichrome staining of collagen deposition (G) and quantitative analysis of percentage area of collagen deposition (H) in injured carotid arteries of Cyp1b1 +/+ mice treated with vehicle or Tempol (collagen: blue; smooth muscle cell: red). Bar=50 μm. (Average areas of images from 3 different sections of each mouse; n=4 for vehicle group, and n=3 for Tempol group). Representative images of superoxide production, as indicated by fluorescence of 2‐hydroxyethidium (2‐OHE) (red) (I) and quantitative analysis of 2‐OHE fluorescence intensity (J) in injured carotid arteries of Cyp1b1 +/+ mice treated with vehicle or Tempol (n=5 for each group). Bar=50 μm. Data are expressed as mean±SEM. *P<0.05, **P<0.01, ***P<0.001.
CYP1B1 Inhibitor, 2,3′,4,5′‐Tetramethoxystilbene, Minimized Neointimal Growth Caused by Wire Injury of Carotid Arteries
Administration of selective CYP1B1 inhibitor 2,3′,4,5′‐tetramethoxystilbene,18 but not its vehicle, attenuated neointima formation after 14 days of carotid artery injury in Cyp1b1 +/+ mice, as observed by reduced intimal area, intima/media ratio, and percentage area of restenosis (Figure S7A through S7E).
Discussion
This study shows, for the first time, that CYP1B1 contributes to PDGF‐BB–induced VSMC migration and proliferation in vitro, and plays a critical role in neointimal growth and restenosis in response to vascular injury via generation of ROS in male mice. Injury during angioplasty/stent placement results in deendothelialization of the vessel wall, leading to platelet activation and inflammation, ultimately leading to migration and proliferation of VSMCs from medial as well as adventitial layers to form the neointimal lesion.2, 19 Among many agents, including cytokines, Ang II, and growth factors, PDGF is considered to be a primary stimulator of VSMC migration and proliferation, acting on the PDGF‐β receptor in medial SMCs and leading to neointimal growth.7, 20 Ang II– and PDGF‐BB–induced VSMC migration and proliferation in vitro are mediated via generation of ROS.6, 21 Ang II stimulates ROS production in VSMCs via activation of nicotinamide adenine dinucleotide phosphate (NADPH oxidase), which is dependent on arachidonic acid (AA).22 Previously, we have reported that Ang II–induced VSMC migration and proliferation are mediated by ROS generated from the metabolism of AA by CYP1B1, resulting in stimulation of ≥1 signaling molecules.6 In the present study, the PDGF‐BB–induced increase in ROS, measured as H2O2 in VSMCs, and their migration and proliferation were attenuated by Cyp1b1 gene disruption and by Tempol, which does not alter CYP1B1 activity6 and inactivates superoxides as well as H2O2 by superoxide dismutase– and catalase‐like actions, respectively.9 This suggested that these effects of PDGF‐BB are also dependent on CYP1B1. In the present study, PDGF‐BB did not alter CYP1B1 protein expression or its activity, and Cyp1b1 gene disruption also did not alter expression of PDGF‐B in VSMCs. CYP1B1 is constitutively active and requires substrate to generate ROS.6 Therefore, the demonstration that PDGF‐BB activates cPLA2 (cytosolic phospholipase A2) and releases AA, which participates in VSMC proliferation23 and migration,24 suggests that PDGF‐BB most likely also generates ROS via CYP1B1‐catalyzed AA metabolism, similar to Ang II.6 The mechanism by which AA metabolism by CYP1B1 leads to the activation of NADPH oxidase still remains to be elucidated and is one of the limitations in our study. In human lens epithelial B3 cells, PDGF via AA metabolites, generated by 5‐ or/and 15‐lipoxygenase pathway, has been proposed to translocate protein kinase C to NADPH oxidase complex and results in ROS production.25 However, Ang II–induced release of AA and its subsequent metabolism by CYP1B1 is associated with increased generation of ROS in VSMCs, which, in turn, activates extracellular signal‐regulated kinase (ERK) 1/2 and p38 mitogen‐activated protein kinase.6 Our preliminary data show that extracellular signal‐regulated kinase 1/2 and p38 mitogen‐activated protein kinase, activated by CYP1B1‐generated ROS, stimulate NADPH oxidase in VSMCs (C.Y.S. and K.U.M., unpublished data). Whether AA released by PDGF also stimulates NADPH oxidase via CYP1B1 in VSMCs remained to be determined.
To determine the contribution of CYP1B1‐dependent VSMC migration and proliferation and ROS production in vivo to neointimal growth and its pathogenesis, we used the carotid artery wire‐injury mouse model. The vascular injury model is widely used to study the mechanism of restenosis, because it closely resembles the angioplasty procedure in humans that results in endothelial cell denudation, VSMC proliferation, and other pathological features of restenosis.11 After 14 days of wire injury of carotid artery, the total intimal area, intima/media ratio, and percentage area of restenosis were markedly reduced in Cyp1b1 −/− compared with Cyp1b1 +/+ mice, suggesting that CYP1B1 is required for neointimal growth caused by vascular injury.
ECM remodeling is a pathological hallmark of injury‐induced neointimal growth.2 Vascular injury is also associated with structural changes in elastin and its degradation.26, 27 In the present study, the extensive disorganization and degradation of elastic fibers in the injured carotid arteries of Cyp1b1 +/+ mice was minimized in the Cyp1b1 −/− gene disrupted mice. Strikingly, the most prominent difference in elastin remodeling between Cyp1b1 +/+ and Cyp1b1 −/− mice was observed in grades 3 and 4 of the elastin degradation index, implying that a breakage of elastin fibers in Cyp1b1 +/+ mice might have permitted medial SMCs to breach the ECM and migrate to form neointima. These results, coupled with our in vitro findings of attenuated migration of VSMCs by Cyp1b1 gene disruption, along with our previous work in mouse models of atherosclerosis and abdominal aortic aneurysms,28, 29 indicate that CYP1B1 plays an important role in elastin metabolism/degradation.
It is well established that inflammation is a major component of restenosis.2, 30 Our demonstration that the infiltration of CD68+ macrophages and CD3+ T cells in injured carotid artery was attenuated by Cyp1b1 gene disruption suggests that CYP1B1 also plays a crucial role in inflammation associated with restenosis. Because CYP1B1 is expressed in extrahepatic tissues, including VSMCs,6, 31 macrophages,32 and T cells,33 further studies using VSMC‐, macrophage‐, and T‐cell–specific conditional knockout mouse models are required to determine the contribution of CYP1B1 in these cell types to injury‐induced neointimal growth.
An interesting finding in the present study was the significant amount of adventitial fibrosis and/or ECM formation in the injured carotid arteries of Cyp1b1 +/+ mice, as indicated by extensive deposition of collagen in the adventitia, similar to “neoadventitia” formation during vascular remodeling.34 The neoadventitia formation is one of the earliest events after vascular injury34 and hence can be used as an alternative predictor of carotid artery disease.35 We have reported that CYP1B1 plays a critical role in collagen deposition and fibrosis in several tissues in hypertension, and in atherosclerosis and abdominal aortic aneurysms.28, 29, 36 Adventitial fibrosis associated with vascular injury also requires CYP1B1, and Cyp1b1 gene disruption decreased adventitial collagen deposition in injured mouse carotid arteries. Adventitia is well known to play a dynamic role in vascular inflammation.37 Migration of adventitial38 and circulating myofibroblasts39 and an early recruitment of inflammatory cells, such as macrophages,19, 30, 39 in the adventitia are considered to be the major contributors to neointima formation. In the present study, expression of CD68+ macrophages in the adventitia of carotid arteries of Cyp1b1 −/− mice was significantly reduced compared with those in Cyp1b1 +/+ mice. These observations also implicate CYP1B1 in the inflammation and collagen production that contribute to the pathogenesis of neointimal growth caused by vascular injury.
Increased ROS generation is one of the key pathological features of vascular injury, which acts as a signaling mechanism for neointimal growth.8, 40, 41 We have previously reported that ROS generated via CYP1B1 contribute to the development of hypertension in different animal models, atherosclerosis, and abdominal aortic aneurysms.5, 28, 29 In the current study, wire injury of the carotid arteries increased superoxide production, as detected by 2‐hydroxyethidium fluorescence in Cyp1b1 +/+ mice, which was reduced in Cyp1b1 −/− mice. Moreover, treatment with Tempol attenuated neointimal growth and associated pathophysiological changes in injured carotid arteries. Although ROS measured by 2‐hydroxyethidium staining was reduced in the carotid arteries of Cyp1b1 −/− mice and by Tempol in Cyp1b1 +/+ mice, it did not reach the level of statistical significance. This could be because of rapid conversion of superoxide into H2O2, which is not detected by the 2‐hydroxyethidium fluorescence method. However, an effective in vivo method for measuring H2O2 in mouse carotid arteries is still lacking. Therefore, these observations, together with our finding that PDGF‐BB induced migration, proliferation, and H2O2 production, were attenuated in VSMCs derived from Cyp1b1 −/− mice, or by treatment with Tempol of VSMCs from Cyp1b1 +/+ mice, indicate that neointimal growth and associated pathological changes are mediated most likely by superoxides generated via CYP1B1. However, further studies are required to confirm the measurement of superoxides using high‐performance liquid chromatography–based assay or electron spin resonance spectroscopy.42 Moreover, further studies are required to establish the contribution of ROS, generated by CYP1B1, to endogenous PDGF‐stimulated neointimal growth during vascular injury.
One of the limitations of our study that remains to be addressed is the relationship between CYP1B1 and other AA‐metabolizing enzymes in neointimal growth caused by vascular injury. The major AA‐metabolizing enzymes cyclooxygenase 2, via production of prostaglandin E2 acting on EP3 receptor,43 12/15 lipoxygenase, and CYP4A1/A2, most likely through generation of AA metabolites 12‐, 15‐, and 20‐hydroxyeicosatetraenoic acids, respectively, also contribute to neointimal growth.44, 45, 46, 47 Whether these enzymes and CYP1B1 act in a sequential or parallel and redundant manner is not known. Previously, we have reported that Cyp1b1 gene disruption does not affect the expression of other AA‐metabolizing enzymes examined in the VSMCs and the kidney.6, 48 However, the cross talk between 12/15‐lipoxygenase and cyclooxygenase 2 pathways, where they can activate each other, has been reported in mesangial cells.49 Because AA metabolism via these enzymes is also associated with generation of ROS,50, 51, 52 it is possible that ROS and/or AA metabolites generated via different enzyme systems stimulate neointimal formation caused by vascular injury, by activating ≥1 common signaling pathways, including extracellular signal‐regulated kinase 1/2 and p38 mitogen‐activated protein kinase.53, 54
The present study provides novel insight into the role of CYP1B1 in neointimal growth and associated pathogenesis via superoxide production in wire‐injured carotid artery of male mice. However, CYP1B1 is known to metabolize 17β‐estradiol into 2‐hydroxyestradiol, and that is subsequently metabolized into 2‐methoxyestradiol, which minimizes Ang II–induced hypertension and oxidative stress in female mice55; both estradiol and 2‐methoxyestradiol reduce neointimal growth in response to balloon angioplasty.56, 57 Therefore, it would be important to determine if Cyp1b1 gene disruption or 2,3′,4,5′‐tetramethoxystilbene treatment enhances neointimal growth and associated pathophysiological changes in response to vascular injury by increasing oxidative stress in female mice.
In conclusion, results of the present study suggest that CYP1B1 is a unique molecule that plays a critical role in neointimal growth caused by vascular injury in male mice. Our finding that the CYP1B1 inhibitor 2,3′,4,5′‐tetramethoxystilbene minimizes hypertension, atherosclerosis, and abdominal aneurysms,5, 28, 29 while also attenuating neointimal growth in wire‐injured carotid artery of Cyp1b1 +/+ male mice, further supports our hypothesis that CYP1B1 is required for neointimal growth and suggests an important translational implication of this work. Thus, CYP1B1 could serve as a potential target for the development of novel agents for the treatment of restenosis after balloon angioplasty, or to coat drug‐eluting stents as antiproliferative agents in male mice.
Sources of Funding
This work was supported by the National Institutes of Health, National Heart, Lung, and Blood Institute grants R01HL‐079109‐09 and R01HL‐19134‐42 (Malik), and by The Neuroscience Institute, University of Tennessee Health Science Center Postdoctoral/Research Associate Award (Mukherjee). The contents of this article are solely the responsibility of the authors and do not necessarily represent the official views of the National Heart, Lung, and Blood Institute.
Disclosures
None.
Supporting information
Figure S1. Cytochrome P450 1B1 (Cyp1b1) gene disruption prevents platelet‐derived growth factor‐BB (PDGF‐BB)‐induced migration of vascular smooth muscle cells (VSMCs). Representative images of migration after 0 and 24 hours of scratch wound healing assay in VSMCs from (A) VSMCs from Cyp1b1 +/+ and Cyp1b1 −/− mice treated with vehicle or PDGF‐BB (10 ng/mL) and (B) VSMCs from Cyp1b1 +/+ mice treated with vehicle or PDGF‐BB (10 ng/mL) in presence or absence of Tempol (2 mmol/L). (n=15 [5 scratch wound areas per well for 3 wells]) for each treatment condition, for each experiment, with experiments repeated at least 3 times).
Figure S2. Treatment with platelet‐derived growth factor‐BB (PDGF‐BB) does not alter expression of cytochrome P450 1B1 (CYP1B1) in vascular smooth muscle cells (VSMCs) of Cyp1b1 +/+ mice, and PDGF‐B expression is not altered in VSMCs from Cyp1b1 +/+ and Cyp1b1 −/− mice. A, Representative Western blot image of protein expression of CYP1B1 and β‐actin in VSMCs from Cyp1b1 +/+ and Cyp1b1 −/− mice treated with vehicle or PDGF‐BB (10 and 20 ng/mL) for 24 hours. C, Representative Western blot image of protein expression of PDGF‐B and β‐actin in VSMCs from Cyp1b1 +/+ and Cyp1b1 −/− mice. Quantitation of (B) CYP1B1 and (D) PDGF‐B protein levels normalized against β‐actin, which was used as a loading control. Values are the mean±SEM density of bands of CYP1B1 and PDGF‐B and β‐actin from 4 different experiments.
Figure S3. Treatment with platelet‐derived growth factor‐BB (PDGF‐BB) does not alter activity of cytochrome P450 1B1 (CYP1B1) in vascular smooth muscle cells (VSMCs). CYP1B1 activity as measured by luminescence using luciferin detection reagent in VSMCs from Cyp1b1 +/+ and Cyp1b1 −/− mice treated with vehicle or PDGF‐BB (20 ng/mL) for 24 hours. Experiments were repeated 6 times and data are expressed as mean±SEM.
Figure S4. Wire injury causes denudation of endothelial layer of carotid artery in mice. Representative images of immunohistochemical detection of von Willebrand factor (vWF) expression (brown) in uninjured and injured carotid arteries of (A) Cyp1b1 +/+ and (B) Cyp1b1 −/− mice, after 1 day of injury. Scale bars represent 200 μm (upper panel) and 50 μm (lower panel).
Figure S5. Cytochrome P450 1B1 (Cyp1b1) gene disruption attenuates smooth muscle actin deposition in wire‐injured carotid artery of mice. Representative images of immunohistochemical detection of smooth muscle α‐actin expression (brown) in uninjured and injured carotid arteries of (A) Cyp1b1 +/+ and (B) Cyp1b1 −/− mice, after 14 days of injury. Scale bars represent 50 μm.
Figure S6. Cytochrome P450 1B1 (Cyp1b1) gene disruption minimizes reactive oxygen species generation in wire‐injured carotid artery of mice. Representative images of reactive oxygen species production as indicated by fluorescence of 2‐hydroxyethidium (2‐OHE) (red) in uninjured and injured carotid arteries of (A) Cyp1b1 +/+ and (B) Cyp1b1 −/− mice, after 14 days of injury. Scale bars represent 200 μm (upper panel) and 50 μm (lower panel). C, Quantitative analysis of 2‐OHE fluorescence intensity in uninjured and injured carotid arteries of Cyp1b1 +/+ and Cyp1b1 −/− mice; n=4 for Cyp1b1 +/+ uninjured, n=5 for Cyp1b1 +/+ injured, n=5 for Cyp1b1 −/− uninjured, n=4 for Cyp1b1 −/− uninjured. Data are expressed as mean±SEM.
Figure S7. Treatment with 2,3′,4,5′‐tetramethoxystilbene (TMS) prevents neointimal growth and associated pathophysiology in wire‐injured carotid artery of Cytochrome P450 1B1 (Cyp1b1) +/+ mice. Representative images of Hematoxylin and Eosin (H&E) staining in injured carotid arteries of Cyp1b1 +/+ mice treated with (A) vehicle (Dimethyl sulfoxide, DMSO) or (B) TMS (300 μg/kg; i.p., every third day) after 14 days of injury. Scale bars represent 200 μm (upper panel) and 50 μm (lower panel). Quantitative analysis of neointimal growth in injured carotid arteries of Cyp1b1 +/+ mice treated with vehicle or TMS, as represented by (C) total intimal area, (D) intima‐to‐media ratio and (E) percentage area of restenosis (n=10 for vehicle group and n=9 for TMS group).
Acknowledgments
We thank Dr Richard Redfearn of the Office of Scientific Writing in the Office of Research at University of Tennessee Health Science Center for editorial assistance.
(J Am Heart Assoc. 2018;7:e010065 DOI: 10.1161/JAHA.118.010065.)
References
- 1. Schwartz SM, Stemerman MB, Benditt EP. The aortic intima, II: repair of the aortic lining after mechanical denudation. Am J Pathol. 1975;81:15–42. [PMC free article] [PubMed] [Google Scholar]
- 2. Er OB, Ma X, Simard T, Pourdjabbar A, Hibbert B. Pathogenesis of neointima formation following vascular injury. Cardiovasc Hematol Disord Drug Targets. 2011;11:30–39. [DOI] [PubMed] [Google Scholar]
- 3. Sata M, Saiura A, Kunisato A, Tojo A, Okada S, Tokuhisa T, Hirai H, Makuuchi M, Hirata Y, Nagai R. Hematopoietic stem cells differentiate into vascular cells that participate in the pathogenesis of atherosclerosis. Nat Med. 2002;8:403–409. [DOI] [PubMed] [Google Scholar]
- 4. Wessely R. New drug‐eluting stent concepts. Nat Rev Cardiol. 2010;7:194–203. [DOI] [PubMed] [Google Scholar]
- 5. Jennings BL, Sahan‐Firat S, Estes AM, Das K, Farjana N, Fang XR, Gonzalez FJ, Malik KU. Cytochrome P450 1B1 contributes to angiotensin II‐induced hypertension and associated pathophysiology. Hypertension. 2010;56:667–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yaghini FA, Song CY, Lavrentyev EN, Ghafoor HU, Fang XR, Estes AM, Campbell WB, Malik KU. Angiotensin II‐induced vascular smooth muscle cell migration and growth are mediated by cytochrome P450 1B1‐dependent superoxide generation. Hypertension. 2010;55:1461–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Raines EW. PDGF and cardiovascular disease. Cytokine Growth Factor Rev. 2004;15:237–254. [DOI] [PubMed] [Google Scholar]
- 8. Azevedo LC, Pedro MA, Souza LC, de Souza HP, Janiszewski M, da Luz PL, Laurindo FR. Oxidative stress as a signaling mechanism of the vascular response to injury: the redox hypothesis of restenosis. Cardiovasc Res. 2000;47:436–445. [DOI] [PubMed] [Google Scholar]
- 9. Wilcox CS, Pearlman A. Chemistry and antihypertensive effects of tempol and other nitroxides. Pharmacol Rev. 2008;60:418–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ellmark SH, Dusting GJ, Fui MN, Guzzo‐Pernell N, Drummond GR. The contribution of Nox4 to NADPH oxidase activity in mouse vascular smooth muscle. Cardiovasc Res. 2005;65:495–504. [DOI] [PubMed] [Google Scholar]
- 11. Lindner V, Fingerle J, Reidy MA. Mouse model of arterial injury. Circ Res. 1993;73:792–796. [DOI] [PubMed] [Google Scholar]
- 12. Buters JT, Sakai S, Richter T, Pineau T, Alexander DL, Savas U, Doehmer J, Ward JM, Jefcoate CR, Gonzalez FJ. Cytochrome P450 CYP1B1 determines susceptibility to 7, 12‐dimethylbenz[a]anthracene‐induced lymphomas. Proc Natl Acad Sci USA. 1999;96:1977–1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wilcox CS. Effects of tempol and redox‐cycling nitroxides in models of oxidative stress. Pharmacol Ther. 2010;126:119–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Miller FJ Jr, Gutterman DD, Rios CD, Heistad DD, Davidson BL. Superoxide production in vascular smooth muscle contributes to oxidative stress and impaired relaxation in atherosclerosis. Circ Res. 1998;82:1298–1305. [DOI] [PubMed] [Google Scholar]
- 15. Mugabe BE, Yaghini FA, Song CY, Buharalioglu CK, Waters CM, Malik KU. Angiotensin II‐induced migration of vascular smooth muscle cells is mediated by p38 mitogen‐activated protein kinase‐activated c‐Src through spleen tyrosine kinase and epidermal growth factor receptor transactivation. J Pharmacol Exp Ther. 2010;332:116–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Parmentier JH, Pavicevic Z, Malik KU. Ang II stimulates phospholipase D through PKCzeta activation in VSMC: implications in adhesion, spreading, and hypertrophy. Am J Physiol Heart Circ Physiol. 2006;290:H46–H54. [DOI] [PubMed] [Google Scholar]
- 17. Yaghini FA, Li F, Malik KU. Expression and mechanism of spleen tyrosine kinase activation by angiotensin II and its implication in protein synthesis in rat vascular smooth muscle cells. J Biol Chem. 2007;282:16878–16890. [DOI] [PubMed] [Google Scholar]
- 18. Chun YJ, Kim S, Kim D, Lee SK, Guengerich FP. A new selective and potent inhibitor of human cytochrome P450 1B1 and its application to antimutagenesis. Cancer Res. 2001;61:8164–8170. [PubMed] [Google Scholar]
- 19. Chaabane C, Otsuka F, Virmani R, Bochaton‐Piallat ML. Biological responses in stented arteries. Cardiovasc Res. 2013;99:353–363. [DOI] [PubMed] [Google Scholar]
- 20. Ferns GA, Raines EW, Sprugel KH, Motani AS, Reidy MA, Ross R. Inhibition of neointimal smooth muscle accumulation after angioplasty by an antibody to PDGF. Science. 1991;253:1129–1132. [DOI] [PubMed] [Google Scholar]
- 21. Sundaresan M, Yu ZX, Ferrans VJ, Irani K, Finkel T. Requirement for generation of H2O2 for platelet‐derived growth factor signal transduction. Science. 1995;270:296–299. [DOI] [PubMed] [Google Scholar]
- 22. Zafari AM, Ushio‐Fukai M, Minieri CA, Akers M, Lassegue B, Griendling KK. Arachidonic acid metabolites mediate angiotensin II‐induced NADP/NADPH oxidase activity and hypertrophy in vascular smooth muscle cells. Antioxid Redox Signal. 1999;1:167–179. [DOI] [PubMed] [Google Scholar]
- 23. Yellaturu CR, Rao GN. Cytosolic phospholipase A2 is an effector of Jak/STAT signaling and is involved in platelet‐derived growth factor BB‐induced growth in vascular smooth muscle cells. J Biol Chem. 2003;278:9986–9992. [DOI] [PubMed] [Google Scholar]
- 24. Kanayasu‐Toyoda T, Morita I, Murota S. Arachidonic acid pretreatment enhances smooth muscle cell migration via increased Ca2+ influx. Prostaglandins Leukot Essent Fatty Acids. 1998;58:25–31. [DOI] [PubMed] [Google Scholar]
- 25. Zhang W, Wang Y, Chen CW, Xing K, Vivekanandan S, Lou MF. The positive feedback role of arachidonic acid in the platelet‐derived growth factor‐induced signaling in lens epithelial cells. Mol Vis. 2006;12:821–831. [PubMed] [Google Scholar]
- 26. Shi HT, Wang Y, Jia LX, Qin YW, Liu Y, Li HH, Qi YF, Du J. Cathepsin s contributes to macrophage migration via degradation of elastic fibre integrity to facilitate vein graft neointimal hyperplasia. Cardiovasc Res. 2014;101:454–463. [DOI] [PubMed] [Google Scholar]
- 27. Indolfi C, Torella D, Coppola C, Stabile E, Esposito G, Curcio A, Pisani A, Cavuto L, Arcucci O, Cireddu M, Troncone G, Chiariello M. Rat carotid artery dilation by PTCA balloon catheter induces neointima formation in presence of IEL rupture. Am J Physiol Heart Circ Physiol. 2002;283:H760–H767. [DOI] [PubMed] [Google Scholar]
- 28. Song CY, Ghafoor K, Ghafoor HU, Khan NS, Thirunavukkarasu S, Jennings BL, Estes AM, Zaidi S, Bridges D, Tso P, Gonzalez FJ, Malik KU. Cytochrome P450 1B1 contributes to the development of atherosclerosis and hypertension in apolipoprotein E‐deficient mice. Hypertension. 2016;67:206–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Thirunavukkarasu S, Khan NS, Song CY, Ghafoor HU, Brand DD, Gonzalez FJ, Malik KU. Cytochrome P450 1B1 contributes to the development of angiotensin II‐induced aortic aneurysm in male ApoE(‐/‐) mice. Am J Pathol. 2016;186:2204–2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Simon DI. Inflammation and vascular injury: basic discovery to drug development. Circ J. 2012;76:1811–1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kerzee JK, Ramos KS. Constitutive and inducible expression of Cyp1a1 and Cyp1b1 in vascular smooth muscle cells: role of the Ahr bHLH/PAS transcription factor. Circ Res. 2001;89:573–582. [DOI] [PubMed] [Google Scholar]
- 32. Ward JM, Nikolov NP, Tschetter JR, Kopp JB, Gonzalez FJ, Kimura S, Siegel RM. Progressive glomerulonephritis and histiocytic sarcoma associated with macrophage functional defects in CYP1B1‐deficient mice. Toxicol Pathol. 2004;32:710–718. [DOI] [PubMed] [Google Scholar]
- 33. Maecker B, Sherr DH, Vonderheide RH, von Bergwelt‐Baildon MS, Hirano N, Anderson KS, Xia Z, Butler MO, Wucherpfennig KW, O'Hara C, Cole G, Kwak SS, Ramstedt U, Tomlinson AJ, Chicz RM, Nadler LM, Schultze JL. The shared tumor‐associated antigen cytochrome P450 1B1 is recognized by specific cytotoxic T cells. Blood. 2003;102:3287–3294. [DOI] [PubMed] [Google Scholar]
- 34. Labinaz M, Pels K, Hoffert C, Aggarwal S, O'Brien ER. Time course and importance of neoadventitial formation in arterial remodeling following balloon angioplasty of porcine coronary arteries. Cardiovasc Res. 1999;41:255–266. [DOI] [PubMed] [Google Scholar]
- 35. Zhang Y, Guallar E, Qiao Y, Wasserman BA. Is carotid intima‐media thickness as predictive as other noninvasive techniques for the detection of coronary artery disease? Arterioscler Thromb Vasc Biol. 2014;34:1341–1345. [DOI] [PubMed] [Google Scholar]
- 36. Jennings BL, Anderson LJ, Estes AM, Fang XR, Song CY, Campbell WB, Malik KU. Involvement of cytochrome P‐450 1B1 in renal dysfunction, injury, and inflammation associated with angiotensin II‐induced hypertension in rats. Am J Physiol Renal Physiol. 2012;302:F408–F420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Maiellaro K, Taylor WR. The role of the adventitia in vascular inflammation. Cardiovasc Res. 2007;75:640–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Haurani MJ, Cifuentes ME, Shepard AD, Pagano PJ. Nox4 oxidase overexpression specifically decreases endogenous Nox4 mrna and inhibits angiotensin II‐induced adventitial myofibroblast migration. Hypertension. 2008;52:143–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wilcox JN, Okamoto EI, Nakahara KI, Vinten‐Johansen J. Perivascular responses after angioplasty which may contribute to postangioplasty restenosis: a role for circulating myofibroblast precursors? Ann N Y Acad Sci. 2001;947:68–90; discussion 90–92. [DOI] [PubMed] [Google Scholar]
- 40. Shi Y, Niculescu R, Wang D, Patel S, Davenpeck KL, Zalewski A. Increased NAD(P)H oxidase and reactive oxygen species in coronary arteries after balloon injury. Arterioscler Thromb Vasc Biol. 2001;21:739–745. [DOI] [PubMed] [Google Scholar]
- 41. Haurani MJ, Pagano PJ. Adventitial fibroblast reactive oxygen species as autacrine and paracrine mediators of remodeling: bellwether for vascular disease? Cardiovasc Res. 2007;75:679–689. [DOI] [PubMed] [Google Scholar]
- 42. Dikalov SI, Harrison DG. Methods for detection of mitochondrial and cellular reactive oxygen species. Antioxid Redox Signal. 2014;20:372–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhang J, Zou F, Tang J, Zhang Q, Gong Y, Wang Q, Shen Y, Xiong L, Breyer RM, Lazarus M, Funk CD, Yu Y. Cyclooxygenase‐2‐derived prostaglandin E2 promotes injury‐induced vascular neointimal hyperplasia through the E‐prostanoid 3 receptor. Circ Res. 2013;113:104–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gu JL, Pei H, Thomas L, Nadler JL, Rossi JJ, Lanting L, Natarajan R. Ribozyme‐mediated inhibition of rat leukocyte‐type 12‐lipoxygenase prevents intimal hyperplasia in balloon‐injured rat carotid arteries. Circulation. 2001;103:1446–1452. [DOI] [PubMed] [Google Scholar]
- 45. Deliri H, Meller N, Kadakkal A, Malhotra R, Brewster J, Doran AC, Pei H, Oldham SN, Skaflen MD, Garmey JC, McNamara CA. Increased 12/15‐lipoxygenase enhances cell growth, fibronectin deposition, and neointimal formation in response to carotid injury. Arterioscler Thromb Vasc Biol. 2011;31:110–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yaghini FA, Zhang C, Parmentier JH, Estes AM, Jafari N, Schaefer SA, Malik KU. Contribution of arachidonic acid metabolites derived via cytochrome P450 4A to angiotensin II‐induced neointimal growth. Hypertension. 2005;45:1182–1187. [DOI] [PubMed] [Google Scholar]
- 47. Orozco LD, Liu H, Perkins E, Johnson DA, Chen BB, Fan F, Baker RC, Roman RJ. 20‐Hydroxyeicosatetraenoic acid inhibition attenuates balloon injury‐induced neointima formation and vascular remodeling in rat carotid arteries. J Pharmacol Exp Ther. 2013;346:67–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Jennings BL, Anderson LJ, Estes AM, Yaghini FA, Fang XR, Porter J, Gonzalez FJ, Campbell WB, Malik KU. Cytochrome P450 1B1 contributes to renal dysfunction and damage caused by angiotensin II in mice. Hypertension. 2012;59:348–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Xu ZG, Li SL, Lanting L, Kim YS, Shanmugam N, Reddy MA, Natarajan R. Relationship between 12/15‐lipoxygenase and COX‐2 in mesangial cells: potential role in diabetic nephropathy. Kidney Int. 2006;69:512–519. [DOI] [PubMed] [Google Scholar]
- 50. Kukreja RC, Kontos HA, Hess ML, Ellis EF. PGH synthase and lipoxygenase generate superoxide in the presence of NADH or NADPH. Circ Res. 1986;59:612–619. [DOI] [PubMed] [Google Scholar]
- 51. Roman RJ. P‐450 metabolites of arachidonic acid in the control of cardiovascular function. Physiol Rev. 2002;82:131–185. [DOI] [PubMed] [Google Scholar]
- 52. Ishizuka T, Cheng J, Singh H, Vitto MD, Manthati VL, Falck JR, Laniado‐Schwartzman M. 20‐Hydroxyeicosatetraenoic acid stimulates nuclear factor‐kappaB activation and the production of inflammatory cytokines in human endothelial cells. J Pharmacol Exp Ther. 2008;324:103–110. [DOI] [PubMed] [Google Scholar]
- 53. Izumi Y, Kim S, Namba M, Yasumoto H, Miyazaki H, Hoshiga M, Kaneda Y, Morishita R, Zhan Y, Iwao H. Gene transfer of dominant‐negative mutants of extracellular signal‐regulated kinase and c‐Jun NH2‐terminal kinase prevents neointimal formation in balloon‐injured rat artery. Circ Res. 2001;88:1120–1126. [DOI] [PubMed] [Google Scholar]
- 54. Ohashi N, Matsumori A, Furukawa Y, Ono K, Okada M, Iwasaki A, Miyamoto T, Nakano A, Sasayama S. Role of p38 mitogen‐activated protein kinase in neointimal hyperplasia after vascular injury. Arterioscler Thromb Vasc Biol. 2000;20:2521–2526. [DOI] [PubMed] [Google Scholar]
- 55. Jennings BL, George LW, Pingili AK, Khan NS, Estes AM, Fang XR, Gonzalez FJ, Malik KU. Estrogen metabolism by cytochrome P450 1B1 modulates the hypertensive effect of angiotensin II in female mice. Hypertension. 2014;64:134–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Foegh ML, Asotra S, Howell MH, Ramwell PW. Estradiol inhibition of arterial neointimal hyperplasia after balloon injury. J Vasc Surg. 1994;19:722–726. [DOI] [PubMed] [Google Scholar]
- 57. Barchiesi F, Jackson EK, Fingerle J, Gillespie DG, Odermatt B, Dubey RK. 2‐Methoxyestradiol, an estradiol metabolite, inhibits neointima formation and smooth muscle cell growth via double blockade of the cell cycle. Circ Res. 2006;99:266–274. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Cytochrome P450 1B1 (Cyp1b1) gene disruption prevents platelet‐derived growth factor‐BB (PDGF‐BB)‐induced migration of vascular smooth muscle cells (VSMCs). Representative images of migration after 0 and 24 hours of scratch wound healing assay in VSMCs from (A) VSMCs from Cyp1b1 +/+ and Cyp1b1 −/− mice treated with vehicle or PDGF‐BB (10 ng/mL) and (B) VSMCs from Cyp1b1 +/+ mice treated with vehicle or PDGF‐BB (10 ng/mL) in presence or absence of Tempol (2 mmol/L). (n=15 [5 scratch wound areas per well for 3 wells]) for each treatment condition, for each experiment, with experiments repeated at least 3 times).
Figure S2. Treatment with platelet‐derived growth factor‐BB (PDGF‐BB) does not alter expression of cytochrome P450 1B1 (CYP1B1) in vascular smooth muscle cells (VSMCs) of Cyp1b1 +/+ mice, and PDGF‐B expression is not altered in VSMCs from Cyp1b1 +/+ and Cyp1b1 −/− mice. A, Representative Western blot image of protein expression of CYP1B1 and β‐actin in VSMCs from Cyp1b1 +/+ and Cyp1b1 −/− mice treated with vehicle or PDGF‐BB (10 and 20 ng/mL) for 24 hours. C, Representative Western blot image of protein expression of PDGF‐B and β‐actin in VSMCs from Cyp1b1 +/+ and Cyp1b1 −/− mice. Quantitation of (B) CYP1B1 and (D) PDGF‐B protein levels normalized against β‐actin, which was used as a loading control. Values are the mean±SEM density of bands of CYP1B1 and PDGF‐B and β‐actin from 4 different experiments.
Figure S3. Treatment with platelet‐derived growth factor‐BB (PDGF‐BB) does not alter activity of cytochrome P450 1B1 (CYP1B1) in vascular smooth muscle cells (VSMCs). CYP1B1 activity as measured by luminescence using luciferin detection reagent in VSMCs from Cyp1b1 +/+ and Cyp1b1 −/− mice treated with vehicle or PDGF‐BB (20 ng/mL) for 24 hours. Experiments were repeated 6 times and data are expressed as mean±SEM.
Figure S4. Wire injury causes denudation of endothelial layer of carotid artery in mice. Representative images of immunohistochemical detection of von Willebrand factor (vWF) expression (brown) in uninjured and injured carotid arteries of (A) Cyp1b1 +/+ and (B) Cyp1b1 −/− mice, after 1 day of injury. Scale bars represent 200 μm (upper panel) and 50 μm (lower panel).
Figure S5. Cytochrome P450 1B1 (Cyp1b1) gene disruption attenuates smooth muscle actin deposition in wire‐injured carotid artery of mice. Representative images of immunohistochemical detection of smooth muscle α‐actin expression (brown) in uninjured and injured carotid arteries of (A) Cyp1b1 +/+ and (B) Cyp1b1 −/− mice, after 14 days of injury. Scale bars represent 50 μm.
Figure S6. Cytochrome P450 1B1 (Cyp1b1) gene disruption minimizes reactive oxygen species generation in wire‐injured carotid artery of mice. Representative images of reactive oxygen species production as indicated by fluorescence of 2‐hydroxyethidium (2‐OHE) (red) in uninjured and injured carotid arteries of (A) Cyp1b1 +/+ and (B) Cyp1b1 −/− mice, after 14 days of injury. Scale bars represent 200 μm (upper panel) and 50 μm (lower panel). C, Quantitative analysis of 2‐OHE fluorescence intensity in uninjured and injured carotid arteries of Cyp1b1 +/+ and Cyp1b1 −/− mice; n=4 for Cyp1b1 +/+ uninjured, n=5 for Cyp1b1 +/+ injured, n=5 for Cyp1b1 −/− uninjured, n=4 for Cyp1b1 −/− uninjured. Data are expressed as mean±SEM.
Figure S7. Treatment with 2,3′,4,5′‐tetramethoxystilbene (TMS) prevents neointimal growth and associated pathophysiology in wire‐injured carotid artery of Cytochrome P450 1B1 (Cyp1b1) +/+ mice. Representative images of Hematoxylin and Eosin (H&E) staining in injured carotid arteries of Cyp1b1 +/+ mice treated with (A) vehicle (Dimethyl sulfoxide, DMSO) or (B) TMS (300 μg/kg; i.p., every third day) after 14 days of injury. Scale bars represent 200 μm (upper panel) and 50 μm (lower panel). Quantitative analysis of neointimal growth in injured carotid arteries of Cyp1b1 +/+ mice treated with vehicle or TMS, as represented by (C) total intimal area, (D) intima‐to‐media ratio and (E) percentage area of restenosis (n=10 for vehicle group and n=9 for TMS group).