

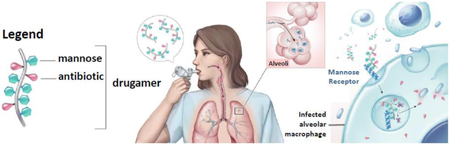
Abstract
Intracellular bacterial infections localized to the lung alveolar macrophage (AM) remain one of the most challenging settings for antimicrobial therapy. Current systemic antibiotic treatment fails to deliver sustained doses to intracellular bacterial reservoirs, which necessitates prolonged treatment regimens. Herein, we demonstrate a new intracellular enzyme-cleavable polymeric prodrug with tailored ciprofloxacin release profiles in the lungs and AM. The targeted polymeric prodrug, termed “drugamers”, incorporates (1) hydrophilic mannose residues to solubilize the antibiotic cargo and to target and enhance AM uptake and intracellular delivery, and (2) enzyme-cleavable linkage chemistry to provide high and sustained intracellular AM drug dosing. Prodrug monomers, derived from the antibiotic ciprofloxacin, were synthesized with either an intracellular protease cleavable dipeptide linker or a hydrolytic phenyl ester linker. RAFT polymerization was used to copolymerize the prodrug monomers and mannose monomer to synthesize well-defined drugamers without requiring a post-polymerization conjugation step. In addition to favorable in vivo safety profiles following intratracheal administration, a single dose of the drugamers sustained ciprofloxacin dosing in lungs and AMs above the minimum inhibitory concentration (MIC) over at least a 48 h period. The enzyme-cleavable therapeutic achieved a greater than 10-fold increase in sustained ciprofloxacin in AM, and maintained a significantly higher whole lung PK as well. Ciprofloxacin dosed in identical fashion displayed rapid clearance with a half-life of approximately 30 min. Notably, inhalation of the mannose-targeted ciprofloxacin drugamers achieved full survival (100%) in a highly lethal mouse model of pneumonic tularemia, contrasted with 0% survival using free ciprofloxacin. These findings demonstrate the versatility of the drugamer platform for engineering the intracellular pharmacokinetic profiles and its strong therapeutic activity in treating pulmonary intracellular infections.
Keywords: prodrug, ciprofloxacin, drug conjugate, enzyme-cleavable linker, Francisella, tularemia
Graphical abstract
1. Introduction
Pulmonary infections are major contributors to the global burden of disease. Non-tuberculous pulmonary infections killed 2.74 million people in 2015, and as a consequence they constitute the leading infectious cause of death and the fifth leading cause of death overall [1], Mycobacterium tuberculosis latently infected 1.7 billion of the world’s population in 2014 [2] and caused the disease tuberculosis (TB) in 10 million people in 2015; of these, 1.3 million died [3], TB is therefore the ninth leading cause of death worldwide [4], Combined, over 4 million people die annually from pulmonary infections. Intracellular infections including TB, legionellosis, tularemia, and melioidosis present therapeutic challenges. The causative bacteria for those diseases have evolved to inhabit mammalian cells, especially phagocytic alveolar macrophages (AMs), an important host cell for intracellular bacteria invading the lung [5-8], This intracellular localization provides a protective niche for those bacteria in the presence of humoral immunity and antibiotic therapy, thus reducing the antibacterial efficacy of pulmonary host defenses.
Current antibiotic therapy for pulmonary intracellular infections usually relies on rigorous oral and IV administration for a prolonged period to ensure therapeutic success and reduce relapse rate (e.g., 26 weeks for TB, 2-3 weeks for pulmonary tularemia, 3-6 months for melioidosis) [9-11], However, systemic delivery of antibiotics results in limited biodistribution and short retention in AMs, and the prolonged dosing regimen potentially promotes the emergence of drug resistance. In addition to the challenges associated with the delivery methods, the intrinsic properties of antibiotics also affects their efficacy against intracellular bacteria [12,13], For example, aminoglycosides fail to penetrate efficiently into eukaryotic cells, while fluoroquinolones exhibit poor retention inside the cells. The resulting off-target drug distribution induces side effects, including aminoglycoside-associated ototoxicity and fluoroquinolone-associated tendon rupture [14,15].
Delivery vehicles, such as liposomes, have been used as “drug pharmacokinetic modifiers” for achieving spatially and/or temporally pre-defined drug delivery goals [16], A prominent clinicalstage example is inhalable liposomal ciprofloxacin (Lipoquin®, Aradigm Corp.) that reports a lung clearance half-life of ~7.4 h (compared with ~1 h for free ciprofloxacin) in mice, leading to superior efficacy to free ciprofloxacin in a murine model of lethal pulmonary Fraticisella talarensis infection [17,18], These important studies have demonstrated up through human trials that the pharmacokinetics (PK) of antibiotics in the lung can be modulated by the carrier. These dispersal based formulation approaches can also suffer from various drawbacks including instability in biological fluids and tissues, initial burst release profiles, challenges in drug loading and difficulties in combination drug formulations [19].
As an alternative approach to the classical dispersal based particle formulations, our group recently developed an inhalable macromolecular prodrug platform, termed “drugamers,” that provides sustained lung delivery of ciprofloxacin with controlled dosing profiles [20,21], This macromolecular prodrug platform has tunable hydrolysis kinetics mediated by drug linkage chemistry (slower-releasing alkyllic vs. faster-releasing phenolic esters). In highly lethal F. novicida mouse challenge models where the mice are inoculated via bacterial aerosolization, a faster-releasing ciprofloxacin macromolecular prodrug (with phenolic ester linker) provided higher cure efficiencies (75% survival rate) than a similar but slower-releasing prodrug system (0% survival with alkyl ester linker). Further PK analysis demonstrated that this efficacy resulted from the sustained and higher level of ciprofloxacin maintained in the lung with a higher drug concentration achieved over minimum inhibitory concentration (MIC).
These results motivated us to further improve the drugamer system in an attempt to provide more specific and higher dosing in the AM intracellular environment. To this end, we herein integrated two new strategies into a new drugamer therapeutic design (Figure 1): (1) a protease cleavable valine-citrulline (VC) linker to facilitate intracellular release while maintaining extracellular stability in the lung, and (2) multivalent mannose ligands to target and enhance internalization by AMs [22], The VC dipeptide linker with a self-immolative spacer has been used in a commercialized antibody-drug conjugate (e.g., ADCETRIS®) to provide serum stability and efficient intracellular drug release [23], Genentech Inc. has also recently reported an antibody-antibiotic conjugate (THIOMAB™ DSTA4637S) with this protease-cleavable valine-citrulline dipeptide linker for systemic infection therapy [5,24], The original polymer-drug conjugates with protease cleavable linkers were pioneered by Duncan and Kopecek and progressed to phase II oncology clinical trials [25,26].
Figure 1.

(A) Ciprofloxacin-derived polymerizable prodrug monomers with cleavable linkers. VC monomer: methacrylate-based monomer linking ciprofloxacin through intracellular protease-cleavable valine-citrulline dipeptide linker. A self-immolative space situates ciprofloxacin from the enzymatic cleavage site (top red dash line). CTM monomer: methacrylate-based monomer linking ciprofloxacin through hydrolytic ester linker. (B) Synthetic strategy for the preparation of polymeric ciprofloxacin prodrugs using reversible addition-fragmentation transfer (RAFT) polymerization. Mannosylated methacrylate was used as a biocompatible hydrophilic comonomer.
Here, the in vitro conditional release characteristics, in vivo pharmacokinetics (PK) drug releasing profiles and therapeutic efficacy of the mannose-targeted, VC drugamer were compared to a drugamer linked through hydrolytic phenol esters. We found that the VC drugamer led to significantly higher maximum intracellular ciprofloxacin concentration (Cmax) and area under curve (AUC) of the PK drug release profiles in the whole lung and in the AM compartment, compared to the drugamers with the hydrolytic ester linker. Both drugamers demonstrate markedly improved survival (100%) compared to free ciprofloxacin (0%) in a lethal mouse model of pneumonic tularemia, indicating the therapeutic potential of this drugamer platform in treating pulmonary intracellular infections.
2. Materials and methods
2.1. Materials
Materials were purchased from Sigma Aldrich (St Louis, MO, USA) unless otherwise specified. 4-Cyano-4-(ethylsulfanylthiocarbonyl) sulfanylpentanoic acid (ECT) was synthesized and fully characterized as described previously [27], Spectra/Por regenerated cellulose dialysis membranes were purchased from Spectrum Laboratories (Houston, TX, USA). Sephadex G-25 prepacked PD-10 columns were obtained from GE Healthcare Life Sciences (Pittsburg, PA, USA). Acetonitrile (ACN) and ultrapure H2O were HPLC grade and purchased from Thermo Fisher Scientific (Waltham, MA, USA).
2.2. Synthesis of ciprofloxacin prodrug monomers
Synthetic procedures illustrated in supplementary information (Schemes S1-S3) were followed to obtain ciprofloxacin prodrug monomers carrying enzyme sensitive VC or phenyl ester linker and mannose ethylmethacrylate for AM targeting. All the synthesized monomers as well as intermediates were purified by precipitation and/or silica gel column chromatography techniques. The successful synthesis and purity of the monomers were confirmed and characterized by 1H NMR spectroscopy (Bruker Avance spectrometers 300 MHz or 500 MHz) and Electrospray Ionization-Mass spectrometry (Bruker Esquire ion trap mass spectrometer).
2.3. Reversible addition–fragmentation chain-transfer (RAFT) synthesis of VC drugamer
RAFT polymerization of mannose monomer and VC monomer was conducted in dimethyl sulfoxide (DMSO) under a nitrogen atmosphere using 4-cyano-4-[(ethylsulfanylthiocarbonyl)sulfanyl]pentanoic acid (ECT) as the chain-transfer agent (CTA) and 4,4’-Azobis(4-cyanovaleric acid) (ABCVA) as the radical initiator (Figure 1). The reaction was conducted with monomer feed ratios shown in Table 1. The initial monomer/CTA ratio ([M]0/[CTA]0) was 40:1 with a CTA/initiator ratio ([CTA]0/[I]0) of 10:1. Initial monomer wt% was about 27%. To a 5 mL round bottom flask was added mannose monomer (0.20 g, 0.67 mmol), VC monomer (0.12 g, 0.12 mmol), ECT (5.17 mg, 0.020 mmol), ABCVA (0.55 mg, 0.002 mmol), and DMSO (0.787 mL/0.869 g). The round bottom flask was then sealed with a rubber septa and purged with nitrogen for 0.5 h. After this time the polymerization solution was transferred to a preheated oil bath at 70 °C and allowed to react for 4 h. The resultant drugamer was isolated by precipitation in diethyl ether to remove DMSO. The precipitated polymer was dialyzed against phosphate buffer (100 mM Na2HP04, pH 7.2, 3 days) and distilled water (dH2O, 3 days) using dialysis tubing (MWCO = 3500 Da) to remove unreacted monomers and exchange trifluoroacetate salt with phosphate salt. The drugamers were then frozen and lyophilized before further purification using PD-10 desalting column followed by lyophilization for an additional 48 h. Purified polymers were characterized using 1H-NMR spectroscopy and size exclusion chromatography (supplementary information).
Table 1.
Summary of compositions, molecular weights (Mn), and molar mass dispersity (Đ) for the polymeric ciprofloxacin prodrugs with either a protease cleavable linker (Man-co-VC) or a hydrolytic ester linker (Man-co-CTM). Mannose monomer: mannosylated ethyl methacrylate. Drug monomer: ciprofloxacin prodrug monomer with Val-Cit linker or phenol ester linker.
| Polymer | Mannose monomer (feed) mol.% |
Mannose Monomera (exp) mol.% |
Drug monomer (feed) mol.% |
Drug monomera (exp) mol.% |
Mnb (kDa) |
Đb |
Druga wt% |
|---|---|---|---|---|---|---|---|
| Man-co-VC | 85 | 90.2 ± 0.8 | 15 | 9.8 ± 0.8 | 25.8 ± 1.2 | 1.10 ± 0.08 | 9.3 ± 0.7 |
| Man-co-CTM | 89 | 90.2 ± 0.6 | 11 | 9.8 ± 0.6 | 26.7 ± 6.8 | 1.02 ± 0.02 | 9.8 ± 0.5 |
As determined by 1H-NMR spectrum in DMSO-d6 using a Boc-protected ciprofloxacin as an internal standard.
As determined by size exclusion chromatography.
2.4. RAFT synthesis of CTM drugamer
RAFT polymerization of mannose monomer and CTM monomer was conducted in DMSO under a nitrogen atmosphere using 4-cyano-4-(phenylcarbonothioylthio)pentanoic acid (CTP) as the CTA and ABCVA as the radical initiator (Figure 1). The reaction was conducted with monomer feed ratios shown in Table 1. The initial monomer/CTA ratio ([M]0/[CTA]0) was 50:1 with a CTA/initiator ratio ([CTA]0/[I]0) of 5:1. Initial monomer wt% was about 24 wt%. To a 5 mL round bottom flask was added mannose monomer (0.362 g, 1.24 mmol), CTM monomer (0.120 g, 0.154 mmol), CTP (7.78 mg, 0.028 mmol), ABCVA (1.56 mg, 0.0056 mmol), and DMSO (1.39 mL/1.53 g). The round bottom flask was then sealed with a rubber septa and purged with nitrogen for 0.5 h. After this time the polymerization solution was transferred to a preheated oil bath at 70 °C and allowed to react for 3 h. The resultant drugamers were then purified and characterized using the same methods as for VC drugamer.
2.5. Enzyme-mediated ciprofloxacin release
Lysosomal protease induced ciprofloxacin release from the drugamers were conducted at conditions mimicking the lysosomal medium [28], Human liver cathepsin B (Enzo Life Sciences, Farmingdale, USA) was activated for 15 minutes in a solution of 0.151 mg/mL cathepsin B, 30 mM DTT, and 15 mM EDTA at 37 °C. Monomers or drugamers were then solubilized in reaction buffer [25 mM sodium acetate, 1 mM EDTA, pH 5.0, or phosphate buffered saline (10 mM, lx PBS), 1 mM EDTA pH 7.4, 37 °C] and added to the enzyme solution for a final concentration of 2.1 μg/mL cathepsin B and 0.3 mg/mL drugamer (equivalent to 0.1 mM ciprofloxacin/peptide). At various timepoints, enzymatic activity was halted by addition of a thioprotease inhibitor [E-64 (Thermo Scientific), 1 mg/mL, 10 μL], 100% ciprofloxacin release was defined by incubating monomers/drugamers in 0.1 N NaOH for 24 h at room temperature to hydrolyze all ester bonds, thus releasing all ciprofloxacin from polymer backbone. The released ciprofloxacin concentration was quantified by liquid chromatography-electrospray ionization mass spectrometry (LC-ESI MS) as shown in the supplementary information.
To evaluate the drugamer stability in the presence of esterases, recombinant human butyrylcholine esterase (BChE, Novoprotein, Summit, NJ, USA) was used as a model esterase. Drugamer solution (concentration ~ 3 mg/mL, equivalent to 1 mM ciprofloxacin, 10 μL) was added to lx PBS (90 μL, containing 0.5 μg BChE) to a final ciprofloxacin concentration of 100 μM and incubated at 37 °C. At various time points, samples were diluted with ACN 5 times to denature BChE and halt the reaction. The samples were centrifuged at 13,000 rpm for 4 min to collect supernatants. The supernatants were subsequently concentrated 4 times by speedvac and analyzed by LC-ESI MS. 100% ciprofloxacin release was defined by incubating drugamers (concentration ~ 3 mg/mL, equivalent to 1 mM ciprofloxacin, 20 μL) in 0.1 N NaOH (90 μL) for 24 h at room temperature to hydrolyze all ester bonds, thus releasing all ciprofloxacin from the polymer backbone.
2.6. Quantification of released ciprofloxacin inside macrophages.
RAW 264.7 cells were seeded in a 48-well plate with antibiotic-free DMEM supplemented with 10% FBS and allowed to adhere overnight. The cells were washed with PBS and drugamers (equivalent to 250 μg/mL ciprofloxacin) or free ciprofloxacin (250 μg/mL) diluted in DMEM were added to each well (0.25 mL). After 2 h incubation, the cells were washed three times with PBS, and 0.25 mL of DMEM with 10% FBS was added to each well. The cells were then incubated for the indicated time points. At each time point, cells were trypsinized and lysed by acetonitrile (ACN) addition (3x dilution). The obtained cell lysates were centrifuged at 14,000×g for 15 min at 4°C to collect supernatants. Concentrations of the released ciprofloxacin were quantified using LC-MS/MS. The LC-MS/MS method was described in the supplementary information.
2.7. Cell viability assay
Viability of RAW 264.7 cells treated with VC or CTM drugamer was evaluated using MTS assay as per manufacturer’s instructions. Cells were seeded into a 96-well plate (5 × 104 cells/well) and incubated for 18 h at 37°C with 5% CO2. Subsequently, drugamers at predetermined concentration (2.5 – 0.039 mg/mL) were added to each well and incubated for 22 h, same as the incubation time used in the subsequent co-culture assay. After incubation, cell media was aspirated and cells were washed with 1x PBS prior to addition of Cell Titer 96®Aqueous One Solution Reagent (Promega). Cells were incubated for 2 h in MTS reagent before absorbance was read at 490 nm. Cell viability was calculated by (Abssample — Absblank)/(Abscontrol — Absblank) × 100%.
2.8. Bacterial preparation
F. novicida was used as a laboratory surrogate for F. tularensis, due to its low virulence for humans and high degree of genetic similarities with F. tularensis (−95%) [29], F. novicida U112 was originally obtained from Francis Nano (University of Victoria, Victoria, Canada). A stock was prepared from overnight growth in trypticase soy broth with 0.1% L-cysteine at 37 °C with aeration. Bacteria were harvested in stationary phase, diluted in 20% glycerol, aliquoted, flash-frozen, and stored at −80 °C, yielding a post-thaw titer of 109 CFU/mL.
2.9. Francisella-macrophage co-culture assay
RAW 264.7 cells were seeded (700,000 cells/mL, 250 μL/well) into a 48 well plate with antibiotic free DMEM containing 10% FBS, and incubated at 37°C with 5% CO2. After 18 h, cells were infected with F. novicida U112 at early log phase of growth (OD600=0.2) at a multiplicity of infection of 50, and then incubated for 1 h. Subsequently, growth media was replaced with fresh DMEM containing 10% FBS and 250 μg/mL kanamycin to eliminate extracellular bacteria not internalized by the cells; cells were then incubated for another hour. After 1 h, cells were washed with 1x PBS and incubated with fresh media containing free ciprofloxacin or drugamers (equivalent to 250 μg/mL ciprofloxacin) for 2 h. Following incubation, cells were washed with 1x PBS and growth media was replaced with fresh DMEM containing 10% FBS. Cells were then incubated for another 22 h (24 h post-infection). After incubation, cells were washed three times with 1x PBS and lysed with 100 μL of PBS containing 0.1% [v/v] Triton X-100. Lysates were serially diluted and plated onto triplicate TSB agar plates, and incubated at 37°C for 24 h. CFUs were counted when individual bacterial colonies were distinguishable.
2.10. Animals and ethics statement
Female C57B1/6 mice, 6 to 8 weeks old, were purchased from The Jackson Laboratory (Bar Harbor, ME), maintained at the University of Washington under specific pathogen-free conditions. All animal procedures and handling were conducted under protocols (4047-02 and 2671-10) approved by the Institutional Animal Care and Use Committee at the University of Washington.
2.11. In vivo lung safety study
C57B1/6 mice (Female, 6-8 weeks old, n=5) were anesthetized with 5% isoflurane for 5 min before administration of 50 μL of PBS (negative control) or drugamer solution in PBS (0.2 μm filtered, equivalent to 20 mg/kg ciprofloxacin dose) via intratracheal aerosolization using a MicroSprayer® Aerosolizer–Model IA-1C (Penn-Century. Wyndmoor Inc., PA, USA). Mice received one daily dose for three consecutive days and their body weight changes were monitored each day before dosing. Twenty-four h after the last dose, the mice were weighed and then euthanized by CO2 asphyxiation. Bronchoalveolar lavage (BAL) was conducted with 1 mL of PBS flush followed by three 0.8 mL flushes. Lungs were harvested and placed into 1 mL PBS on ice. Lung tissue in 1 mL of PBS was subsequently mixed with 1 mL of PBS containing protease inhibitor cocktail (Pierce™ Protease Inhibitor, Thermo Fisher Scientific) before mechanical homogenization using a TissueRuptor (Qiagen, Valencia, CA, USA). Lung tissue homogenates (LTH) were then stored at 4°C for 30 min followed by centrifuge at 5000xg for 15 minutes to collect the supernatant for TNF-α quantification.
BAL fluid was spun at 400 g for 15 min to pellet lavage cells. Cell-free BAL fluid was retained and stored at −80°C. BAL cells were resuspended into 0.5 mL RPMI 1640 supplemented with 10% FBS. BAL cells were mounted onto microscopy slides with a Cytospin centrifuge at 500 rpm for 5 min, and then stained with Hemacolor (EMD Millipore, Billerica, MA, USA) prior to cytology analysis. Stained slides were analyzed for macrophage to neutrophil ratio with a minimum of 200 cells per slide counted. TNF-α concentrations in the supernatant of LTH and cell free BAL fluid were assayed using mouse TNF-α ELISA MAXTM Standard kit (BioLegend, San Diego, CA, USA) as per manufacturer’s instructions.
2.12. In vivo pharmacokinetics (PK) and biodistribution studies of drugamer-released ciprofloxacin
In vivo PK and biodistribution properties of free-form ciprofloxacin and drugamer-released ciprofloxacin were compared using C57BL/6 mice (female, 6-8 weeks old, n=4). Mice were treated with free ciprofloxacin solution (5% dextrose in water, 20 mg/kg) or drugamer solution (PBS, equivalent to 20 mg/kg ciprofloxacin) via intratracheal aerosolization using the MicroSprayer® Aerosolizer. Specifically, mice were anesthetized with 5% isoflurane with O2 at 1 L/min exposure after filling the chamber for one min. Using a mouse laryngoscope (Penn-Century LS-2, LS-2M, PA, USA), and an intratracheal MicroSprayer®, 50 μL of free ciprofloxacin, VC drugamer, or CTM drugamer was administered. At various time points post drug administration (0.5, 1, 2, 4, 8, 18, 24 and 48 h), mice were euthanized and the blood was then collected by cardiac puncture. The left mainstem bronchus was tied off and the right lung was lavaged with 0.6 mL followed by three 0.5 mL flushes of 0.85% NaCl with 0.6 mM EDTA. The left lung, liver, spleen and kidney were then surgically removed and weighed fresh. To determine the actual deposited dose, mice were euthanized to collect tissue samples right after dosing. Details of the tissue process and ciprofloxacin quantification methods were described in the supplementary information.
2.13. In vivo antibiotic activity studies using a lethal pulmonary Francisella infection model
The antibiotic activity of drugamers was tested in C57Bl/6 mice (female, 8 weeks old, n=8) challenged with aerosolized F. novicida U112. Mice were given three consecutive doses of free ciprofloxacin or VC/CTM drugamers using MicroSprayer®, with 24 h interval between each dose (Figure. 6A). Two challenge studies were conducted with ciprofloxacin dosing at either 20 or 10 mg/kg. Lethal bacterial infection was conducted 1 h after the first dose using a method described previously by our lab [21]. Briefly, a bacterial aerosol was generated from 6 mL of bacteria suspension (2×107 CFU/mL) using a Heart Mini-hi-flow nebulizer (Westmed, Arizona, USA). Mice from all three treatment groups were simultaneously exposed to aerosolized bacteria using a Biaera whole-body exposure chamber (Biaera Technologies, Maryland, USA), with total airflow through the chamber maintained at 19.5 L/min during a 20 min exposure.
Figure 6. In vivo therapeutic efficacy of polymeric ciprofloxacin prodrugs against lethal pulmonary infections of Francisella novicida in mice.

(a) Workflow schematic. Free ciprofloxacin or polymeric ciprofloxacin prodrugs with protease-cleavable linker (Man-co-VC) or hydrolytic ester linker (Man-co-CTM) were intratracheally administered (50 μL aerosolization) at 0, 1, and 2 days using a Micro Sprayer® (n = 8 for each treatment group). All bacterial inoculation occurred 1 h after treatment on day 0. Survival rate and health condition of mice were monitored for 14 days. (B) and (C) survival rate of infected mice treated with 20 or 10 mg/kg ciprofloxacin dose.
To determine bacterial deposition in the lungs, four mice were euthanized immediately postexposure with pentobarbital (300 mg/kg intraperitoneally) followed by exsanguination. The left lungs were harvested, homogenized in PBS, and quantitatively cultured on trypticase soy agar supplemented with 0.1% L-cysteine. The bacteria titers in the two challenge studies were about 10-15x higher than LD50 (105 ± 9 CFU/lung for 20 mg/kg dose; 130 ± 5 CFU/lung for 10 mg/kg dose, Mean ± SEM) [29]. The infected animals were monitored daily for mobility and for deaths from the infection. At day 14 after infection, the number of mice that survived the otherwise lethal infection was recorded. Lungs were harvested from mice that survived 14 days after infection and plated to determine viable bacteria surviving after treatment.
2.14. Statistical analysis
Student’s two-tailed t-tests were performed to compare two groups and analysis of variance (ANOVA) was performed for comparisons of multiple groups, through GraphPad Prism (version 5.0, GraphPad Software Inc.). Unless otherwise stated, data reported denote the means ± SD. Survival analyses were performed using a log-rank test in GraphPad Prism. No specific methods of randomization were applied to group or animal allocation. Investigators were not blinded to group allocation. P < 0.05 was considered statistically significant.
3. Results
3.1. Synthesis and characterization of ciprofloxacin prodrug monomers
The Valine-Citruline (VC) dipeptide sequence was coupled with the self-immolative spacer para-aminobenzyl alcohol (PABOH) to construct the cathepsin B sensitive ciprofloxacin prodrug monomer (Scheme S1, supplementary information). Succinimidyl activated methacrylate monomer was first coupled with VC-PABOH to get SMA-VC-PABOH (6), which was then conjugated to Boc-ciprofloxacin using uronium-based coupling agent HBTU, the base diisopropylethylamine and the catalyst DMAP. Vinyl signals at 5.69 and 6.02 ppm, and aromatic signals of ciprofloxacin at 7.79 and 8.46 ppm in 1H-NMR spectrum confirmed the successful conjugation. Finally, Boc group was deprotected in 20 % trifluoroacetic acid to get the enzyme cleavable monomer (11). To evaluate linker effects on intracellular drug release kinetics, a control monomer CTM (14) containing phenyl ester linker susceptible for nonspecific drug hydrolysis was also synthesized following similar HBTU coupling and deprotection chemistry described above for VC-ciprofloxacin monomer (Scheme S2, supplementary information). 1H-NMR spectrum and ESI-MS spectral characterizations, and protease-mediated ciprofloxacin release kinetics are available in Supplementary Information.
3.2. Optimizing compositions of protease-cleavable drugamer
Ciprofloxacin prodrug monomers were copolymerized with mannose monomers by RAFT polymerization (Figure 1) [20,30], Due to the hydrophobic nature of the dipeptide VC linker and ciprofloxacin, the feed mole ratio of VC monomer and mannose monomer was optimized such that resulting polymers would be sufficiently soluble in PBS to achieve the ciprofloxacin dosing for the subsequent in vivo studies (20 mg/kg ciprofloxacin dose, ~80 mg/mL polymer solution) (Table S1). The optimized drugamer compositions were characterized using size exclusion chromatography (SEC) and 1H-NMR spectrum, and are summarized in Table 1 and Figure S10-S12. Monomer feed ratio and degree of polymerization for drugamers containing the phenol ester linker (CTM) were adjusted to yield a similar ciprofloxacin wt% and molecular weight (MW) as the VC drugamer for proper comparison. For example, the MW of VC and CTM drugamers was found to be 25.8 and 26.7 kDa with ciprofloxacin wt% around 9-10%. Additionally, both drugamers have narrow MW distributions (Đ) with dispersity of 1.10 and 1.02, for VC and CTM, respectively.
3.3. Cathepsin B mediated ciprofloxacin release kinetics
To verify the linker accessibility and enzyme-specific cleavage of ciprofloxacin from the VC linker, drugamers were incubated with human liver cathepsin B enzyme. The released ciprofloxacin was quantified as a function of time using LC-ESI MS. Incubation of VC drugamer with cathepsin B resulted in the rapid release of ciprofloxacin with a t1/2 of ~30 min (Figure 2A). By contrast, drug release from a control drugamer containing a non-cleavable peptide linker (CTM drugamer) was not observed over 4 h in the presence of cathepsin B. Time-dependent ciprofloxacin release from VC drugamer was clearly observed in the LC chromatograms (Figure 2B). The aminobenzyl alcohol side product (Scheme S4) was detectable by LC-MS analysis with similar release rate as ciprofloxacin (Figure S13).
Figure 2. Lysosomal protease-mediated drug release from polymeric ciprofloxacin prodrug.

(A) Ciprofloxacin release kinetics of polymeric ciprofloxacin prodrugs with either a protease-cleavable dipeptide linker (Man-co-VC) or a hydrolytic ester linker (Man-co-CTM) in the presence of a human lysosomal protease, cathepsin B (CatB). (B) Liquid chromatograms showing the time-dependent ciprofloxacin released from Man-co-VC incubated with CatB. (C) Ciprofloxacin release kinetics of Man-co-VC in the presence of CatB (pH 5.0 or 7.4), and in human serum or acetate buffer (pH 5.0). (D) Ciprofloxacin release kinetics of polymeric ciprofloxacin prodrugs incubated with a model esterase, human butyrylcholinesterase. Error bars represent means ± SD from triplicate samples. Cipro: Ciprofloxacin.
As cathepsin B could be translocated to the cell surface or secreted into the extracellular space under certain physiological and pathological conditions (e.g., inflammation) [31], we also evaluated cathepsin B-mediated release at neutral pH, which demonstrated minimal ciprofloxacin release (~10%) after 4 h incubation (Figure 2C). To investigate if ciprofloxacin release from VC drugamer is primarily attributed to the cleavage by cathepsin B, and not spontaneous hydrolysis, the release profile was evaluated in various aqueous conditions, including in an acetate buffer mimicking the lysosomal pH (pH 5.0) and human serum. No detectable ciprofloxacin release from VC drugamer was observed in 4 h under those aqueous conditions tested. To examine whether the ester bond in both drugamers is susceptible to nonspecific esterase cleavage, we probed ciprofloxacin release from drugamers in the presence of butyrylcholinesterase (BChE), a human serum esterase involved in detoxification and metabolism of ester-containing drugs [32], Accumulation of the released ciprofloxacin was monitored for up to 14 days; only ~2.7% and —15.1% of the active ciprofloxacin was released from VC and CTM drugamers, respectively (Figure 2D).
3.4. In vitro intracellular ciprofloxacin release and efficacy against intracellular infections
To compare the intracellular ciprofloxacin release kinetics of VC and CTM drugamers, tandem LC-MS (LC-MS/MS) was used to quantify ciprofloxacin concentrations in RAW 264.7 macrophages incubated with drugamers (0.25 mg/mL equivalent ciprofloxacin, ~2.5 mg/mL durgamer) over 4 h. At predetermined time points, drugamer treatment solutions were removed, and cells were washed several times with DPBS in order to remove all extracellular (non-bound or internalized) drugamers. Cells were then lysed to extract the intracellular ciprofloxacin and subjected to LC-MS/MS analysis to quantify drug concentrations. Notably, VC drugamer demonstrates a time-dependent intracellular release profile with significantly higher intracellular ciprofloxacin concentrations than CTM drugamer after 1-4 h incubations (Figure 3A). With the knowledge of the intracellular ciprofloxacin release profiles of the drugamers, we further investigated the ability of the drugamers to clear intracellular bacterial infections. Using an in vitro macrophage-bacteria co-culture assay, RAW 264.7 cells were infected with F novicida, a laboratory surrogate for F. tularensis [29](Figure 3B). The two drugamers were compared for their effectiveness in decreasing intracellular bacterial levels. Whereas CTM drugamer demonstrated modest reduction in the intracellular bacteria, VC drugamer displayed antimicrobial activity indistinguishable from free ciprofloxacin. This significant difference in antibacterial potency between VC drugamer and CTM drugamer corresponds with the faster intracellular release profile of VC drugamer shown in Figure 3A. Notably, drugamers displayed limited cytotoxicity to RAW 264.7 at concentration used in in vitro assays (~2.5 mg/mL, Figure 3C).
Figure 3. In vitro properties of polymeric ciprofloxacin prodrugs.

(A) Mass spectrometric quantification of released antibiotic inside macrophages incubated with polymeric ciprofloxacin prodrugs made with Val-Cit dipeptide linker (protease cleavable, Man-co-VC) or ester linker (Man-co-CTM). (B) Intracellular antibacterial activity of polymeric prodrugs in RAW 264.7 macrophages infected with Francisella novicida. Surviving bacteria were enumerated 24 h after infection. NT: untreated control. (C) Cell viability of RAW 264.7 cells incubated with Man-co-VC or Man-co-CTM for 22 h. Cell viability numbers were relative to untreated controls as determined by MTS. Error bars represent means ± SD from triplicate wells. One-way ANOVA analysis with a Dunnett test was utilized to establish statistical significance (**p < 0.01, ***p < 0.001, ****p< 0.0001). Cipro: ciprofloxacin.
3.5. In vivo lung safety of polymeric ciprofloxacin prodrugs
To examine the in vivo utility of the drugamer system, intratracheal aerosolization of drugamer solutions (50 μL) or PBS as a control were delivered to mouse lungs at a ciprofloxacin dose of 20 mg/kg once daily for three-consecutive days. Respiratory rate and effort were not noted to differ among mice treated with drugamers compared to PBS control mice. Lung, bronchoalveolar lavage fluid (BALF), and alveolar cells were then collected 24 h after the last dose. Acute lung inflammation was evaluated using the metrics of animal weight change, infiltrating neutrophil in the bronchoalveolar space, and tumor necrosis factor alpha (TNF-α) concentration in lung tissue homogenate (LTH) and BALF (Figure 4). These metrics were selected based on the understanding that neutrophils are the earliest immune cells to be recruited to the site of lung injury or inflammation [33] and TNF-α is an early response cytokine in acute lung injury and inflammation [34,35], As shown in Figure 4, all observed toxicity markers for the VC and CTM drugamers are indistinguishable from the PBS control (P > 0.1). The body weight of mice gradually increased with time to match typical growth curves and the neutrophil percentages were comparable to the reported values of healthy mice (~3%) [33].
Figure 4. Pulmonary safety evaluation of polymeric prodrugs dosed at 20 mg/kg ciprofloxacin.

Polymeric antibiotic prodrugs were intratracheally administered to uninfected C57BL/6 mice once a day for 3 days, and tissues were harvested 24 h after the last dose. (A) Animal weights measured prior to treatment and 24 h after the final dose were ratioed to calculate percent weight change per mouse. (B) Percentage of neutrophils was determined by differential cell counting of cells from bronchoalveolar lavage fluid (BALF). Values were calculated as [neutrophil/(neutrophil + macrophage)] × 100%. Tumor necrosis factor alpha (TNF-α) levels were measured by ELISA from (C) lung tissue homogenates (LTH) and (D) BALF. Concentration values of TNF-α from LTH were adjusted to mass of the harvested lungs, while BALF TNF-α levels are presented as absorbance values corrected to total volume of harvested fluid. No statistically significant differences were observed using one-way ANOVA and a Dunnett post hoc test between polymer treated and PBS treated mice (n = 5). Error bars represent means ± SEM. VC: polymer with protease-cleavable peptide linker. CTM: polymer with hydrolytic ester linker.
3.6. In vivo pharmacokinetics and biodistribution of polymeric ciprofloxacin
In vivo PK and biodistribution of ciprofloxacin from drugamer-dosed or free-drug dosed animals were evaluated using an analytical LC–MS/MS technique after a single intratracheal administration at 20 mg/kg ciprofloxacin dose. All data were normalized to mass of tissue, volume of blood, or intracellular volume. To ensure proper comparisons across the three treatment groups, the actual deposited ciprofloxacin dose was determined from the ciprofloxacin concentration in the lung at about 10 min post-administration (minimal time required for animal euthanasia and tissue collection) (Figure S14). Similar deposited doses in right and left lungs were observed with VC and CTM drugamers. The single dose of free ciprofloxacin resulted in a relatively high initial concentration and rapid elimination from the lungs over 2 h, consistent with the reported PK profiles of free ciprofloxacin [18] (Figure 5A & 5B). In contrast, the concentration of ciprofloxacin in whole lung released from both drugamers remained detectable and higher than MIC (0.065 μg/g, see Table 2) over a 48 h period. Additionally, the measurement of total drug exposure in the whole lung (i.e. the area-under-the-curve, AUC) and maximum concentration (Cmax) following single administration of VC drugamer were 1.8- and 2.3-fold higher, respectively, than those of CTM drugamer (Table 2).
Figure 5. In vivo pharmacokinetics analysis of polymer-released ciprofloxacin.

Time-course of ciprofloxacin concentration was evaluated in lung (A & B), alveolar macrophage (AM) (C & D) and plasma (E & F). All figures, except for Figure 5C, present the free/released ciprofloxacin concentrations; Figure 5C shows concentrations of the drugamer depot in AMs. Figure 5A and E were expanded along y-axis to yield Figure 5B & F. After intratracheal administration of each drugamer or free ciprofloxacin solution to mice, at predetermined time points, single bronchoalveolar lavage was conducted to collect AMs from the right lung and the left lung was collected intact. Ciprofloxacin concentration was determined using LC-MS/MS with deuterated ciprofloxacin as an internal standard. In all cases, released drug was quantified by comparison to an authentic drug standard curve prepared in the same matrix. Each value represents the mean ± SEM (n=3-4). Cipro: ciprofloxacin.
Table 2.
Pharmacokinetic parameters of ciprofloxacin in lung, alveolar macrophage (AM), and plasma after intratracheal administration of polymeric prodrugs to mice. Parameters were obtained from data shown in Figure 5.
| Treatment | Tissue | Tmaxa (h) |
Cmaxb (μg/g or μg/mL) |
AUC0-24c (μg*h/g or μg*h/mL) |
Cmax/MICd | AUC0-24/MICd (h) |
|---|---|---|---|---|---|---|
| VC | 4 | 25 ± 5 | 321 | 386 | 4945 | |
| CTM | Lung | 8 | 11 ± 2 | 180 | 165 | 2766 |
| Free | 0.17 | 83 ± 18 | 38 | 1275 | 585 | |
| VC | 4 | 2914 ± 772 | 33971 | 97149 | 1132351 | |
| CTM | AM | 18 | 85 ± 19 | 1326 | 2821 | 44213 |
| Free | 0.5 | 447 ± 48 | 454 | 14885 | 15141 | |
| VC | 1 | 0.15 ± 0.04 | 0.79 | 5.10 | 26.43 | |
| CTM | Plasma | 1 | 0.11 ± 0.03 | 0.88 | 3.60 | 29.47 |
| Free | 0.17 | 3.54 ± 1.13 | 4.21 | 117.99 | 140.34 |
The time to reach maximum concentration (Cmax) after administration.
Cmax are represented as mean ± SEM. Values are normalized to lung mass (g) or intracellular/plasma volume (mL).
Area under the curve are presented as mean value from 0 to 24 h (AUC0-24) and determined by GraphPad Prism 5.
Both the mannose-targeted VC and CTM parent drugamers were internalized and remained concentrated in AMs at similar levels throughout the 48 h time frame of the experiment (Figure 5C). The most striking effect of the VC linker was the order of magnitude increase in intracellular AM ciprofloxacin concentrations and their maintenance over the observed 48 h time period (Figure 5D). The time of maximum concentration observed (Tmax) also appeared earlier in mice treated with VC drugamer compared to the Tmax for the CTM drugamer (Table 2). Quantitatively, the VC drugamer in the AM compartment achieved a higher Cmax compared to free ciprofloxacin (6.5x) and CTM drugamer (34.4x). The sustained delivery of ciprofloxacin from the VC drugamer resulted in intracellular ciprofloxacin concentrations more than 3 magnitudes higher than the MIC over the entire 48 h period. This unique intracellular PK profile of the VC drugamer led to superior AUC compared to free ciprofloxacin (74.8x) and CTM drugamer (25.6x). Although the VC drugamer provided higher intracellular ciprofloxacin dosing, the released ciprofloxacin concentration from the VC drugamer in plasma and other organs (liver, spleen and kidney) was minimal compared to free ciprofloxacin (Figure 5E, 5F & S15, Table S2). In comparison to the CTM drugamer, the VC drugamer resulted in higher released ciprofloxacin concentrations in all the tested organs (see supplementary information), but similar ciprofloxacin concentrations in plasma.
3.7. Therapeutic efficacy of polymeric ciprofloxacin prodrug against lethal pneumonic tularemia.
With significant improvement in the PK profiles of the drugamer-released ciprofloxacin, we next investigated the antibacterial efficacy of drugamers in a lethal mouse model of airborne F. novicida infection. Drugamers were administered using an intratracheal microsprayer with a ciprofloxacin dose of 20 mg/kg or 10 mg/kg at 0 (1 h prior to infection), +1, and +2 days (Figure 6A). Animal health condition and survival were monitored until mice qualified for euthanasia or reached the experimental end-point (14 days post-infection). In the first challenge study where mice were dosed at 20 mg/kg ciprofloxacin, none of the mice receiving free ciprofloxacin survived to the experimental end-point. In contrast, VC and CTM drugamers provided full protection against the lethal infection of aerosolized F. nocivida (Figure 6B).
In an attempt to distinguish the efficacies of VC and CTM drugamers, we further reduced the ciprofloxacin dose to 10 mg/kg with the assumption that the high AUC and Cmax of PK profiles in AM would allow the VC drugamer to lower the effective dose. The transition from 20 to 10 mg/kg dose led to the survival rate of mice treated with VC or CTM drugamer decreased from 100% to 62.5% (n= 5/8) and 50% (n= 4/8), respectively (Figure 6C). The body weight change and the number of bacteria in the lung of survived mice reflected the better health conditions of mice treated with the VC drugamer. For example, body weights of mice treated with the VC drugamer started recovering at 3 days post infection with maximum ~13% body weight loss, whereas, mice treated with free ciprofloxacin or CTM drugamer kept dropping to a minimum of 30% and 20%, respectively (Figure S16). Additionally, the bacteria titers in the surviving mice treated with VC and CTM drugamers were 7.6 ± 3.2 CFU/lung and 253.5 ± 243.2 CFU/lung, respectively.
4. Discussion
This study demonstrates the versatility of this polymeric antibiotic prodrug platform by adding mannose-targeting and an intracellular enzyme-cleavable linker to dramatically increase the alveolar macrophage drug PK and corresponding antibacterial efficacy. This prodrug strategy is needed to address significant limitations associated with conventional ciprofloxacin therapy. Free ciprofloxacin is rapidly cleared from the site of infection, and its clinical significance can be compromised due to adverse reactions, including tendon rupture and irreversible nerve damage [37-39].
The new mannose-targeted ciprofloxacin drugamers reported here provide two advantageous features compared to prior pegylated ciprofloxacin drugamers. First, mannose-targeting affords increased alveolar macrophage uptake and internalization. Second, the drugamer with a protease-cleavable peptide linker (VC drugamer) displays a sustained 10-fold higher intracellular dosing of ciprofloxacin compared to the hydrolytic phenolyic ester drugamer. Both the mannose-targeted hydrolytic ester drugamer and the VC drugamer achieved full cohort survival in the aerosolized pulmonary intracellular Francisella infections, and at lower ciprofloxacin doses compared to prior pegylated phenyl ester linked drugamers [21]. The unique PK profile with the intracellular enzyme-cleavable linker may yield therapeutic advantages over current antibiotic therapy, especially in disease settings where bacteria are resident in AM, such as pneumonic tularemia and tuberculosis [6,40]. Additionally, the drug release mechanism and kinetics can be encoded into the polymer by choosing various linkers, such as the peptide linker and phenyl ester linker in the present study (Figure 1). The synthetic approach also allows facile design of multivalent mannose ligands that have been shown to actively target AMs [22,41,42]. The resulting targeted drugamers with a peptide linker (VC drugamer) or a hydrolytic ester (CTM drugamer) have a high degree of uniformity and reproducibility of molecular weight and compositions (Table 1 & Figure S10), which are important for future drug development.
The Val-Cit dipeptide linker with a self-immolative spacer has been preciously shown to be cleavable by lysosomal proteases in mice and human [5,23,43]. Consistent with previous findings of antibody-drug conjugates, the VC drugamer were stable under physiological conditions but was rapidly hydrolyzed by lysosomal protease cathepsin B particularly at lysosomal pH (Figure 2). This demonstrates that the dipeptide portion of the full linker is accessible within the context of the mannose monomer surrounding environment. Notably, no detectable cathepsin B-mediated drug release was identified from CTM drugamer and only limited release resulting from the model extracellular esterase BChE over 14 days. The faster intracellular release of VC drugamers than CTM drugamer was also demonstrated using in vitro RAW cells without and with the F. novicida infection (Figure 3A&B). The VC drugamer showed similar antibacterial potency as free ciprofloxacin in a representative macrophage-bacteria assay, whereas CTM drugamer do not. This observation confirms the rapid intracellular drug release of VC drugamer in cells infected with F. novicida.
This inhalable drugamer platform shows high activity and favorable toxicity profiles. The prodrug-based drugamer therapeutic avoids burst release and its favorable safety profile could result from minimizing the dosing burst of active antibiotics compared to free drug formulations. The release profiles of prodrugs combined with active AM targeting could result in lowered drug contact with the airway during inhalation, a major reason for the respiratory adverse events (e.g, cough and dyspnea) associated with an inhalable antibiotic product [44]. Although the mannosylated drugamer systems did not cause any acute lung toxicity/injury, future studies are needed to characterize potential issues of the mannose-targeted ciprofloxacin in distributed organs and the reticuloendothelial system (RES) such as Kupffer cells in the liver. The ability to control the PK profile allows the design of dosing profiles to match antibiotic dosing requirements, e.g. by maximizing Cmax and AUC for concentration-dependent antibiotics (e.g., fluoroquinolones and aminoglycoside) and extending T > MIC for time-dependent antibiotics (e.g., β-lactam and macrolides) [45,46], The lung and alveolar macrophage PK profiles of both VC and CTM drugamers show greater AUC and extended T > MIC than free drug (Figure 5 & Table 2)
Though in this disease model the CTM was good enough to effect full cohort survival, the PK profiles of the VC drugamers were highly superior to the CTM drugamer and free drug in all the three PK parameters- Cmax, AUC, and T > MIC. This prominent difference between the cellular PK profiles of VC and CTM drugamers further demonstrates the modularity of the drugamer platform in targeting and enhancing intracellular release and dosing profiles. It is notable for future therapeutic and prophylactic studies that the dosing provided by the VC drugamer was sustained at the longest timepoint monitored of 48 h with intracellular ciprofloxacin concentration three magnitudes higher than MIC (0.03 μg/mL) [36,47], While the intracellular Cmax level of VC drugamer was found to be comparable to that of a liposomal ciprofloxacin formulation reported previously [41], the VC drugamer demonstrated markedly longer retention of the released ciprofloxacin in the AMs (24 h for liposomal ciprofloxacin, > 48 h for VC drugamer), highlighting the sustained release characteristics of the drugamer platform. Future studies with a further optimized quantitative method will be conducted to evaluate the time frame that drugamers keep the released drug concentrations higher than MIC and also the subsequent duration when the concentrations is below MIC.
The superior antibiotic efficacy of the mannose-targeted drugamers was demonstrated in a highly lethal murine model of pulmonary F. novicida infection (Figure 6). The full protection provided by both mannose-targeted drugamers (VC and CTM) compared to past pegylated drugamers in this model can be attributed to their increased internalization and sustained intracellular dosing achieved for both linkers [21], The mannose-targeted CTM drugamer exhibits an excellent dosing profile that is sufficient to achieve full cohort survival in this Francisella challenge model and treatment regime. Thus it is not possible for the VC drugamer to show improvement in survival but the PK analysis clearly shows the strong improvement in dosing profile and extended intracellular dosing levels. We anticipate that these increases in the PK properties (Cmax, AUC, and T > MIC) of the VC drugamer will provide future opportunities for prophylactic therapy or for treatment of more advanced disease presentations. Additionally, the PK profiles of the VC drugamer could potentially fulfill the dosing requirements of both concentration- and time-dependent antibiotic classes. Taken together, the unique PK profiles and high activity may allow the mannose-targeted prodrug system (especially the VC drugamer) to be a useful PK modifier for improving the dosing regimens (e.g., reduce dosage and frequency) and for prophylactic application of different classes of antibiotics against pulmonary intracellular infections.
5. Conclusion
Pulmonary intracellular infections, such as F. tularensis and other CDC-classified Tier 1 pathogens (e.g. Burkholderia pseudomallei, the causative agent of melioidosis), require immediate medical intervention with localized and sustained intracellular delivery of concentrated drugs. We have demonstrated that a polymeric antibiotic prodrug platform can be engineered to dramatically enhance intracellular PK drug release profiles by incorporating mannose-targeting ligands and enzymatic drug release. Both targeted drugamers (VC & CTM) provided higher AUC and persistent ciprofloxacin dosing above the minimum inhibitory concentration over a 48 h period in the lung and alveolar macrophages, resulting in full protection in a lethal Francisella murine challenge model. The overall enhancement of the intracellular PK properties (Cmax, AUC, and T > MIC) of the protease-responsive drugamer suggests the potential utility of this platform for prophylaxis or for treatment of more advanced disease presentations. Collectively, this drugamer platform creates new opportunities to construct effective antimicrobial systems with modular control of intracellular PK properties that match the dosing requirements of specific drug classes.
Supplementary Material
Highlights.
Enzyme-cleavable prodrug increases intracellular dosing profile >10x
Antibiotic produgs achieve full survival in lethal Francisella model
Drugamers achieve unique pharmacokinetics/efficacy for pulmonary infection therapy
6. Acknowledgements
The authors thank LC-MS/MS support provided by School of Pharmacy’s Mass Spectrometry Center at University of Washington. This work was supported by the Defense Threat Reduction Agency (Grant #HDTRA1-13-1-0047) and by the NM (R01AI134729). F.Y.S. is an international student research fellow of the Howard Hughes Medical Institute and J.C. was a graduate research fellow of the National Science Foundation.
Footnotes
Competing financial interests
The authors declare no competing financial interests.
Appendix A. Supplementary information
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
9. Reference
- [1].Estimates of the global, regional, and national morbidity, mortality, and aetiologies of lower respiratory tract infections in 195 countries: a systematic analysis for the Global Burden of Disease Study 2015, Lancet Infect. Dis 17 (2017) 1133–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Houben RMGJ, Dodd PJ, The global burden of latent tuberculosis infection: A re estimation using mathematical modelling, PLoS Med. 13 (2016) el002152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].The global burden of tuberculosis: results from the Global Burden of Disease Study 2015, Lancet Infect. Dis 18 (2018) 261–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].World Health Organization, Global Tuberculosis Report 2017, 2017. [Google Scholar]
- [5].Lehar SM, Pillow T, Xu M, Staben L, Kajihara KK, Vandlen R, et al. , Novel antibody-antibiotic conjugate eliminates intracellular S. aureus, Nature. 527 (2015) 323–328. [DOI] [PubMed] [Google Scholar]
- [6].Russell DG, Mycobacterium tuberculosis: here today, and here tomorrow, Nat. Rev. Mol. Cell Biol. 2 (2001) 569–577. [DOI] [PubMed] [Google Scholar]
- [7].Hall JD, Woolard MD, Gunn BM, Craven RR, Taft-Benz S, Frelinger JA, et al. , Infected-host-cell repertoire and cellular response in the lung following inhalation of Francisella tularensis Schu S4, LVS, or U112, Infect. Immun. 76 (2008) 5843–5852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Kubica M, Guzik K, Koziel J, Zarebski M, Richter W, Gajkowska B, et al. , A potential new pathway for Staphylococcus aureus dissemination: the silent survival of S. aureus phagocytosed by human monocyte-derived macrophages, PLoS ONE. 3 (2008) el409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].World Health Organization, Treatment of tuberculosis, World Health Organization, 2010. [Google Scholar]
- [10].Dennis DT, Inglesby TV, Henderson DA, Bartlett JG, Ascher MS, Eitzen E, et al. , Tularemia as a biological weapon, Jama. 285 (2001) 2763. [DOI] [PubMed] [Google Scholar]
- [11].Dance D, Treatment and prophylaxis of melioidosis, Int. J. Antimicrob. Agents. 43 (2014) 310–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Carryn S, Chanteux H, Serai C, Mingeot-Leclercq M-P, Van Bambeke F, Tulkens PM, Intracellular pharmacodynamics of antibiotics, Infect. Dis. Clin. North Am 17 (2003) 615–634. [DOI] [PubMed] [Google Scholar]
- [13].Van Bambeke F, Barcia-Macay M, Lemaire S, Tulkens PM, Cellular pharmacodynamics and pharmacokinetics of antibiotics: current views and perspectives, Curr. Opin. Drug Discov. Devel 9 (2006) 218–230. [PubMed] [Google Scholar]
- [14].de Jager P, van Altena R, Hearing loss and nephrotoxicity in long-term aminoglycoside treatment in patients with tuberculosis, Int. J. Tuberc. Lung Dis 6 (2002) 622–627. [PubMed] [Google Scholar]
- [15].Aschenbrenner DS, The FDA revises boxed warning for fluoroquinolones-again, Am. J. Nurs 116 (2016)22–23. [DOI] [PubMed] [Google Scholar]
- [16].Hamidi M, Azadi A, Rafiei P, Ashrafi H, A pharmacokinetic overview of nanotechnology-based drug delivery systems: an ADME-oriented approach, Crit. Rev. Ther. Drug Carrier Syst 30 (2013) 435–467. [DOI] [PubMed] [Google Scholar]
- [17].Hamblin KA, Armstrong SJ, Barnes KB, Davies C, Wong JP, Blanchard JD, et al. , Liposome encapsulation of ciprofloxacin improves protection against highly virulent Francisella tularensis strain Schu S4, 58 (2014) 3053–3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Cipolla D, Blanchard J, Gonda I, Development of liposomal ciprofloxacin to treat lung infections, Pharmaceutics. 8 (2016) 1–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Chang M, Zhang F, Wei T, Zuo T, Guan Y, Lin G, et al. , Smart linkers in polymer–drug conjugates for tumor-targeted delivery, J. Drug Target. 24 (2015) 475–491. [DOI] [PubMed] [Google Scholar]
- [20].Das D, Srinivasan S, Kelly AM, Chiu DY, Daugherty BK, Ratner DM, et al. , RAFT polymerization of ciprofloxacin prodrug monomers for the controlled intracellular delivery of antibiotics, Polym. Chem 7 (2016) 826–837. [Google Scholar]
- [21].Das D, Chen J, Srinivasan S, Kelly AM, Lee B, Son H-N, et al. , Synthetic macromolecular antibiotic platform for inhalable therapy against aerosolized intracellular alveolar infections, Mol. Pharmaceutics. 14 (2017) 1988–1997. [DOI] [PubMed] [Google Scholar]
- [22].Chen J, Son H-N, Hill JJ, Srinivasan S, Su F-Y, Stayton PS, et al. , Nanostructured glycopolymer augmented liposomes to elucidate carbohydrate-mediated targeting, Nanomedicine. 12 (2016) 2031–2041. [DOI] [PubMed] [Google Scholar]
- [23].Doronina SO, Toki BE, Torgov MY, Mendelsohn BA, Cerveny CG, Chace DF, et al. , Development of potent monoclonal antibody auristatin conjugates for cancer therapy, Nat. Biotech 21 (2003) 778–784. [DOI] [PubMed] [Google Scholar]
- [24].Zhou C, Lehar S, Gutierrez J, Rosenberger CM, Ljumanovic N, Dinoso J, et al. , Pharmacokinetics and pharmacodynamics of DSTA4637A: A novel THIOMAB antibody antibiotic conjugate against Staphylococcus aureus in mice, MAbs. 8 (2016) 1612–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Kopecek J, Rejmanova P, Strohalm J, Ulbrich K, Rihova B, Chytry V, et al. , Synthetic polymeric drugs, US patent 5037883. [Google Scholar]
- [26].Kopecek J, Duncan R, Targetable polymeric prodrugs J. Controlled Release. 6 (1987) 315–327. [Google Scholar]
- [27].Convertine AJ, Benoit DSW, Duvall CL, Hoffman AS, Stayton PS, Development of a novel endosomolytic diblock copolymer for siRNA delivery, J. Controlled Release. 133 (2009)221–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Dubowchik GM, Firestone RA, Padilla L, Willner D, Hofstead SJ, Mosure K, et al. , Cathepsin B-labile dipeptide linkers for lysosomal release of doxorubicin from internalizing immunoconjugates: Model studies of enzymatic drug release and antigen-specific in vitro anticancer activity, Bioconjug. Chem 13 (2002) 855–869. [DOI] [PubMed] [Google Scholar]
- [29].Kingry LC, Petersen JM, Comparative review of Francisella tularensis and Francisella novicida, Front. Cell. Infect. Microbiol. 4 (2014) 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Moad G, Chiefari J, Chong YK, Krstina J, Mayadunne R, Postma A, et al. , Living free radical polymerization with reversible addition-fragmentation chain transfer (the life of RAFT), Polymer International. 49 (2000) 993–1001. [Google Scholar]
- [31].Fonović M, Turk B, Cysteine cathepsins and extracellular matrix degradation, matrix-mediated cell behaviour and properties. Biochim. Biophys. Acta. 1840 (2014) 2560–2570. [DOI] [PubMed] [Google Scholar]
- [32].Lockridge O, Review of human butyrylcholinesterase structure, function, genetic variants, history of use in the clinic, and potential therapeutic uses, Pharmacol. Ther 148 (2015) 34–46. [DOI] [PubMed] [Google Scholar]
- [33].Liu W, Wan J, Han J-Z, Li C, Feng D-D, Yue S-J, et al. , Antiflammin-1 attenuates bleomycin-induced pulmonary fibrosis in mice, Respir. Res 14 (2013) 101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Malaviya R, Laskin JD, Laskin DL, Anti-TNFalpha therapy in inflammatory lung diseases, Pharmacol. Ther 180 (2017) 90–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Mazzon E, Cuzzocrea S, Role of TNF-α in lung tight junction alteration in mouse model of acute lung inflammation, Respir. Res 8 (2007) 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Johansson A, Berglund L, Gothefors L, Sjostedt A, Tamvik A, Ciprofloxacin for treatment of tularemia in children, Pediatr. Infect. Dis J 19 (2000) 449–453. [DOI] [PubMed] [Google Scholar]
- [37].Cohen JS, Peripheral neuropathy associated with fluoroquinolones, Ann. Pharmacother 35 (2001)1540–1547. [DOI] [PubMed] [Google Scholar]
- [38].Tennyson LE, Averch TD, An update on fluoroquinolones: the emergence of a multisystem toxicity syndrome, Urology Practice. 4 (2017) 383–387. [DOI] [PubMed] [Google Scholar]
- [39].Lowes DA, Wallace C, Murphy MP, Webster NR, Galley HF, The mitochondria targeted antioxidant MitoQ protects against fluoroquinolone-induced oxidative stress and mitochondrial membrane damage in human Achilles tendon cells, Free Radic. Res 43 (2009) 323–328. [DOI] [PubMed] [Google Scholar]
- [40].Oyston PCF, Sjostedt A, Titball RW, Tularaemia: bioterrorism defence renews interest in Francisella tularensis, Nat. Rev. Microbiol 2 (2004) 967–978. [DOI] [PubMed] [Google Scholar]
- [41].Chono S, Tanino T, Seki T, Morimoto K, Efficient drug targeting to rat alveolar macrophages by pulmonary administration of ciprofloxacin incorporated into mannosylated liposomes for treatment of respiratory intracellular parasitic infections, J. Controlled Release. 127 (2008) 50–58. [DOI] [PubMed] [Google Scholar]
- [42].Su F-Y, Chen J, Son H-N, Kelly AM, Convertine AJ, West TE, et al. , Polymer-augmented liposomes enhancing antibiotic delivery against intracellular infections, Biomater. Sci 6 (2018) 1976–1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Senter PD, Sievers EL, The discovery and development of brentuximab vedotin for use in relapsed Hodgkin lymphoma and systemic anaplastic large cell lymphoma, Nat. Biotechnol 30 (2012) 631–637. [DOI] [PubMed] [Google Scholar]
- [44].Barker AF, Couch L, Fiel SB, Gotfried MH, Ilowite J, Meyer KC, et al. , Tobramycin solution for inhalation reduces sputum Pseudomonas aeruginosa density in bronchiectasis, Am. J. Respir. Crit. Care Med 162 (2000) 481–485. [DOI] [PubMed] [Google Scholar]
- [45].Jacobs MR, Optimisation of antimicrobial therapy using pharmacokinetic and pharmacodynamic parameters, Clinical Microbiology and Infection. 7 (2001) 589–596. [DOI] [PubMed] [Google Scholar]
- [46].Leekha S, Terrell CL, Edson RS, General principles of antimicrobial therapy, Mayo. Clin. Proc 86 (2011) 156–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Urich SK, Petersen JM, In vitro susceptibility of isolates of Francisella tularensis types A and B from north America, Antimicrob. Agents Chemother. 52 (2008) 2276–2278. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.