

Abstract
Perinatal ethanol (EtOH) exposure disrupts a variety of developmental processes in neurons important for establishing a healthy brain. These EtOH-induced impairments known as fetal alcohol spectrum disorder (FASD) are not fully understood, and currently, there is no effective treatment. Further, growing evidence suggests that adult females are more susceptible to EtOH with the effects of perinatal EtOH exposure also being sexually divergent. Female models have been historically underutilized in neurophysiological investigations, but here, we used a third trimester binge-EtOH model of FASD to examine changes to basal forebrain (BF) physiology and behavior in female Sprague-Dawley rats. We also tested varenicline as a potential cholinomimetic therapeutic. Rat pups were gavage-treated with binge-like EtOH, varenicline and EtOH, and varenicline alone. Using patch clamp electrophysiology in BF slices, we observed that binge-EtOH exposure increased spontaneous post-synaptic current (sPSC) frequency. Varenicline exposure alone also enhanced sPSC frequency. Varenicline plus EtOH co-treatment prevented the sPSC frequency increase. Changes in BF synaptic transmission persisted into adolescence after binge-EtOH treatment. Behaviorally, binge-EtOH treated females displayed increased anxiety (thigmotaxis) and demonstrated learning deficits in the water maze. Varenicline / EtOH co-treatment was effective at reducing these behavioral deficits. In the open field, EtOH treated rats displayed longer distances traveled and spent less time in the center of the open field box. Co-treated rats displayed less anxiety demonstrating a possible effect of varenicline on this measure. In conclusion, EtOH-induced changes in both BF synaptic transmission and behavior were reduced by varenicline in female rats supporting a role for cholinergic therapeutics in FASD treatment.
Keywords: binge ethanol, sIPSC, FASD, water maze, varenicline, basal forebrain
INTRODUCTION
Fetal Alcohol Spectrum Disorder (FASD) is the leading cause of preventable cognitive impairment in the U.S., with an estimated 40,000 infants affected each year, according to the National Organization on Fetal Alcohol syndrome (NOFAS) (2014). In the United States, prevalence ranges from 0.5 – 2.0 cases per 1000 births with approximately 12% of pregnant women reporting drinking within the last month and up to 2% binge drinking according to the (NIH FASD Fact Sheet) (2010). It is this binge drinking behavior that appears to be the major cause of damage to the developing fetus because it results in peak blood EtOH concentrations (BECs) that are especially harmful (West and Goodlett 1990, Maier and West 2001). Women who binge drink have the greatest number of FASD infants (May, Gossage et al. 2009, May and Gossage 2011). Binge drinking during the third trimester has been shown to result in particularly severe behavioral and cognitive dysfunctions (Larsson, Bohlin et al. 1985, Smith, Coles et al. 1986, Coles, Brown et al. 1991). These behavioral deficits are the result of EtOH exposure to developing neurons and synapses. Many of the consequences of perinatal EtOH intoxication can be modeled in animals and in vitro (Driscoll, Streissguth et al. 1990, West, Chen et al. 1994, Becker 1996, Mattson and Riley 1998, Hannigan and Berman 2000). These studies have shown that brain injury may involve neuronal loss, fiber tract disruption, aberrant proliferation, migration, synaptogenesis, transmission and signaling (West, Chen et al. 1994, Luo and Miller 1998, Costa, Savage et al. 2000, Hannigan and Berman 2000, Tanner, Githinji et al. 2004). EtOH exposure is capable of altering the formation of appropriate synapses and neurocircuits in many brain areas including the cholinergic system of the BF (Arendt, Allen et al. 1989, Floyd, Young-Seigler et al. 1997, Savage, Candon et al. 2000, Wang, Dubois et al. 2013) and the hippocampus (West 1993, Berman, Hannigan et al. 1996, Yanni and Lindsley 2000, Lindsley, Comstock et al. 2002). Mechanisms responsible for distortion of synaptic formation and maturation by EtOH are not completely understood but may represent a convergence of several EtOH-sensitive factors such as neurotransmitter receptors, voltage gated ion channels, or growth and neurotrophic factors (West, Chen et al. 1994, Costa, Savage et al. 2000, Thomas, Fleming et al. 2002, Heaton, Moore et al. 2003) differentially expressed across maturing neurons during the third trimester.
The complexity of the perinatal effects of EtOH is also reflected by the variability of injury severity, which may be sex specific (Streissguth 1996, Hellemans, Verma et al. 2010, Uban, Sliwowska et al. 2010, Uban, Comeau et al. 2013, Paolozza, Munn et al. 2015, Tesche, Kodituwakku et al. 2015, Fontaine, Patten et al. 2016, Paolozza, Treit et al. 2017). In 1996, Streissguth et al. observed sexual dimorphisms in the epidemiology of mood disorders in the FASD population (Streissguth 1996). More recently, Paolozza et al. reported sexually dimorphic deficits in FASD children in both saccade eye movements (Paolozza, Munn et al. 2015) and imaging of brain white matter (Paolozza, Treit et al. 2017). Sex-specific differences have been noted also in brain dynamics during auditory processing using magnetoencephalography (Tesche, Kodituwakku et al. 2015). In animal models, sex differences have been observed in several behavioral indices such as levels of hyperactivity (Grant, Choi et al. 1983, Thomas, Garrison et al. 2004), processing of social cues, stress responsiveness (Kelly et al. 2009; Weinberg et al. 2008), spatial memory (Kelly, Goodlett et al. 1988, Thomas, Biane et al. 2007, Banuelos 2011), and anxiety (Hellemans, Verma et al. 2010). In addition, sex specific differences have been observed in neurogenesis after just one day of postnatal EtOH treatment (Coleman, Oguz et al. 2012). In extended EtOH exposure models, changes in hypothalamic-pituitary-adrenal (HPA) axis activity (Weinberg, Sliwowska et al. 2008), and hippocampal synaptic plasticity were also sex-dependent (Helfer, White et al. 2012, Sickmann, Patten et al. 2014, Fontaine, Patten et al. 2016). Furthermore, the increase in alcohol use by females over the last three decades (Mancinelli, Vitali et al. 2009, Hoeppner, Paskausky et al. 2013, Graziani, Nencini et al. 2014) plus the evidence suggesting that females may be more susceptible to EtOH injury (Frezza, di Padova et al. 1990, Baraona, Abittan et al. 2001, Hommer, Momenan et al. 2001, Mancinelli, Vitali et al. 2009, Ceylan-Isik, McBride et al. 2010, Graziani, Nencini et al. 2014) makes the examination of females in EtOH exposure models critical.
Although there has been much interest in the promise of neurotrophic agents, neuroactive peptides, dietary antioxidants, and NMDA receptor antagonists (Idrus and Thomas 2011), these agents have yet to lead to an effective, FDA approved treatment for FASDs. There has been great enthusiasm surrounding the use of cholinergic / cholinomimetic therapeutics in ameliorating the effects of perinatal EtOH exposure on the developing brain. For example, choline supplementation significantly limits many of EtOH’s effects on development in rats (Thomas, Biane et al. 2007, Thomas, Abou et al. 2009). The approval of use patents for acetylcholinesterase inhibitors for the treatment of FASDs (Martinez and Egea 2007) also indicates the utility of cholinergic therapeutics. Varenicline, an agonist at nicotinic receptors (nAChRs) and FDA approved for smoking cessation, reduces EtOH drinking behavior (McKee, Harrison et al. 2009) and improves EtOH-induced learning deficits in adult mice (Gulick and Gould 2008). Acting as a cholinomimetic, varenicline is likely to share in the potential ‘Cholinergic Therapy’ benefits of agents such as choline and acetylcholinesterase inhibitors. In fact, recent publications have demonstrated that nicotine, choline, and varenicline mitigated EtOH-induced depression of microRNA expression levels in a second trimester, neurosphere model of neurogenesis (Balaraman, Winzer-Serhan et al. 2012, Tsai, Bake et al. 2014, Balaraman, Idrus et al. 2017). Varenicline also has enhanced prospects as a FASD treatment over other cholinergic therapies because it could also limit damage to the developing brain by reducing maternal EtOH consumption (McKee, Harrison et al. 2009, Mineur, Eibl et al. 2009, Rollema, Hajos et al. 2009).
Previously, we have reported that binge-EtOH exposure on postnatal day (PD) 4–9 alters the development of BF GABA receptors (GABARs) and miniature inhibitory postsynaptic current (mIPSC) physiology (Hsiao, Acevedo et al. 2001, Hsiao, DuBois et al. 2004, Wang, Dubois et al. 2013). During development, GABA plays a dual role as an excitatory neurotrophic signal that ultimately transitions to its primary role as an inhibitory neurotransmitter in adulthood. In the hippocampus, early, endogenous nicotinic signaling drives this transition in GABAergic development by changing chloride transporter expression and promoting the maturation of the chloride gradient (Zhang and Berg 2007). This interplay of nicotinic and GABAergic signaling could serve as a major influence shaping neuronal development (Liu, Neff et al. 2006, Zhang and Berg 2007). Interestingly, acute EtOH (25 −100 mM) has been shown to block cytosolic Ca2+ responses triggered by activation of excitatory GABAARs in cultured proliferating rat neuroepithelial cells (Ma, Pancrazio et al. 2001). However, it is unknown whether more long term EtOH exposure such as binge-like treatments affect this developmental switch (Sanderson, Donald Partridge et al. 2009). Although we did not test this mechanism directly in this study, we hypothesize that by promoting neuronal development, early nicotinic signaling can act to ameliorate EtOH’s disruptive, deleterious effects. Based on this supporting evidence, we tested varenicline as a potential therapy for third trimester equivalent, binge-EtOH exposure in female Sprague-Dawley rats by measuring variables known to change as a result of perinatal binge-EtOH exposure. We used whole-cell patch-clamp electrophysiology to examine synaptic transmission in the BF, a region known to play a role in EtOH’s impact on learning and memory (Arendt, Allen et al. 1989, Arendt 1994, Mitchell, Paiva et al. 2000, Hsiao, DuBois et al. 2004, Ehlers, Criado et al. 2011, Vetreno, Hall et al. 2011, Wang, Dubois et al. 2013, Fernandez and Savage 2017). We used a water maze spatial memory task to assess cognitive deficits and the open field to compare anxiety and hyperactivity between treated groups.
MATERIALS & METHODS
Animals and Treatments
Timed-pregnant Sprague-Dawley rats (Harlan) were maintained in the AAALAC-accredited vivarium at the Texas A&M University Health Science Center under controlled conditions (22–25°C; lights 0700–1900 h; rat chow and water ad lib) in accordance with policies of the Texas A&M University Laboratory Animal Care Committee and NIH guidelines. Postnatal EtOH intubation of rat pups was performed as previously described (Hsiao, DuBois et al. 2004, Banuelos 2011, Wang, Dubois et al. 2013) (Fig. 1). Gestational day 22 was designated as PD 0. Litters were culled to 8 pups per litter on PD 2. Pups remained with the dam except for ~10 min periods on a covered heating pad for intubation. For postnatal varenicline or EtOH treatment on PD 4, female pups were randomly assigned to control, varenicline alone, EtOH alone, or varenicline plus EtOH groups. Varenicline (1 mg/kg/day in water) and EtOH (5.25 mg/kg/day, EtOH 11.9% v/v-95% w/v in Enfamil with iron (Mead Johnson), to offset intoxication reduced nursing, was given via gastric intubation for the PD 4–9 treatment window (Hsiao, DuBois et al. 2004, Banuelos 2011, Wang, Dubois et al. 2013). Sham intubation without milk was given to control animals on the same schedule because their ability to nurse was unimpaired by EtOH intoxication. On PD 9, all pups were tattooed with non-toxic permanent black India ink for future identification. Pups stayed with the dam until used or weaned on PD 25. Blood samples (10 µl from the tip of the tail) were collected in heparinized Unipets (Beckon Dickinson) on PD 6. Samples were collected 90 min after the second EtOH treatment, and BECs were determined using gas chromatography (Wang, Dubois et al. 2013). For this model, blood EtOH levels have been shown to peak 90 min after treatment at levels of ~250 mg% (Hsiao, DuBois et al. 2004, Banuelos 2011, Wang, Dubois et al. 2013) and then decline to zero by 24 hr. This pattern and magnitude of BECs meets the Centers for Disease Control definition of binge drinking (Centers for Disease Control and Prevention, 2010). Mean BEC levels in this study were 264 ± 16 mg% for EtOH treated pups and 270 ± 20 mg% for varenicline + EtOH pups. Female rats were smeared daily at 8 a.m. beginning 1 week prior to water maze testing, and continued throughout. Cell sample analysis demonstrated that females cycled normally throughout experimentation. Due to the limited scope of the study, there was not enough statistical power to evaluate estrus cycle with rat behavior. Additional cohorts would be required for that analysis. Data in this study were collected across as many as 20 litters of pups.
Figure 1:
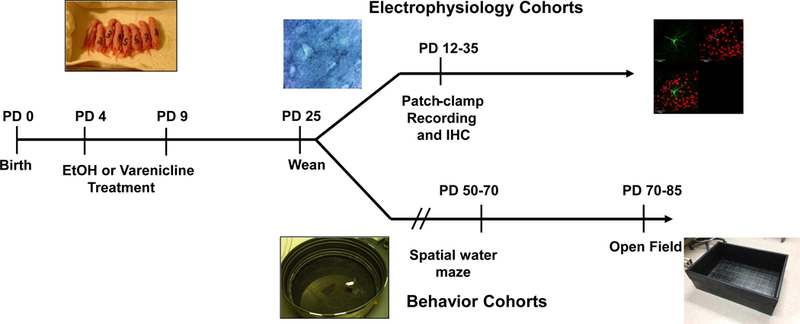
Experimental timeline of treatments, electrophysiology, and behavioral experiments. Male and female Sprague-Dawley rat pups were treated PD 4 – 9 with EtOH, varenicline alone, or varenicline plus EtOH. For electrophysiology experiments, animal ages ranged from PD 12 – 35. For behavioral testing, animals were separated post weaning and allowed to age to approximately PD 50.
Electrophysiology
For whole-cell recordings, brain tissue was harvested after lethal isoflurane inhalation and was cooled by immersion in 3 – 4°C artificial c erebrospinal fluid (aCSF) solution containing (mM): NaCl, 120; KCl, 3; MgCl2, 6; CaCl2, 0.5; D-(+)-glucose, 11; NaHCO3, 26; HEPES, 5; kynurenic acid, 0.3 (Sigma); bubbled with 95/5% O2/CO2; pH 7.4; ~340 mosM. Coronal slices (300 μm) were prepared using a refrigerated (Microm CU65, Thermo Scientific) vibrating microtome (Microm HM650 V, Thermo Scientific) in the same solution. Slices were transferred to a holding chamber (Scientific Systems Design, Inc.) of oxygenated (95/5% O2/CO2), ‘standard’ aCSF solution containing, (mM): NaCl, 124; KCl, 3; MgSO4, 1.2; CaCl2, 2.4; NaH2PO4, 1.25; D-(+)-glucose, 10; NaHCO3, 26; pH 7.4; ~320 mosM, gradually warmed to ~32° C and incubated for ~30 min then allowed to cool to ~22° C prior to experimentation. Individual slices were placed in a recording chamber, submerged using a slice anchor (Warner Instruments Corp.), and continuously perfused with oxygenated standard aCSF. Conventional patch-clamp techniques were used as described previously (DuBois, Parrish et al. 2004, Hsiao, DuBois et al. 2004, Wang, Dubois et al. 2013). Patch pipettes were pulled from glass capillary tubing (KG-33, 1.5mm OD, Garner Glass Co.) on a Brown and Flaming P-97 pipette puller (Sutter Instr.) with resistance 2–8 MΩ when filled with a pipette solution (mM): CsCl, 130; ethylene glycol-bis (β-aminoethyl ether) N,N,N’,N’-tetraacetic acid (EGTA), 10; MgCl2, 2; HEPES, 10; Mg-ATP, 4; GTP, 0.1; 0.1% Neurobiotin (Vector, Burlingame CA); pH 7.2 with CsOH; 295–300 mosM. Neurons were visualized in the BF using water immersion, differential interference contrast, video-enhanced optics on the stage of an upright Olympus BX51WI microscope. Voltage-clamp recordings were collected / digitized with a Multiclamp 700B amplifier (Axon Instruments/Molecular Devices), Digidata 1440A interface and pClamp 10 software (Molecular Devices). Membrane capacitance (pF) was obtained by digital cancellation of whole-cell capacitance transients. Membrane currents were recorded at −60 mV, low-pass filtered at 2 kHz and digitized at 20 kHz. To isolate GABAAR-mediated postsynaptic currents, D(−)2-amino-5-phosphonovaleric acid (APV, 40 µM, Sigma) and 6,7-dinitroquinoxaline-2,3-dione (DNQX, 10 µM, Sigma) were added to the perfusion solution. Spontaneous synaptic currents were analyzed using MiniAnalysis 6.0.3 (Synaptosoft) as described previously (DuBois, Parrish et al. 2004, DuBois, Damborsky et al. 2013, Wang, Dubois et al. 2013, Griffith, Dubois et al. 2014). Analysis of the electrophysiological data was conducted blind with respect to treatment. Cumulative probability curves were compared with a Kolmogorov-Smirnov (K-S) test, whereas mean data were compared using a one-way ANOVA or t-test (Prism 5, GraphPad, San Diego, CA). For sIPSC decay analysis, low noise traces and non-overlapping events were used to generate an ensemble averaged sIPSC by aligning currents on the rising phase. The 10–90% decay phase of this average for each neuron was fitted with a biexponential function:
where A1 and A2 were the fraction of the fast and slow decay components, respectively, As was the steady-state current, and τ1 and τ2 were the fast and slow decay time constants, respectively. Values are expressed as means ± S.E.M.
Immunohistochemistry
For imaging and identifying recorded BF neurons, slices were fixed 12–24 hr in 4% paraformaldehyde / 0.1 M phosphate buffered saline (PBS) before being transferred to a sterile solution of 0.1 MPBS (pH 7.4) until processing. Slices were rinsed in 0.1 M PBS (3 × 15 min) before blocking with 0.1 M PBS / 5% NDS / 1% Triton X-100 for 1 h at room temperature. Slices were then permeabilized with 1% Triton X-100 / 0.1 M PBS for 2 hours at room temperature. Next, slices were incubated for 48–72 h at 4°C with goat anti-ChAT (choline acetyltransferase, a cholinergic cell-marker) primary antibody (1:2000; Millipore). Slices were then incubated 24–48 h at 4°C with both AlexaFluor-488 conjugated donkey anti-goat (1:1000; Life Technologies) and AlexaFluor-568 conjugated streptavidin (1:2000; Life Technologies). Slices were mounted and cover slipped using ProLong Gold anti-fade reagent (Life Technologies). In order to identify neurobiotin filled cells, fluorescent z-stack images were acquired with a laser scanning Olympus Fluoview 1200 confocal microscope. The confocal microscope was equipped with an UPlanFL N 10× 0.30 numerical aperture (NA) and an UPlanSApo 60× 1.35 NA objectives and excitation lasers at 488 and 568 nm.
Throughout this study, neurons were randomly selected for patch clamping based on visual inspection. Using immunohistochemistry, we were able to identify 64 ChAT+, 46 non-ChAT, and 46 unidentified neurons across all treatment groups. In some cases, inadequate Neurobiotin filling, poor ChAT+ staining, or tissue damage resulted in an inability to identify neurons. We observed no significant difference in sPSC frequency across ChAT+ and non-ChAT neurons. Thus, for each treatment group, data was pooled across a mixed population of neuronal phenotypes.
Behavioral Assessment
Animals:
Rat behavior was assessed at 2 to 4 months across as many as 12 litters with one animal from each treatment group for spatial learning using the Morris water maze and anxiety / hyperactivity using the open field. Sample sizes varied from 10 to 12 animals per treatment groups. Subjects that experienced video tracking issues were excluded, and in some cases, outliers beyond three standard deviations of the cohort were also excluded. Rats tested in the water maze comprised of 10 controls, 12 EtOH, 10 varenicline, and 10 varenicline + EtOH treated rats. Rats tested in the open field were 12 controls, 11 EtOH, 10 varenicline, and 11 varenicline + EtOH treated rats.
Morris Water maze
Apparatus.
Spatial learning abilities were assessed on the Morris water maze task using a modified protocol described previously (Banuelos 2011). The maze, a black circular tank measuring 183 cm in diameter with a wall height of 58 cm, was filled with water (27°C). A retractable escape platform (clear plastic, 12 cm diameter, HVS Image, UK) was submerged 2 cm below the water surface near the center of the southwest quadrant of the maze. White curtains, on which were affixed large black geometric shapes (extra-maze cues), surrounded the maze. Data were analyzed using a computer-based video tracking system (Water 2020, HVS Image).
Spatial reference memory trials.
Spatial reference memory was assessed as described previously (Banuelos 2011). Briefly, rats received three daily training trials with a 30 s inter-trial interval (ITI) over six consecutive days. In each trial, rats were placed into the water facing the wall of the maze at one of four equally spaced start positions (north, south, east, or west). The start positions were varied in a pseudorandom manner, such that all rats started from each of the locations approximately the same number of times. Once in the water, rats were allowed to swim until they found the hidden platform or until 90 s elapsed, at which time the rats were guided to the escape platform by the experimenter. Rats remained on the platform for 30 s and then were placed in a holding chamber for 30 s before the next trial. Every sixth trial was a probe trial in which the platform was lowered to the bottom of the maze for the first 30 s of the trial, after which it was raised to allow the rats to escape.
Cued (visible platform) task.
After spatial reference memory training, rats were given one session with six trials of cue training. For cue training, rats were trained to escape to a visible platform (painted black and protruding 2 cm above the surface). The start position and platform location were varied in each trial, making the extra-maze cues explicitly irrelevant to the platform location. In each trial, rats were allowed to search for the platform for 90 s and then were allowed to remain there for 30 s before a 30 s ITI.
Statistical analyses.
For each task, data files were created by the Water 2020 software and were exported to Microsoft Excel, Statview (version 5.01) and GraphPad Prism 7.02 for analysis and graphing. In all statistical comparisons, p-values less than 0.05 were considered significant. Training trial data were averaged into days consisting of the trials excluding the probe trial, and performance was quantified using latency (in seconds) for regular learning trials. Cumulative search error and % time in target quadrant were used for probe trials. Thigmotaxis, which is the percent time spent in the maze periphery, was assessed as the time the rat spent swimming in the outer 10% of the maze (Mendez, Montgomery et al. 2008). Additional measures of performance [e.g. path length, defined as the distance (cm) it took the rat to reach the platform, and swim speed, defined as the rat’s swim speed (cm/s) averaged across the entire trial] also were recorded. Interpolated probe-trial data (i.e. every sixth trial) were analyzed using the mean search error. This measure was derived by dividing the cumulative search error in these trials by the duration of the probe trial (30 s). Measures not reported were not significant. Repeated measures ANOVA were used for training and probe trials and Tukey-Kramer tests were performed post hoc.
Open Field
Apparatus.
Approximately one week after behavioral assessment in the water maze, rats were tested for anxiety and hyperactivity in the open field. The box was 90 cm x 60 cm, and a camera was attached above the box. The videos were tracked and processed post hoc with EthoVision XT software from Noldus.
Procedure.
Rats were allowed to acclimate to the room for 10 minutes before the single trial. They were then placed in the center of the maze, and allowed to freely explore for 10 minutes. Only the first 5 minutes of the trial were used in the analysis.
Statistical analysis.
Data will be acquired and analyzed with a computerized video tracking system (Ethovision XT, Noldus). The perimeter of the box was defined as the 10 cm closest to the walls of the box. When the center point of the animal was detected outside this perimeter, the animal was consider to be located at the center of the box. T-tests were performed between ethanol and the other groups using Prism 5 (GraphPad, San Diego, CA). Values are expressed as means ± S.E.M.
RESULTS
Pup Growth and Viability
Pups were weighed the first day of treatment (PD 4) through PD 26 (Fig. 2A). There were no significant differences in initial pup weights on PD 4 across all treatment groups. Female pups demonstrated daily weight gain through PD 26. We saw a significant effect of treatment (F(3, 111) = 12.406, p < 0.0001), and by PD 5, groups exposed to EtOH gained less weight when compared to controls (sham intubated) (Tukey’s HSD; EtOH, p < 0.05; var + EtOH, p < 0.05). Groups exposed to varenicline alone showed no delay in growth (Tukey’s HSD; p > 0.05). Post hoc analyses showed that EtOH and var + EtOH treatment groups weighed significantly less than varenicline only treated rats and never caught up to control weights at PD 25 (Tukey’s HSD; p < 0.05). There were no pup mortalities related to binge-EtOH, varenicline alone, or var + EtOH treatments.
Figure 2:
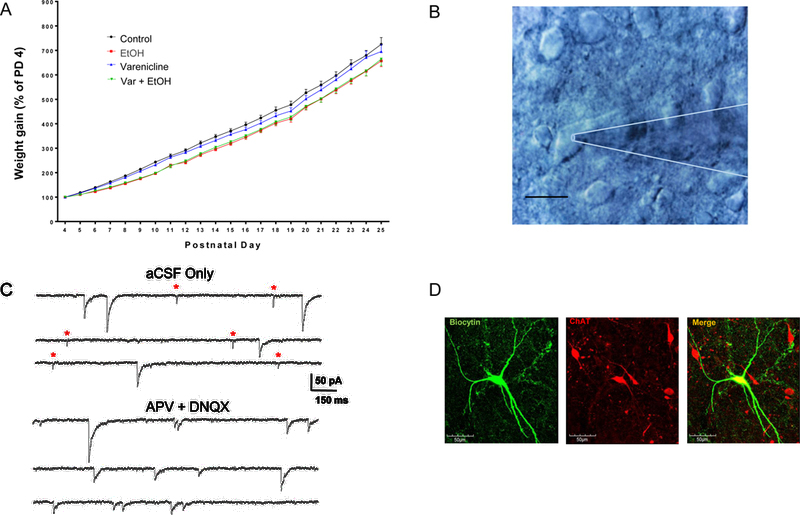
A. Female pup weight gain normalized as a percentage of PD 4 weight. By PD 5, pups exposed to binge-EtOH began to lag in growth and never caught up to control rats as late as PD 25. B. Image of a BF slice featuring a large cholinergic neuron with attached patch pipette, scale bar = 10 μm. The patch pipette (out of focus) is outlined in white. C. Representative traces from a BF cholinergic neuron illustrating a mixture of excitatory (represented by red *) and inhibitory sPSCs recorded in aCSF only. The addition of APV + DNQX was used to pharmacologically isolate GABAA-R sIPSCs. D. Confocal images of neurobiotin filled, ChAT+ BF neuron from a female control rat.
Electrophysiology
Postnatal basal forebrain synaptic transmission in female rats
Whole-cell voltage-clamp experiments were performed on both ChAT+ and non-ChAT+ BF neurons (~PD 12 – 35; Fig. 1) in acute coronal brain slices (Fig. 2B). Traces in figure 2C (top) illustrate mixed spontaneous postsynaptic currents (sIPSCs + sEPSCs) recorded in aCSF. These currents can be distinguished as GABAergic IPSCs (slow decaying) or as glutamatergic EPSCs (fast decaying). GABAergic sIPSCs were isolated also with the glutamate blockers, APV and DNQX (Fig. 2C, bottom). Although BF neurons receive both excitatory and inhibitory inputs, BF synaptic transmission is dominated by GABAergic IPSCs as reported previously (Griffith, Dubois et al. 2014). In these experiments, approximately 90% of all synaptic transmission was GABAAR-mediated. In contrast to the large GABAAR-mediated inhibitory tone, we observed that only 10% of synaptic transmission on to BF neurons was characterized by fast decaying, APV + DNQX sensitive currents, presumed to be AMPAR-mediated spontaneous excitatory postsynaptic currents (sEPSCs). In female controls, 70% of neurons (14/20) displayed sEPSCs (Fig 3C, top) with a mean frequency of 0.19 ± 0.02 Hz (Fig. 3C, bottom) and amplitude of 14.4 ± 0.9 pA (not shown).
Figure 3:
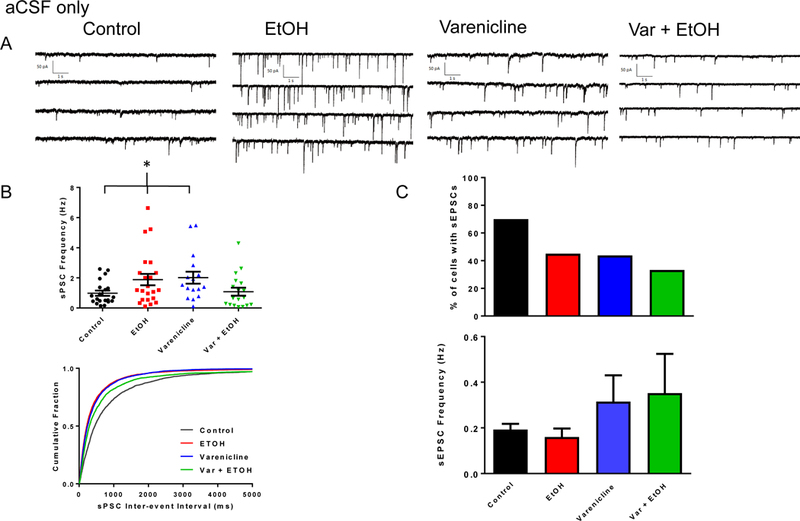
Binge-EtOH and varenicline treatments affect BF synaptic transmission in female rats. A. Representative traces of sPSCs from female rats across control, EtOH, varenicline alone, and var + EtOH treatments. B. (Top) Scatter plots of sPSC frequencies from recorded BF neurons across all treatment groups. Data are mean ± S.E.M. (Bottom) Cumulative probability plots of sPSC inter-event intervals from recorded BF neurons. (*) indicates statistical significance. C. (Top) Percent of BF neurons exhibiting sEPSCs across all treatment groups. (Bottom) Bar graph representation of sEPSCs across treatment groups. Data are mean ± S.E.M.
Postnatal binge ethanol exposure increases inhibition in BF neurons that is prevented by varenicline
Binge-EtOH treatment moderately increased mean sPSC frequency in BF neurons of female rats (Fig. 3B; control, 0.98 ± 0.17 Hz, 20 neurons from 5 offspring from 5 litters; EtOH, 1.89 ± 0.38 Hz, 22 neurons from 5 offspring from 5 litters; ANOVA, p = 0.03, K-S test, p < 0.0001). Varenicline prevented the EtOH-induced increase and returned sPSC frequency to near control levels (Fig. 3B; var + EtOH, 1.09 ± 0.27 Hz, 18 neurons from 4 offspring from 4 litters). Varenicline alone also increased sPSC frequency (Fig. 3B; varenicline, 2.00 ± 0.40 Hz, 16 neurons from 4 offspring from 4 litters) in BF neurons of females. There were no significant differences across treatment groups for sPSC amplitudes in females (control, 30.6 ± 3.1 pA; EtOH, 41.4 ± 6.1 pA; varenicline, 29.4 ± 2.6 pA; var + EtOH, 40.4 ± 7.5 pA; ANOVA, p = 0.26; not shown).
Approximately 45% of EtOH treated neurons (10/22) exhibited sEPSCs (Fig 3C, top) with a mean frequency of 0.16 ± 0.04 Hz (Fig. 3C, bottom) and amplitude of 14.1 ± 2.1 pA (not shown). Varenicline plus EtOH treatment decreased the percentage of neurons having sEPSCs to 33% (6/18) (Fig. 3C, top) and had a mean frequency of 0.35 ± 0.17 Hz (Fig. 3C, bottom) and an amplitude of 11.4 ± 4.0 pA (not shown). In females treated with varenicline alone, 44% of neurons (7/16) had sEPSCs (Fig 3C, top) with a mean frequency of 0.32 ± 0.12 Hz (Fig. 3C, bottom) and an amplitude of 12.0 ± 4.1 pA (not shown). There were no significant differences across treatment groups for sEPSC frequency (ANOVA, p = 0.27) or amplitude (ANOVA, p = 0.15) (Fig. 3C, bottom).
In the presence of APV + DNQX, drug treatment effects mirrored those seen when recordings were made in aCSF, but some statistical significance was lost (Fig. 4; control sIPSC frequency, 1.02 ± 0.17 Hz; EtOH, 2.30 ± 0.65 Hz; varenicline, 2.52 ± 0.47 Hz; var + EtOH, 1.40 ± 0.44 Hz; ANOVA, p = 0.08; K-S test, p < 0.0001). There also were no significant differences across treatment groups with sIPSC amplitudes (control, 39.6 ± 3.2 pA; EtOH, 36.3 ± 2.8 pA; varenicline, 35.0 ± 2.6 pA; var + EtOH, 32.6 ± 4.7 pA; ANOVA, p = 0.53, not shown). Exponential fits of sIPSC 10–90 decay kinetics did not reveal any significant treatment effects (τ1 (ms); control, 16.3 ± 1.1; EtOH, 13.2 ± 1.0; Varenicline, 14.8 ± 1.1; Var + EtOH, 15.4 ± 2.0; ANOVA, p = 0.28; τ2 (ms); control, 46.5 ± 4.9; EtOH, 34.1 ± 4.2; Varenicline, 41.8 ± 4.4; Var + EtOH, 40.0 ± 9.4; not shown). When compared to aCSF, the frequency of sIPSCs was increased in some BF neurons recorded in APV + DNQX. This increase was greater in the EtOH-treated group compared to the control group (Fig. 4D; mean % change in sIPSC frequency in APV + DNQX; control = −6.59%; EtOH = 32.34%; t-test, p = 0.02). This differential response in the presence of APV + DNQX suggest that excitatory upstream BF circuitry may be particularly sensitive to binge-EtOH.
Figure 4:
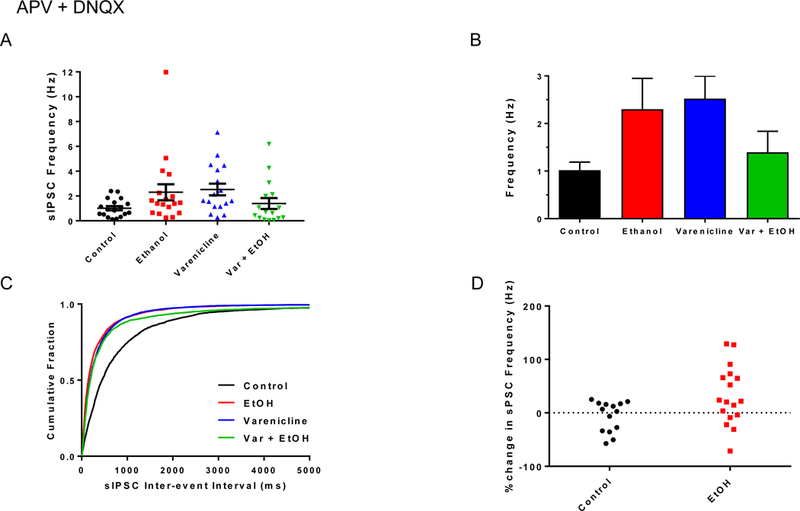
Binge-EtOH and varenicline treatments affect BF inhibitory synaptic transmission in female rats. A. Scatter plots of sIPSC frequencies from recorded BF neurons across all treatment groups. Data are mean ± S.E.M. B. Bar graph representation of data in A. C. Cumulative probability plots of sIPSC inter-event intervals from recorded BF neurons. D. Scatter plot distribution of percent change in female sIPSC frequency in the presence of APV + DNQX compared to control sPSC frequency in aCSF.
In summary, third trimester binge-EtOH increased inhibition in female BF, an effect which could be prevented by concurrent varenicline treatment. It should be noted that varenicline alone caused changes in BF synaptic transmission, which could be problematic for its use as a therapeutic. More sophisticated, circuit based analysis is needed and will be the focus of future studies.
Behavioral Characterization
In order to identify any pre-training differences in the rats’ ability to learn the position of the hidden platform in the water maze, latency to reach the platform (s) was assessed for the initial swim. No differences were found as a function of treatment (F(3,79) = 1.131, p = 0.3419). Previously, we observed a swim speed difference with binge-EtOH treated rats swimming faster than controls (Banuelos 2011). In the current study, there were no differences in swim speed due to treatment during visible cue training (F(3,79) = 4.595, p > 0.05).
Varenicline improves learning and anxiety in binge-EtOH treated females.
We did not observe a treatment difference in latencies during cue training (F(3,38) = 2.421, p = 0.08, Fig. 5A). There were also no thigmotaxis differences between the treatment groups during cue training (F(3,38) = 2.036, p = 0.13). However, as shown in Figure 5B, there was a significant effect of EtOH treatment during hidden platform training (F(3,38) = 5.069, p = 0.0047) with EtOH-exposed females spending more time looking for the platform than controls, the varenicline alone group, and the var + EtOH (Tukey’s HSD; p < 0.05). Although all treatment groups reduced latency to the platform as training progressed, the EtOH group never achieved the performance of controls. Increased thigmotaxis is often considered to represent an increase in anxiety and EtOH has been reported to increase this behavior (Banuelos 2011). There was a significant difference in thigmotaxis between treatment groups during hidden platform training (Fig. 5C; F(3,38) = 3.364, p = 0.03) with EtOH-exposed females showing more thigmotaxic behavior throughout testing. When we compared percent thigmotaxis time on the last day of hidden platform (day 6), EtOH treated and control groups were still different (t(21) = −2.506, p = 0.021), demonstrating the persistent detrimental effect of EtOH on anxiety. These effects of EtOH were ameliorated by co-treatment with varenicline (varenicline + EtOH vs. EtOH (t(19) = 2.907, p = 0.009). EtOH treatment did not impair performance in the initial or interpolated probe trials for search error (F(3,38) = 0.733, p = 0.54) or for percent time in quadrant (F(3,38) = 0.657, p = 0.58).
Figure 5:
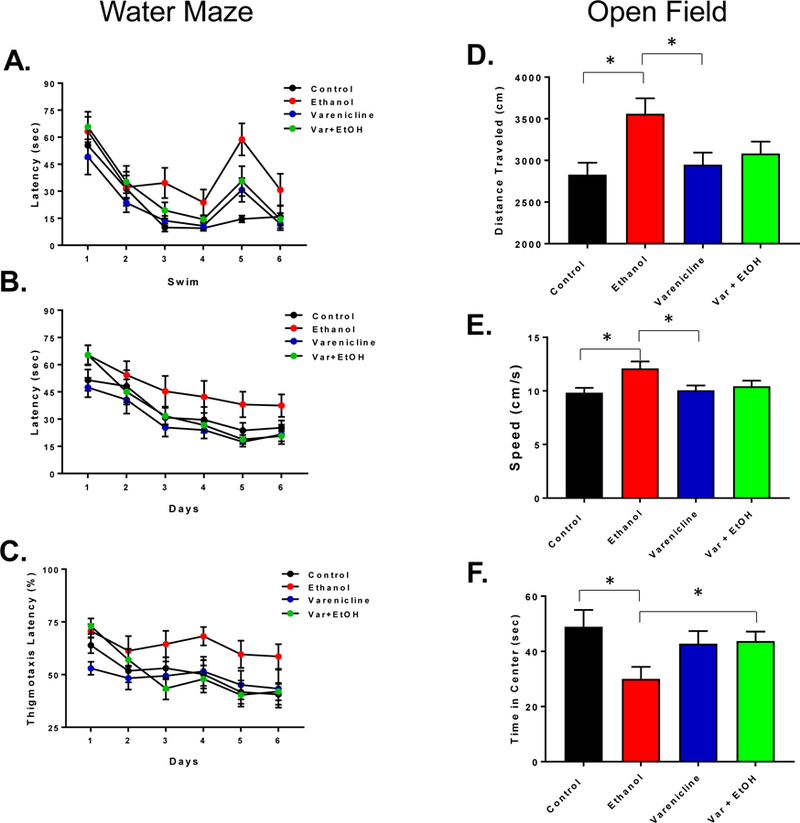
Spatial reference memory performance on Morris water maze and behavior in the open field. Latency (s) of control mice (black), binge-ethanol (red), varenicline (blue) and varenicline-ethanol treated (green) during cue trials in the water maze (A), latency (s) during training trials over 6 days (B) and thigmotaxis (% time spent in the outer 10% of the tank during training) (C). In (D), total distance travelled (cm) in the open field, average speed (cm/s) in (E) and average time spent in the center during 5 minutes in the open field (F). Data are mean ± S.E.M. Significance in ANOVA (p < 0.05) is demonstrated by *.
In the open field, EtOH treated rats showed increased locomotor activity compared to controls (Fig. 5D, t(21) = −3.129, p = 0.005) and the varenicline alone group (t(19) = 2.549, p = 0.02), but varenicline co-treatment did not attenuate this effect (EtOH + Var: t(22) = 1.783, p = 0.05). When we assessed the speed of the animals, EtOH-treated rats were faster than controls (Fig. 5E, t(21) = −2.920, p = 0.0082), and the varenicline-treated rats (t(19) = 2.557, p = 0.019), and close to significance when compared to the varenicline plus EtOH group (t(20) = 2.004, p = 0.05).
Similar to the water maze, EtOH treatment resulted in increased thigmotaxic behavior, with rats spending less total time in the center of the box when compared to controls (Fig. 5F; t(21) = 2.480, p = 0.02) and trending when compared to varenicline alone (t(19) = −2.023, p = 0.057). Varenicline did seem to attenuate this effect with rats treated with EtOH showing increased time in the center of the box (t(19) = −2.023, p = 0.057).
DISCUSSION
EtOH exposure during brain development can result in cognitive impairment in humans (Larsson, Bohlin et al. 1985, Brown, Coles et al. 1991, Coles, Brown et al. 1991, Mattson and Riley 1998). Rodent studies have been useful for modeling the symptoms of FASD both molecularly and behaviorally (Becker, Diaz-Granados et al. 1996, Diaz, Jotty et al. 2014, Atalar, Uzbay et al. 2016). Deficits in learning, inhibition, attention and motor activity have been reported in both rodents and children (Driscoll, Streissguth et al. 1990). Furthermore, the severity and type of teratogenic effects appear to be sex dependent. The literature on human sex differences in FASD is limited, but sex specific effects of perinatal EtOH exposure have been observed in a variety of behaviors in rodents. Prenatal EtOH exposure has been shown to affect females more severely in many cognitive and metabolic domains. For example, females prenatally exposed to EtOH are more susceptible to high-fat metabolic syndrome as adults (He, Li et al. 2015). Exposure to EtOH both prenatally and postnatally also results in sex specific social memory deficits in rats. In a study by Kelly et al., exposed female rats showed a deficit in memory encoding in a social recognition task along with lowered oxytocin receptor binding in the amygdala, while males showed a decrease in social memory retention (Kelly, Leggett et al. 2009). For these reasons, we chose to limit the scope of the study to the effects on female rats. Four treatment groups and two sexes would have reduced the statistical power of each litter. Nevertheless, a careful examination of mixed sex litters will be the focus of future studies.
Neuronal nicotinic acetylcholine receptors (nAChRs) have become a target for cholinergic / cholinomimetic therapeutics as a potential treatment for a variety of conditions including addiction, Alzheimer’s and Parkinson’s diseases, pain, schizophrenia, anxiety and depression (Newhouse, Singh et al. 2004, Jensen, Frolund et al. 2005, Taly, Corringer et al. 2009). Such therapeutics have shown promise in ameliorating the effects of perinatal EtOH exposure on the developing rodent brain. For example, choline supplementation significantly reduces many of EtOH’s effects on development in rats, such as brain weight and behavioral measures like surface righting reflex (Thomas, Abou et al. 2009), spatial learning deficits in females (Thomas, Biane et al. 2007), and working memory (Schneider and Thomas 2016). It also attenuates alterations in hippocampal microRNA after postnatal EtOH exposure (Balaraman, Idrus et al. 2017). Varenicline, an agonist at nAChRs and FDA approved for smoking cessation, has been shown to decrease EtOH consumption in humans and in animal models of addiction (McKee, Harrison et al. 2009, Rollema, Hajos et al. 2009), enhance mood (Mineur, Eibl et al. 2009), and reduce acute EtOH-induced learning deficits in adult mice (Gulick and Gould 2008). In order to test the potential of cholinergic therapeutics in EtOH exposure, we treated female neonatal (PD 4–9) rat pups with third trimester equivalent, binge-EtOH and assessed the effectiveness of the cholinomimetic, varenicline, in preventing the effects of EtOH on BF synaptic transmission and behavior.
Binge-EtOH and BF synaptic transmission
Similar to sex hormones, the impact of perinatal EtOH exposure on the developing brain also depends on the stage of development in which the exposure occurs. This interplay of gonadal hormones, EtOH, and critical exposure windows, very likely accounts for the variety of sex specific cognitive impairments related to third trimester EtOH exposure. In the hippocampus, for example, changes in synaptic plasticity induced by perinatal EtOH exposure that could result in learning and memory deficiencies may involve alterations in estradiol levels (He 2012, Sickmann, Patten et al. 2014, Fontaine, Patten et al. 2016). Like the hippocampus, the BF is sensitive to the influence of perinatal EtOH (Mitchell, Paiva et al. 2000, DuBois, Parrish et al. 2004, Hsiao, DuBois et al. 2004, Wang, Dubois et al. 2013). In addition, cholinergic neurons of the BF are also sensitive to the influence of periadolescent EtOH exposure (Ehlers, Criado et al. 2011, Fernandez and Savage 2017). The BF provides the main cholinergicinput to several brain regions such as the neocortex, hippocampal formation (Dutar, Bassant et al. 1995, Gielow and Zaborszky 2017) and amygdala (Unal, Pare et al. 2015, Gielow and Zaborszky 2017). While septal neurogenesis is complete by embryonic day (ED) 20, projections to the hippocampus increase through PD 10 (Bayer 1979, Leranth and Frotscher 1989, Linke and Frotscher 1993). Thus, synaptogenesis of BF circuitry is occurring during the PD 4–9 critical postnatal window when our animals were exposed to binge-EtOH. This synaptogenesis gives rise to a complex network that includes a combination of cholinergic and GABAergic projecting neurons as well as local GABAergic and glutamatergic interneurons (Colom, Castaneda et al. 2005, Manseau, Danik et al. 2005) along with peptidergic neurons and fibers (Zaborszky and Duque 2000).
Cholinergic BF neurons are surrounded by large numbers of GABAergic varicosities (Zaborszky, Heimer et al. 1986). The predominance of inhibitory GABAergic currents (~90%) that we recorded in BF slices is consistent with this anatomy. The GABA neurotransmitter system is a primary target of EtOH in the adult and during development (Valenzuela, Puglia et al. 2011). Because GABA signaling plays a vital trophic role as neurons differentiate, develop neurites, and form synapses (Ben-Ari 2001, Owens and Kriegstein 2002), even transient distortions of GABAergic signaling by EtOH may disrupt maturation of septal synapses and important neurocircuits. We found that binge-EtOH (PD 4–9) enhanced spontaneous synaptic transmission on to BF neurons, and because GABAergic tone dominates in this region, much of EtOH’s effect appears to be focused on altering inhibitory inputs. However, our data also suggest that there may be some impact of EtOH exposure on excitatory transmission in the BF, perhaps presynaptic to local GABAergic interneurons, as there were some differences in results obtained in APV and DNQX compared to results in aCSF.
In a recent report, rabies virus techniques were used to map BF cholinergic circuits (Gielow and Zaborszky 2017). They discovered that many of the afferent inputs to BF cholinergic neurons derive from a large component of putatively GABAergic inputs from the striatum with a smaller component from the lateral septum. How exactly these connections are formed during development and how postnatal EtOH alters them is not well understood. Nevertheless, in adults exposed to repeated bouts of EtOH exposure and withdrawal, the overall impact on to the projecting, GABAergic medium spiny neurons of the striatum is a net increase in glutamatergic synaptic transmission and decreased inhibition (Lovinger and Alvarez 2017), a mechanism that could elevate GABAergic transmission into the BF in EtOH treated animals. Our model of third trimester, binge-EtOH directly affects circuitry that develops late in infancy. Other impacted circuits are prefrontal cortex (Louth, Bignell et al. 2016) and the basolateral amygdala (BLA) (Baculis, Diaz et al. 2015). The changes we found in synaptic transmission likely result from the effect of EtOH on network activity. Interneuronopathies due to EtOH damage have been described previously (Skorput, Gupta et al. 2015), and dysfunction of GABAergic neurons can lead to altered excitatory / inhibitory (E/I) balance in important neurocircuits that may underlie neurobehavioral deficits seen in FASD (Sadrian, Wilson et al. 2013). Altered E/I balance on to BF cholinergic neurons, could alter the efflux of acetylcholine (Moore, Sarter et al. 1995) to brain regions important for FASD behaviors like the prefrontal cortex and amygdala (Moore, Sarter et al. 1995). Recently, using optogenetic techniques and patch-clamp recordings, Unal et. al. described the effects of cholinergic inputs to BLA neurons (Unal, Pare et al. 2015). It is possible that altered E/I control of BF cholinergic neurons could have detrimental downstream consequences in amygdala circuits and related behaviors. Because of the limited scope of the study, the number of treatment groups, and the occasional inability to identify neurons, further circuit specific / cell specific studies are needed to examine the impact on BF cholinergic circuitry. These studies will be the focus of future experiments.
FASD, spatial learning and memory, and synaptic physiology
As reported previously in Banuelos et al. also using the water maze task (Banuelos 2011), EtOH-treated females took longer to find the hidden platform than controls. While the EtOH exposed females learned the task, their performance was impaired relative to that of the other test groups. Thigmotaxic behavior was significantly greater in the binge-EtOH females also. As the behavior of the female test groups was not different in probe trials, our results suggest that binge-EtOH exposure impacts learning processes more than spatial memory. Hyperactivity, attention, learning and sensory processing deficits are observed in humans exposed to EtOH during development (Malisza, Buss et al. 2012, Mattson, Roesch et al. 2013, Gautam, Nuñez et al. 2015, O’Conaill, Malisza et al. 2015, Tesche, Kodituwakku et al. 2015, Kingdon, Cardoso et al. 2016). It is interesting that varenicline / binge-EtOH co-treated females’ behavioral performance was not different from controls. However, it is unclear whether the effects of varenicline on behavior are directed at learning processes per se or at attentional processes as supported by the results on BF synaptic transmission. Likewise, thigmotaxic behavior has been used to identify anxiety-like behavior in rodents (Banuelos 2011). However, without other anxiety behavioral assessments, we cannot exclude that the thigmotaxic behavior in binge-EtOH treated females is due to impaired learning or attention, which also could result in more time in the outer edge of the tank. The open field data suggests that hyperactivity may be affecting performance of binge-EtOH treated rats, as observed by higher distances travelled by the EtOH treated rats. Surprisingly, treatment with varenicline did seem to ameliorate anxiety some, as observed in more time spent in the center of the open field by the varenicline plus EtOH treated rats. These results suggest that the effects of binge-EtOH on female rats can be ameliorated by treatment with varenicline.
Although we cannot directly correlate water maze behavior and BF physiology in this study because data were collected from separate cohorts, it is possible that there are sex specific differences in the development of BF synaptic transmission that underlies differences in water maze performance. The organizational-activational hypothesis of hormone action on the developing brain establishes that during critical perinatal periods, the organization of neuronal architecture (i.e. the masculinization of specific brain regions) is under the influence of gonadal steroid hormones (Arnold 2009). In rodents, the critical perinatal testosterone surge occurs late in gestation (~embryonic day 18) and declines shortly after birth (Weisz and Ward 1980). This surge in hormonal activity is responsible for many of the sex differences that have been observed in neuronal developmental processes such as cell survival, morphology, and synaptogenesis. Using the preoptic area as a case study, Lenz et al. described how gonadal hormones establish region specific sex differences in neuronal development that are responsible for sex specific behavior (Lenz, Nugent et al. 2012). Further, Kight and McCarthy describe how sex differences in the developing brain also impact epileptic behaviors (Kight and McCarthy 2014). Related to our work, an important sex difference exists in the critical developmental period when GABAARs shift from depolarizing to hyperpolarizing. In multiple brain regions, this shift occurs later in development in males (Kyrozis, Chudomel et al. 2006, Nunez and McCarthy 2007). It is likely that third trimester binge-EtOH exposure alters these developmental processes resulting in persistent synaptic changes in BF that may contribute to sex-specific behavioral differences. Ongoing studies in our lab using male subjects may shed light on these important sex differences.
Cholinergic therapy and varenicline’s impact on BF circuitry and behavior
In addition to its various roles in the adult brain, spontaneous nicotinic excitation is widespread in early CNS development. This early, endogenous nicotinic signaling plays an important role in guiding neuronal development specifically the development and maturation of GABAergic neurons (Liu, Neff et al. 2006, Zago, Massey et al. 2006, Campbell, Fernandes et al. 2010). Zago and colleagues found that α7 nAChRs appeared to co-localize with GABAARs in early hippocampal development (Zago, Massey et al. 2006), and Zhang and Berg also found that nicotine acting through α7 nAChRs directly regulated GABAAR-mediated currents (Zhang and Berg 2007). More profoundly, nAChRs demonstrated an important role in determining the timing of the GABAergic signaling conversion from depolarizing to hyperpolarizing (Liu, Neff et al. 2006) likely by changing the Cl− transporter levels. Recently, nAChRs have also been shown to promote the maturation and integration of adult-born neurons in the hippocampus (Campbell, Fernandes et al. 2010). In that study, α7 nAChR knockout mice displayed GABAergic postsynaptic currents with immature kinetics and prolonged periods of GABA depolarization characteristic of immature GABAergic neurons (Campbell, Fernandes et al. 2010). Varenicline is a nAChR agonist. It is a low affinity full agonist at α7 and a high affinity partial agonist at α4β2* nAChRs. In a recent publication by Miranda et. al., they demonstrated that nicotine and varenicline ameliorated the effects of EtOH-induced depression of microRNA expression levels in a 2nd trimester in vitro neurosphere model of neurogenesis (Tsai, Bake et al. 2014). Early, endogenous nicotinic signaling could provide an avenue for varenicline to intervene in EtOH’s actions by promoting the normal development and maturation of GABAergic neurons especially in brain regions important for learning and memory. In fact, varenicline has recently been shown to target epigenetic mechanisms in cortical GABAergic neurons increasing GAD67 mRNA and protein and decreasing cortical DNA methyltransferase 1 mRNA (Maloku, Kadriu et al. 2011).
Our results show the potential for varenicline and other cholinergic therapies to intervene perinatally in suspected cases of FASD. Such therapeutics could prevent or mitigate physiological or behavioral deficits associated with fetal EtOH exposure, perhaps in a sex specific manner. Continued drug innovations targeting developmental cholinergic mechanisms could offer substantial relief to the challenges surrounding FASD without the non-specific side effects associated with varenicline.
Highlights.
Third trimester equivalent, binge-EtOH exposure increased spontaneous post-synaptic current (sPSC) frequency in basal forebrain (BF) neurons from female Sprague-Dawley rats.
Varenicline exposure also enhanced sPSC frequency in BF neurons.
Varenicline plus binge-EtOH co-treatment prevented the increase in sPSC frequency.
Binge-EtOH treated females displayed increased anxiety (thigmotaxis), and demonstrated learning deficits in the water maze. Varenicline / EtOH co-treatment was effective at reducing these behavioral deficits.
In the open field, EtOH treated rats displayed longer distances traveled and spent less time in the center of the open field box. Co-treated rats displayed less anxiety demonstrating a possible effect of varenicline on this measure.
Binge-EtOH induced changes in both BF synaptic transmission and water maze performance were reduced by varenicline in female rats, supporting a role for cholinergic therapeutics in FASD treatment.
ACKNOWLEDGEMENT
We thank Dr. William H. Griffith for his comments and consultation, and Dr. Jennifer Bizon and Ryan J. Gilbert for technical expertise related to the water maze experiments.
Grant Sponsors: NIH-NIAAA/ORWH 1R56AA021844-01A1
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- (2010). “Fetal Alcohol Spectrum Disorders Fact Sheet” From https://report.nih.gov/nihfactsheets/Pdfs/FetalAlcoholSpectrumDisorders(NIAAA).pdf.
- (2014). “National Organization on Fetal Alcohol Syndrome Fact Sheet” From https://www.nofas.org/factsheets/.
- Arendt T (1994). “Impairment in memory function and neurodegenerative changes in the cholinergic basal forebrain system induced by chronic intake of ethanol.” J Neural Transm Suppl 44: 173–187. [DOI] [PubMed] [Google Scholar]
- Arendt T, Allen Y, Marchbanks RM, Schugens MM, Sinden J, Lantos PL and Gray JA (1989). “Cholinergic system and memory in the rat: effects of chronic ethanol, embryonic basal forebrain brain transplants and excitotoxic lesions of cholinergic basal forebrain projection system.” Neuroscience 33(3): 435–462. [DOI] [PubMed] [Google Scholar]
- Arnold AP (2009). “The organizational-activational hypothesis as the foundation for a unified theory of sexual differentiation of all mammalian tissues.” Horm Behav 55(5): 570–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atalar EG, Uzbay T and Karakas S (2016). “Modeling Symptoms of Attention-Deficit Hyperactivity Disorder in a Rat Model of Fetal Alcohol Syndrome.” Alcohol Alcohol 51(6): 684–690. [DOI] [PubMed] [Google Scholar]
- Baculis BC, Diaz MR and Valenzuela CF (2015). “Third trimester-equivalent ethanol exposure increases anxiety-like behavior and glutamatergic transmission in the basolateral amygdala.” Pharmacol Biochem Behav 137: 78–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balaraman S, Idrus NM, Miranda RC and Thomas JD (2017). “Postnatal choline supplementation selectively attenuates hippocampal microRNA alterations associated with developmental alcohol exposure.” Alcohol 60: 159–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balaraman S, Winzer-Serhan UH and Miranda RC (2012). “Opposing actions of ethanol and nicotine on microRNAs are mediated by nicotinic acetylcholine receptors in fetal cerebral cortical-derived neural progenitor cells.” Alcohol Clin Exp Res 36(10): 1669–1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banuelos C, Gilbert RJ, Montgomery KS, Fincher AS, Wang H, Frye GD, Setlow B, Bizon JL (2011). “Altered spatial learning and delay discounting in a rat model of human third trimester binge ethanol exposure.” Behav Pharm [DOI] [PMC free article] [PubMed]
- Baraona E, Abittan CS, Dohmen K, Moretti M, Pozzato G, Chayes ZW, Schaefer C and Lieber CS (2001). “Gender differences in pharmacokinetics of alcohol.” Alcohol Clin Exp Res 25(4): 502–507. [PubMed] [Google Scholar]
- Bayer SA (1979). “The development of the septal region in the rat. I. Neurogenesis examined with 3H-thymidine autoradiography.” J Comp Neurol 183(1): 89–106. [DOI] [PubMed] [Google Scholar]
- Becker HC (1996). Effects of ethanol on the central nervous system: Fetal damage - neurobehavioral effects Boca Raton, CRC Press. [Google Scholar]
- Becker HC, Diaz-Granados JL and Randall CL (1996). “Teratogenic actions of ethanol in the mouse: a minireview.” Pharmacol Biochem Behav 55(4): 501–513. [DOI] [PubMed] [Google Scholar]
- Ben-Ari Y (2001). “Developing networks play a similar melody.” Trends Neurosci 24(6): 353–360. [DOI] [PubMed] [Google Scholar]
- Berman RF, Hannigan JH, Sperry MA and Zajac CS (1996). “Prenatal alcohol exposure and the effects of environmental enrichment on hippocampal dendritic spine density.” Alcohol 13(2): 209–216. [DOI] [PubMed] [Google Scholar]
- Brown RT, Coles CD, Smith IE, Platzman KA, Silverstein J, Erickson S and Falek A (1991). “Effects of prenatal alcohol exposure at school age. II. Attention and behavior.” Neurotoxicol Teratol 13(4): 369–376. [DOI] [PubMed] [Google Scholar]
- Campbell NR, Fernandes CC, Halff AW and Berg DK (2010). “Endogenous signaling through alpha7-containing nicotinic receptors promotes maturation and integration of adult-born neurons in the hippocampus.” J Neurosci 30(26): 8734–8744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceylan-Isik AF, McBride SM and Ren J (2010). “Sex difference in alcoholism: who is at a greater risk for development of alcoholic complication?” Life Sci 87(5– 6): 133–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman LG Jr., Oguz I, Lee J, Styner M and Crews FT (2012). “Postnatal day 7 ethanol treatment causes persistent reductions in adult mouse brain volume and cortical neurons with sex specific effects on neurogenesis.” Alcohol 46(6): 603–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coles CD, Brown RT, Smith IE, Platzman KA, Erickson S and Falek A (1991). “Effects of prenatal alcohol exposure at school age. I. Physical and cognitive development.” Neurotoxicol Teratol 13(4): 357–367. [DOI] [PubMed] [Google Scholar]
- Colom LV, Castaneda MT, Reyna T, Hernandez S and Garrido-Sanabria E (2005). “Characterization of medial septal glutamatergic neurons and their projection to the hippocampus.” Synapse 58(3): 151–164. [DOI] [PubMed] [Google Scholar]
- Costa ET, Savage DD and Valenzuela CF (2000). “A review of the effects of prenatal or early postnatal ethanol exposure on brain ligand-gated ion channels.” Alcohol Clin Exp Res 24(5): 706–715. [PubMed] [Google Scholar]
- Diaz MR, Jotty K, Locke JL, Jones SR and Valenzuela CF (2014). “Moderate Alcohol Exposure during the Rat Equivalent to the Third Trimester of Human Pregnancy Alters Regulation of GABAA Receptor-Mediated Synaptic Transmission by Dopamine in the Basolateral Amygdala.” Front Pediatr 2: 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driscoll CD, Streissguth AP and Riley EP (1990). “Prenatal alcohol exposure: comparability of effects in humans and animal models.” Neurotoxicol Teratol 12(3): 231–237. [DOI] [PubMed] [Google Scholar]
- DuBois DW, Damborsky JC, Fincher AS, Frye GD and Winzer-Serhan UH (2013). “Varenicline and nicotine enhance GABAergic synaptic transmission in rat CA1 hippocampal and medial septum/diagonal band neurons.” Life Sci 92(6–7): 337–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DuBois DW, Parrish AR, Trzeciakowski JP and Frye GD (2004). “Binge ethanol exposure delays development of GABAergic miniature postsynaptic currents in septal neurons.” Brain Res Dev Brain Res 152(2): 199–212. [DOI] [PubMed] [Google Scholar]
- Dutar P, Bassant MH, Senut MC and Lamour Y (1995). “The septohippocampal pathway: structure and function of a central cholinergic system.” Physiol Rev 75(2): 393–427. [DOI] [PubMed] [Google Scholar]
- Ehlers CL, Criado JR, Wills DN, Liu W and Crews FT (2011). “Periadolescent ethanol exposure reduces adult forebrain ChAT+IR neurons: correlation with behavioral pathology.” Neuroscience 199: 333–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez GM and Savage LM (2017). “Adolescent binge ethanol exposure alters specific forebrain cholinergic cell populations and leads to selective functional deficits in the prefrontal cortex.” Neuroscience 361: 129–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floyd EA, Young-Seigler AC, Ford BD, Reasor JD, Moore EL, Townsel JG and Rucker HK (1997). “Chronic ethanol ingestion produces cholinergic hypofunction in rat brain.” Alcohol 14(1): 93–98. [DOI] [PubMed] [Google Scholar]
- Fontaine CJ, Patten AR, Sickmann HM, Helfer JL and Christie BR (2016). “Effects of pre-natal alcohol exposure on hippocampal synaptic plasticity: Sex, age and methodological considerations.” Neuroscience & Biobehavioral Reviews 64: 12–34. [DOI] [PubMed] [Google Scholar]
- Frezza M, di Padova C, Pozzato G, Terpin M, Baraona E and Lieber CS (1990). “High blood alcohol levels in women. The role of decreased gastric alcohol dehydrogenase activity and first-pass metabolism.” N Engl J Med 322(2): 95–99. [DOI] [PubMed] [Google Scholar]
- Gautam P, Nuñez SC, Narr KL, Mattson SN, May PA, Adnams CM, Riley EP, Jones KL, Kan EC and Sowell ER (2015). “Developmental Trajectories for Visuo-Spatial Attention are Altered by Prenatal Alcohol Exposure: A Longitudinal FMRI Study.” Cerebral Cortex (New York, NY) 25(12): 4761–4771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gielow MR and Zaborszky L (2017). “The Input-Output Relationship of the Cholinergic Basal Forebrain.” Cell Rep 18(7): 1817–1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant KA, Choi EY and Samson HH (1983). “Neonatal ethanol exposure: effects on adult behavior and brain growth parameters.” Pharmacol Biochem Behav 18 Suppl 1: 331–336. [DOI] [PubMed] [Google Scholar]
- Graziani M, Nencini P and Nistico R (2014). “Genders and the concurrent use of cocaine and alcohol: Pharmacological aspects.” Pharmacol Res 87: 60–70. [DOI] [PubMed] [Google Scholar]
- Griffith WH, Dubois DW, Fincher A, Peebles KA, Bizon JL and Murchison D (2014). “Characterization of age-related changes in synaptic transmission onto F344 rat basal forebrain cholinergic neurons using a reduced synaptic preparation.” J Neurophysiol 111(2): 273–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulick D and Gould TJ (2008). “Varenicline ameliorates ethanol-induced deficits in learning in C57BL/6 mice.” Neurobiol Learn Mem 90(1): 230–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannigan JH and Berman RF (2000). “Amelioration of fetal alcohol-related neurodevelopmental disorders in rats: exploring pharmacological and environmental treatments.” Neurotoxicol Teratol 22(1): 103–111. [DOI] [PubMed] [Google Scholar]
- He F-Q, Zhang J, Guo X, 2012 (2012). “Prenatal Ethanol Exposure IncreasesDepressive-like behavior and central estrogen receptor and oxytocinexpressions in adult female mandarin voles.” Zool Stu 51(1): 1–11. [Google Scholar]
- He Z, Li J, Luo H, Zhang L, Ma L, Chen L and Wang H (2015). “Sex-specific increase in susceptibility to metabolic syndrome in adult offspring after prenatal ethanol exposure with post-weaning high-fat diet.” Scientific Reports 5: 17679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heaton MB, Moore DB, Paiva M, Madorsky I, Mayer J and Shaw G (2003). “The role of neurotrophic factors, apoptosis-related proteins, and endogenous antioxidants in the differential temporal vulnerability of neonatal cerebellum to ethanol.” Alcohol Clin Exp Res 27(4): 657–669. [DOI] [PubMed] [Google Scholar]
- Helfer JL, White ER and Christie BR (2012). “Enhanced deficits in long-term potentiation in the adult dentate gyrus with 2nd trimester ethanol consumption.” PLoS One 7(12): e51344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellemans KG, Verma P, Yoon E, Yu WK, Young AH and Weinberg J (2010). “Prenatal alcohol exposure and chronic mild stress differentially alter depressive- and anxiety-like behaviors in male and female offspring.” Alcohol Clin Exp Res 34(4): 633–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeppner BB, Paskausky AL, Jackson KM and Barnett NP (2013). “Sex differences in college student adherence to NIAAA drinking guidelines.” Alcohol Clin Exp Res 37(10): 1779–1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hommer D, Momenan R, Kaiser E and Rawlings R (2001). “Evidence for a gender-related effect of alcoholism on brain volumes.” Am J Psychiatry 158(2): 198–204. [DOI] [PubMed] [Google Scholar]
- Hsiao SH, Acevedo JL, DuBois DW, Smith KR, West JR and Frye GD (2001). “Early postnatal ethanol intubation blunts GABA(A) receptor up-regulation and modifies 3alpha-hydroxy-5alpha-pregnan-20-one sensitivity in rat MS/DB neurons.” Brain Res Dev Brain Res 130(1): 25–40. [DOI] [PubMed] [Google Scholar]
- Hsiao SH, DuBois DW, Miranda RC and Frye GD (2004). “Critically timed ethanol exposure reduces GABAAR function on septal neurons developing in vivo but not in vitro.” Brain Res 1008(1): 69–80. [DOI] [PubMed] [Google Scholar]
- Idrus NM and Thomas JD (2011). “Fetal alcohol spectrum disorders: experimental treatments and strategies for intervention.” Alcohol Res Health 34(1): 76–85. [PMC free article] [PubMed] [Google Scholar]
- Jensen AA, Frolund B, Liljefors T and Krogsgaard-Larsen P (2005). “Neuronal nicotinic acetylcholine receptors: structural revelations, target identifications, and therapeutic inspirations.” J Med Chem 48(15): 4705–4745. [DOI] [PubMed] [Google Scholar]
- Kelly SJ, Goodlett CR, Hulsether SA and West JR (1988). “Impaired spatial navigation in adult female but not adult male rats exposed to alcohol during the brain growth spurt.” Behav Brain Res 27(3): 247–257. [DOI] [PubMed] [Google Scholar]
- Kelly SJ, Leggett DC and Cronise K (2009). “Sexually Dimorphic Effects of Alcohol Exposure during Development on the Processing of Social Cues.” Alcohol and Alcoholism 44(6): 555–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kight KE and McCarthy MM (2014). “Using sex differences in the developing brain to identify nodes of influence for seizure susceptibility and epileptogenesis.” Neurobiol Dis 72 Pt B: 136–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingdon D, Cardoso C and McGrath JJ (2016). “Research Review: Executive function deficits in fetal alcohol spectrum disorders and attention-deficit/hyperactivity disorder – a meta-analysis.” Journal of Child Psychology and Psychiatry 57(2): 116–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyrozis A, Chudomel O, Moshe SL and Galanopoulou AS (2006). “Sex-dependent maturation of GABAA receptor-mediated synaptic events in rat substantia nigra reticulata.” Neurosci Lett 398(1–2): 1–5. [DOI] [PubMed] [Google Scholar]
- Larsson G, Bohlin AB and Tunell R (1985). “Prospective study of children exposed to variable amounts of alcohol in utero.” Arch Dis Child 60(4): 316–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenz KM, Nugent BM and McCarthy MM (2012). “Sexual differentiation of the rodent brain: dogma and beyond.” Front Neurosci 6: 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leranth C and Frotscher M (1989). “Organization of the septal region in the rat brain: cholinergic-GABAergic interconnections and the termination of hippocampo-septal fibers.” J Comp Neurol 289(2): 304–314. [DOI] [PubMed] [Google Scholar]
- Lindsley TA, Comstock LL and Rising LJ (2002). “Morphologic and neurotoxic effects of ethanol vary with timing of exposure in vitro.” Alcohol 28(3): 197–203. [DOI] [PubMed] [Google Scholar]
- Linke R and Frotscher M (1993). “Development of the rat septohippocampal projection: tracing with DiI and electron microscopy of identified growth cones.” J Comp Neurol 332(1): 69–88. [DOI] [PubMed] [Google Scholar]
- Liu Z, Neff RA and Berg DK (2006). “Sequential interplay of nicotinic and GABAergic signaling guides neuronal development.” Science 314(5805): 1610–1613. [DOI] [PubMed] [Google Scholar]
- Louth EL, Bignell W, Taylor CL and Bailey CD (2016). “Developmental Ethanol Exposure Leads to Long-Term Deficits in Attention and Its Underlying Prefrontal Circuitry.” eNeuro 3(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovinger DM and Alvarez VA (2017). “Alcohol and basal ganglia circuitry: Animal models.” Neuropharmacology [DOI] [PMC free article] [PubMed]
- Luo J and Miller MW (1998). “Growth factor-mediated neural proliferation: target of ethanol toxicity.” Brain Res Brain Res Rev 27(2): 157–167. [DOI] [PubMed] [Google Scholar]
- Ma W, Pancrazio JJ, Andreadis JD, Shaffer KM, Stenger DA, Li BS, Zhang L, Barker JL and Maric D (2001). “Ethanol blocks cytosolic Ca2+ responses triggered by activation of GABA(A) receptor/Cl-channels in cultured proliferating rat neuroepithelial cells.” Neuroscience 104(3): 913–922. [DOI] [PubMed] [Google Scholar]
- Maier SE and West JR (2001). “Drinking patterns and alcohol-related birth defects.” Alcohol Res Health 25(3): 168–174. [PMC free article] [PubMed] [Google Scholar]
- Malisza KL, Buss JL, Bolster RB, de Gervai PD, Woods-Frohlich L, Summers R, Clancy CA, Chudley AE and Longstaffe S (2012). “Comparison of spatial working memory in children with prenatal alcohol exposure and those diagnosed with ADHD; A functional magnetic resonance imaging study.” Journal of Neurodevelopmental Disorders 4(1): 12–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maloku E, Kadriu B, Zhubi A, Dong E, Pibiri F, Satta R and Guidotti A (2011). “Selective alpha4beta2 nicotinic acetylcholine receptor agonists target epigenetic mechanisms in cortical GABAergic neurons.” Neuropsychopharmacology 36(7): 1366–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancinelli R, Vitali M and Ceccanti M (2009). “Women, alcohol and the environment: an update and perspectives in neuroscience.” Funct Neurol 24(2): 77–81. [PubMed] [Google Scholar]
- Manseau F, Danik M and Williams S (2005). “A functional glutamatergic neurone network in the medial septum and diagonal band area.” J Physiol 566(Pt 3): 865–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez SE and Egea G (2007). “Novel molecular targets for the prevention of fetal alcohol syndrome.” Recent Pat CNS Drug Discov 2(1): 23–35. [DOI] [PubMed] [Google Scholar]
- Mattson SN and Riley EP (1998). “A review of the neurobehavioral deficits in children with fetal alcohol syndrome or prenatal exposure to alcohol.” Alcohol Clin Exp Res 22(2): 279–294. [DOI] [PubMed] [Google Scholar]
- Mattson SN, Roesch SC, Glass L, Deweese BN, Coles CD, Kable JA, May PA, Kalberg WO, Sowell ER, Adnams CM, Jones KL, Riley EP and Cifasd (2013). “Further Development of a Neurobehavioral Profile of Fetal Alcohol Spectrum Disorders.” Alcoholism: Clinical and Experimental Research 37(3): 517–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May PA and Gossage JP (2011). “Maternal risk factors for fetal alcohol spectrum disorders: not as simple as it might seem.” Alcohol Res Health 34(1): 15–26. [PMC free article] [PubMed] [Google Scholar]
- May PA, Gossage JP, Kalberg WO, Robinson LK, Buckley D, Manning M and Hoyme HE (2009). “Prevalence and epidemiologic characteristics of FASD from various research methods with an emphasis on recent in-school studies.” Dev Disabil Res Rev 15(3): 176–192. [DOI] [PubMed] [Google Scholar]
- McKee SA, Harrison EL, O’Malley SS, Krishnan-Sarin S, Shi J, Tetrault JM, Picciotto MR, Petrakis IL, Estevez N and Balchunas E (2009). “Varenicline reduces alcohol self-administration in heavy-drinking smokers.” Biol Psychiatry 66(2): 185–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez IA, Montgomery KS, LaSarge CL, Simon NW, Bizon JL and Setlow B (2008). “Long-term effects of prior cocaine exposure on Morris water maze performance.” Neurobiol Learn Mem 89(2): 185–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mineur YS, Eibl C, Young G, Kochevar C, Papke RL, Gundisch D and Picciotto MR (2009). “Cytisine-based nicotinic partial agonists as novel antidepressant compounds.” J Pharmacol Exp Ther 329(1): 377–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell JJ, Paiva M and Heaton MB (2000). “Effect of neonatal ethanol exposure on parvalbumin-expressing GABAergic neurons of the rat medial septum and cingulate cortex.” Alcohol 21(1): 49–57. [DOI] [PubMed] [Google Scholar]
- Moore H, Sarter M and Bruno JP (1995). “Bidirectional modulation of cortical acetylcholine efflux by infusion of benzodiazepine receptor ligands into the basal forebrain.” Neurosci Lett 189(1): 31–34. [DOI] [PubMed] [Google Scholar]
- Newhouse P, Singh A and Potter A (2004). “Nicotine and nicotinic receptor involvement in neuropsychiatric disorders.” Curr Top Med Chem 4(3): 267–282. [DOI] [PubMed] [Google Scholar]
- Nunez JL and McCarthy MM (2007). “Evidence for an extended duration of GABA-mediated excitation in the developing male versus female hippocampus.” Dev Neurobiol 67(14): 1879–1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Conaill CR, Malisza KL, Buss JL, Bolster RB, Clancy C, de Gervai PD, Chudley AE and Longstaffe S (2015). “Visual search for feature conjunctions: an fMRI study comparing alcohol-related neurodevelopmental disorder (ARND) to ADHD.” Journal of Neurodevelopmental Disorders 7(1): 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owens DF and Kriegstein AR (2002). “Is there more to GABA than synaptic inhibition?” Nat Rev Neurosci 3(9): 715–727. [DOI] [PubMed] [Google Scholar]
- Paolozza A, Munn R, Munoz DP and Reynolds JN (2015). “Eye movements reveal sexually dimorphic deficits in children with fetal alcohol spectrum disorder.” Frontiers in Neuroscience 9(76). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paolozza A, Treit S, Beaulieu C and Reynolds JN (2017). “Diffusion tensor imaging of white matter and correlates to eye movement control and psychometric testing in children with prenatal alcohol exposure.” Hum Brain Mapp 38(1): 444–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rollema H, Hajos M, Seymour PA, Kozak R, Majchrzak MJ, Guanowsky V, Horner WE, Chapin DS, Hoffmann WE, Johnson DE, McLean S, Freeman J and Williams KE (2009). “Preclinical pharmacology of the alpha4beta2 nAChR partial agonist varenicline related to effects on reward, mood and cognition.” Biochem Pharmacol 78(7): 813–824. [DOI] [PubMed] [Google Scholar]
- Sadrian B, Wilson DA and Saito M (2013). “Long-lasting neural circuit dysfunction following developmental ethanol exposure.” Brain Sci 3(2): 704–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanderson JL, Donald Partridge L. and Valenzuela CF (2009). “Modulation of GABAergic and glutamatergic transmission by ethanol in the developing neocortex: an in vitro test of the excessive inhibition hypothesis of fetal alcohol spectrum disorder.” Neuropharmacology 56(2): 541–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savage LM, Candon PM and Hohmann HL (2000). “Alcohol-induced brain pathology and behavioral dysfunction: using an animal model to examine sex differences.” Alcohol Clin Exp Res 24(4): 465–475. [PubMed] [Google Scholar]
- Schneider RD and Thomas JD (2016). “Adolescent Choline Supplementation Attenuates Working Memory Deficits in Rats Exposed to Alcohol During the Third Trimester Equivalent.” Alcohol Clin Exp Res 40(4): 897–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sickmann HM, Patten AR, Morch K, Sawchuk S, Zhang C, Parton R, Szlavik L and Christie BR (2014). “Prenatal ethanol exposure has sex-specific effects on hippocampal long-term potentiation.” Hippocampus 24(1): 54–64. [DOI] [PubMed] [Google Scholar]
- Skorput AG, Gupta VP, Yeh PW and Yeh HH (2015). “Persistent Interneuronopathy in the Prefrontal Cortex of Young Adult Offspring Exposed to Ethanol In Utero.” J Neurosci 35(31): 10977–10988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith IE, Coles CD, Lancaster J, Fernhoff PM and Falek A (1986). “The effect of volume and duration of prenatal ethanol exposure on neonatal physical and behavioral 1001 development.” Neurobehav Toxicol Teratol 8(4): 375–381. [PubMed] [Google Scholar]
- Streissguth AB, HM.; Kogan J; Bookstein FL (1996). Understanding the occurence 1003 of secondary disabilities in clients with fetal alcohol syndrome (FAS) and fetal alcohol 1004 effects (FAE). Final report to the Centers for Disease Control and Prevention on Grant No. RO4/CCR008515 (Tech. Report No. 96–06) University of Washington, Fetal 1006 Alcohol and Drug Unit; Seattle, WA. [Google Scholar]
- Taly A, Corringer PJ, Guedin D, Lestage P and Changeux JP (2009). “Nicotinic 1008 receptors: allosteric transitions and therapeutic targets in the nervous system.” Nat Rev 1009 Drug Discov 8(9): 733–750. [DOI] [PubMed] [Google Scholar]
- Tanner DC, Githinji AW, Young EA, Meiri K, Savage DD and Perrone-Bizzozero NI (2004). “Fetal alcohol exposure alters GAP-43 phosphorylation and protein 1012 kinase C responses to contextual fear conditioning in the hippocampus of adult rat 1013 offspring.” Alcohol Clin Exp Res 28(1): 113–122. [DOI] [PubMed] [Google Scholar]
- Tesche CD, Kodituwakku PW, Garcia CM and Houck JM (2015). “Sex-related 1015 differences in auditory processing in adolescents with fetal alcohol spectrum disorder: A 1016 magnetoencephalographic study.” NeuroImage: Clinical 7: 571–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas JD, Abou EJ and Dominguez HD (2009). “Prenatal choline 1018 supplementation mitigates the adverse effects of prenatal alcohol exposure on 1019 development in rats.” Neurotoxicol Teratol 31(5): 303–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas JD, Biane JS, O’Bryan KA, O’Neill TM and Dominguez HD (2007). “Choline supplementation following third-trimester-equivalent alcohol exposure attenuates behavioral alterations in rats.” Behav Neurosci 121(1): 120–130. [DOI] [PubMed] [Google Scholar]
- Thomas JD, Fleming SL and Riley EP (2002). “Administration of low doses of MK-801 during ethanol withdrawal in the developing rat pup attenuates alcohol’s teratogenic effects.” Alcohol Clin Exp Res 26(8): 1307–1313. [DOI] [PubMed] [Google Scholar]
- Thomas JD, Garrison M and O’Neill TM (2004). “Perinatal choline supplementation attenuates behavioral alterations associated with neonatal alcohol exposure in rats.” Neurotoxicol Teratol 26(1): 35–45. [DOI] [PubMed] [Google Scholar]
- Tsai PC, Bake S, Balaraman S, Rawlings J, Holgate RR, Dubois D and Miranda RC (2014). “MiR-153 targets the nuclear factor-1 family and protects against teratogenic effects of ethanol exposure in fetal neural stem cells.” Biol Open 3(8): 741–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uban KA, Comeau WL, Ellis LA, Galea LA and Weinberg J (2013). “Basal regulation of HPA and dopamine systems is altered differentially in males and females by prenatal alcohol exposure and chronic variable stress.” Psychoneuroendocrinology 38(10): 1953–1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uban KA, Sliwowska JH, Lieblich S, Ellis LA, Yu WK, Weinberg J and Galea LA (2010). “Prenatal alcohol exposure reduces the proportion of newly produced neurons and glia in the dentate gyrus of the hippocampus in female rats.” Horm Behav 58(5): 835–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unal CT, Pare D and Zaborszky L (2015). “Impact of basal forebrain cholinergic inputs on basolateral amygdala neurons.” J Neurosci 35(2): 853–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenzuela CF, Puglia MP and Zucca S (2011). “Focus on: neurotransmitter systems.” Alcohol Res Health 34(1): 106–120. [PMC free article] [PubMed] [Google Scholar]
- Vetreno RP, Hall JM and Savage LM (2011). “Alcohol-related amnesia and dementia: animal models have revealed the contributions of different etiological factors on neuropathology, neurochemical dysfunction and cognitive impairment.” Neurobiol Learn Mem 96(4): 596–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Dubois DW, Tobery AN, Griffith WH, Brandt P and Frye GD (2013). “Long-lasting distortion of GABA signaling in MS/DB neurons after binge-like ethanol exposure during initial synaptogenesis.” Brain Res 1520: 36–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg J, Sliwowska JH, Lan N and Hellemans KG (2008). “Prenatal alcohol exposure: foetal programming, the hypothalamic-pituitary-adrenal axis and sex differences in outcome.” J Neuroendocrinol 20(4): 470–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisz J and Ward IL (1980). “Plasma testosterone and progesterone titers of pregnant rats, their male and female fetuses, and neonatal offspring.” Endocrinology 106(1): 306–316. [DOI] [PubMed] [Google Scholar]
- West JR (1993). “Use of pup in a cup model to study brain development.” J Nutr 123(2 Suppl): 382–385. [DOI] [PubMed] [Google Scholar]
- West JR, Chen WJ and Pantazis NJ (1994). “Fetal alcohol syndrome: the vulnerability of the developing brain and possible mechanisms of damage.” Metab Brain Dis 9(4): 291–322. [DOI] [PubMed] [Google Scholar]
- West JR and Goodlett CR (1990). “Teratogenic effects of alcohol on brain development.” Ann Med 22(5): 319–325. [DOI] [PubMed] [Google Scholar]
- Yanni PA and Lindsley TA (2000). “Ethanol inhibits development of dendrites and synapses in rat hippocampal pyramidal neuron cultures.” Brain Res Dev Brain Res 120(2): 233–243. [DOI] [PubMed] [Google Scholar]
- Zaborszky L and Duque A (2000). “Local synaptic connections of basal forebrain neurons.” Behav Brain Res 115(2): 143–158. [DOI] [PubMed] [Google Scholar]
- Zaborszky L, Heimer L, Eckenstein F and Leranth C (1986). “GABAergic input to cholinergic forebrain neurons: an ultrastructural study using retrograde tracing of HRP and double immunolabeling.” J Comp Neurol 250(3): 282–295. [DOI] [PubMed] [Google Scholar]
- Zago WM, Massey KA and Berg DK (2006). “Nicotinic activity stabilizes convergence of nicotinic and GABAergic synapses on filopodia of hippocampal interneurons.” Mol Cell Neurosci 31(3): 549–559. [DOI] [PubMed] [Google Scholar]
- Zhang J and Berg DK (2007). “Reversible inhibition of GABAA receptors by alpha7-containing nicotinic receptors on the vertebrate postsynaptic neurons.” J Physiol 579(Pt 3): 753–763. [DOI] [PMC free article] [PubMed] [Google Scholar]