

SUMMARY
Inducers of satiety are drug targets for weight loss to mitigate obesity-associated diseases. Nucleobindin-2 (Nucb2) is thought to be post-translationally processed into bioactive nesfatin-1 peptide, which reportedly induces satiety, causes weight loss, and thus improves insulin sensitivity. Here, we show that deletion of Nucb2 did not affect food intake or adiposity and, instead, caused insulin resistance in mice fed a high-fat diet. In addition, ablation of Nucb2 in orexigenic hypothalamic Agrp neurons did not affect food intake, and nesfatin-1 was detectable in serum, despite global deletion of Nucb2 protein. Upon high-fat diet feeding, the loss of Nucb2 exacerbated metabolic inflammation in adipose tissue macrophages in an NFκB-dependent manner without inducing classical M1 or alternative M2-like macrophage polarization. Furthermore, the loss of Nucb2 in myeloid cells but not in adipocytes mediated the insulin resistance in response to a high-fat diet. Our study reveals that Nucb2 links metabolic inflammation to insulin resistance without affecting weight gain and food intake.
In Brief
Obesity induces Nucb2 expression in adipose macrophages as an endogenous brake to restrain metabolic inflammation. Ravussin et al. show that ablation of Nucb2 globally or knockdown in orexigenic neurons and adipocytes did not affect food intake or adiposity. The loss of Nucb2 in myeloid cells drive inflammation and insulin resistance upon high-fat feeding.
Graphical Abstract
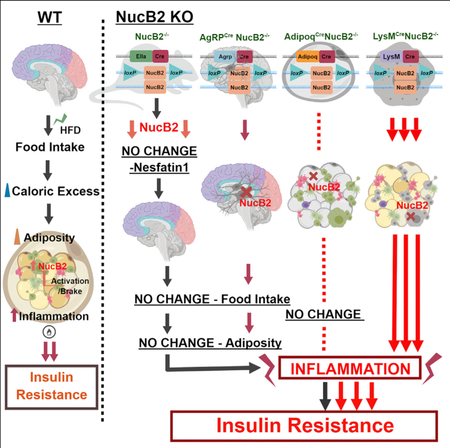
INTRODUCTION
Loss of appetite as a result of inflammation, as well as the ability of immune cells to sense changes in circulating hormones and nutrient levels, suggests the existence of shared signaling pathways between immune and neuroendocrine systems (Besedovsky and del Rey, 1996; Dixit, 2008). Accordingly, orexigenic peptides inhibit (Dixit et al., 2004; Cheyuo et al., 2012), while anorexigenic hormones promote, inflammation (Matarese, 2000; Procaccini et al., 2014). First identified in a B cell line, nucleobindin-2 (NUCB2)/non-esterified fatty acids (NEFA) (for DNA-binding/EF-hand/acidic protein), is ubiquitously expressed in peripheral tissues and immune cells and is a calcium-binding protein that contains nuclear targeting signals, two helix-loop-helix regions with a concurrent EF-hand, and a leucine zipper motif (Miura et al., 1992; Lin et al., 1998). NUCB2 is encoded on chromosome 7F1, containing 14 exons in mice (chromosome 11 in human), with a genomic sequence of 36,186 bp. Post-transcription modifications lead to an mRNA sequence of 1,697 bp with a polypeptide 420 amino acids long. This protein then undergoes posttranslational modifications and is thought to be cleaved into three peptides, termed nesfatin-1, nesfatin-2, and nesfatin-3. Nesfatin-1, the 82-amino-acid peptide derived from Nucb2 protein, was found to reduce food intake by acting on an unknown receptor in the hypothalamus (Oh-I et al., 2006; Dore et al., 2017). Consistent with this, the vast majority of published studies on Nucb2 suggest that nesfatin-1-mediated anorexigenic effects on the hypothalamus are key drivers of the metabolic action of Nucb2.
In addition to the brain, NUCB2/nesfatin-1 is ubiquitously expressed in peripheral tissues, including the pancreas, adipose tissue, liver, kidney, and the gut (Dore et al., 2017). Such distribution implies that NUCB2/nesfatin-1 has important metabolic as well as immunologic functions. Pharmacological studies using administration of nesfatin-1 peptide suggest effects on glucose homeostasis, gastrointestinal function, water intake, temperature regulation, and sleep (Shimizu et al., 2009). Importantly, the presence of nesfatin-1 has been reported using commercial antibodies that have not been rigorously tested to discriminate full-length Nucb2 from its cleaved peptides (Stengel et al., 2009). Although evidence from multiple studies supports that administration of nesfatin-1 peptide is biologically active, the validity of such a mechanism downstream of Nucb2 in vivo has been not been established in a physiological setting. Thus, the function of Nucb2 in various cell types remains unknown, and its mechanism of action in regulation of whole-body metabolism is also unclear. Here, through global, Agrp-neuron-specific, macrophage-specific, and adipocyte-specific deletion of Nucb2, we provide evidence that this protein regulates metabolic inflammation and insulin action but does not affect satiety.
RESULTS AND DISCUSSION
Given current evidence that Nucb2 inhibits feeding (Oh-I et al., 2006), we hypothesized that the anorexigenic protein Nucb2 might link the metabolic and immune systems by regulating inflammation. First, to understand whether Nucb2 expression was regulated by age and/or diet, we analyzed Nucb2 mRNA expression in 3-month-old and 6-month-old wild-type mice on either a low-fat diet or a high-fat diet (HFD). These analyses revealed that, upon HFD feeding, the hypothalamus, liver, and visceral adipose tissue (VAT) displayed no change in Nucb2 expression (Figures S1A–S1C). Interestingly, 3 and 6 months of HFD feeding led to a significant increase in Nucb2 expression in adipose tissue macrophages (ATMs) (Figure 1A) and in adipose T cells (Figure S1D) isolated from the VAT. These data suggest a potential role of Nucb2 in integrating host immunometabolic responses in conditions of chronic positive energy balance.
Figure 1. Nucleobindin-2 Is Not a Physiological Regulator of Food Intake.

(A) Real-time PCR analysis of Nucb2 mRNA in F4/80+ visceral ATMs derived from mice fed a 60% HFD for 3 and 6 months (n = 6 per group; *p<0.05, **p<0.01.) (Figure S1D).
(B) Immunoblot analysis of Nucb2 protein from different tissues of control and global Nucb2-deficient mice. The arrowhead on the left indicates the molecular weight of the Nucb2-specific band. NS, non-specific band. (See Figures S2A and S2B for mRNA expression in cells and organs of control and Nucb2−/− mice.)
(C) Representative confocal microscopy images of mouse embryonic fibroblasts (MEFs) from WT (top) and Nucb2−/− (bottom) mice with (right) and without (left) treatment of 1 μg/mL LPS for 1 hr, labeled for Nucb2 (green), F-actin (red), and nucleus (blue). Scale bars, 50 μM.
(D) ELISA of nesfatin-1 in plasma from WT and Nucb2-deficient mice and measurement of nesfatin-1 after the addition of 1,000 pg/mL peptide in plasma.
(E) Body weight (left) and food intake (right) of WT and AgRPCreNucb2−/− mice.
(F) Food intake over 24 hr after fasting and refeeding (left) and blood glucose levels after fasting (right) in control and Agrp-neuron-specific Nucb2-deficient mice (n = 12 per group).
See also Figures S1D and S2.
To gain definitive insights into the physiological roles of Nucb2 in the regulation of food intake and immune-metabolic interaction, we generated mice harboring a floxed allele of the Nucb2 gene (Figures S1E–S1H). Global deletion of Nucb2 was achieved by breeding Nucb2 floxed mice to those expressing Cre recombinase under the control of the EIIA promoter. This resulted in efficient loss of Nucb2 protein and mRNA in hypothalamus, hippocampus, cortex, striatum, cerebellum, pituitary, and brown adipose and various white adipose tissues, as well as pancreas, liver, spleen, and thymus (Figures 1B and S2A), without any overt phenotypes in knockout mice (data not shown) maintained on regular chow diet. In terms of cell-type specificity, the Nucb2 was highly expressed in fibroblasts, adipose tissue T cells, B cells, and macrophages; and, as expected, the Nucb2 mRNA was not detectable in the cells derived from the knockout (KO) mice (Figures 1B and S2B). These data demonstrate that Nucb2 is widely expressed and that loss of Nucb2 does not induce any developmental defects in mice.
As Nucb2 contains nuclear targeting signals, we investigated the localization of Nucb2 in unstimulated and lipopolysaccharide (LPS)-activated murine embryonic fibroblasts (MEFs). The immunostaining confocal microscopy analyses revealed that Nucb2 protein was localized in the cytoplasm and did not translocate into nucleus upon LPS stimulation (Figure 1C). Prior studies suggest that nesfatin-1 is the primary endocrine mediator of Nucb2’s biological effects. Surprisingly, despite efficient deletion of the transcript and Nucb2 protein in mice, the commercial ELISAs used to detect nesfatin-1 appear to be non-specific, as no change in serum nesfatin-1 levels were detected in wild-type (WT) or global Nucb2-deficient mice (Figure 1D). Further validation of the commercial nesfatin-1 ELISA using three different commercially available ELISA kits did not accurately detect nesfatin-1 peptide (Figures S2C–S2F). These results question the specificity of reagents used to ascribe the physiological function of endogenous nesfatin-1 peptide that is derived from Nucb2. Consistent with this, we found that global deletion of Nucb2 had no effect on body weight and food intake in mice (Figures S2G and S2H), with no change in the orexigenic neuropeptide Agouti-related protein (AgRP) mRNA in the hypothalamus (Figure S2I). To definitively answer whether Nucb2 is a regulator of food intake, we also deleted Nucb2 from the AgRP orexigenic neurons in the hypothalamus. The deletion of Nucb2 in AgRP neurons also had no effect on body weight and food intake (Figure 1E) upon fasting and re-feeding and did not affect fasting blood glucose (Figure 1F). It is plausible that previous studies ascribing anorexigenic function to Nucb2 might result from supraphysiological doses of intracerebroventricularly injected synthetic nesfatin-1 peptide or short hairpin RNA injection (Nakata et al., 2016), which may, indeed, be bioactive, but based on our data, this anorexigenic action appears irrelevant to Nucb2’s physiological function and biology. Taken together, these results demonstrate that Nucb2 is regulated by HFD in ATMs and T cells and that Nucb2 protein or its post-translationally processed peptides do not control food intake or body weight in mice.
Given that HFD increases Nucb2 expression in ATMs, we next investigated the role of this gene in a model of diet-induced obesity (DIO). The control and Nucb2−/− mice fed an HFD displayed no significant difference in adipocyte size or fat mass (Figures S2J–S2M) and also had similar circulating levels of leptin and adiponectin (Figures S2N and S2O). When fed a chow diet and compared to control animals, Nucb2−/− mice showed no differences in whole-body insulin sensitivity, as measured by glucose infusion rate to maintain euglycemia during the entirety of the hyperinsulinemic-euglycemic clamp (Figures S3A–S3F) or plasma free fatty acids (Figures S3H and S3I). Despite high expression of Nucb2 in pancreas (Figures 1B and S2A), control and Nucb2−/− mice showed no difference in insulin production in both basal and clamped states in the chow-fed condition (Figure S3G). Interestingly, Nucb2−/− mice fed an HFD displayed significant whole-body insulin resistance, as reflected by an almost 50% decrease in the glucose infusion rate (21.3 ± 2.1 and 11.0 ± 0.9 for WT and Nucb2−/−, respectively; p = 0.002) required to maintain euglycemia during the hyperinsulinemic-euglycemic clamp (Figures 2A and 2B) without affecting insulin secretion (Figure 2C). On the other hand, there was a significant increase in endogenous hepatic glucose output (EGP) in the obese Nucb2−/− clamped state but not in the basal state (Figure 2D). Furthermore, compared to DIO WT mice, the Nucb2−/− animals fed an HFD had a significant increase in insulin-stimulated plasma free fatty acids (FFAs) (Figure 2E). This resulted in an approximately 57% FFA suppression in WT mice and only a 2% suppression in the Nucb2−/− mice under clamped conditions, suggesting adipocyte dysfunction in the absence of Nucb2 (Figure 2F). Overall, these data show that the primary function of Nucb2 is to sense a hyper-caloric HFD and control insulin sensitivity in conditions of chronic caloric excess.
Figure 2. Elimination of Nucleobindin-2 Causes Insulin Resistance during Diet-Induced Obesity.

(A–F) Shown here: (A) the time course of glucose infusion rate in DIO WT and Nucb2−/− during hyperinsulinemic euglycemic clamp, (B) total glucose infusion rate, (c) basal and clamped plasma insulin levels, (D) basal and clamped endogenous hepatic glucose output, (E) basal and clamped plasma fatty acids, and (F) plasma fatty acid suppression during hyperinsulinemic euglycemic clamp in control and Nucb2-deficient mice fed an HFD for 6 months. All clamp data are represented as mean ± SEM (WT, n = 11; Nucb2−/− n = 8).
(G and H) Fluorescence-activated cell sorting (FACS) quantification of (G) macrophage and (H) lymphocyte cell population frequencies in VAT from HFD-fed WT and Nucb2−/− mice.
(I and J) FACS quantification of (I) CD4+ naive and effector and memory (EM) cell subsets and (J) CD8+ naive and effector and memory cell subsets in VAT from 8-month-old, HFD-fed WT and Nucb2−/− mice (WT, n = 5; Nucb2−/− n = 8).
*p<0.05, **p<0.01, ***p<0.005. See also Figure S4.
To determine the mechanism of how Nucb2 deficiency causes insulin resistance, we next examined the adipose tissue immune cells that are implicated in inducing inflammation and impairing insulin signaling (Hotamisligil, 2006; Shoelson, 2006; Lumeng and Saltiel, 2011; Lee et al., 2018). Surprisingly, despite severe insulin resistance in obese Nucb2-deficient mice, there was no increase in the frequency of ATMs and naive or effector and memory CD4 and CD8 cells in the VAT (Figures 2G–2J; Figures S4A–S4F). Therefore, we next determined whether Nucb2 controls macrophage activation and inflammation. The bone-marrow-derived macrophages (BMDMs) deficient in Nucb2 were differentiated in vitro using either LPS and interferon (IFN) γ or interleukin (IL)-4 to obtain pro-inflammatory (former “M1-like”) or anti-inflammatory (former “M2-like”) activation states, respectively. Interestingly, compared to WT control cells, the BMDMs lacking Nucb2 displayed increased expression of the pro-inflammatory cytokines IL-1β, tumor necrosis factor alpha (TNF-α), and MIP-1α (Figures 3A–3C) in the IFNγ-activated macrophages, while no changes were detected in IL-4-activated cells. We confirmed these data in vivo, as the F4/80+ ATMs isolated from VAT of Nucb2−/− mice also displayed higher expression of IL-1β, TNFα, and MIP-1α (Figures 3D–3F). Furthermore, Nucb2-deficient BMDMs produced increased levels of active IL-1β (p17) protein (Figure 3G) with higher nuclear factor κB (NFκB) activation (Figure 3H) in response to Nlrp3 inflamma-some-dependent stimuli such as ATP, ceramides, or silica. Consistent with these data, inhibition of the NFκB pathway in Nucb2-deficient BMDMs abrogated the LPS-induced increased expression of IL-1β, TNFα, and MIP-1α (Figures 3I–3K). Together, these results demonstrate that loss of Nucb2 increases macrophage inflammation in an NFκB-dependent mechanism.
Figure 3. Ablation of Nucleobindin-2 Promotes Inflammation in Macrophages in an NFκB-Dependent Manner.

Primary bone-marrow-derived macrophages (BMDMs) from WT and Nucb2−/− mice were treated with media alone for unpolarized macrophages; 1 μg/mL LPS plus 4 ng/mL interferon gamma IFNγ) to generate proinflammatory, former classically activated “M1-like” macrophages; or 1 ng/ml IL-4 to obtain anti-inflammatory, former alternatively activated “M2-like” macrophages.
(A–C) Relative gene expression levels of (A) IL-1β, (B) TNFα, and (C) MIP-1α in control and Nucb2-deficient BMDMs. *p<0.05, **p<0.01, ***p<0.005. ****(D–F) Relative gene expression levels of (D) IL-1β, (E) TNFtα, and (F) MIP-1α in F4/80+ visceral adipose tissue sorted macrophages from obese, HFD-fed WT and Nucb2−/− mice. All data are represented as mean ± SEM (n = 4 per group). *p < 0.05, compared to WT.
(G and H) Representative western blot analyses of (G) IL-1β (active p17) in the supernatant and (H) NFκB (p65) and phospho-NFκB in the cell lysates of BMDMs primed with LPS for 4 hr and stimulated with either ATP for 1 hr, ceramide for 6 hr, or silica for 5 hr in WT- or Nucb2−/−-derived cells.
(I–K) Primary BMDMs from WT mice and Nucb2−/− mice were treated with media and 1 μg/mL LPS plus 4 ng/mL interferon gamma (IFNγ) to generate proinflammatory macrophages. Some macrophages were pretreated with Bay-11-7085 NFκB inhibitor. Displayed are the relative gene expression levels of (I) IL-1β, (J) TNFα, and (K) MIP-1α. All data are represented as mean ± SEM (n = 6 per group). *p < 0.05, compared to WT; **p<0.01, ***p<0.005, ****p<0.001.
We next sought to understand the mechanism of macrophage-mediated inflammatory response in obese mice that lack Nucb2. The F4/80+ macrophages from the VAT of obese WT and Nucb2−/− mice were sorted for whole transcriptome analysis by RNA sequencing (workflow in Figure 4A). Principal-component analysis (PCA) revealed separate clusters of Nucb2−/− and WT ATMs (Figure 4B), which was further supported by the hierarchical cluster analysis of the top 1,000 variable or differentially expressed (DE) genes (Figure 4C and 4D). To further investigate the activation status of ATMs, linear support vector regression (SVR) was used to map the previously established multi-dimensional model of macrophage activation, reflected through 28 different activation states (Xue et al., 2014; Camell et al., 2017), onto the ATMs isolated from obese Nucb2-deficient and WT mice. These unbiased analyses revealed a surprisingly high heterogeneity of signatures in both WT and Nucb2−/− ATMs with no clear trend toward previously described pro-inflammatory (“M1-like”) or anti-inflammatory (“M2-like”) activation states (Figure 4E). Moreover, the SVR analyses also demonstrate that when the transcriptome of macrophages activated by 28 different stimuli was overlaid with visceral ATMs in vivo, there were only minor differences between WT and Nucb2−/− macrophages in conditions of obesity (Figure 4E). Together, these data demonstrate that, in conditions of metabolic inflammation, the models of macrophage activation and polarization induced in the presence of high levels of IFNγ and IL-4 produced during an active immune response do not inform about the status of ATM dysfunction seen in Nucb2-dependent insulin resistance.
Figure 4. Macrophage Intrinsic Nucb2 Controls Whole-Body Insulin Resistance in Obesity by Regulating Inflammatory Tone in Adipose Tissue.

(A)The workflow of RNA-seq data analysis in (B)–(F).
(B) Principal-component analysis (PCA) based on 14,905 present genes displaying ATMs derived from control and Nucb2−/− mice fed an HFD.
(C) Heatmap depicting the top 1,000 most variable genes in Nucb2-deficient ATMs compared to control littermates. Z score of expression is scaled to a minimum of –2 and a maximum of 2. Rows and columns were ordered based on hierarchical clustering.
(D) Scatterplot of log2 average expression (x axis) versus log2 fold change (y axis) between WT ATMs and ATMs lacking Nucb2. Genes being significantly up- or downregulated (unadjusted p < 0.05) are indicated in red and blue, respectively. In addition, the top 20 up- or downregulated genes, ranked according to either fold change or expression in Nucb2−/− mice, were specifically labeled in black or brown, respectively.
(E) Relative fractions of 29 different human in-vitro-activated macrophage subset gene signatures represented in WT ATMs or ATMs lacking Nucb2.
(F) Pathways (blue indicates downregulated, and red indicates upregulated) in Nucb2-deficient ATMs.
(G) Scatterplot depicting log2 expression values in WT mice(x axis), compared to Nucb2−/− mice (y axis), for NFκB-regulated genes (Table S1) significantly altered upon deletion of Nucb2 in ATMs that promote inflammation.
(H) Total body weight and glucose excursion and area under the curve after i.p. injection.
(I–L) Glucose tolerance test (GTT; glucose levels in I and area under the curve in J) and insulin tolerance test (ITT; glucose levels in K and area under the curve in L) in control (Nucb2fl/fl), macrophage-specific (Nucb2Mac−/−), and adipocyte-specific (Nucb2Adip−/−) Nucb2-deficient mice fed an HFD for 6 weeks. n = 6–8 per group, *p < 0.05.
See also Table S1.
It is now recognized that, depending upon the tissue of residence, macrophages display unique transcriptional signatures that reflect the local microenvironment and functionality of a particular organ (Lavin et al., 2014; Geissmann and Mass, 2015; Camell et al., 2017). Consistent with these data, whole transcriptome analyses further revealed that, compared to control cells, the Ncub2-deficient ATMs display an enrichment of metabolic Gene Ontology (GO) terms, including upregulation of pentose phosphate pathway (PPP), glycolysis, one-carbon metabolism, and branched-chain amino-acid degradation (Figure 4F). Given our prior findings that Nucb2 controls pro-inflammatory cytokines via NFκB, we further mined the RNA sequencing (RNA-seq) data to determine whether NFκB-regulated inflammatory mediators were impacted by Nucb2 deficiency in ATMs. This analysis, together with the search of existing databases, revealed that multiple pro-inflammatory NFκB regulatory genes (Table S1) were upregulated in Nucb2−/−, compared to WT, ATMs (Figure 4G). These included Wdfy1, Fcna, and genes of the paired immunoglobulin (Ig)-like receptor A family (Pira) (Figure 4G; Table S1). To determine the cell-autonomous regulation of insulin action by Nucb2 in obesity, we specifically ablated Nucb2 in myeloid-lineage cells (Nucb2Mac−/−) and adipocytes (Nucb2Adip−/−). Compared to control mice fed an HFD, ablation of Nucb2 on macrophages or adipocytes had no impact on total body weight (Figure 4H). Interestingly, compared to control mice fed an HFD, the loss of Nucb2 in macrophages, but not adipocytes, significantly worsened the insulin action, as measured by glucose tolerance (Figures 4I and 4J) and an insulin tolerance test (Figures 4K and 4L). Taken together, these data demonstrate that nucleobindin-2 does not impact hypothalamic satiety mechanisms, as was previously reported, but instead controls macrophage-mediated metabolic inflammation to control insulin resistance in obesity.
Our studies demonstrate that the physiological function of NucB2 is to regulate insulin sensitivity in obesity. The Nucb2-deficient mice on an HFD demonstrate severe insulin resistance. Importantly, this defect in insulin action is not a reflection of increased obesity in Nucb2-deficient mice. The HFD has been shown to be associated with increased inflammation and inflammatory cytokine production (Hotamisligil, 2006; Vandanmagsar et al., 2011). It is, therefore, plausible that macrophages and T cells respond by increasing production of Nucb2 to try to dampen this inflammatory response. Our data suggest that macrophage Nucb2 deficiency increases metabolic inflammation and insulin resistance and reveals a homeostatic role of Nucb2 to regulate inflammatory cytokine production.
It has been previously shown that the EF-hand of Nucb2, as well as Nucb1, may also influence G protein signaling by binding to the Gα motif (Garcia-Marcos et al., 2011). Whether Nucb2 may influence G protein and calcium-dependent signaling in immune cells warrants further study. Also, given that Nucb2 is highly expressed in plasmacytoid dendritic cells (Immgen) and endocrine glands like pituitary and pancreas, future studies are required to determine the role of this protein in integrating host defense with hormone secretion.
Importantly, using multiple Cre-Lox-based mouse models, our results establish that Nucb2 is not involved in regulation of food intake. Thus, the prior results assigning function to Nucb2 should be re-evaluated, because of the limitations of approaches such as pharmacologic peptide/shRNA-based targeting to brain via ICV injections. In summary, our findings show that obesity-induced upregulation of Nucb2 in macrophages may serve to sense energy excess that links control of inflammatory response to the regulation of insulin resistance.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact Vishwa Deep Dixit (vishwa.dixit@yale.edu)
EXPERIMENTAL MODEL SYSTEMS
All studies were approved by the Institutional Animal Care and Use Committees of Yale University and performed in accordance with NIH guidelines. Mice were generated within the transgenic core at Pennington Biomedical Research Center kept in a pathogen-free barrier facility maintained at 22–24°C with a 12:12-h dark-light cycle (lights on at 0700 h). Genetic targeting approach was used to create Nucb2 floxed allele by recombination of a PL253-loxP-frt-neo cassette to exon 3 in mouse embryonic stem cells. Once our vector was correctly designed and screened against neomycin for initial enrichment of the targeted clone, we used these embryonic stem cells for injection into blastocysts for the generation of heterozygous Nucb2-loxP floxed mice. These positive ES cells were used. Using polymerase chain reaction to genotype these neo-founder mice, we then removed the neomycin resistant drug marker by crossing to a recombinase FLP derived mouse which recognizes the flippase recognition target (FRT) for mediated cleavage and generation of our founder mice. We initially created a global KO by crossing floxed Nucb2 founder C57BL/6 mice with a global Ella-Cre driver to remove the floxed sequence.
Mice were housed up to 5 per cage and given ad-libitum access to normal chow (#5002, at least 4.5% kcal crude fat, LabDiet), low fat diet (LFD; #D12450B, 10% kcal as fat, Research Diets, New Brunswick, NJ) or a high fat diet (HFD; #D12492i, 60% kcal as fat; Research Diets, New Brunswick, NJ) and sterilized water (Hydropac Alternative Watering System: Seaford, DE, USA). The mice were placed on the LFD or HFD at 6 weeks of age and maintained on it until death. All transgenic and WT mice were cross-fostered to parent cohorts in our colony and littermate controls males were randomly allocated to the different groups. The sentinel mice in our animal rooms were negative for currently tested standard murine pathogens (ectromelia, epizootic diarrhea of infant mice [EDIM], lymphocytic choriomeningitis [LCMV], Mycoplasma pulmonis, mouse hepatitis virus [MHV], murine norovirus [MNV], mouse parvovirus [MPV], murine minute virus [MVM], pneumonia virus of mice [PVM], reovirus type 3 [REO3], Theiler’s murine encephalomyelitis virus [TMEV], and Sendai virus) at various times while the studies were performed. Changes in body weights were measured during the life of the animals. Fat mass (FM) and fat free mass were assessed by time-domain-nuclear magnetic resonance (Minispec Analyst AD; Bruker Optics, Silberstreifen, Germany) at different time points.
METHOD DETAILS
Hyperinsulinemic-euglycemic clamp
Clamp studies were performed according to recommendations of the Mouse Metabolic Phenotyping Center Consortium (Ayala et al., 2010). The clamp studies were performed on 14 week old, chow-fed mice, and on 12 week old mice (n = 8 WT, n = 7 Nucb2−/−) on a high fat diet for 6 weeks (n = 11 WT, n = 7 Nucb2−/−). After surgical implantation of an indwelling catheter in the right jugular vein, mice were allowed to recover for 7 days prior to the clamp experiments. Following an overnight 14-h fast, mice were infused with 3-[3H] glucose (HPLC purified; PerkinElmer Life Sciences, Waltham, Massachusetts) at a rate of 0.05 μCi/min for 120 min to determine basal glucose turnover. Next, a primed infusion of insulin and 3-[3H] glucose was administered for 4 min at rates of 7.14 milliunits·kg–1·min–1 and 0.24 μCi/min, respectively, after which the rates were reduced to 2.5 milliunits·kg–1·min–1 insulin for chow fed mice/3 milliunits·kg–1·min–1 insulin for HFD mice (Novolin; Novo Nordisk, Bagsværd, Denmark) and 0.1 μCi/min 3-[3H] glucose for the remainder of the experiment. Plasma samples were obtained from the tip of the tail at 0, 25, 45, 65, 80, 90, 100, 110, 120, 130, and 140 min for plasma glucose, insulin, and tracer levels. A variable infusion of 20% dextrose was given to maintain euglycemia (100-120 mg/dL). Also, mice received an i.v. albumin-containing solution mimicking artificial plasma (115 mM NaCl, 5.9 mM KCl, 1.2 mM MgCl2-6H2O, 1.2 mM NaH2PO4-H2O, 1.2 mM Na2SO4, 2.5 mM CaCl-2H2O, 25 mM NaHCO3, and 4% BSA [pH 7.4]) at a rate of 4.2 μL/min, during the insulin-stimulated period of the clamp to compensate for volume loss secondary to blood sampling.
Glucose turnover was calculated as the ratio of the 3-[3H] glucose infusion rate to the specific activity of plasma glucose at the end of the basal infusion and during the last 40 min of the hyperinsulinemic-euglycemic clamp study. Hepatic glucose production represents the difference between the glucose infusion rate and the rate of glucose appearance. A 10-μCi bolus injection of 2-deoxy-d-[14C]-glucose was given at 90 min to determine tissue-specific glucose uptake, which was calculated from the area under the curve of 2-deoxy-d-[14C]-glucose detected in plasma and the tissue content of [14C]2-deoxyglucose-6-phosphate, as previously described. Following collection of the final blood sample, mice were anesthetized with an intravenous injection of 150 mg/kg pentobarbital, and tissues were harvested and frozen with aluminum forceps in liquid nitrogen. All tissues were stored at −80°C until later use.
Tissue digestion and positive selection of T cells and macrophages
Adipose tissue was digested using enzymatic digestion as previously described (Vandanmagsar et al., 2011; Grant et al., 2013; Camell et al., 2017) Adipose tissue stromal vascular fraction cells were then collected, and T cells and macrophages were positively selected using CD3 and F4/80 labeled antibodies (Dynabeads, Life Technologies).
Quantitative PCR
RNA from tissue and cells was isolated using an RNeasy Plus mini and micro kit (QIAGEN; 74106 and 74034) according to the manufacturer’s instructions. DNA digestion was performed on the columns to remove DNA using RNase-Free DNase according to manufacturer’s instructions (79254; QIAGEN). Following RNA purification, samples were then used for iScript cDNA synthesis using a reverse transcriptase PCR kit (BIO-RAD; Hercules, CA, USA). Quantitative PCR was performed with the LightCycler 480 II (Roche Applied Science; Indianapolis, IN, USA) and Power SYBR Green detection reagent (Applied Biosystems by Thermo Fischer Scientific; Woolston Warrington, UK). Primer sequences for transcripts encoding proteins involved in lipid and glucose metabolism were designed with Primer Express Software.
In all qRT-PCR experiments, 25 ng cDNA was used. Fold induction of gene expression with Nucb2 was analyzed with the ΔΔCt method (also known as the comparative Ct method) as determined by the following equation: ΔΔCt = ΔCt treatment (age and diet) - ΔCt control (young/chow). Here, the ΔCt is the Ct value for the sample (Nucb2) normalized to the endogenous housekeeping gene GAPDH transcript.
Western Blotting
Tissues from WT and Nucb2−/− mice were prepared by grinding with mortar and pestle while kept frozen in liquid nitrogen followed by homogenization with RIPA buffer. Bone marrow derived macrophage cell lysates were prepared using RIPA buffer and immediately snap frozen in liquid nitrogen. Samples were vortexed every 10 min for 1 hr. Samples were centrifuged at 14,000 g for 15 min, the supernatant was collected and the protein concentration was determined using the DC Protein Assay (Bio-RAD). Antibodies to Nucb2 (1:1000, N6789; Sigma), IL-1β (1:500, GTX74034; Genetex), NFκB p65 (1:1,000 8242s, Cell Signaling), phosphorylated-NFκB p65 (1:1000 3033S, Cell Signaling), and β-actin (1:1,000 4967L; Cell Signaling) were used at the dilutions specified by the manufacturer. The immune complexes were visualized by incubation with horseradish peroxidase-conjugated anti-rat (PI31470; Pierce) or anti-rabbit secondary antibody (PI31460, Pierce). Immuno-reactive bands were visualized by enhanced chemiluminescence (PI32209; Pierce). Densitometry analysis was performed using the ImageJ Gel Analysis tool, where gel background was also removed individually for each band.
Immunofluorescence microscopy
WT and Nucb2−/− MEFs were plated on chamber slides 1 day before treatment. The following day cells were treated with LPS (1ug/ml) for 1 hr or non-treated for control. Cells were fixed with 4% paraformaldehyde, permeabilized with 0.1% Triton X-100. After blocking with 10% BSA, the cells were stained with Nucb2 (AF6895; R&D Systems) followed by Donkey anti-Sheep IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 (Thermo Fisher Scientific), Alexa Fluor 594 Phalloidin (Thermo Fisher Scientific) for F-actin and DAPI (D9542; Sigma-Aldrich) for nucleus. The images were acquired on Leica TCS SP5 confocal microscope with 63x objective.
Leukocyte quantification in adipose tissue
Leukocytes were quantified in inguinal and epididymal fat pads using flow cytometry analysis as previously described by our laboratory (Vandanmagsar et al.,2011; Grant et al., 2013; Camell et al., 2017). Cells were labeled using antibodies described below, and cells were run on a FACSCalibur (BD Biosciences, San Jose, CA, USA). Macrophage subsets were identified using F4/80, CD206 and CD11c antibodies (eBiosciences, San Diego, CA, USA and Biolegend, San Diego, CA, USA). T cell subpopulations were identified by CD3, CD4 and CD8 antibodies, while B cell populations were identified by B220 antibodies for B220, IgM and CD19 (eBiosciences). FACS data were analyzed by post-collection compensation using FlowJo software (Treestar Inc., Ashland, OR).
Tissue Hematoxylin and Eosin (H&E) staining
The adipose tissue and liver were collected from mice, fixed in 4% (vol/vol) buffered paraformaldehyde, embedded in paraffin and optimal cutting temperature (OCT) compound, then cut into 5 μm thick sections. Tissue sections were stained with hematoxylin and eosin (H&E). Fluorescent images of H&E-stained paraffin-sections were photographed using the Texas Red filter cube on an Axioplan 2 imaging microscope (Carl Zeiss Microscopy; Thornwood, NY, USA). Quantification of adipocyte size was measured using ImageJ image analysis software. The averages were found by measuring 15 adipocytes from 3 different sections from 4 different mice per group.
Enzyme-linked immunosorbent assay (ELISA)
The sera from mice were collected and stored at −80°C and used to quantify the concentration of Nucleobindin-2(Nucb2) in serum using the CUSABIO Mouse Nucb2 Elisa kit (CSB-EL016146MO). ELISAs were read on the Tecan Infinite M200 i-control. Nesfatin-1 was measured using nesfatin-1 (1-82aa) (rat) EIA kit (EK-003-22; Phoenix Pharmaceuticals; Burlingame, CA, USA) with a detectable range of 0.1-1000ng/ml. Adiponectin was measured using Mouse Adiponectin ELISA kit (EMD Millipore; St. Charles, Missouri, USA). Leptin was measured using Mouse/Rat Leptin Quantikine ELISA (R&D Systems; Minneapolis, MN, USA).
RNA-Sequencing data generation
Adipose tissue stromal vascular fraction cells were then collected, and macrophages were positively selected using F4/80 labeled antibodies (Dynabeads, Life Technologies). Optical density values of extracted RNA were measured using NanoDrop (Thermo Scientific) to confirm an A260:A280 ratio above 1.9. RNA integrity number (RIN) was measured using BioAnalyzer (Agilent) RNA 6000 Pico Kit to confirm RIN above 7.
Samples were run by the Yale Center for Genome Analysis using Clontech ultra low-input Library Prep Kit V2 (#634899, Clontech Laboratories, Inc. Mountain View, CA, USA) and RNA-seq was performed as described previously (Camell et al., 2017). Samples were sequenced using single-end 1×75 on the Illumina HiSeq2500 (Illumina Biotechnology, San Diego, CA, USA). Read counts were normalized using DESeq2 (R-package) in collaboration with Life & Medical Sciences Institute at the University of Bonn. To avoid spurious fold changes during differential expression analysis, all normalized counts were set to a value of at least 1.
Raw fastq-files have been deposited at the Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/) and are available under the accession number GSE97309.
Alignment and mRNA quantification of RNA-Seq data
Raw fastq-files were aligned against the murine genome version mm10 using HISAT version 0.1.7-β (Kim et al., 2015) with all default options. The resulting BAM files were imported into Partek® Genomics Suite® software, version 6.6 Copyright©; 2017 (PGS). mRNA quantification was performed against the mm10-RefSeq Transcripts database version 2016-02-02 to obtain read counts for each individual RefSeq gene.
Primary analysis of RNA-Seq data
The obtained table containing read counts for 18,939 genes was normalized using DESeq2 and then imported back into PGS. Before further analyses, all normalized count values smaller than 1 were set to 1 to avoid spurious Fold-Changes later on. Then, the dataset was filtered down to expressed genes, by keeping those having an average normalized read count of at least 10 in at least one of the two investigated groups. The resulting 14,905 present genes were used as input for principal component analysis. In addition, the top 1000 most variable genes of those were displayed within a heatmap, where rows and columns were ordered by hierarchical clustering using Euclidean distance and average linkage.
Visualization of differentially expressed genes
Within a scatterplot, the log2-average expression across Nucb2 deficient and WT mice versus the log2-Fold-Change between Nucb2 deficient and WT mice was displayed for each of the 14,905 present genes. In addition, genes being significantly up- or downregulated determined using one-way ANOVA and requiring an unadjusted p value < 0.05, were colored in red or blue, respectively.
Comparison of Nucb2−/− and WT macrophages to human in vitro activated monocyte-derived cells
To link the Nucb2−/− and WT macrophages to in vitro activated macrophages, the 14,905 present genes were used as input for CIBERSORT. As signature matrix, 29 macrophage subsets including inactivated human macrophages were used (Xue et al., 2014). To obtain a corresponding murine signature file, human symbols were translated to murine ones based on one-to-one orthologs, where non-matching genes were excluded from the signature. Based on linear support vector regression (SVR) using all default options, the relative fractions of the 29 macrophage subsets were determined for the Nubc2 deficient and WT ATMs.
Detection of differentially expressed genes and pathways between Nucb2−/− and WT macrophages
Using a one-way ANOVA and requiring an unadjusted p value < 0.05 and a Fold-Change > 2 or < –2, 236 genes were identified to be up- and 83 to be downregulated in murine Nucb2−/− compared to WT macrophages, respectively. Using those genes separately as input for Partek Pathway, 22 pathways were identified to be up- and 8 to be downregulated in Nucb2−/− compared to WT mice, respectively using Fisher’s Exact test.
QUANTIFICATION AND STATISTICAL ANALYSIS
We used a two-tailed Student’s t test to examine differences between genotypes or treatments (age and feeding) with a p < 0.05 considered statistically significant. The results are expressed as the arithmetic means ± SEM. Differences between means and different genetic model were determined by one-way ANOVA using Tukey’s test, using SigmaStat or Graphpad Prism software, which protects the significance (p < 0.05) of all pair combinations.
DATA AND SOFTWARE AVAILABILITY
Data Resources
The accession number for the raw fastq-files for the RNA-seq analysis reported in this paper is GEO: GSE97309.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-Mouse: Nucb2 (C-terminal) | Sigma | N6789 RRID:AB_1854684 |
| Anti- Mouse/Rat Nesfatin-1/Nucleobindin-2 | R&D Systems | AF6895 RRID:AB_10972964 |
| Anti-Mouse: IL-1β | Genetex | GTX74034 RRID:AB_378141 |
| Anti-Mouse: NFκB p65 | Cell Signaling | 8242s RRID:AB_10859369 |
| Anti-Mouse: phosphorylated-NFκB p65 | Cell Signaling | 3033s RRID:AB_331284 |
| Anti-Mouse: β-actin | Cell Signaling | 4967L RRID:AB_330288 |
| F4/80 | eBiosciences | 48-4801 RRID:AB_1548747 |
| CD206 | Biolegend | 141720 RRID:AB_2562248 |
| CD11b | Biolegend | 45-0112 RRID:AB_953560 |
| CD3 | eBiosciences | 13-0037 RRID:AB_1234956 |
| CD4 | Biolegend | 100548 RRID:AB_2563054 |
| CD8 | eBiosciences | 12-0081 RRID:AB_465530 |
| B220 | eBiosciences | 13-0452 RRID:AB_466450 |
| Alexa Fluor 488 | Thermo Fisher Scientific |
S11223 RRID:AB_2336881 |
| Alexa Fluor 594 Phalloidin (F-actin) | Thermo Fisher Scientific | A12381 RRID:AB_2315633 |
| DAPI | Sigma-Aldrich | D9542 |
| horseradish peroxidase-conjugated anti-rat | Pierce | PI31470 |
| horseradish peroxidase-conjugated anti-rabbit | Pierce | PI31460 |
| Biological Samples | ||
| Mouse: Tissues and cells | This Paper/our animal models | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Glucose | Pierce | PI32209 |
| Insulin | Eli Lilly | Humulin R |
| Chemiluminescence | Pierce | PI32209 |
| Bay-11-7085 NFκB inhibitor | Sigma-Aldrich | B5681 |
| recombinant nesfatin-1 | Phoenix Pharmaceuticals | 003-22A |
| Recombinant nesfatin-1 (1-29) | Phoenix Pharmaceuticals | 009-76 |
| recombinant nesfatin-2 | Phoenix Pharmaceuticals | 009-73 |
| recombinant nesfatin-3 | Phoenix Pharmaceuticals | 003-28 |
| Critical Commercial Assays | ||
| Mouse Nucb2 Elisa kit | CUSABIO | CSB-EL016146MO |
| nesfatin-1 (1-82aa) ELISA kit | Phoenix Pharmaceuticals | EK-003-22 |
| Mouse/Rat Leptin Quantikine ELISA | R&D Systems | MOB00 |
| Mouse Adiponectin ELISA kit | EMD Millipore | EZMADP-60K |
| DC Protein Assay | BIO-RAD | 5000111 |
| Deposited Data | ||
| Raw data files for RNA sequencing | NCBI Gene Expression Omnibus | GEO: GSE97309 |
| Experimental Models: Cell Lines | ||
| Mouse: Primary BMDM cells | This Paper | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6 | The Jackson Laboratory | 000664 |
| Mouse: Nucb2 fl/fl | This Paper | Generated by PBRC transgenic core |
| Mouse: E11a-Cre | The Jackson Laboratory | 011104 |
| Mouse: Adipoq-Cre | The Jackson Laboratory | 024671 |
| Mouse: LysM-Cre | The Jackson Laboratory | 004781 |
| Mouse: AgRP-Cre | The Jackson Laboratory | 012899 |
| Oligonucleotides | ||
| Nucb2 | This Paper | N/A |
| forward primer sequence | ||
| 5′-AAAACCTTGGCCTGTCTGAA-3′ | ||
| reverse primer | ||
| 5′-CATCGATAGGAACAGCTTCCA-3′ | ||
| CCL4 | This paper | N/A |
| forward primer sequence | ||
| 5′-GAAACAGCAGGAAGTGGGAG-3′ | ||
| reverse primer sequence | ||
| 5′-CATGAAGCTCTGCGTGTCTG-3′ | ||
| IL-1β | This Paper | N/A |
| forward primer sequence | ||
| 5′-GGTCAAAGGTTTGGAAGCAG-3′ | ||
| reverse primer sequence | ||
| 5′-TGTGAAATGCCACCTTTTGA-3′ | ||
| IL-6 | This paper | N/A |
| forward primer sequence | ||
| 5′-ACCAGAGGAAATTTTCAATAGGC-3′ | ||
| reverse primer sequence | ||
| 5′-TGATGCACTTGCAGAAAACA-3′ | ||
| TNFα | This paper | N/A |
| forward primer sequence | ||
| 5′-AGGGTCTGGGCCATAGAACT-3′ | ||
| reverse primer sequence | ||
| 5′-CCACCACGCTCTTCTGTCTAC-3′ | ||
| MIP-1α | This paper | N/A |
| forward primer sequence | ||
| 5′-ACCATGACACTCTGCAACCA-3′ | ||
| reverse primer sequence | ||
| 5′-GTGGAATCTTCCGGCTGTAG-3′ | ||
| Software and Algorithms | ||
| ImageJ | BMC Bioinformatics | https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-017-1934-z |
| FLOWJO | FlowJo LLC | https://www.flowjo.com/solutions/flowjo |
| SigmaPlot | Systat Software | www.systatsoftware.com |
| SigmaSTAT | Systat Software | www.systatsoftware.com |
Highlights.
Ablation of Nucb2 does not control food intake and adiposity
Loss of Nucb2 increases inflammation and insulin resistance
Nucb2 does not induce leukocytosis in obesity
Macrophage-expressed Nucb2 drives insulin resistance
ACKNOWLEDGMENTS
We thank J. Zhang, B. Vandanmagsar, and R. Mynatt in the PBRC transgenic core for preparation of Nucb2 targeting vector and for expert technical assistance with genotyping and floxed mouse production services and the Yale Center on Genomic Analysis (YCGA) core for services in performing the RNA-seq. J.L.S. was funded by the German Research Foundation (SFB704 and SFB645) and by the ImmunoSensation Cluster of Excellence Bonn. The Dixit lab is supported in part by NIH grants P01AG051459, AI105097, AG051459, AR070811; the Glenn Foundation on Aging Research; and Cure Alzheimer’s Fund.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes four figures and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.06.112.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Ayala JE, Samuel VT, Morton GJ, Obici S, Croniger CM, Shulman GI, Wasserman DH, and McGuinness OP; NIH Mouse Metabolic Pheno-typing Center Consortium (2010). Standard operating procedures for describing and performing metabolic tests of glucose homeostasis in mice. Dis. Model. Mech 3, 525–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besedovsky HO, and del Rey A (1996). Immune-neuro-endocrine interactions: facts and hypotheses. Endocr. Rev 17, 64–102. [DOI] [PubMed] [Google Scholar]
- Camell CD, Sander J, Spadaro O, Lee A, Nguyen KY, Wing A, Goldberg EL, Youm YH, Brown CW, Elsworth J, et al. (2017). Inflammasome-driven catecholamine catabolism in macrophages blunts lipolysis during ageing. Nature 550, 119–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheyuo C, Jacob A, and Wang P (2012). Ghrelin-mediated sympathoinhibition and suppression of inflammation in sepsis. Am. J. Physiol. Endocrinol. Metab 302, E265–E272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixit VD (2008). Adipose-immune interactions during obesity and caloric restriction: reciprocal mechanisms regulating immunity and health span. J. Leukoc. Biol 84, 882–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixit VD, Schaffer EM, Pyle RS, Collins GD, Sakthivel SK, Palaniappan R, Lillard JW Jr., and Taub DD (2004). Ghrelin inhibits leptin- and activation-induced proinflammatory cytokine expression by human monocytes and T cells. J. Clin. Invest 114, 57–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dore R, Levata L, Lehnert H, and Schulz C (2017). Nesfatin-1: functions and physiology of a novel regulatory peptide. J. Endocrinol 232, R45–R65. [DOI] [PubMed] [Google Scholar]
- Garcia-Marcos M, Kietrsunthorn PS, Wang H, Ghosh P, and Farquhar MG (2011). G protein binding sites on Calnuc (nucleobindin 1) and NUCB2 (nucleobindin 2) define a new class of G(alpha)i-regulatory motifs. J. Biol. Chem 286,28138–28149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geissmann F, and Mass E (2015). A stratified myeloid system, the challenge of understanding macrophage diversity. Semin. Immunol 27, 353–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant R, Youm YH, Ravussin A, and Dixit VD (2013). Quantification of adipose tissue leukocytosis in obesity. Methods Mol. Biol 7040, 195–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotamisligil GS (2006). Inflammation and metabolic disorders. Nature 444, 860–867. [DOI] [PubMed] [Google Scholar]
- Kim D, Langmead B, and Salzberg SL (2015). HISAT:a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavin Y, Winter D, Blecher-Gonen R, David E, Keren-Shaul H, Merad M, Jung S, and Amit I (2014). Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell 159, 1312–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YS, Wollam J, and Olefsky JM (2018). An integrated view of immuno-metabolism. Cell 172, 22–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin P, Le-Niculescu H, Hofmeister R, McCaffery JM, Jin M, Hennemann H, McQuistan T, De Vries L, and Farquhar MG (1998). The mammalian calcium-binding protein, nucleobindin (CALNUC), is a Golgi resident protein. J. Cell Biol 141, 1515–1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lumeng CN, and Saltiel AR (2011). Inflammatory links between obesity and metabolic disease. J. Clin. Invest 121, 2111–2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matarese G (2000). Leptin and the immune system: how nutritional status influences the immune response. Eur. Cytokine Netw 11, 7–14. [PubMed] [Google Scholar]
- Miura K, Titani K, Kurosawa Y, and Kanai Y (1992). Molecular cloning of nucleobindin, a novel DNA-binding protein that contains both a signal peptide and a leucine zipper structure. Biochem. Biophys. Res. Commun 187, 375–380. [DOI] [PubMed] [Google Scholar]
- Nakata M, Gantulga D, Santoso P, Zhang B, Masuda C, Mori M, Okada T, and Yada T (2016). Paraventricular NUCB2/nesfatin-1 supports oxytocin and vasopressin neurons to control feedingb and fluid balance in male mice. Endocrinology 157, 2322–2332. [DOI] [PubMed] [Google Scholar]
- Oh-I S, Shimizu H, Satoh T, Okada S, Adachi S, Inoue K, Eguchi H, Yamamoto M, Imaki T, Hashimoto K, et al. (2006). Identification of nesfatin-1 as a satiety molecule in the hypothalamus. Nature 443, 709–712. [DOI] [PubMed] [Google Scholar]
- Procaccini C, Pucino V, De Rosa V, Marone G, and Matarese G (2014). Neuro-endocrine networks controlling immune system in health and disease. Front. Immunol 5, 143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu H, Oh-I S, Okada S, and Mori M (2009). Nesfatin-1: an overview and future clinical application. Endocr. J 56, 537–543. [DOI] [PubMed] [Google Scholar]
- Shoelson SE (2006). Banking on ATM as a new target in metabolic syndrome. Cell Metab 4, 337–338. [DOI] [PubMed] [Google Scholar]
- Stengel A, Goebel M, Yakubov I, Wang L, Witcher D, Coskun T, Taché Y, Sachs G, and Lambrecht NW (2009). Identification and characterization of nesfatin-1 immunoreactivity in endocrine cell types of the rat gastric oxyntic mucosa. Endocrinology 150, 232–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandanmagsar B, Youm YH, Ravussin A, Galgani JE, Stadler K, Mynatt RL, Ravussin E, Stephens JM, and Dixit VD (2011). The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat. Med 17, 179–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue J, Schmidt SV, Sander J, Draffehn A, Krebs W, Quester I, De Nardo D, Gohel TD, Emde M, Schmidleithner L, et al. (2014). Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity 40, 274–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.