

Summary
Telomerase adds telomeric repeats to chromosome ends to balance incomplete replication. Telomerase regulation is implicated in cancer, aging and other human diseases, but progress towards telomerase clinical manipulation is hampered by the lack of structural data. Here we present the cryo-electron microscopy structure of substrate-bound human telomerase holoenzyme at subnanometer resolution, describing two flexibly RNA-tethered lobes: the catalytic core with telomerase reverse transcriptase (TERT) and conserved motifs of telomerase RNA (hTR), and an H/ACA ribonucleoprotein (RNP). In the catalytic core, RNA encircles TERT, adopting a well-ordered tertiary structure with surprisingly limited protein-RNA interactions. The H/ACA RNP lobe comprises two sets of heterotetrameric H/ACA proteins and one Cajal body protein, TCAB1, representing a pioneering structure of a large eukaryotic family of ribosome and spliceosome biogenesis factors. Our findings provide a structural framework for understanding human telomerase disease mutations and represent an important step towards telomerase-related clinical therapeutics.
Linear eukaryotic chromosomes are capped by telomeric repeat tracts that recruit the telomere-binding proteins essential to distinguish telomeres from DNA breaks and thus avoid telomere end-resection and interchromosomal fusions1,2. Due to inherently incomplete genome replication, telomeres are progressively shortened in each cell cycle3. Critically short telomeres result in genome instability and cell death2,4,5. To compensate for this sequence loss, a specialized reverse transcriptase, telomerase, adds telomeric repeats to the chromosome 3’ end, using the telomerase reverse transcriptase (TERT), and an integral telomerase RNA subunit (TER) with an internal template for repeat synthesis6. Human telomerase activation in embryogenesis and its repression in somatic tissues govern cellular renewal capacity, with telomerase deficiency imposing hematopoietic and epithelial failures and aberrant telomerase activation enabling tumorigenesis7.
Despite its medical relevance, efforts to design drugs targeting telomerase are hampered by limited structural information. Endogenous human telomerase holoenzyme is scarce. Most studies of human telomerase use cellular overexpression of limiting TER (hTR in human) and TERT8. Two hTR domains are critical for activity9,10: a domain with the template and adjacent pseudoknot (PK, together t/PK) and the conserved regions 4 and 5 (CR4/5) consisting of a branched junction of stems P5, P6, and the activity-critical stem-loop P6.1 (Fig. 1a). Together with TERT, they are sufficient to reconstitute telomerase activity with the repeat addition processivity characteristic of cellular holoenzyme11. TERT is composed of four domains: the N-terminal (TEN) domain, the high-affinity RNA binding domain (TRBD), the reverse transcriptase domain (RT), and a C-terminal extension (CTE) analogous to a polymerase thumb11 (Fig. 1a). The high-resolution structure of a streamlined TERT from the flour beetle Tribolium castaneum, which lacks the TEN domain, established that the latter three domains form a ring, referred to as the TERT ring12. The TERT ring alone can bind a template-primer duplex13, and, accordingly, the human TERT ring has been shown to support single-repeat synthesis14. Purified or overexpressed hTR and TERT form heterogeneous complexes, only some of which are catalytically active15. It remains controversial whether active enzyme requires an hTR and TERT dimer16,17 or only a monomer of each subunit18,15. The landmark 9 Å cryo-electron microscopy (cryo-EM) structure of the ciliate Tetrahymena thermophila telomerase holoenzyme revealed its overall architecture, including a monomeric TERT-TER catalytic core19. However, the subunit composition of Tetrahymena telomerase holoenzyme is distinct from its human counterpart, and available structural information on human telomerase holoenzyme is limited to a 30 Å negative-stain EM reconstruction17. Therefore, both structure and composition of human telomerase holoenzyme remain poorly characterized.
Figure 1 |. Telomerase holoenzyme reconstitution and characterization.
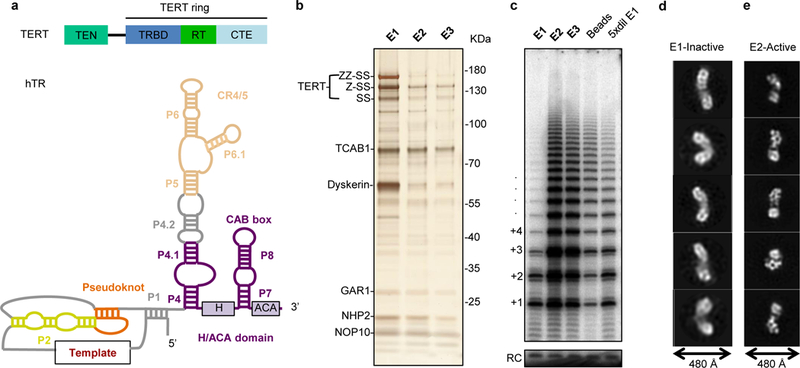
a, Domain architecture of TERT (top) and secondary structure of hTR. Paired stems (P) are shown compacted in length. b, Silver-stained SDS-PAGE gel showing fractionation of purified, heterogeneously active TERT RNP. Proteins were detected by mass spectrometry and immunoblotting (Extended Data Fig. 1a and Extended Data Table 1). c, In vitro telomerase assays performed on the three elution fractions shown in b, the beads after elution, and five-fold diluted E1. A 12 nucleotide (nt) oligo was used as recovery control (RC). Processive repeat additions are numbered. d, e, Representative negative-stain 2D class averages for fractions E1 and E2, respectively. We also observed a small fraction of E2-like particles in E1 (data not shown), while E2 contained predominantly the particles shown in e. Experiments in b and c were performed three times and twice, respectively, with similar results. For gel source data, see Supplementary Figure 1.
Telomerase assembly initiates in human cells with nascent hTR binding to H/ACA proteins and their chaperones20. Human H/ACA proteins are shared by telomerase and the small nucleolar (sno) and small Cajal body (sca) RNP families that catalyze ribosomal and spliceosomal RNA modifications21,22. Eukaryotic H/ACA snoRNPs contain the pseudouridine synthase dyskerin (Cbf5), NOP10, NHP2, and GAR1, assembled on each of two hairpin stems; and sca RNPs also contain TCAB1/WDR79 bound to loop CAB-box motifs. The hTR H/ACA 3’ hairpin loop has a CAB box but the 5’ hairpin instead extends into CR4/5 (Fig. 1a). All eukaryotic H/ACA RNPs, including telomerase, require both hairpins of the H/ACA motif for RNP biogenesis in cells22, unlike their single-hairpin archaeal counterparts21, but it remains controversial whether active human telomerase holoenzyme has one or two sets of H/ACA proteins18, only dyskerin23, or only dyskerin and NOP10 (ref. 17). Syndromes of human telomerase deficiency have been traced to mutations in dyskerin, NOP10, NHP2 and their chaperone NAF15. While structure of the related single-hairpin archaeal RNP is known24, there is no high-resolution structure of any complete eukaryotic H/ACA RNP.
Here, to address the structure and composition of human telomerase holoenzyme, we optimized active telomerase purification and used cryo-EM to determine its structure at subnanometer resolution, which allowed us to fit available high-resolution structures and homology models of all components. Our structure reveals in detail the organization of 10 protein subunits and hTR, illuminating the structural basis of hTR motif functions and human disease mutation causes of telomerase deficiency.
Different activities for distinct structural states of purified human telomerase
We reconstituted human telomerase holoenzyme by transient transfection of HEK 293T cells with vectors expressing TERT and hTR8. After affinity purification steps to enrich for hTR and TERT, the predominant additional proteins in the sample were TCAB1 and the H/ACA proteins (dyskerin, NOP10, NHP2 and GAR1), each of which was assigned by immunoblotting and, excepting glycine-arginine rich GAR1, also confirmed by mass spectrometry (Fig. 1b, Extended Data Fig. 1a, Extended Data Table 1). Furthermore, protein identities were confirmed by subunit drop-out purifications (see below). Direct primer-extension assays of eluted fractions showed that the amount of TERT did not fully correlate with the amount of telomerase activity; also, dilution of the less active fraction E1 increased rather than decreased activity, (Fig. 1c). Visualization by negative-stain EM of E1 and E2, which we refer to as inactive and active fractions, respectively, revealed morphologically distinct populations of particles. Particles from the inactive fraction were more elongated (typically 400 Å or more in length) and more heterogeneous than those in the active fraction (Fig. 1d and e). Particles from the active fraction consisted of two asymmetric lobes, nearly 300 Å in length (Fig. 1e). Previous studies have shown that cellular human telomerase reconstitution yields complexes both monomeric and dimeric for TERT15,17, explaining the observed particle heterogeneity. For cryo-EM studies, we modified our purification protocol to recover only active fractions (Extended Data Fig. 1b and c), added a DNA substrate (T14AG3) during purification, and confirmed the presence of the bound substrate in the purified RNP (Extended Data Fig. 1c). We also demonstrated that our tagged TERT was active in vivo (Extended Data Fig. 2a-e).
Overall architecture and flexibility of human telomerase
We prepared ΔTCAB1 telomerase with a hTR CAB box mutant that disrupted TCAB1 binding25,26 and TERT-hTRmin27,28, in which hTRmin only contains the activity-essential t/PK and CR4/5 domains of hTR. By comparing their negative-stain 2D class averages to those of intact holoenzyme, we established the particle regions corresponding to TCAB1, the H/ACA RNA motif and proteins, and TERT with the two activity-essential domains of hTR (Extended Data Fig. 1d-h). This revealed that one lobe of the holoenzyme contained TERT and its associated hTR motifs (henceforth the catalytic core) and the other contained H/ACA RNP components and TCAB1 (henceforth the H/ACA lobe). We next collected cryo-EM data to determine the structure of the substrate-bound human telomerase holoenzyme (Extended Data Fig. 3a and b, Extended Data Table 2a). Initial image processing revealed extensive conformational flexibility between the two lobes, as well as within each lobe, which limited the quality of the overall reconstruction (Extended Data Fig. 3c and 4a). We performed focused classification with signal subtraction29 to obtain more homogeneous subsets of particles for each lobe separately. This procedure yielded reconstructions at 7.7 Å and 8.2 Å resolution for the catalytic core and H/ACA lobes, respectively (Extended Data Fig. 3c, 4b-e). At these resolutions, densities for protein α-helices, β-barrels, and double-stranded RNA are discernible, allowing us to unambiguously fit available protein and RNA structures or homology models, and ultimately account for the majority of the density, except for flexible regions of single-stranded RNA and protein extensions (Fig. 2a, 2b and Extended Data Fig. 4f). We also obtained a reconstruction of the entire holoenzyme at 10.2 Å resolution (Extended Data Fig. 3c).
Figure 2 |. Cryo-EM structure of the substrate-bound human telomerase holoenzyme.
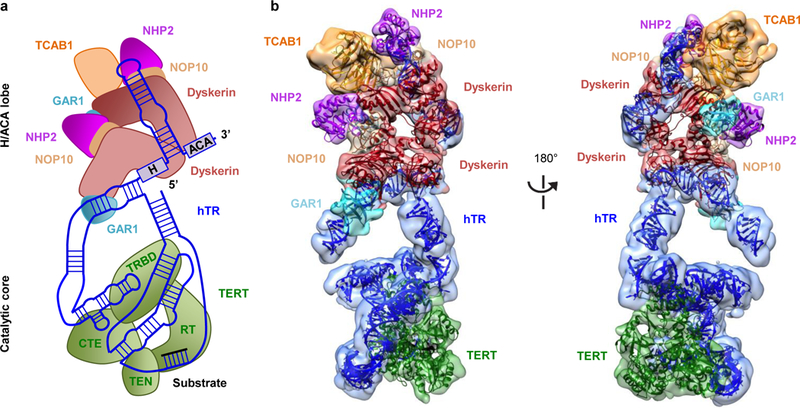
a, Schematic of subunit arrangements. b, Front (left panel) and back (right panel) views of the cryo-EM reconstructions for the H/ACA lobe at 8.2 Å and the catalytic core at 7.7 Å, with fitted subunits color-coded as indicated.
Subunit distribution across the two lobes of human telomerase
The bilobal human telomerase includes one TERT molecule and one hTR (Fig. 2a and b), in agreement with suggestions of an evolutionarily conserved single-TERT catalytic core for telomerases from ciliate and yeast model systems30,31 to human15,18,32. The complex also contains one TCAB1 and two sets of the four mature H/ACA RNP proteins. The previous human telomerase negative-stain EM reconstruction also had a bilobal architecture, interpreted as dimeric TERT, although gold-labeling experiments indicated the presence of a mixture of RNPs containing both one (50%) and two (36.5%) TERT molecules17. Incomplete separation of active from inactive TERT assemblies may account for the previous study mixture of complexes. Our finding of separate lobes for the catalytic core and the H/ACA RNP gives a structural explanation for why telomerase holoenzyme RNP biogenesis strategies have such plasticity across eukaryotes11.
The extended architecture of hTR scaffolds the overall subunit organization across both lobes (Fig. 3a and b). The catalytic core and H/ACA lobes are bridged by stems P1a and P4.2 (Fig. 2a, 2b and 3b). The crystal structure of Tribolium TERT in complex with an RNA-DNA duplex13 could be readily fitted within the catalytic core density (Fig. 4a; Extended Data Fig. 5c, 5d and 6b). As the Oryzias latipes (medaka) TRBD structure33 includes vertebrate TERT motifs not present in the streamlined Tribolium TERT (Extended Data Fig. 7e), we removed extensions from the medaka TRBD structure that did not fit our density and used this model to replace the Tribolium TRBD (Extended Data Fig. 5b). Adjacent to the TERT ring, there is globular density with approximately the dimensions of the Tetrahymena TEN domain crystal structure34 (Fig. 4b, Extended Data Fig. 5a). The Tetrahymena and human TEN domains share only 20% sequence identity and have differences in predicted secondary structure35 (Extended Data Fig. 6a); thus we removed regions of the former that are likely to be less structurally congruent with the human structure for tentative fitting into our map. The NMR structure for the human PK36 and the crystal structure of the medaka CR4/5 in the conformation bound to TRBD33 were fitted in one side of the catalytic core (Fig. 2b, 3a, 3b, 4a, and Extended Data Fig. 5h). The PK (P3+P2b) is connected to the template region located in the TERT active site by a bent helical RNA density that we attributed to J2a/b-P2a-J2a/a.1-P2a.1 (ref. 10, 37, 38) (Fig. 4a).
Figure 3 |. hTR structure in human telomerase holoenzyme.
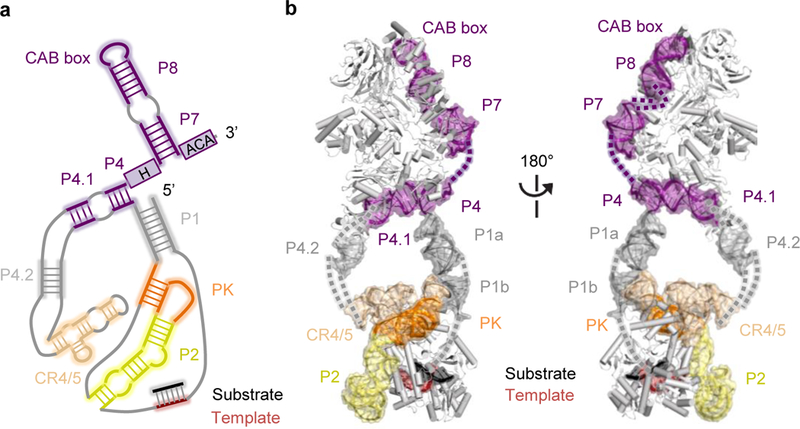
a, Schematic of secondary structure of hTR, with domains arranged based on the cryo-EM reconstruction. Regions modeled are highlighted with soft-colored background. b, Front and back views of the structure of hTR highlighted within the human telomerase holoenzyme structure, with domains color-coded as in a and unmodelled connections shown as dashes.
Figure 4 |. Structure of the catalytic core.
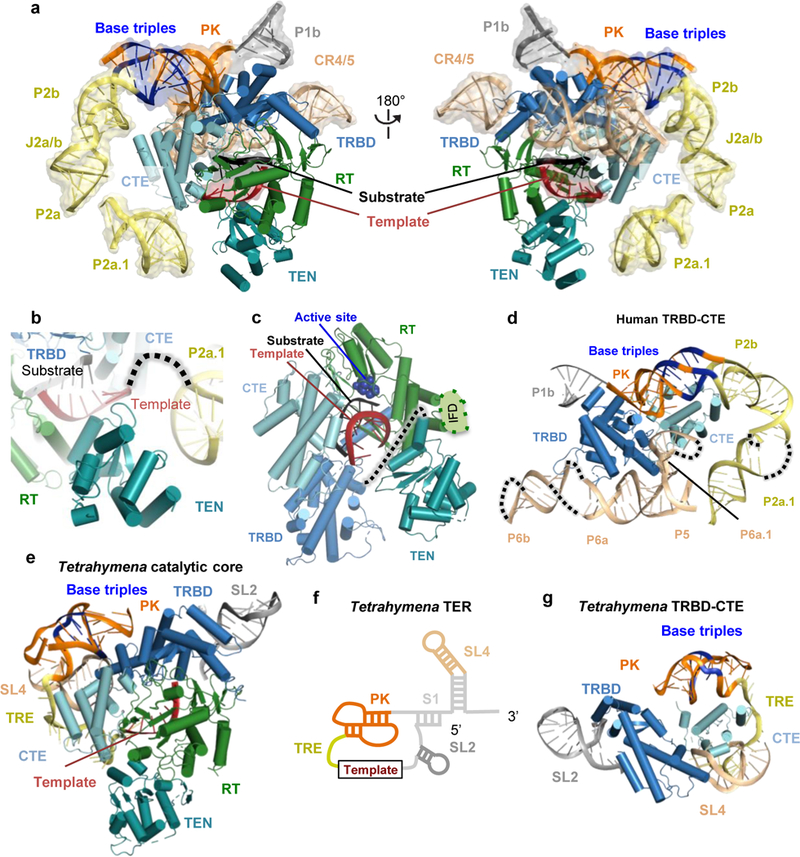
a, Front (left panel) and back (right panel) views of the catalytic core. Domains of human TERT and hTR are color-coded as in Fig. 1a. hTR is in semi-transparent space-filling representation, and base-triples within the PK are indicated in blue. b, Route of single-stranded RNA connecting hTR P2a.1 and the template, which threads near the TEN domain. c, TERT interactions with the substrate-template duplex. Active site residues are shown as blue spheres. The human TERT IFD insertion missing in the Tribolium TERT is indicated between helices α10 and α11 of the Tribolium TERT13 (Extended Data Fig. 6b). A dotted line depicts the linker connection of TEN and TRBD domains. d, The human TRBD and CTE domains are encircled by hTR P2 (yellow) and PK (orange) and CR4/5 (wheat) domains. e, f, Architecture of Tetrahymena TERT domains with TER motif colors as indicated19: PK, orange; template recognition element (TRE), yellow; SL2, dark grey; and SL4, wheat. Base-triples within the PK are indicated in blue. For clarity, domains of TER colored in light grey in f are not shown in the structure in e. g, The Tetrahymena TRBD and CTE domains are encircled by t/PK (orange), TRE (yellow), SL2 (grey) and part of SL4 (wheat). Base-triples with the PK are indicated in blue.
We docked two copies of the crystal structure of an archaeal H/ACA RNP24, each consisting of Cbf5 (orthologous to dyskerin), GAR1, NOP10 and L7Ae (structurally related to NHP2) and a single-hairpin RNA, into the H/ACA lobe (Fig. 2a-c, 5a, 5b and Extended Data Fig. 5e and 5f, 7f, 8a-d). The single-hairpin RNA was truncated to a 9 bp stem flanked by the ACA motif to mimic P4 and P7 stems flanked by box H and ACA, respectively, in hTR. The hTR segments bound by each archaeal RNP equivalent can be unambiguously assigned based on their connectivity with the rest of the RNA densities. The P4-P4.1 stem is connected to the CR4/5 domain in the catalytic core via the P4.2 stem, while P7 is connected to the P8 stem-loop (Fig. 3a, 3b, 5a and 5b and Extended Data Fig. 5i). Density in the regions of P1, P4.1 and P4.2 was fitted with A-form RNA duplexes. TCAB1 harbors a WD40 domain, and its homology model fit uniquely at the top of the H/ACA lobe (Fig. 2b, 5a and 5b and Extended Data Fig. 5g and 7g), consistent with the missing density from hTR CAB-box mutation (Extended Data Fig. 1h). This assignment of TCAB1 places it adjacent to an RNA-hairpin-like density that was fitted with the NMR structure of the P8 stem-loop39.
Figure 5 |. The H/ACA domain and disease mutations.
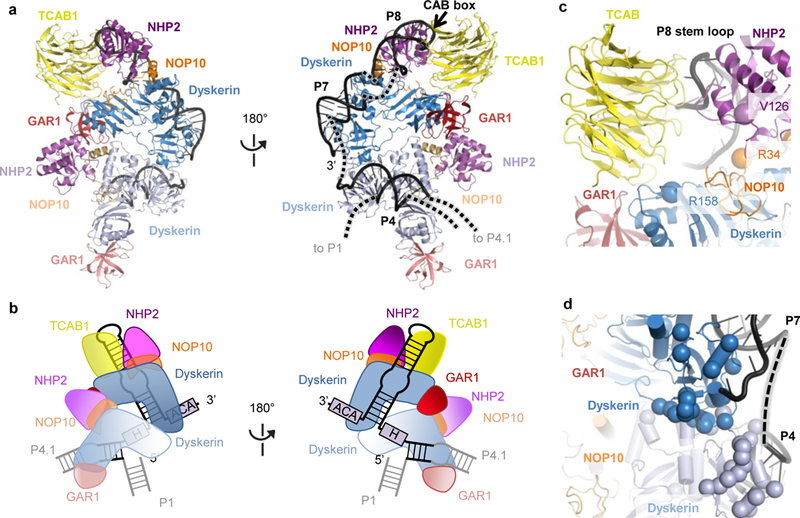
a, Front and back views of the H/ACA lobe within the human telomerase structure, with color-coded subunits: TCAB1, yellow; dyskerin, blue; GAR1, red; NOP10, orange; NHP2, magenta; hTR, black. To distinguish the two sets of the H/ACA proteins, the first set bound to the 5’ hairpin (P4 stem) is in lighter shade. b, Schematics of subunit arrangements within the H/ACA lobe. c, Interactions of TCAB1 with hTR and H/ACA proteins. The backbone amides of disease-associated mutations in dyskerin, NOP10 and NHP2 that mapped near TCAB1 and P8 interaction surfaces are highlighted as spheres. d, Mapping of a cluster of disease mutations (backbone amides shown as spheres) in dyskerin near the dyskerin-dyskerin and dyskerin-RNA interfaces.
The catalytic core
The t/PK and CR4/5 domains of hTR wrap around TERT to form the catalytic core (Fig. 4a). In turn, the human TERT ring encircles the fitted Tribolium TERT RNA-DNA bound to the TERT active site (Fig. 4a and 4c), which shifts the template position compared to that in the substrate-free Tetrahymena telomerase holoenzyme (Extended Data Fig. 7i). The majority of the TERT RNA binding surface is contributed by the TRBD and CTE, which are sandwiched between the PK (P2b-P3) and CR4/5 (Fig. 4d). The human PK (P2b-P3) is connected to P2a by the J2a/b bulge, and P2a connects to the less conserved helical extension P2a.1 before the transition to the single-stranded 3’-template-flanking region (Fig. 1a, 3a and 4a). The t/PK forms a rigid, arc-like structure (Fig. 1a and 4a), as predicted by modeling using NMR structures of its subdomains10,36,38. The less conserved end of the PK P3 stem approaches the TRBD close to its interface with the CTE and co-axially stacks with P2b (Fig. 4d). The unexpectedly limited interaction between the hTR PK and TERT favors a model for indirect PK function in the co-folding of TERT and hTR30,40. The location of P2a.1 positions the immediately 5’ single-stranded 3’-template-flanking RNA such that it could thread between the TEN domain and the TERT ring (Fig. 4b and Extended Data Fig. 7c), similar to the path of the 3’-template-flanking TER in the Tetrahymena telomerase holoenzyme 9,10,19 (Fig. 4e), despite major changes in the remainder of t/PK secondary and tertiary structure (Fig. 1a and 4f and Extended Data Fig. 7j). The TEN domain proximity to the template 3’ end further reinforces its proposed role in stabilization of RNA-DNA duplex at the template 3’ end14,27. The position of the Tetrahymena 5’ template-boundary element, stem-loop 2 (SL2), relative to the TRBD, is roughly similar to that of human P1b (Fig. 4d and g), consistent with their common function in 5’ template boundary determination41.
The CR4/5 domain, which consists of P5, P6 and P6.1 stems connected as a three-way junction, adopts a similar conformation to that in the medaka CR4/5-TRBD crystal structure33 (Fig. 4d and Extended Data Fig. 7d), except that human P6 is longer and composed of two stems, P6a and P6b. The P6a stem binds along TRBD (Fig. 4d), while the P6.1 hairpin emanates from the three-way junction almost perpendicularly to P6a, inserting into the interface between the TRBD and CTE. Clear helical density is observed for P6b, which makes no RNA-RNA or RNA-protein interactions. P5 co-axially stacks with P6a, but like P6b, it does not interact with TERT (Fig. 4d and Extended Data Fig. 7d). The P6.1-TERT interaction is critical for TERT association with hTR and telomerase catalytic activity42,43. The P6.1 apical loop was disordered in the TRBD-CR4/5 crystal structure33 and, although our resolution is insufficient for de novo building, its density is visible in our cryo-EM map, appearing packed against the CTE (Extended Data Fig. 7d). Our structure suggests that interaction with the CTE stabilizes this loop, agreeing with previous crosslinking studies44. In the Tetrahymena telomerase cryo-EM structure, the stem-terminus element (SL4) is bound at the interface between the TRBD and CTE of TERT19, similar to P6.1 in our human telomerase structure (Fig. 4d and 4g). Whether or not they are related evolutionarily, P6.1 and SL4 serve a similar structural role in telomerase catalytic core architecture. Finally, the catalytic core has extra density that likely corresponds to TERT and hTR regions that were not possible to model (Extended Data Fig. 4f), such as the “insertion in fingers” (IFD) subdomain of the RT domain (residues 734-802) that is missing in Tribolium TERT (Extended Data Fig. 6b, 7a and 7b), and the non-conserved flexible linker between the TEN domain and the TERT ring (Fig. 4c).
The H/ACA RNP and human disease mutations
Our structure offers the first glimpse of the subunit architecture of an intact eukaryotic H/ACA RNP, with two sets of H/ACA heterotetramers that bind two RNA hairpins (Fig. 5a and b). As expected, the 3’ hairpin loop associates with the single holoenzyme TCAB1 subunit (Fig. 5a-c). We observe an interaction between the two H/ACA protein heterotetramers, predominantly mediated by the two dyskerin molecules (Fig. 5d and Extended data Fig. 7h). The first set of H/ACA proteins contacts the 5’ hairpin P4 stem exclusively via dyskerin (Fig. 5a and b, Extended Data Fig. 5e). In contrast, the second set forms more extensive interactions with the 3’ hairpin P7 stem and P8 stem-loop. The P7 stem is bound by dyskerin, similarly to the 5’ hairpin P4, and continues to P8, which interacts with NOP10, NHP2 and additionally TCAB1 (Fig. 5a and b, Extended Data Fig. 5f and 5i). That binding of dyskerin at the base of the 5’ hairpin is sufficient for anchoring the entire core heterotetramer, and accounts for the evolutionary divergence of the vertebrate TER 5’ H/ACA hairpin45 and for the hTR 5’ hairpin tolerance of changes in stem and pocket structure46. Loss of hTR 5’ hairpin contacts to H/ACA proteins is compensated by an hTR-specific increase in protein binding to the 3’ hairpin18. The position of dyskerin defined by our structure explains why H/ACA RNP subunit exchange occurs in cell extracts for GAR1, NOP10 and NHP2, but not dyskerin47. The surprisingly limited protein-RNA interaction of the hTR 5’ hairpin implies that a wider breadth of RNA structures can assemble H/ACA proteins than has been predicted from the snoRNAs and scaRNAs, which conserve not only H/ACA protein binding but also requirements for dyskerin catalytic function21.
The single subunit of TCAB1 in human telomerase contacts both the 3’ hairpin CAB box and dyskerin in the second set of H/ACA proteins (Fig. 5c; Extended Data Fig. 7g), giving a unifying explanation for its simultaneous identification as an RNA-independent dyskerin binding protein and a sequence-specific, direct RNA binding protein48,49. Unexpectedly, TCAB1 also contacts GAR1 (Fig. 5c; Extended Data Fig. 7g). Protein-protein interactions by TCAB1 account for why such a limited amount of CAB-box sequence can stabilize its association with H/ACA RNPs. Structure fitting of the archaeal H/ACA RNP, combined with sequence homology between the archaeal and human proteins, allowed mapping of human dyskeratosis congenita and Hoyerall-Hreidarsson disease mutations in our structure50 (Extended Data Fig. 8a-d). Interestingly, a large number of dyskerin mutations cluster at the dyskerin-dyskerin interface, which is also close to its RNA binding surface (Fig. 5d; Extended Data Fig. 7f and 7h). Additionally, dyskerin R158 is near TCAB1 and NOP10 R34 and NHP2 V126 are located near the P8 stem (Fig. 5c). Mutations at any of these newly evident interfaces would be expected to have more severe impact on hTR than snoRNAs or scaRNAs because (a) crippled protein-RNA interaction of the hTR 5’ hairpin makes 5’ hairpin assembly more dependent on cross-hairpin dyskerin-dyskerin interactions, (b) hTR is uniquely reliant on enhanced H/ACA-protein affinity for the 3’-hairpin P8 stem-loop, and (c) hTR has one rather than two CAB box locations for TCAB1 association18,46. Our ability to map disease mutations to H/ACA RNP structure expands the scope of telomerase deficiency mechanisms beyond decreased assembly of core heterotrimer (dyskerin, NOP10, NHP2) (ref. 22) to include decreased dyskerin interaction with itself and with TCAB1 as well as with hTR.
Conclusions
The structure presented here provides the first architectural characterization of a telomerase holoenzyme from a multicellular species, addressing the longstanding question of vertebrate telomerase holoenzyme subunit content and arrangement. Our structure opens new opportunities for the design and screening of inhibitors targeting hTR folding and TERT interaction that will hold promise as general anti-cancer therapeutics7.
Methods
Telomerase reconstitution
Human telomerase was reconstituted in cells with the TERT expression plasmid pcDNA3.1-ZZ-Tev-TwinStrep-TERT and the hTR expression plasmid pcDNA3.1-U3-hTR-HDV. HDV is the Hepatitis Delta Virus ribozyme, which was previously shown to increase hTR accumulation46,28. Initially we used suspension HEK 293 cells lacking N-acetylglucosaminyltransferase I (GnTI)51 and transfected the cells using polyethylenimine (PEI)52. These cells were cultured in FreeStyle 293 Expression medium (ThermoFisher). After 48 hours of transfection, whole cell-extract was prepared by CHAPS lysis53. We obtained populations of inactive and active telomerase RNP purified from these extracts. To enrich for the active telomerase RNP, we used adherent HEK 293T cultured in DMEM with GlutaMAX (ThermoFisher) supplemented with 10% FBS, 100 units/mL penicillin and 100 μg/ml Streptomycin. The cells were transfected with TERT and hTR expression plasmids by calcium phosphate. After 48 hours, the cells were harvested and resuspended in hypotonic lysis buffer (HLB) (20 mM HEPES NaOH pH 8, 2 mM MgCl2, 0.2 mM EGTA, 10% glycerol, 0.1% NP40, 1 mM DTT and 0.1 mM PMSF). Whole-cell extract was prepared by three freeze-thaw cycles and adjusted to 300 mM before being cleared by centrifugation. The clarified extract was adjusted to 150 mM NaCl. For ΔTCAB1 telomerase and TERT-hTRmin, we used a previously characterized CAB-box mutant hTR (G414C)25 and LhTRmin28. HEK 293 cell lines are long-term lab stocks and had the expected morphology and growth. They have not been authenticated or tested for mycoplasma recently.
Telomerase purification
The prepared whole-cell extract (from 120 plates of adherent cells or 6L of suspension cells) was incubated with streptavidin agarose resin (Sigma-Aldrich) pre-bound to a 5’-biotinylated oligonucleotide15 at room temperature for 3 hours. The resin was washed with wash buffer (20 mM HEPES NaOH pH 8.0, 150 mM NaCl, 2 mM MgCl2, 0.2 mM EGTA, 10% glycerol, 0.1% Igepal CA-630, 1 mM DTT) and eluted with a competitor oligonucleotide15. The eluate was subsequently incubated with MagStrep XT resin (IBA LifeSciences) for 2 hours at 4°C. The resin was washed with wash buffer and eluted in batch with biotin elution buffer (100 mM Tris HCl pH 8.0, 150 mM NaCl, 1 mM EDTA, 5-10 mM biotin, 2 mM MgCl2, 0.1% Igepal CA-630, 10% glycerol, 1 mM DTT). We collected three sequential batch elution fractions for which the elution buffer was incubated with the resin for 30 minutes each time. For mass spectrometry, the eluate was combined and precipitated using trichloroacetic acid (TCA). The pellet was washed extensively with acetone and resuspended in water. The sample was subjected by MudPIT to identify the protein components present in the sample54. For cryo-EM studies, the final eluate was concentrated and buffer-exchanged with cryo-EM buffer (20 mM HEPES NaOH pH 8.0, 150 mM NaCl, 2 mM MgCl2, 0.05% Igepal CA-630, 1% trehalose, 1 mM DTT). To prepare DNA-substrate bound telomerase, oligonucleotide T14AG3 (1 μM) was added in the MagStrep binding step, and excess DNA substrate was washed away. Substrate-bound telomerase was eluted as described above.
Immunoblotting
The purified telomerase samples (E1 and E2) from the CHAPS lysis protocol were resolved by bis-Tris SDS-PAGE. Proteins were transferred to a nitrocellulose membrane and blocked with 4% nonfat milk (Carnation) in TBS buffer (50 mM Tris pH 7.5, 150 mM NaCl) for 1 hour at room-temperature. The membranes were incubated with primary antibodies overnight at 4°C and washed with TBS before being probed with secondary antibodies (Invitrogen) in 4% nonfat milk in TBS for 1 hour at room-temperature. We blotted for TERT, TCAB1, dyskerin and GAR1 on the same membrane, and NHP2 and NOP10 on a different membrane. The small sizes of NHP2 and NOP10 required a shorter protein transfer time than the other proteins. The membranes were washed extensively with TBS and visualized on a LI-COR Odyssey imager. The primary antibodies used were rabbit anti-dyskerin (Santa Cruz Biotechnology), mouse anti-TCAB1 (Sigma), rabbit anti-GAR1 (Novus), rabbit anti-NHP2 (Proteintech), rabbit anti-NOP10 (Abcam), mouse anti-Strep (Abcam), mouse anti-tubulin (Abcam) and rabbit IgG (Sigma-Aldrich).
Direct telomerase activity assays
Direct primer extension assays were performed using 1 μl of sample in a 20 μl reaction volume containing 50 mM Tris-acetate pH 8.0, 4 mM MgCl2, 5 mM DTT, 250 μM dTTP, 250 μM dATP, 5 μM unlabeled dGTP, 0.1 μM α−32P dGTP (3000 Ci/mmole, 10 mCi/ml, Perkin-Elmer), 500 nM 5’-(T2AG3)3-3’ primer unless primer was pre-bound. Reactions were incubated at 30°C for 40 min and stopped with TES buffer (50 mM Tris HCl pH 7.5, 20 mM EDTA, 0.2% SDS). The products were extracted and precipitated together with a 12-nt 32P-end-labeled oligonucleotide as a recovery control and resolved on a denaturing polyacrylamide gel. The dried gels were exposed to a phosphorimager screen and imaged on a Typhoon Trio system (GE Healthcare).
HCT116 cell culture and genome editing
HCT116 cells were cultured in DMEM with GlutaMax (ThermoFisher) supplemented with 10% FBS and 100 μg/ml Primocin (Invivogen). Clonal cell lines with homozygous TERT disruption were generated previously28. Transgene integration at the AAVS1 safe harbor locus was conducted as previously described55 using CAGGS promoter for TERT expression and a neomycin resistance cassette for selection. Genome engineering in HCT116 cells was performed using the manufacturer’s transfection protocol (Lipofectamine 3000, Thermo) with media exchange at 24 hours into selection with 200 μg/ml of G418 and 100 μg/ml of hygromycin. Cells were passaged continuously with selection media for >3 months after selection for cells with the integrated TERT transgenes. HCT116 cells were obtained from Molecular and Cell Biology Department Tissue Culture Facility (UC Berkeley) and were tested for mycoplasma.
Telomeric repeat amplification assays
Cell extract was prepared using the HLB method as described above and normalized to 2 mg/ml, which was quantified by the Bio-Rad Bradford protein assay. Dilutions were made of the whole cell extract, which were used for the PCR-based telomeric repeat amplification protocol (TRAP) as previously described55. Two μl with 400 ng total protein was used per 20 μl QTRAP reaction, consisting of 0.1 μg/μl TS primer (5’-AATCCGTCGAGCAGAGTT-3’), 0.1 μg/μl ACX primer (5’-GCGCGGCTTACCCTTACCCTTACCCTAACC-3’), and iTaq Universal Green Supermix (BioRad). Samples were incubated on a CFX96 Touch Real-Time PCR Detection System (Bio-Rad, Hercules, CA) at 30°C for 30 min, followed by a hot start of 95°C for 2 min. The samples were then cycled 35 times between 95°C for 20 seconds and 61°C for 90 seconds. Relative telomerase activity was calculated by a ΔCt normalized to the parental sample. Serial dilutions of 10, 30, and 100 ng were used for the hotTRAP protocol. The TS and ACX primers were used to amplify telomeric repeats labeled by incorporation of α−32P-dGTP. TSNT (5’-AATCCGTCGAGCAGAGTTAAAAGGCCGAGAAGCGAT-3’) was used as the internal control (IC) for all samples, and NT (5’- ATCGCTTCTCGGCCTTTT-3’) was the dedicated TSNT reverse primer. Samples were resolved on a native gel. The gel was dried, exposed on a phosphorimager screen and imaged on a Typhoon scanner (GE Healthcare).
Southern blotting
Genomic DNA preparation was conducted as previously described28. Four-8 ug of DNA was digested 8 hours to overnight with AluI and MboI at 37°C before electrophoresis on a 0.7% agarose gel (Seakem ME agarose). The gel was dried and denatured in 1.5 M NaCl and 0.5 M NaOH for 1 hour at 50°C. The gel was then washed twice with 4X SSC + 0.1% SDS before prehybridization with Church’s buffer (1% BSA, 1 mM EDTA, 0.5 M Na3PO4 pH 7.2, 7% SDS) for 30 minutes at 50°C. 32P-end-labeled probes were made of the telomeric repeat (T2AG3)3 as well as the O’Generuler 1 kb plus ladder (Thermo). These probes were both added and hybridized overnight at 50°C. The gel was washed with 4X SSC + 0.1% SDS at 40°C before exposing on a phosphorimager screen and imaging on a Typhoon scanner (GE Healthcare).
Negative-stain electron microscopy and data processing
For negative-stain grid preparation, 4 μL of telomerase sample was incubated on glow-discharged 400-mesh copper grids, which had been previously coated with a homemade carbon film on nitrocellulose, for 90 s and stained with 2% w/v uranyl formate. Micrographs for the inactive and active fractions of telomerase were collected on an FEI Tecnai 12 transmission electron microscope operated at 120 kV and equipped with a TVIPS TemCam F416. Data was collected using the Leginon package56 and data processing was done in RELION 1.4 (ref. 57) unless otherwise indicated. Initial particle picking of ~1,500 particles was performed manually, particles were extracted with a 2202 pixel box at a pixel size of 2.18 Å/pixel and subjected to reference-free 2D classification to obtain 2D initial references for auto-picking within RELION58. A total of 51,423 and 26,350 particles were picked for the inactive and active fractions, respectively. We performed two rounds of reference-free 2D classification for each dataset.
Micrographs for the enriched active fraction of intact holoenzyme, ΔTCAB1 telomerase, and TERT-hTRmin samples were collected on an FEI Tecnai F20 transmission electron microscope operated at 120 kV and equipped with a Gatan US4000 CCD camera. Data collection was done using the Leginon software package56. Initial particle picking for intact telomerase holoenzyme was done using DOG PICKER59 within the APPION pipeline60. This yielded approximately 16,000 particles, which were extracted using a 4002 pixel box at a pixel size of 1.5 Å/pixel and subjected to reference-free 2D classification to obtain initial 2D class averages. These 2D class averages were subsequently used as references for automatic picking within RELION58 to yield 15,293 and 23,922 particles for intact telomerase holoenzyme and ΔTCAB1 mutant, respectively. Reference-free 2D classification was again performed on these particles. For TERT-hTRmin complex, which is a much smaller RNP than telomerase holoenzyme, DOG PICKER was initially used to pick approximately 48,000 particles, which were extracted using a 1802 pixel box at a pixel size of 1.5 Å/pixel. 2D class averages resulting from reference-free 2D classification of these particles were used as references for automatic picking in RELION to give 50,720 particles, which were subjected to another round of reference-free 2D classification. A comparison of the 2D class averages of intact telomerase holoenzyme, ΔTCAB1 mutant and TERT-hTRmin is shown in Extended Data Fig. 1e-h.
Cryo-EM electron microscopy
For cryo-EM analysis, 3 μl of the purified telomerase was applied to C-flat CF 4/2 grids (Protochips) that had been previously coated with a 6-nm-thick layer of homemade continuous carbon film and glow-discharged in amylamine61 to overcome the preferred orientation of the telomerase holoenzyme on untreated carbon film. The grids were blotted for 4-5 s at 100% humidity and 4°C and plunged into a mixture of liquid ethane and propane cooled by liquid nitrogen62 using an FEI Vitrobot MK IV. The grids were loaded onto an FEI Titan Krios transmission electron microscope operated at 300 kV and equipped with a Gatan K2 Summit direct electron detector camera mounted behind a GIF Quantum energy filter. Images were collected automatically using the Serial-EM package63 in super-resolution counting mode at a calibrated magnification of 43477x (physical pixel size of 1.15 Å/pixel), with a total electron dose of 42 electrons per Å2 during a total exposure time of 7 s, dose-fractionated into 28 movie frames. We used a slit width of 25 eV on the energy filter and a defocus range of 1.5-3.5 μm. A total of 11,654 micrographs were collected from two separate data collection sessions.
Image processing
Most steps of data processing were done in RELION 2.0 (ref. 64) unless otherwise stated (Extended Data Fig. 3c). The movie stacks for each micrograph were corrected for drift and dose-weighted using MOTIONCOR2 (ref. 65) running within Focus66 during data collection. Contrast transfer function (CTF) parameters were estimated for the drift-corrected micrographs using GCTF67. Initial particle picking was performed on a subset of 350 micrographs using Gautomatch (obtained from Kai Zhang, MRC-LMB, Cambridge) and templates from a previous dataset collected on substrate-free telomerase at a different magnification (data not used and not shown in this study), resulting in approximately 32,000 particles. These particles were extracted with a 4002 pixel box for reference-free 2D classification to obtain 2D references for automatic picking of all micrographs by Gautomatch. A total of 1,091,177 particles from 11,450 micrographs were picked, binned by 2 and extracted with a box size of 2002 pixel. The data were initially processed in two batches, one for each data collection session. 3D classification into five classes was run for 30 iterations with an angular sampling of 7.5°, a regularization parameter T of 4 and a 60 Å-low-pass filtered initial model obtained from the previous dataset of substrate-free telomerase (data not used and not shown in this study). The initial model used for the substrate-free telomerase was that of the published 30 Å negative stain reconstruction of human telomerase17. For dataset 1, a subset of 121,116 particles from one class with the best features was selected and un-binned. For dataset 2, we selected two 3D classes and subjected them to one round of reference-free 2D classification to remove more particle images showing contaminants or damaged telomerase complexes. A subset of 214,157 particles from this dataset was unbinned and combined with the subset of particles selected from dataset 1. The combined subset of 335,283 particles was refined and yielded a reconstruction that was hard to interpret due to the conformational heterogeneity between the two lobes and within each lobe. The angular assignments resulting from this refinement were used for focused 3D classification with signal subtraction for each lobe to resolve the conformational heterogeneity within each lobe29. The two lobes are henceforth referred to as the H/ACA lobe and the catalytic core, according to our subunit assignments using negative-stain EM (Extended Data Fig. 1e-h). The best class consisting of 103,363 particles for the catalytic lobe was refined to 7.8 Å resolution while that for the H/ACA lobe was refined to 8.8 Å resolution from 104,330 particles. These reconstructions show clear secondary structure features as expected for this resolution range. To further reduce conformational heterogeneity, we performed another round of alignment-free 3D classification, using the alignment parameters from the refinement run for each lobe, and refined the resulting best class. We obtained a 7.7 Å resolution reconstruction from 39,860 particles for the catalytic core and 8.2 Å resolution reconstruction from 36,210 particles for the H/ACA lobe. These reconstructions were used for model fitting (see below).
We also performed alignment-free 2D classification on the original (unsubtracted) particles using the orientations determined by the 3D refinement of each lobe to assess the relative orientation of the other lobe (Extended Data Fig. 4a). For both cases, the other lobe adopts a wide range of conformations relative to the lobe that is aligned, clearly showing that the quality of any reconstruction for the whole structure is limited by both the continuous movements between the two lobes relative to each other and the conformational heterogeneity within each lobe. To obtain a better reconstruction encompassing both lobes, we performed global 3D classification for the subset of 335,283 particles before signal subtraction (25 iterations, T=8, angular sampling of 7.5°) (Extended Data Fig. 3c). As expected for a particle population exhibiting continuous motion, it resulted in four classes with nearly equal distribution and the two lobes adopting different conformations relative to each other. We refined each of these classes and obtained 10.2 Å resolution for the best class.
All refinements were performed using fully independent data half-sets (gold-standard refinement) and resolutions are reported based on the Fourier shell correlation (FSC) = 0.143 criterion68,69 (Extended Data Fig. 4b). FSC curves were calculated with a soft mask. For visualization, maps were corrected for the modulation transfer function of the detector and sharpened by applying a negative B-factor as determined by the post-processing function of RELION. Local resolution (Extended Data Fig. 4d and 4e) was calculated within RELION 2.0. For model versus map FSC curves calculations, we converted the catalytic core and H/ACA RNP models into density using EMAN70. Due to small regions of unmodelled density in our maps (Extended Data Fig. 4f), we prepared a soft mask (6-pixel soft edge) from the model and extracted the cryo-EM maps of each model using this mask. FSC model versus map was calculated using Xmipp71 (Extended Data Fig. 4c).
Model fitting
Initial fitting of all X-ray structures, NMR ensembles and homology models was performed manually by visual inspection of the best maps of the individual two lobes, followed by fit optimization in Chimera72 (Extended Data Table 2b, Supplementary Data 1 and Supplementary Video 1). The fitted structure for each lobe was subsequently rigid-body fitted into the overall map resulting from refinement of the best 3D class from global 3D classification. A homology model for TCAB1 WD40 domain was obtained using Phyre73 based on the structure of coatomer subunit alpha in the COPI coat linkage structure74 (PDB 5A1V) and fitted into the density located by comparisons of the 2D class averages for WT and ΔTCAB1 telomerase (Extended Data Fig. 1e-h). The Tetrahymena TEN domain and the TRBD, RT and CTE domains of the Tribolium TERT13 (PDB 3KYL) were fitted by rigid-body fitting into the map separately. We compared the fittings of the Tribolium TRBD and the medaka TRBD (PDB 4O26) (Extended Data Fig. 7e). The former has a truncated TRBD, while the latter is closely related to the human TRBD in sequence. Some of the medaka TRBD extensions close to the RNA binding surface fit our map, while those in the periphery of the domain (including residues 403-425 and residues 545-572) did not. We removed extensions that did not fit from the medaka TRBD and replace the Tribolium TRBD with this truncated version. Our substrate can form a 6bp duplex with hTR template region. Thus we truncated the DNA/RNA duplex model in the duplex-bound Tribolium TERT (PDB 3KYL) to 6 bp. Overall, the relative orientations of the domains and the DNA/RNA duplex are very similar to that of the crystal structure. For the H/ACA RNP, the RNA used in the single-hairpin archaeal crystal structure (PDB 2HVY) was truncated to a 9 bp hairpin flanked by the 3’ ACA motif. We used this model to fit both H/ACA copies because no structure of H/ACA proteins bound to the H box is available. Fitted structures of available hTR domains include the human P2b-P3 pseudoknot36 (PDB 2K95), P2a-J2a/b-P2b37 (PDB 2L3E), P2a.1 (ref. 38), P8 stem (or CR7) (ref. 39) (PDB 2QH2), and the medaka TRBD-bound CR4/5 (ref. 33) (PDB 4O26). The P4 and P7 stems bound by the H/ACA proteins were adapted from the truncated archaeal hairpin RNA model, as mentioned above. We used COOT75 to place idealized A-form RNA helices in the map regions assigned to P1, P4.2, P4.2 and P6b stems.
Map and model visualization
Maps were visualized in Chimera72 and all model illustrations were prepared using either Pymol (www.pymol.org) or Chimera.
Extended Data
Extended Data Figure 1 |. Protein identification by immunoblotting, enriching active telomerase, substrate pre-binding, and comparison of wild-type (WT), ΔTCAB1, and TERT-hTRmin RNPs.
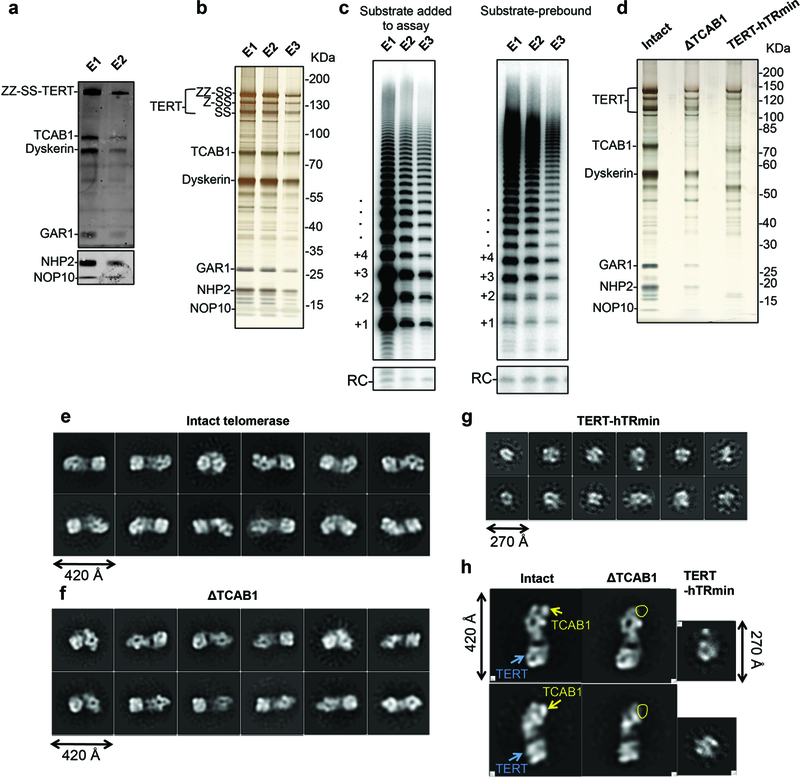
a, Immunoblotting of TERT, TCAB1, dyskerin, GAR1, NHP2 and NOP10 in telomerase purified after CHAPS lysis protocol as shown in Fig. 1b. We used primary antibodies against each protein, except ZZ-SS-TERT, for which we used rabbit IgG. Due to the wide range of the molecular weights of the proteins in our sample, TERT, TCAB1, dyskerin and GAR1 were detected in one blot, while NHP2 and NOP10 were detected in a separate blot. The use of the same sample to probe all proteins was performed only once, but TERT, dyskerin and TCAB1 were also probed individually twice. b, Silver-stained SDS-PAGE gel of purified telomerase fractions obtained from adherent cells lysed using the hypotonic lysis method that enriches active telomerase. This experiment was repeated over five times with similar results. c, Direct primer-extension assays of the purified telomerase fractions shown in b, confirming that E1 is no longer inactive (left panel), and of the substrate-bound purified telomerase fractions with additional DNA substrate omitted from the assays (right panel). The activity observed confirmed that purified telomerase contains the DNA substrate. The activity assays with substrate added were repeated over five times and the activity assays with substrate pre-bound were repeated twice. All repeats showed similar results. d, Silver-stained SDS-PAGE gel of purified intact and ΔTCAB1 telomerase and TERT-hTRmin telomerase prepared for subunit assignments. This experiment was done only once to provide a direct comparison between these different purified telomerase complexes. e, f, g, Negative-stained 2D class averages of intact and ΔTCAB1 telomerase and TERT-hTRmin, respectively. h, Comparison of representative 2D class averages intact and ΔTCAB1 and TERT-hTRmin showing the inferred localization of TCAB1 and TERT. For gel source data, see Supplementary Figure 1.
Extended Data Figure 2 |. Cellular function of the tagged TERT used for structural analysis.
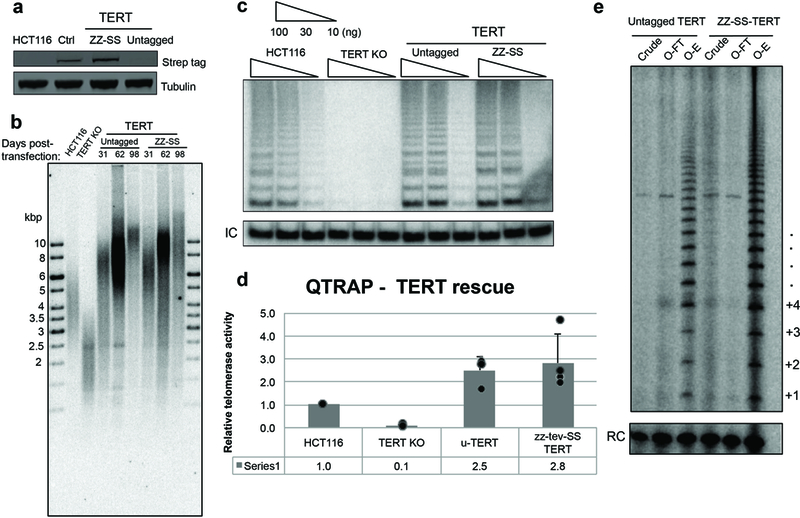
a, Western blot detection of ZZ-SS-TERT in TERT knock-out (KO) cells rescued by untagged TERT or ZZ-SS-TERT expression. Whole-cell extracts were probed using Strep antibody. HCT116 is the parental cell line. Lysate prepared from HEK 293T cells transiently transfected with ZZ-SS-TERT and hTR was used as a positive control (Ctrl). Tubulin was detected as a loading control. This experiment was performed only once to confirm the success of ZZ-SS-TERT incorporation into the HCT116 TERT KO cells. b, Telomeric restriction fragment analysis of HCT116 parental cells, TERT KO cells (before senescence), and TERT KO cells rescued with untagged or ZZ-SS-TERT transgene. Transgene-expressing cells were sampled at 31, 62 and 98 days post-transfection with transgene vectors. This experiment was performed twice with similar results. c, TRAP assay detection of telomerase activity in HCT116 parental cells, TERT KO cells, and TERT KO cells rescued with untagged TERT or ZZ-SS-TERT transgene. Whole-cell extracts were normalized by total protein concentration and assayed at 100, 30 or 10 ng of total protein per reaction. IC is an internal control. This experiment was repeated four times with similar results. d, Quantification of Q-TRAP assay detection of telomerase activity in HCT parental cells, TERT KO cells and TERT KO cells rescued with untagged TERT or ZZ-SS-TERT transgene. Error bars were calculated by taking the standard deviation of the average ΔCt from four different time points. Data points were shown as overlays. e, Direct primer-extension assay of telomerase after template-complementary oligonucleotide purification from extracts of TERT KO cells rescued by untagged TERT or ZZ-SS-TERT transgene. Assays were performed on clarified cell lysate (crude), flow-through (O-FT) and elution (OE) using equivalent amounts of cell extract. This experiment was performed only once to re-confirm the results in c. For gel source data, see Supplementary Figure 1.
Extended Data Figure 3 |. Image processing procedures.
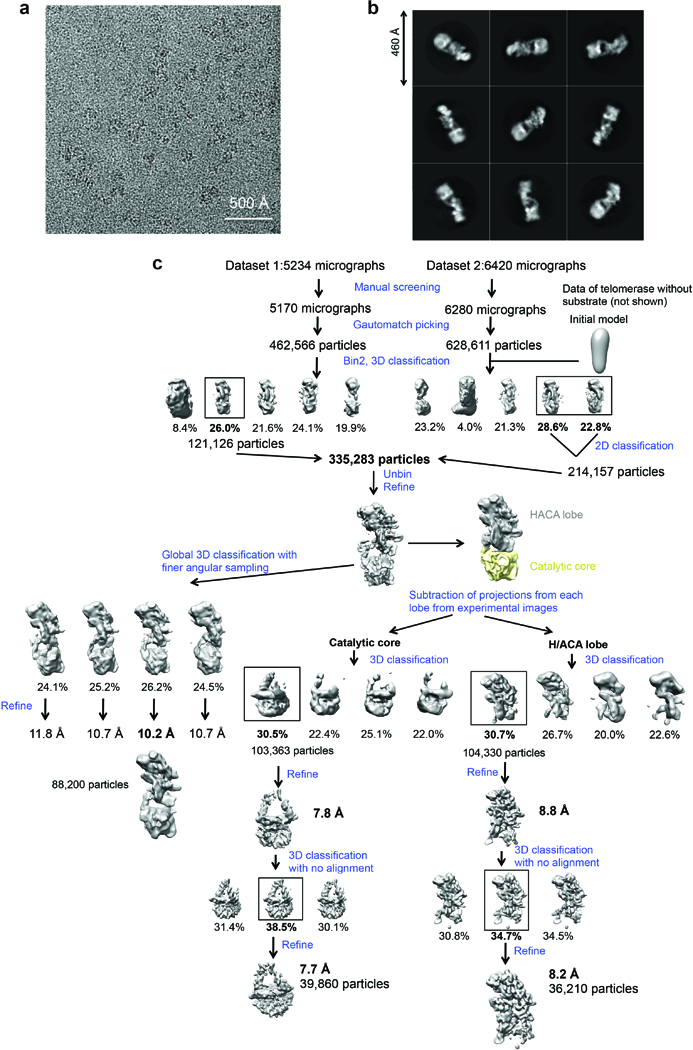
a, Representative raw micrograph. We collected a total of 11,654 micrographs for this study. b, Representative 2D class averages obtained from reference-free 2D classification. c, Data processing strategy used in this study.
Extended Data Figure 4 |. Resolution estimation and analysis of the flexibility of the complex.
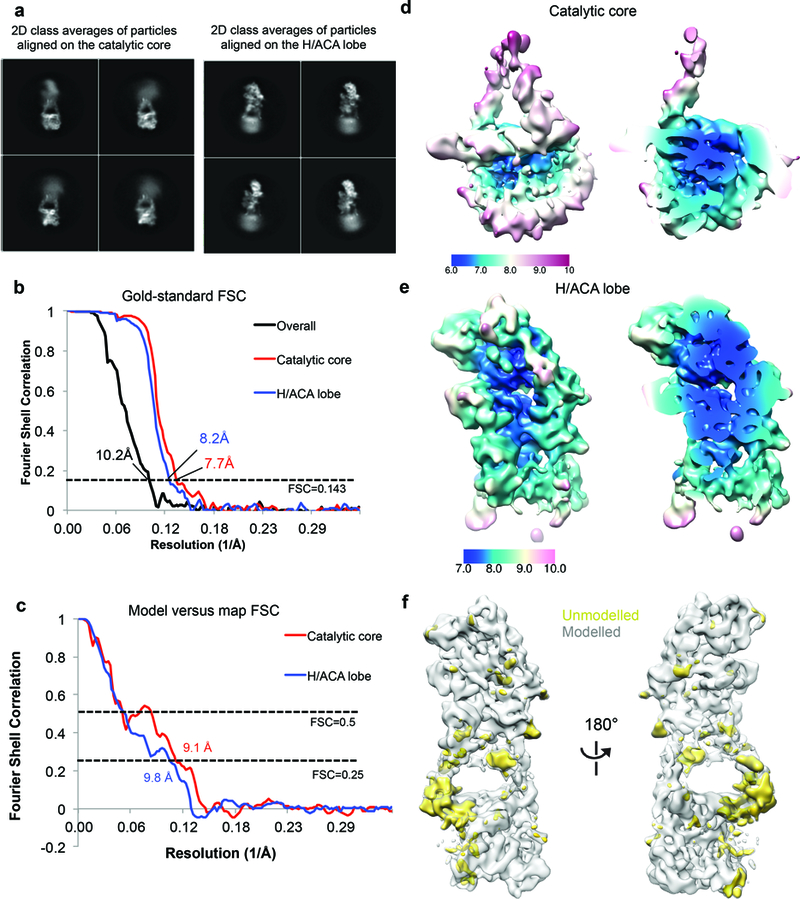
a, Representative 2D class averages obtained from 2D classification without alignment of particles that were aligned on either the catalytic core or the H/ACA lobe. For both cases, the other lobe adopts a wide range of conformations, as illustrated by the blurriness of the density. b, FSC curves for the overall map and the maps of the catalytic core and H/ACA lobe resulting from focused classification with signal subtraction and gold-standard refinement. c, Model versus map FSC curves for the catalytic core and the H/ACA RNP. We fitted only homology models as rigid bodies into the map and did not perform model coordinate refinement due to the limited resolutions of the maps. Therefore, we used a lower FSC threshold of 0.25 for resolution estimates. d, e, Local resolution for the catalytic lobe (d) and the H/ACA lobe (e) estimated by RELION 2.0 (ref. 64). Most of the catalytic core is resolved at 6-8 Å while most of the H/ACA lobe is resolved at 7-9 Å. f, Front (left panel) and back (right panel) views of the reconstruction showing modeled (grey) and unmodelled (gold) density. Most of the unmodelled density corresponds to single stranded RNA regions or RNA bulges, and human protein extensions that cannot be built de novo at this resolution.
Extended Data Figure 5 |. Fittings of proteins and RNA into the cryo-EM map.
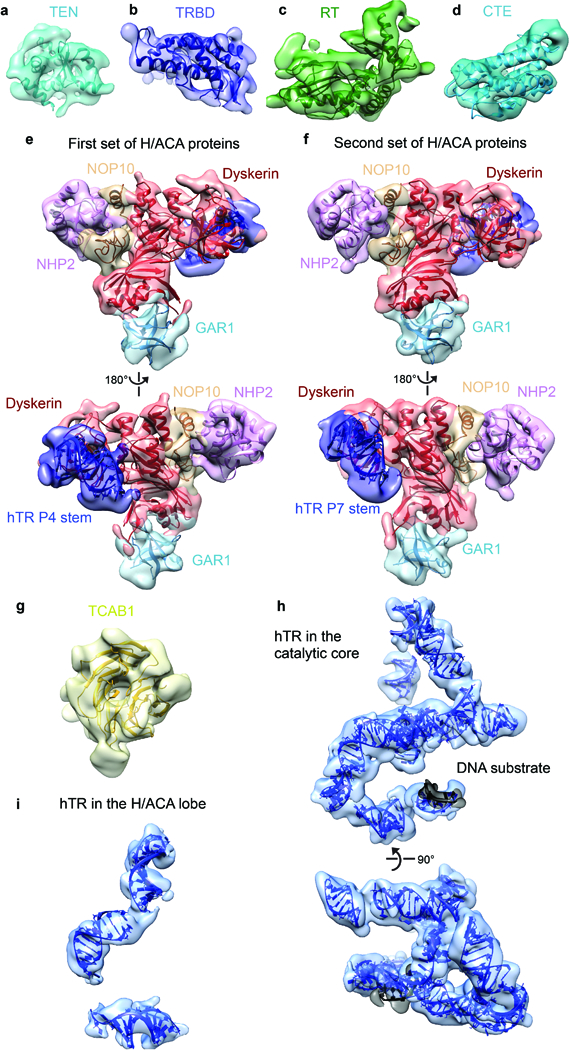
a-d, Domains of TERT. a, The TEN domain from Tetrahymena34 (PDB 2B2A). b, The truncated medaka TRBD domain33 (PDB 4O26). c, d, The RT and CTE domains from Tribolium13 (PDB 3KYL). e, Front (top) and back (bottom) views of the 5’ hairpin set of H/ACA proteins (dyskerin, red; GAR1, cyan; NOP10, wheat; NHP2, pink) bound to P4 stem (dark blue) fit by the archaeal H/ACA RNP24 (PDB 2HVY). f, Front (top) and back (bottom) views of the 3’ hairpin set of H/ACA proteins using the same model and color scheme as e. g, Homology model of TCAB1 WD40 domain. h, Front (top) and bottom (bottom) views of hTR in the catalytic core. i, hTR in the H/ACA lobe.
Extended Data Figure 6 |. Sequence alignment of TERT with secondary structure assignments based on known structures.
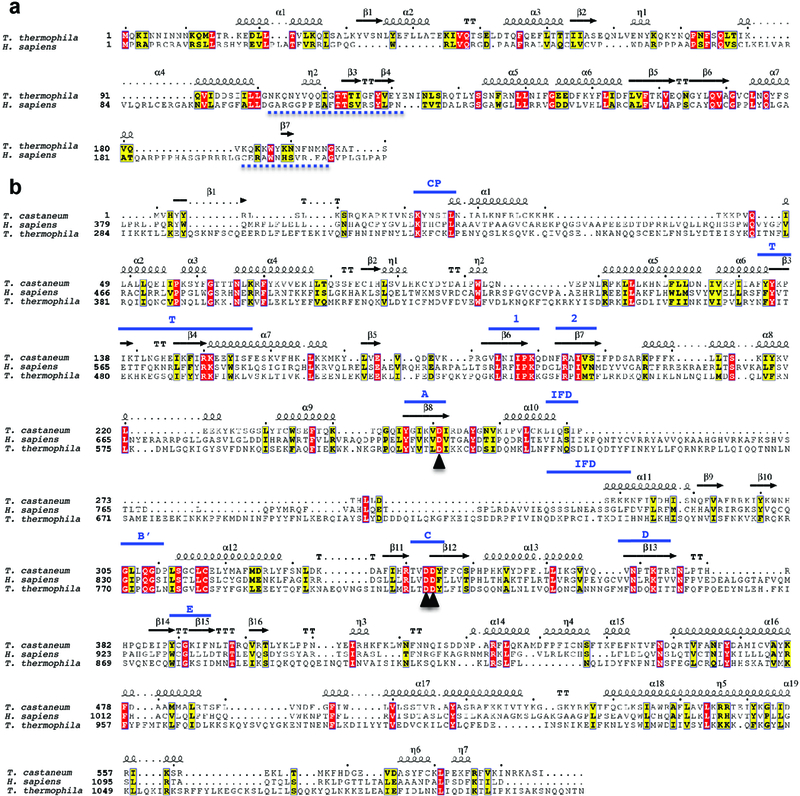
a, Sequence alignment of Tetrahymena and human TEN domains. The secondary structure assignments of the Tetrahymena TEN domain34 (PDB 2B2A) are shown above the aligned sequences. Regions removed prior to fitting are indicated with dashed lines below the sequences. b, Sequence alignment of the Tribolium, human and Tetrahymena TERT, with the latter two N-terminally truncated to match Tribolium. Secondary structure assignments of the Tribolium TERT are shown on top, with conserved motifs labeled in blue. Throughout the figure, the η symbol refers to a 310-helix. Strict β-turns and strict α-turns are displayed as TT and TTT. The three catalytic aspartic acids are indicated with black arrowheads. ESpript was used to generate this figure76.
Extended Data Figure 7 |. Selected protein-protein and protein-RNA interactions in telomerase holoenzyme and comparisons between human and Tetrahymena TERT.
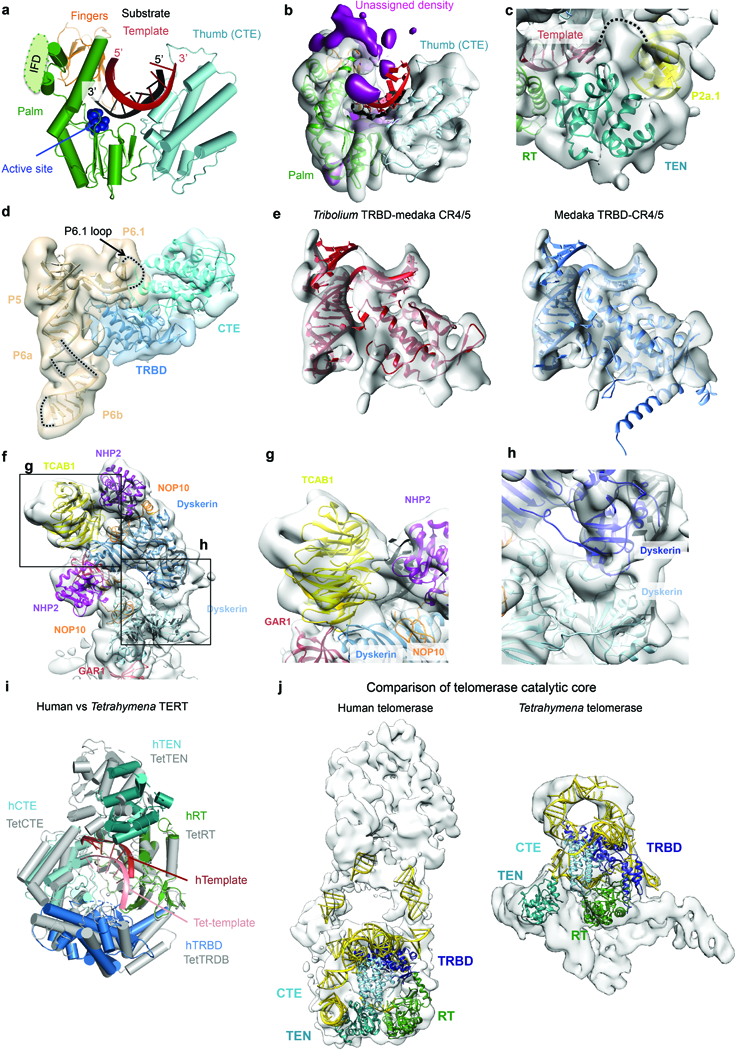
a, Interactions between the RT and CTE domains of TERT and the substrate-template duplex. The RT domain is divided into two subdomains, the palm (green) and fingers (orange) that are commonly observed in retroviral reverse transcriptases. The CTE (cyan) is the putative thumb. The IFD insertion that is missing in the Tribolium TERT is indicated. b, Region of the cryo-EM reconstruction shown in a. Unassigned density close to the IFD insertion is highlighted in magenta. c, Cryo-EM density of the TEN domain in the same view as that in Fig. 4b. Connecting density is observed between the template region and the P2a.1 stem. d, Map of the CR4/5 three-way junction (wheat) and the nearby TERT domains highlighting the position of the P6.1 loop near the interface of the CTE (cyan) and TRBD (blue) domains of TERT. This loop was not ordered in medaka CR4/5 bound to the TRBD alone33. e, Comparison of the Tribolium (left panel) and medaka TRBD (right panel) with the medaka CR4/5 domain of hTR13,33. Extensions of the medaka TRBD that did not fit the map were truncated for visualization. f, Cryo-EM map with H/ACA components fitted. Detailed views of regions boxed in f show TCAB1 interactions with dyskerin, GAR1 and the P8 stem-loop (g); and interactions between the two dyskerin molecules (h), where a cluster of DC mutations are found (Fig. 5d). i, Comparison of the human and Tetrahymena TERT superposed on the RT domain. Domains of human TERT are colored as in Fig. 1a, while Tetrahymena TERT is colored in grey. The bound human and Tetrahymena templates are colored in dark and light red, respectively. j, Comparison of human and Tetrahymena19 catalytic core fitted into the corresponding cryo-EM maps. Domains of TERT were colored as in Fig. 1a and TER is colored as yellow. We used the catalytic core and H/ACA lobe densities resulting from our focused classification/refinement for the human telomerase and the overall 9.4 Å Tetrahymena telomerase map (EMD-6442).
Extended Data Figure 8 |. Sequence alignments of H/ACA proteins with secondary structure assignments based on known structures.
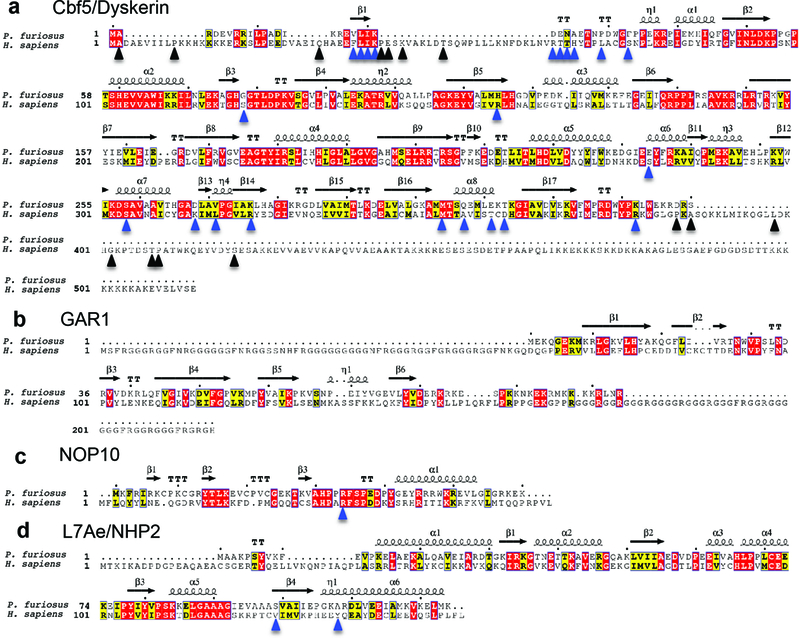
a-d, Sequence alignments of Pyrococcus furiosus (archaeal) and human Cbf5/dyskerin (a), GAR1 (b), NOP10 (c), and L7Ae/NHP2 (d). Secondary structure assignments displayed on the top are from the archaeal H/ACA RNP structure24 (PDB 2HVY). The η symbol refers to a 310-helix. Strict β-turns and strict α-turns are displayed as TT and TTT. Known human dyskeratosis congenita and Hoyeraal-Hreidarsson disease mutations50 in H/ACA proteins are indicated with arrowheads. Blue arrowheads indicate residues that can be mapped onto the archaeal structure and black arrowheads indicate residues that were not mapped. ESpript was used to generate this figure76.
Extended Data Table 1 |. Mass spectrometry analysis of the purified telomerase sample.
Proteins are ranked according to their percentage of sequence coverage eliminating keratins, histones, and abundant cytoskeletal proteins. Only proteins with sequence coverages of above 5% or known telomerase-associated proteins (highlighted in red background) with any level of detection are shown.
| Protein name | Sequence count | Sequence coverage (%) | Length (aa) | MW (KDa) |
|---|---|---|---|---|
| NHP2 (H/ACA subunit 2) | 2 | 19 | 153 | 17.2 |
| NOP10 (H/ACA subunit 3) | 1 | 17.2 | 64 | 7.7 |
| Nuclease-sensitive element binding protein | 4 | 13.6 | 324 | 35.9 |
| Methylosome subunit pICIn | 2 | 13.5 | 237 | 26.2 |
| TERT | 11 | 10.6 | 1132 | 127 |
| Insulin-like growth factor 2 | 3 | 8.6 | 548 | 60.1 |
| TCAB1/WDR79/WRAP53beta | 3 | 8.6 | 548 | 59.3 |
| squamous cell carcinoma antigen recognised by T-cells 3 (SART3) | 6 | 8 | 963 | 110 |
| Importin subunit a-1 | 3 | 7.8 | 529 | 57.9 |
| Dyskerin (H/ACA subunit 4) | 5 | 7.2 | 514 | 57.7 |
| hnRNP A2/B1 | 2 | 7.7 | 313 | 33.9 |
| hnRNP A3 | 1 | 5.8 | 378 | 39.6 |
| Protein arginine N-methyltransferase 5 | 1 | 5.6 | 531 | 61.1 |
| ATP-dependent RNA helicase DHX36 | 2 | 2.7 | 1008 | 114.8 |
Extended Data Table 2 |. Summary of cryo-EM data collection and modelling of protein and RNA subunits.
| a. Cryo-EM data collection |
| Catalytic Core (EMDB-7518) |
H/ACA lobe (EMDB-7519) |
Overall (EMDB-7521) |
|
|---|---|---|---|
| Data collection and processing | |||
| Magnification | 300 | 300 | 300 |
| Voltage (kV) | |||
| Electron exposure (e—/Å2) | 42 | 42 | 42 |
| Defocus range (μm) | 1.5-3.5 | 1.5-3.5 | 1.5-3.5 |
| Pixel size (Å) | 1.15 | 1.15 | 1.15 |
| Symmetry imposed | C1 | Cl | Cl |
| Initial particle images (no.) | 1,091,177 | 1,091,177 | 1,091,177 |
| Final particle images (no.) | 39,860 | 36,210 | 88,200 |
| Map resolution (Å) | 7.7 | 8.2 | 10.2 |
| FSC threshold | 0.143 | 0.143 | 0.143 |
| Map resolution range (Å) | 6-9 | 7-9 |
| b. Modelling of protein and RNA subunits |
| Protein/RNA | Total residues | M.W. (KDa) | Domain | PDB code or modelling | Reference |
|---|---|---|---|---|---|
| TERT | 1132 | 127 | TEN | 2B2A (Tetrahymena model) | 34 |
| TRBD | 4026 (medaka model) | 33 | |||
| RT | 3KYL (Tribolium model) | 13 | |||
| CTE | |||||
| TCAB1 | 548 | 59 | β-propeller domain | homology model by Phyre | |
| Dyskerin | 514 | 57 | 2HVY (archaeal model) | 24 | |
| GAR1 | 217 | 21 | |||
| NOP10 | 64 | 8 | |||
| NHP2 | 153 | 17 | |||
| hTR | 451 | 138 | P1 | A-form double helix | |
| P2a.1 | model from Chan et al, 2017 | 10, 38 | |||
| P2a-J2a/b-P2b | 2L3E | 37 | |||
| template | 3KYL | 13 | |||
| P3 | 2K95 | 36 | |||
| P4 | 2HVY as part of the H/ACA RNP | 24 | |||
| P4.1 | A-form double helix | ||||
| P4.2 | A-form double helix | ||||
| P5-P6.1-P6a | 4026 (medaka model) | 33 | |||
| P6b | A-form double helix | ||||
| P7 | 2HVY as part of the H/ACA RNP | 24 | |||
| P8 | 2QH2 | 39 |
Supplementary Material
Acknowledgements
We thank P. Grob, J. Fang, A. Chintangal, P. Tobias and H. Upton for technical support; X. Zhang and J. Vogan for establishing the suspension cell transfection protocol used in early stages of this work and sharing cell lines for in vivo studies and CAB-box mutant hTR; L. Kohlstaedt and QB3 mass spectrometry facility for analysis of purified telomerase; Y. Wang, J. Jiang and J. Feigon for sharing the coordinates of the Tetrahymena catalytic core and the modeled human t/PK; E. Rodina for sharing the Hansenula polymorpha TEN coordinates prior to publication; and H. Upton and A. Deshpande for comments on the manuscript. We thank the National Energy Research Scientific Computing Center supported by the Office of Science of the US Department of Energy for providing computing resources under contract number DE-AC02-05CH11231. This work was funded by N.I.H. grant GM054198 to K.C. T.H.D.N. is a Fellow of the University of California, Berkeley Miller Institute for Basic Research in Science. B.J.G. was supported by fellowships from the Swiss National Science Foundation (projects P300PA_160983, P300PA_174355). E.N. is a Howard Hughes Medical Investigator.
Footnotes
Data availability
Figure source data is provided in Supplementary Figure 1. Cryo-EM maps of the catalytic core, H/ACA lobe and overall human telomerase holoenzyme have been deposited with the Electron Microscopy Database under accession number EMD-7518, EMD-7519 and EMD-7521, respectively. PDB coordinates are included in a Pymol session provided as Supplementary Information.
References
- 1.Arnoult N & Karlseder J Complex interactions between the DNA-damage response and mammalian telomeres. Nat. Struct. Mol. Biol. 22, 859–866 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dosakni Y, de Lange T The role of double-strand break repair pathways at functional and dysfunctional telomeres. Cold Spring Harb. Perspect. Biol. 6, a016576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Levy MZ, Allsopp RC, Futcher AB, Greider CW & Harley CB Telomere end-replication problem and cell aging. J. Mol. Biol. 225, 951–960 (1992). [DOI] [PubMed] [Google Scholar]
- 4.Holohan B, Wright WE & Shay JW Telomeropathies: an emerging spectrum of disorder. J. Cell Biol. 205, 289–299 (2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wegman-Ostrosky T & Savage SA The genomics of inherited bone marrow failure: from mechanism to the clinic. Br. J. Haematol. 177, 526–542 (2017). [DOI] [PubMed] [Google Scholar]
- 6.Blackburn EH & Collins K Telomerase: an RNP enzyme synthesizes DNA. Cold Spring Harb. Perspect. Biol. 3, a003558 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shay JW Role of Telomeres and Telomerase in Aging and Cancer. Cancer Discov 6, 584–593, doi: 10.1158/2159-8290.CD-16-0062 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fu D & Collins K Distinct biogenesis pathways for human telomerase RNA and H/ACA small nucleolar RNAs. Mol. Cell 11, 1361–1372 (2003). [DOI] [PubMed] [Google Scholar]
- 9.Podlevsky JD & Chen JJ Evolutionary perspectives of telomerase RNA structure and function. RNA Biol, 1–13 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chan H, Wang Y & Feigon J Progress in Human and Tetrahymena Telomerase Structure Determination. Annu. Rev. Biophys. 46, 199–225 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wu RA, Upton HE, Vogan JM & Collins K Telomerase Mechanism of Telomere Synthesis. Annu. Rev. Biochem. 86, 439–460,(2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gillis AJ, Schuller AP & Skordalakes E Structure of the Tribolium castaneum telomerase catalytic subunit TERT. Nature 455, 633–637 (2008). [DOI] [PubMed] [Google Scholar]
- 13.Mitchell M, Gillis A, Futahashi M, Fujiwara H & Skordalakes E Structural basis for telomerase catalytic subunit TERT binding to RNA template and telomeric DNA. Nat. Struct. Mol. Biol. 17, 513–518 (2010). [DOI] [PubMed] [Google Scholar]
- 14.Robart AR & Collins K Human telomerase domain interactions capture DNA for TEN domain-dependent processive elongation. Mol. Cell 42, 308–318 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu RA, Dagdas YS, Yilmaz ST, Yildiz A & Collins K Single-molecule imaging of telomerase reverse transcriptase in human telomerase holoenzyme and minimal RNP complexes. eLife 4, 10.7554 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wenz C et al. Human telomerase contains two cooperating telomerase RNA molecules. EMBO J. 20, 3526–3534 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sauerwald A et al. Structure of active dimeric human telomerase. Nat. Struct. Mol. Biol. 20, 454–460 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Egan ED & Collins K Specificity and stoichiometry of subunit interactions in the human telomerase holoenzyme assembled in vivo. Mol. Cell. Biol. 30, 2775–2786 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jiang J et al. Structure of Tetrahymena telomerase reveals previously unknown subunits, functions, and interactions. Science 350, aab4070 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.MacNeil DE, Bensoussan HJ & Autexier C Telomerase regulation from beginning to the end. Genes 7, 64 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hamma T & Ferre-D’Amare AR The box H/ACA ribonucleoprotein complex: interplay of RNA and protein structures in post-transcriptional RNA modification. J. Biol. Chem. 285, 805–809 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yu Y-T & Meier UT RNA-guided isomerization of uridine to pseudouridine-pseudouridylation. RNA Biology 11, 1483–1494 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cohen SB et al. Protein composition of catalytically active human telomerase from immortal cells. Science 315, 1850–1853 (2007). [DOI] [PubMed] [Google Scholar]
- 24.Li L & Ye K Crystal structure of an H/ACA box ribonucleoprotein particle. Nature 443, 302–307 (2006). [DOI] [PubMed] [Google Scholar]
- 25.Fu D & Collins K Purification of human telomerase complexes identifies factors involved in telomerase biogenesis and telomere length regulation. Mol. Cell 28, 773–785 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cristofari G et al. Human telomerase RNA accumulation in Cajal bodies facilitates telomerase recruitment to telomeres and telomere elongation. Mol. Cell 27, 882–889 (2007). [DOI] [PubMed] [Google Scholar]
- 27.Wu RA & Collins J Human telomerase specialization for repeat synthesis by unique handling of primer-template duplex. EMBO J. 33, 921–935 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vogan JM et al. Minimized human telomerase maintains telomeres and resolves endogenous roles of H/ACA proteins, TCAB1, and Cajal bodies. eLife 5, 10.7554 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bai X-C, Rajendra E, Yang G, Shi Y & Scheres SHW Sampling the conformational space of the catalytic subunit of human γ-secretase. eLife 4, e11182 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jiang J et al. The architecture of Tetrahymena telomerase holoenzyme. Nature 496, 187–192 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bajon E, Laterreur N & Wellinger RJ A single templating RNA in yeast telomerase. Cell Rep. 12, 441–448 (2015). [DOI] [PubMed] [Google Scholar]
- 32.Alves D et al. Single-molecule analysis of human telomerase monomer. Nat. Chem. Biol. 4, 287–289 (2008). [DOI] [PubMed] [Google Scholar]
- 33.Huang J et al. Structural basis for protein-RNA recognition in telomerase. Nat. Struct. Mol. Biol. 21, 507–512 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jacobs SA, Podell ER & Cech TR Crystal structure of the essential N-terminal domain of telomerase reverse transcriptase. Nat. Struct. Mol. Biol. 13, 218–225 (2006). [DOI] [PubMed] [Google Scholar]
- 35.Petrova OA et al. Structure and function of the N-terminal domain of the yeast telomerase reverse transcriptase. Nucleic Acids Res. doi: 10.1093/nar/gkx1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim N-K et al. Solution structure and dynamics of the wild-type pseudoknot of human telomerase RNA. J. Mol. Biol. 384, 1249–1261 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang Q, Kim N-K & Feigon J Architecture of human telomerase RNA. Proc. Nat Acad. Sci. USA 108, 20325–20332 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang Y, Yesselman JD, Zhang Q, Kang M & Feigon J Structural conservation in the template/pseudoknot domain of vertebrate telomerase RNA from teleost fish to human. Proc. Nat Acad. Sci. USA 113, E5125–5134 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Theimer CA et al. Structural and functional characterization of human telomerase RNA processing and Cajal body localization signals. Mol. Cell 27, 869–881 (2007). [DOI] [PubMed] [Google Scholar]
- 40.Cash DD & Feigon J Structure and folding of the Tetrahymena telomerase RNA pseudoknot. Nucleic Acids Res. 45, 482–495, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jansson LI, et al. Structural basis of template-boundary definition in Tetrahymena telomerase. Nat. Struct. Mol. Biol. 22, 883–888 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mitchell JR & Collins K Human telomerase activation requires two independent interactions between telomerase RNA and telomerase reverse transcriptase in vivo and in vitro. Mol. Cell 6, 361–371 (2000). [DOI] [PubMed] [Google Scholar]
- 43.Chen J-L, Opperman KK & Greider CW A critical stem-loop structure in the CR4-CR5 domain of mammalian telomerase RNA. Nucleic Acids Res. 30, 592–597 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bley CJ et al. RNA-protein binding interface in the telomerase ribonucleoprotein. Proc. Nat Acad. Sci. USA 108, 20333–20338 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen J-L, Blasco M, & Greider CW A Secondary structure of vertebrate telomerase RNA. Cell 100, 503–514 (2000). [DOI] [PubMed] [Google Scholar]
- 46.Egan ED & Collins K An enhanced H/ACA RNP assembly mechanism for human telomerase RNA. Mol. Cell. Biol. 32, 2428–2439 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kittur N, Darzacq X, Roy S, Singer RH & Meier UT Dynamic association and localization of human H/ACA RNP proteins. RNA 12, 2057–2062 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Venteicher AS et al. A human telomerase holoenzyme protein required for Cajal body localization and telomere synthesis. Science 323, 644–648, (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tycowski KT, Shu MD, Kukoyi A & Steitz JA A conserved WD40 protein binds the Cajal body localization signal of scaRNP particles. Mol. Cell 34, 47–57 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sarek G, Marzec P, Margalef P & Boulton SJ Molecular basis of telomere dysfunction in huma genetic diseas. Nat. Struct. Mol. Biol. 22, 867–874 (2015). [DOI] [PubMed] [Google Scholar]
References
- 51.Reeves PJ, Callewaert N, Contreras R & Khorana HG Structure and function in rhodopsin: High-level expression of rhodopsin with restricted and homogeneous N-glycosylation by a tetracycline-inducible N-acetyleglucosaminyltransferase I-negative HEK293S stable mammalian cell line. Proc. Nat Acad. Sci. USA 99, 13419–13424 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Durocher Y, Perret S & Kamem A High-level and high-throughput recombinant protein production by transient transfection of suspension-growing human 293-EBNA1 cells. Nucleic Acids Res. 30, e9 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kim NW et al. Specific association of human telomerase activity with immortal cells and cancer. Science 266, 2011–2015 (1994). [DOI] [PubMed] [Google Scholar]
- 54.Schirmer EC, Yates JR 3rd & Gerace L MudPIT: a powerful proteomics tool for discovery. Discov. Med. 3, 38–29 (2003). [PubMed] [Google Scholar]
- 55.Sexton AN et al. Genetic and molecular identification of three human TPP1 functions in telomerase action: recruitment, activation, and homeostasis set point regulation. Genes & Dev. 28, 1885–1899 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Suloway C et al. Automated molecular microscopy: the new Leginon system. J. Struct. Biol. 151, 41–60 (2005). [DOI] [PubMed] [Google Scholar]
- 57.Scheres SHW RELION: implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol. 180, 519–530 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Scheres SHW Semi-automated selection of cryo-EM particles in RELION-1.3. J. Struct. Biol. 189, 114–122 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Voss NR, Yoshioka CK, Radermacher M, Potter CS & Carragher B DoG Picker and TiltPicker: software tools to facilitate particle selection in single particle electron microscopy. J. Struct. Biol. 166, 205–213 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lander GC et al. Appion: an integrated, database-driven pipeline to facilitate EM image processing. J. Struct. Biol. 166, 95–102 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nguyen THD et al. The architecture of the spliceosomal U4/U6.U5 tri-snRNP. Nature 523, 47–52 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tivol WF, Briegel A & Jensen GJ An improved cryogen for plunge freezing. Microsc. Microanal. 14, 375–379 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mastronarde DN Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol. 152, 36–51 (2005). [DOI] [PubMed] [Google Scholar]
- 64.Kimanius D, Forsberg BO, Scheres SHW & Lindahl E Accelerated cryo-EM structure determination with parallelisation using GPUs in RELION-2. eLife, e18722 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zheng SQ, Palovcak E, Armache J-P, Cheng Y & Agard DA Anisotropic correction of beam-induced motion for improved single particle electron cryo-microscopy. Nature Methods 14, 331–332 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Biyani N et al. Focus: The interface between data collection and data processing in cryo-EM. J. Struct. Biol. 198, 124–133 (2017). [DOI] [PubMed] [Google Scholar]
- 67.Zhang K Gctf: Real-time CTF determination and correction, J. Struct. Biol. 93, 1–12 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rosenthal PB & Henderson R Optimal determination of particle orientation, absolute hand, and contrast loss in single-particle electron cryomicroscopy. J. Mol. Biol. 333, 721–745 (2003). [DOI] [PubMed] [Google Scholar]
- 69.Scheres SH & Chen S Prevention of overfitting in cryo-EM structure determination. Nature Methods 9, 853–854 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tang G, et al. EMAN2: an extensive image processing suite for electron microscopy. J. Struct. Biol. 157, 38–46 (2007). [DOI] [PubMed] [Google Scholar]
- 71.Scheres SHW, Nuñez-Ramirez R, Sorzano COS, Carazo JM & Marabini R Image processing for electron microscopy single-particle analysis using Xmipp. Nature Protoc. 3, 977–990 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Goddard TD, Huang CC & Ferrin TE Visualizing density maps with UCSF Chimera. J. Struct. Biol. 157, 281–287 (2007). [DOI] [PubMed] [Google Scholar]
- 73.Kelley LA, Mezulis S Yates CM, Wass MN & Sternberg MJE The Phyre2 web portal for protein modeling, prediction and analysis. Nature Protocols 10, 845–858 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dodonova SO et al. The structure of the COPI coat linkage I. Science 349, 195–198. [DOI] [PubMed] [Google Scholar]
- 75.Emsley P, Lohkamp B, Scott WG, & Cowtan K Features and development of Coot. Acta. Cryst. D66, 486–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Robert X & Gouet P Deciphering key features in protein structures with the new ENDscript server. Nucl. Acids Res. 42(W1), W320–W324 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.