

Abstract
In the development of antidotal therapy for treatment of organophosphate exposure from pesticides used in agriculture and nerve agents insidiously employed in terrorism, the alkylpyridinium aldoximes have received primary attention since their early development by I. B. Wilson in the 1950s. Yet these agents, by virtue of their quaternary structure, are limited in rates of crossing the blood-brain barrier, and they require administration parenterally to achieve full distribution in the body. Oximes lacking cationic charges or presenting a tertiary amine have been considered as alternatives. Herein, we examine the pharmacokinetic properties of a lead ionizable, zwitterionic hydroxyiminoacetamido alkylamine in mice to develop a framework for studying these agents in vivo and generate sufficient data for their consideration as appropriate antidotes for humans. Consequently, in vitro and in vivo efficacies of immediate structural congeners were explored as leads or backups for animal studies. We compared oral and parenteral dosing, and we developed an intramuscular loading and oral maintenance dosing scheme in mice. Steady-state plasma and brain levels of the antidote were achieved with sequential administrations out to 10 hours, with brain levels exceeding plasma levels shortly after administration. Moreover, the zwitterionic oxime showed substantial protection after gavage, whereas the classic methylpyridinium aldoxime (2-pyridinealdoxime methiodide) was without evident protection. Although further studies in other animal species are necessary, ionizing zwitterionic aldoximes present viable alternatives to existing antidotes for prophylaxis and treatment of large numbers of individuals in terrorist-led events with nerve agent organophosphates, such as sarin, and in organophosphate pesticide exposure.
Introduction
Treatment of pesticide and nerve agent poisoning (Dolgin, 2013) has relied on a combination of agents, such as antagonists to minimize excessive muscarinic stimulation affecting cardiovascular and respiratory parameters, anticonvulsant agents to avert seizures presumably initiated from central acetylcholine accumulation, and acetylcholinesterase (AChE) reactivating agents as antidotes to reduce the excessive acetylcholine levels through cholinesterase reactivation (Somani et al., 1992; Taylor, 2018). The classic reactivating agents are derivatives of an N-methylpyridinium 2-aldoxime lead compound, 2-pyridinealdoxime methiodide (2-PAM), which was shown over 60 years ago to be site directed to the then-presumed active site of AChE (Wilson, 1951, 1959; Wilson and Ginsburg, 1955). These agents, although intrinsically active in reactivating the organophosphate (OP)–conjugated AChE, are limited to their peripheral activity in being unable to cross the blood-brain barrier in appreciable concentrations to reactivate central AChE (Bodor et al., 1975; Shih et al., 2012; Harvilchuck et al., 2013; Worek and Thiermann, 2013). Since the OPs are typically neutral molecules, and several have high vapor pressures, they easily cross the blood-brain barrier and therefore inhibit central as well as peripheral AChE. Similar to other quaternary amine congeners interacting at cholinergic target sites, quaternary antidotes are poorly and very slowly absorbed when administered orally (Somani et al., 1992; Taylor, 2018).
To circumvent these intrinsic limitations, investigators have explored other alternatives. These alternatives have included administering protopam, a tertiary prodrug that can cross the blood-brain barrier and through reduction in situ form the desired agent 2-PAM (Bodor et al., 1975); attaching 2-PAM or its analogs to various transportable sugars in which the oxime becomes the cargo (Garcia et al., 2010; Bhonsle et al., 2013); attaching extended hydrophobic moieties to classic pyridinium aldoximes (Chambers et al., 2016); employing small neutral oximes, and other tertiary oximes or amidines that may have multiple ionization states (Rajapurkar and Koelle, 1958; de Koning et al., 2011; Kalisiak et al., 2011; Mercey et al., 2011; Renou et al., 2013; Kliachyna et al., 2014). Recognizing the importance of the limited angle of access of the nucleophile within the narrow and tortuous active center gorge, which is further impacted with the conjugated OP, we have developed a series of oximes, the hydroximinoacetamido alkylamines (Sit et al., 2011; Radić et al., 2012). Here the aldoxime is attached to a carbamoyl residue of small steric dimensions compared with the quaternary methylpyridinium moiety, thereby enhancing the angle of attack at the electrophilic, conjugated phosphorus. This minimizes orientation constraints on an SN2 nucleophilic attack by the oxime or oximate in forming a phospho-oxime and/or in cleaving the phosphonyl or phosphoryl serine bond.
The ionization equilibria of such zwitterionic molecules offer the possibility of achieving sufficient neutral species at physiologic pH to cross the blood-brain barrier, as well as presenting a cationic species upon entry to the AChE active center gorge. Oximes, among the candidate nucleophiles, also present an intrinsic capacity to form an oximate anion for nucleophilic attack at the conjugated phosphorus atom. This process may be facilitated by proton abstraction in the active center gorge.
Scheme 1 shows the four species of the hydroxyiminoacetamido alkyl amines that exist in various fractional concentrations at physiologic pH resulting from the protonation state of the amine and loss of a proton from the oxime.
Scheme 1.

Ionization equilibria of amino oximes with two ionizing groups proceed through formation of a neutral or zwitterionic species. CNS-penetrant species are highlighted in light gray, and dominant OP-AChE binding and catalytic species are highlighted in gray.
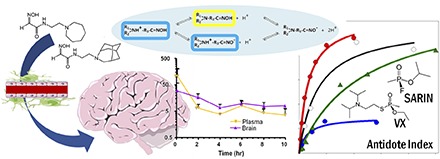
We examine here the ionization equilibria, pharmacokinetic properties, and oral bioavailability of RS194B [N-(2-(azepan-1-yl)ethyl)2-hydroxyimino)acetamide] as a lead compound in the hydroxyiminoacetamido alkylamine series. Furthermore, we designed and synthesized 11 immediate structural congeners of RS194B to rank order the respective reactivation rates. Given its favorable antidote index, defined by the oxime-elicited reactivation rate of OP-conjugated human acetylcholinesterase (hAChE) in vitro, relative to the LD50 toxicity in mice (Radić et al., 2012), we developed a profile of pharmacokinetic studies directed to RS194B effectiveness as an oral and parenteral antidote. In addition, we examine the capacity of a single dose of RS194B to reactivate blood and brain AChE and butyrylcholinesterase (BChE) in mice exposed to sublethal doses of sarin [isopropyl methylphosphonoflouridate] and VX [O-ethyl S-(2-diisopropylaminoethyl) methylphosphonothioate].
Materials and Methods
Chemicals and Enzyme.
Low-toxicity, nonvolatile fluorescent methylphosphonates were used in all in vitro experiments as analogs of the following methylphosphonate nerve agents: sarin, cyclosarin, and VX. These surrogate analogs differ from actual nerve agent OPs, only by the structure of their respective leaving groups (Sit et al., 2011; Radić et al., 2012). Inhibition of hAChE by these fluorescent analogs results in OP-hAChE covalent conjugates identical to the ones formed upon inhibition with the corresponding volatile, nerve agent OPs. Their lack of volatility ensures greater safety in laboratory use, and the reaction can also be monitored by release of the fluorophore. Paraoxon was purchased from Sigma (St. Louis, MO). Nerve agent OPs used in in vivo experiments were purchased from NC Laboratory (Spiez, Switzerland). 2-PAM was purchased from Sigma (St. Louis, MO).
Our previous studies indicated that the tertiary amine-containing region of the zwitterionic oxime molecule is a critical modulator of activity (Sit et al., 2011, 2014; Radić et al., 2012). Herein, we explored the effect of the tertiary amine ring flexibility and dimensions on the rate of reactivation of OP-inhibited AChE. Building on our lead compound (RS194B), we investigate here 11 additional derivatives (Table 1).
TABLE 1.
Kinetic parameters for reactivation of various OP conjugates with hAChE
| Oxime | Oxime Structure | Chemical Name | OP | k2 | KOX | kr |
|---|---|---|---|---|---|---|
| min−1 | mM | M−1 min−1 | ||||
| RS194Ba | 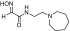 |
N-(2-(azepan-1-yl)ethyl)-2-(hydroxyimino)acetamide | VX | 3.1 | 2.1 | 1600 |
| Sarin | 2.5 | 1.9 | 1300 | |||
| Cyclosarin | 0.88 | 3.9 | 230 | |||
| Paraoxon | 0.38 | 7.4 | 51 | |||
| RS2-138B | 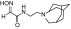 |
N-(2-(4-azatricyclo[4.3.1.13,8]undecan-4-yl)ethyl)-2-(hydroxyimino)acetamide | VX | 2.6 | 1.6 | 1600 |
| Sarin | 2.4 | 1.2 | 2000 | |||
| Cyclosarin | 0.83 | 6.8 | 120 | |||
| Paraoxon | 0.23 | 2.2 | 100 | |||
| RS3-43D | 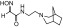 |
N-(2-(3-azabicyclo[3.2.1]octan-3-yl)ethyl)-2-(hydroxyimino)acetamide | VX | 1.6 | 1.3 | 1200 |
| Sarin | 1.1 | 0.8 | 1400 | |||
| Cyclosarin | 0.63 | 3.3 | 190 | |||
| Paraoxon | 0.22 | 11 | 19 | |||
| RS2-150C | 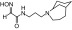 |
N-(3-(4-azatricyclo[4.3.1.13,8]undecan-4-yl)propyl)-2-(hydroxyimino)acetamide | VX | — | — | ≤1600 |
| Sarin | — | — | ≤2000 | |||
| Cyclosarin | — | — | ≤30 | |||
| Paraoxon | — | — | ≤70 | |||
| RS3-36D | 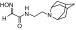 |
N-(2-(2-azaadamantan-2-yl)ethyl)-2-(hydroxyimino)acetamide | VX | 6.7 | 7.7 | 870 |
| Sarin | >10 | >10 | 820 | |||
| Cyclosarin | 0.69 | 5.9 | 120 | |||
| Paraoxon | 0.71 | 29 | 25 | |||
| RS2-148B | 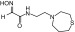 |
N-(2-(1,4-thiazepan-4-yl)ethyl)-2-(hydroxyimino)acetamide | VX | — | — | ≤360 |
| Sarin | — | — | ≤2000 | |||
| Cyclosarin | — | — | ≤30 | |||
| Paraoxon | — | — | ≤8 | |||
| RS2-237D | 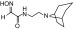 |
N-(2-(8-azabicyclo[3.2.1]octan-8-yl)ethyl)-2-(hydroxyimino)acetamide | VX | 2.1 | 3.6 | 570 |
| Sarin | 2.9 | 4.9 | 590 | |||
| Cyclosarin | — | — | 89 | |||
| Paraoxon | 0.15 | 5.2 | 29 | |||
| RS2-234D | 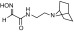 |
N-(2-(7-azabicyclo[2.2.1]heptan-7-yl)ethyl)-2-(hydroxyimino)acetamide | VX | 3.2 | 6.2 | 520 |
| Sarin | 1.7 | 1.8 | 940 | |||
| Cyclosarin | 0.79 | 15 | 51 | |||
| Paraoxon | 0.14 | 4.6 | 30 | |||
| RS2-140B | 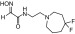 |
N-(2-(4,4-difluoroazepan-1-yl)ethyl)-2-(hydroxyimino)acetamide | VX | — | — | ≤300 |
| Sarin | — | — | ≤600 | |||
| Cyclosarin | — | — | ≤30 | |||
| Paraoxon | — | — | ≤8 | |||
| RS2-245C | 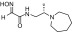 |
(S)-N-(2-(azepan-1-yl)propyl)-2-(hydroxyimino)acetamide | VX | 0.5 | 4.7 | 110 |
| Sarin | 0.41 | 4.1 | 100 | |||
| Cyclosarin | 0.47 | 9.5 | 49 | |||
| Paraoxon | 0.04 | 5.9 | 7 | |||
| RS2-90C | 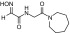 |
N-(2-(azepan-1-yl)-2-oxoethyl)-2-(hydroxyimino)acetamide | VX | — | — | ≤1 |
| Sarin | — | — | ≤1 | |||
| Cyclosarin | — | — | ≤1 | |||
| Paraoxon | — | — | ≤1 | |||
| RS2-292A | 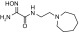 |
N-(2-(azepan-1-yl)ethyl)-2-(hydroxyamino)-2-iminoacetamide | VX | — | — | ≤1 |
| Sarin | — | — | ≤1 | |||
| Cyclosarin | — | — | ≤1 | |||
| Paraoxon | — | — | ≤1 | |||
| 2-PAMa | 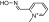 |
2-Pyridinealdoxime methiodide | VX | 0.73 | 0.3 | 2400 |
| Sarin | 1.1 | 0.34 | 3200 | |||
| Cyclosarin | 0.73 | 6.6 | 110 | |||
| Paraoxon | 0.27 | 1.8 | 150 |
Rates of reactivation were studied as a function of oxime concentration and kinetic parameters: KOX and k2 were calculated from the concentration dependencies, and kr is the ratio of k2 and KOX. Dashes indicate a linear concentration dependence, precluding a calculation of constants.
Data are from Radić et al. (2012).
Synthetic schemes and characterizations of each derivative are detailed in the Supplemental Material. Highly purified monomeric hAChE was prepared as described earlier (Sit et al., 2011; Radić et al., 2012).
In Vitro Oxime Reactivation Assays.
In in vitro assays, we employed fluorescent analogs of the toxic methylphosphonates, which are nonvolatile to minimize laboratory risk. These analogs form a conjugate identical to the nerve agent of congeneric structure (Radić et al., 2012) and similar enantiomeric ratio of the racemic nerve agent (Kovarik et al., 2004). hAChE activities were measured using a spectrophotometric assay (Ellman et al., 1961) at 37°C in 0.1 M sodium phosphate buffer, pH 7.4, containing 0.01% bovine serum albumin and substrate acetylthiocholine. OP-hAChE conjugates were prepared, and oxime reactivation was performed and analyzed as described earlier (Radić et al., 2012). A previous study (Kovarik et al., 2004) described evaluation of the dependence of reactivation rates (kobs) on oxime concentrations, as well as the determination of maximal reactivation rate constant k2, an apparent Michaelis-Menten type constant Kox, and the overall second-order reactivation rate constant kr. kobs, obtained from in vitro reactivation of hAChE, was then related to the LD50 in mice to obtain an antidote index, analogous to the therapeutic index, employed in efficacy rankings of pharmacologic agents.
Acute Oxime Toxicity and Oxime Treatment of OP-Exposed Mice.
Male CD-1 mice (25-30 g body weight) were purchased from Rudjer Bošković Institute (Zagreb, Croatia) and maintained as previously described (Radić et al., 2012). For the experimental sequences, mice were randomly divided into groups of four. Mice were treated in accord with approval from the Ethics Committee of the Institute for Medical Research and Occupational Health in Zagreb, Croatia.
Acute toxicity (LD50) was based on 24-hour mortality rates calculated according to Thompson (1947) and Weil (1952). Antidotal activity against OP exposure was tested by intramuscularly administering oximes (at the specified dose) to male CD-1 mice, together with atropine sulfate (10 mg/kg) 1 minute after subcutaneous OP administration. Alternatively, mice were pretreated orally with oximes by gavage (at the specified dose, but without atropine) 15 minutes before subcutaneous OP administration. Concentrated stock solutions of nerve agents were prepared in isopropyl alcohol. Further dilutions were made in saline immediately before use.
Antidotal efficacy of oximes in the intact mouse was expressed as a protective index (PI) with 95% confidence limits and the maximal dose of organophosphate poison (MDP). The PI is the ratio of LD50 values between OP with oxime with atropine and OP given alone. The MDP is the highest multiple dose of the OP LD50 that was fully counteracted by the antidotal treatment.
Blood and Brain AChE and Blood BChE Activity Measurements.
Mice were divided into five groups and administered OP and antidotes as follows: 1) saline control: intramuscular administration with saline only (2 ml/kg); 2a and 2b) separate OP controls: subcutaneous administration with a 0.75 LD50 dose of VX (LD50 = 28.3 μg/kg body weight) or a 0.75 LD50 dose of sarin (LD50 = 864 μg/kg body weight); and 3a and 3b) intramuscular administration with RS194B (125 mg/kg) plus atropine (10 mg/kg) 1 minute after the above subcutaneous OP exposure (Thiermann et al., 2011). All animals were humanely euthanized at 0.5, 1, 3, or 6 hours after the antidotal treatments.
Blood was drawn directly from the heart into heparinized vacutainers (Becton Dickenson, Plymouth, UK), diluted 25 times in 0.1 M Na phosphate buffer, pH 7.4, and stored overnight at –20°C. Prior to activity measurements, samples were centrifuged for 10 minutes at 4°C and 2100g. Brain samples were rinsed with saline, frozen in liquid nitrogen, and stored at –20°C. To prepare 40 mg/ml homogenates, brains were weighed and diluted in a corresponding volume of 1% Triton X-100 in 0.1 M Na phosphate buffer, pH 7.4. After homogenization, samples were sonicated for 10 minutes in an ultrasonic bath filled with ice. The supernatants from 21,255g centrifugation for 30 minutes were assayed for activity promptly after preparation (Ellman at al., 1961). Final sample dilutions were 200 times for blood and 2000 times for brain samples. AChE and BChE activity was determined using selective inhibitors 10 µM BW284C51 [4,4′-(3-oxo-1,5-pentanediyl)bis(N-allyl-N,N-dimethylanilinium)] (Sigma, St. Louis, MO) and 20 µM ethopropazine (Sigma, St. Louis, MO), respectively, as described previously (Katalinić et al., 2015).
Oxime Pharmacokinetics in Mice.
Female CD-1 mice aged 4−8 weeks (22−34 g body weight) were purchased from Harlan (Livermore, CA). Mice were fed Purina Certified Rodent Chow 5002 (Purina, St. Louis MO). Food and purified water were provided ad libitum. Mice were kept in hanging polycarbonate cages at 21−23°C, and light and dark cycles were exchanged every 12 hours. General procedures for animal care and housing were in accordance with the 1996 National Research Council Guide for the Care and Use of Laboratory Animals and the 1991 Animal Welfare Standards (incorporated in U.S. 9 CFR Part 3).
For the pharmacokinetic single dose and multiple loading and maintenance doses, mice were divided into groups of three for parenteral dosing and oral dosing by gavage. In the single-dose pharmacokinetic studies, RS194B was administered by various routes in the absence of OP: 20 mg/kg i.v., 80 mg/kg i.m., and 50 and 200 mg/kg p.o. (gavage). Three animals were injected for every time point analyzed, and blood (approximately 300 μl) was collected at each time point from the retro-orbital sinus of mice under isoflurane anesthesia into tubes containing EDTA, processed to plasma within 30 minutes of collection, and then stored frozen at ≤−80 ± 10°C.
In the sequential loading and maintenance dose studies, mouse brains and plasma were collected and analyzed individually at each time point. Brain weight was documented for each animal before storage on dry ice. Brains were stored at ≤–80 ± 10°C until analysis. Concentration of the oxime in body compartments was determined by liquid chromatography–mass spectrometry using multiple reaction monitoring electrospray ionization detection in positive ion mode (Radić et al., 2012).
Results
In Vitro Reactivation Kinetics by Hydroxyimino Acetamido Alkyl Cyclic Amines.
In searching for efficient reactivators with the RS194B structural scaffold, we tested in vitro reactivity of 11 new congeners (Table 1) in addition to analogs studied previously (Sit et al., 2011; Radić et al., 2012; Kovarik et al., 2013). Several reactivators of moderate potency were identified, in which the azepine ring of RS194B was replaced by bridged azatricyclo undecane (RS2-138B and RS2-150C) or azabicyclo octane (RS3-43D) rings. Substitution by aza adamantane (RS3-36D), azabicyclo octane (RS2-237D), and heptane (RS2-234D), which create larger steric bulk in the immediate vicinity of the heteroatom, resulted in reduced reactivation potential due to a diminished reversible binding component (an increase in Kox). Substitutions of the azepine ring (RS2-140B) or within its linker region (RS2-245C and RS2-90C) reduced reactivation parameters by compromising both the reversible interaction (Kox) and the optimal approach to the reactivation target, the conjugated phosphate (reduction in k2). The most significant compromise of reactivation resulted from steric/electronic distortion of aldoxime (RS2-292A) and alkyl (RS2-90C) substitutions. It is also evident that reactivation parameters for the oxime antidotes vary substantially with the conjugated OP. Through this and prior analyses, we created four nearly equipotent, uncharged acetamido RS194B congeners as potential leads.
Reactivation Capacity Measured as an Antidote Index.
Our previous studies comparing RS194B and RS41A [N-(2-(pyrrolidinyl)ethyl)-2-(hydroxyimino)acetamide] with 2-PAM in terms of reactivation of hAChE showed comparable properties in terms of reactivation rates and their two deconstructed catalytic parameters, Kox and k2, obtained from an analysis of reactivator concentration dependence against hAChE (Sit et al., 2011; Radić et al., 2012). Superiority against monoisonitrosoacetone (MINA) and diacetylmonoxime (DAM), two other neutral oximes, was also demonstrated (Fig. 1).
Fig. 1.

(Top) Rates of reactivation of paraoxon, sarin, cyclosarin, and VX inhibited hAChE measured as a function of the respective oxime concentration: 2-PAM (blue), RS194B (red), RS2-138B, (black), RS41A (green), MINA (light gray), and DAM (light gray) (Rajapurkar and Koelle, 1958). (Bottom) Calculation of antidote indices comparing the in vitro rates of reactivation of recombinant hAChE conjugated with the specified OPs with the toxic doses (LD50) of the respective oximes. DAM, diacetylmonoxime; MINA, monoisonitrosoacetone.
If we relate these kinetic parameters of hAChE to the toxicity of the antidote in the mouse, the superiority of RS194B, as well as RS2-138B, manifests as an antidote index that relates the reactivation rate constant, kobs, for hAChE measured in vitro to the LD50 of the antidote in the mouse (Fig. 1).
These parameters provide justification for a more detailed pharmacokinetic analysis in the mouse to ascertain the rate of crossing the blood-brain barrier of the oxime, its tissue disposition, plasma lifetime, and clearance within the body.
Single-Dose Comparisons of Plasma and Brain Levels in the Mouse.
In this sequence, we examined the plasma and tissue concentrations after single administrations of RS194B after intravenous, intramuscular, and oral dosing (Fig. 2). After intravenous and intramuscular dosing, plasma levels showed maxima within the first 5 minutes and declined thereafter, with a half-life of approximately 1 hour. Upon oral dosing by gavage, the systemic absorption also appeared to be quite rapid, in which a maximal plasma level was seen within 30 minutes with a similar rate of decline of plasma concentrations as oral absorption diminished. A previous comparison of brain and plasma levels of RS194B after intramuscular administration showed rapid penetration into the central nervous system (CNS), where CNS levels exceeded plasma levels after 30 minutes (Radić et al., 2012). However, the crossover of brain and plasma values arises largely from the rapid decline of plasma concentrations with more retention in the brain. Accordingly, a comparison of plasma levels in Figs. 2 and 3 suggests that oral administration as a maintenance dose after a parenteral loading dose affords a means of sustaining sufficiently high levels of antidote in the brain.
Fig. 2.

Plasma levels after single-dose administration of RS194B to mice by intravenous, intramuscular, and oral (gavage) routes: 20 mg/kg i.v. (inverted triangles), 80 mg/kg i.m. (open squares), 50 mg/kg p.o. (filled circles), and 200 mg/kg p.o. (open circles). Measurements were made at 5, 10, 15, 30, 60, 90, 120, 180, and 240 minutes thereafter in separate groups of three mice.
Fig. 3.

Plasma (open circles) and whole-brain (solid circles) pharmacokinetics of RS194B in mice after an intramuscular loading dose of 220 mg/kg and 2-hour subsequent oral maintenance doses of 110 mg/kg by gavage. Concentrations (in micromoles) are measured with respect to plasma volume and brain weight. An apparent steady state is reached with repetitive dosing out to 10 hours.
Loading and Maintenance Dose Profiles.
To examine antidote levels after an intramuscular loading dose and oral maintenance dose sequence, we administered RS194B by gavage at sequential 2-hour intervals after an initial loading dose given intramuscularly (Fig. 3). The study required separate groups of three mice that were sacrificed immediately or 2 hours after the loading dose, but before the first oral maintenance dose. Others in the group received the loading dose followed by maintenance doses at each 2-hour interval. Each group of mice was sacrificed prior to the time of the projected next loading dose, while the remaining mice received the next dose in the sequence. Thus, at the final 10-hour time point, mice in this group received the loading dose as well as subsequent maintenance doses at 2, 4, 6 and 8 hours and were sacrificed at 10 hours after the initial antidote exposure.
The sequential dosing study shows that within the 2-hour time period, brain concentrations exceed the plasma concentrations, as expected from the previous single-dose kinetic studies. Hence, if elimination approximates a first-order decline after a short distribution period, then an apparent steady state in plasma concentrations is obtained with the 2-hour intervals. If larger numbers of mice were used and plasma and brain samples were taken at various times after each administration, then we might anticipate a “saw tooth”–appearing profile within each injection interval. Importantly, our data shown here reveal no evidence for antidote accumulation when administered successively over the 10-hour interval.
Over this interval, mice were monitored for toxicity symptoms of RS194B administration, and we noted no gross evidence of toxicity at this dose level in terms of compromised motor activity and overt symptoms. Hence, these findings warrant higher-dose studies with samples to be taken from other tissues containing cholinesterase targets or presumed to be involved in the disposition of the antidote.
Antidotal Efficacy of RS194B and RS2-138B as Therapy or Pretreatment in OP-Exposed Mice.
To compare oral efficacies of RS194B and 2-PAM, we administered RS194B and 2-PAM by gavage 15 minutes prior to VX exposure of mice and determined their PIs with increasing doses of VX (Table 2). What is plainly evident is that protection by gavage of RS194B was substantial, whereas no protection was afforded by 2-PAM when given by the same route in mice. We also note in this study that both 2-PAM and RS194B conferred comparable protection when administered intramuscularly 1 minute postexposure to VX, when scaled to 25% of the LD50 of the two oximes.
TABLE 2.
Protection Index (PI) determined in mice for RS194B and 2-PAM after oral administration by gavage compared with intramuscular administration
| Oxime | Dose (Route) | PI
(Confidence Limits) |
|
|---|---|---|---|
| 1 min after VX | 15 min before VX | ||
| mg/kg | |||
| RS194B | 125 (i.m.) | 18 (12.4–25.7) | |
| 200 (p.o.) | 40 (17.2–93) | ||
| 2-PAM | 26 (i.m.) | 9.3 (7.3–13) | |
| 42 (p.o.) | 1.1 (–) | ||
Atropine (10 mg/kg) was administered intramuscularly 1 minute after VX. Increasing doses of VX were administered in groups of four mice, and fractional survival was measured at each OP dose.
The oral efficacy of RS194B in the treatment of VX-exposed mice was further examined with a higher dose and repetitive dosing of RS194B (Table 3). However, the PI was only slightly higher when the oxime dose was increased to 400 mg/kg with only minimal improvement in terms of survival and lessened toxicity symptoms compared with the 200 mg/kg dose.
TABLE 3.
Protection of OP-exposed mice by orally administered RS194B either before or after OP administered subcutaneously
| OP | RS194B | Pretreatment Time before OP Exposure | Therapy 1 min after OP Exposure (Route) | PI (MDP) |
|---|---|---|---|---|
| mg/kg | min | |||
| VX | 400 + 200 + 200 | 6, 4, and 2 h | Atropine (i.m.) | 7.9 (<6.3) |
| 200 | 15 | Atropine (i.m.) | 40.0 (20.0) | |
| 400 | 15 | Atropine (i.m.) | 43.3 (31.8) | |
| 200 | None | RS194B (p.o.) + atropine (p.o.) | 3.2 (<2.5) | |
| 200 | None | RS194B (p.o.) + atropine (i.m.) | 3.2 (2.5) | |
| Paraoxon | 200 | 15 | Atropine (i.m.) | 7.3 (5.0) |
| Tabun | 200 | 15 | Atropine (i.m.) | 3.4 (1.6) |
| Soman | 200 | 15 | Atropine (i.m.) | NDa |
The RS194B oxime was administered either as a single dose or multiple doses at the indicated times. Atropine (10 mg/kg) was always administered 1 minute after OP. Increasing doses of OP were administered in groups of four mice, and fractional survival was determined at each OP dose. The PI is the ratio of LD50 for treated OP-exposed animals and LD50 for animals given OP alone. MDP (the highest multiple dose of OP LD50 fully counteracted by the treatment) is given in parentheses. ND, not determined.
The survival was 1 of 4, 0 of 4, 0 of 4, and 0 of 4 on 1, 1.26, 1.59, and 2.0 × LD50 of soman, respectively.
Repetitive oral dosing of RS194B (a total of 800 mg/kg) at 6, 4, and 2 hours pre-exposure afforded less protection of mice than pre-exposure treatment with 200 mg/kg 15 minutes before OP exposure. This result is consistent with the short half-life and rapid clearance of RS194B. Oral administration of 200 mg/kg RS194B in paraoxon, tabun [N,N-dimethylamido-O-ethyl cyanophosphate], and soman [pinacolyl methylphosphonofluoridate] exposure was not as efficient as intramuscular RS194B administration, as reported previously for RS194B (Radić et al., 2012).
Since in vitro experiments indicate that RS2-138B is a potentially effective VX and sarin reactivator, we tested its antidotal efficacy in the case of different OP exposures: VX, sarin, tabun, paraoxon and soman. The acute toxicity of RS2-138B administered intramuscularly to mice was found to be relatively low (283 mg/kg), even though it appeared more toxic than RS194B (LD50 > 500 mg/kg; Radić et al., 2012). Nevertheless, we tested the efficacy of oxime RS2-138B as therapy or pretreatment in the case of OP-exposed mice with a dose roughly equivalent to 25% of RS2-138 LD50. It was evident that mice recovered better if they were pretreated with oxime RS2-138B (intramuscularly) 15 minutes before OP (subcutaneous) and received therapy (RS2-138B plus atropine, intramuscularly) 1 minute after OP exposure (Table 4). However, the lower acute toxicity and higher PIs determined for intramuscularly administered RS194B (Radić et al., 2012) justify its choice for further in vivo studies on mice. RS2-138B can be considered as a backup, zwitterionic compound.
TABLE 4.
Protection of OP-exposed mice by RS2-138B administered intramuscularly either before or after, or both before and after OP administered subcutaneously
| OP | RS2-138B | Pretreatment 15 min before OP Exposure | Therapy 1 min after OP Exposure | PI (MDP) |
|---|---|---|---|---|
| mg/kg | ||||
| VX | 60 | None | RS2-138B + atropine | 10.8 (7.9) |
| 60 + 60 | RS2-138B | RS2-138B + atropine | 31.8 (25.2) | |
| 60 | RS2-138B | None | 3.4 (2.5) | |
| Sarin | 60 | None | RS2-138B + atropine | 1.6 (1.3) |
| 60 + 60 | RS2-138B | RS2-138B + atropine | 4.0 (3.2) | |
| Tabun | 60 | None | RS2-138B + atropine | 2.0 (1.6) |
| 60 + 60 | RS2-138B | RS2-138B + atropine | 2.7 (2.0) | |
| Paraoxon | 60 | None | RS2-138B + atropine | 5.4 (4.0) |
| 60 + 60 | RS2-138B | RS2-138B + atropine | 12.6 (<7.9) | |
| Soman | 60 | None | RS2-138B + atropine | NDa |
| 60 + 60 | RS2-138B | RS2-138B + atropine | NDb |
Atropine (10 mg/kg) was administered intramuscularly 1 minute after OP. Increasing doses of OP were administered in groups of four mice and fractional survival was determined at each OP dose. The LD50 of RS138B was 238.3 mg/kg i.m. The PI is the ratio of LD50 for treated OP-exposed animals and LD50 for animals given OP alone. The MDP (the highest multiple dose of OP LD50 fully counteracted by the treatment) is given in parentheses. ND, not determined.
Survival was 1 of 4, 0 of 4, 0 of 4, and 0 of 4 on 1, 1.26, 1.59, and 2.0 × LD50 of soman, respectively.
Survival was 0 of 4, 0 of 4, 0 of 4, and 0 of 4 on 1, 1.26, 1.59, and 2.0 × LD50 of soman, respectively.
Blood and Brain Cholinesterase Activity in Sarin- and VX-Exposed Mice upon Administration of RS194B.
Mice in separate groups of four were exposed to the following: 1) sublethal doses of sarin or VX (0.75 LD50, s.c.); 2) sublethal doses of sarin or VX (0.75 LD50, s.c.) and treatment with RS194B (125 mg/kg in 10 mg/kg atropine) administered intramuscularly 1 minute after subcutaneous OP exposure; and 3) control treated with saline only (2 ml/kg, i.m.). AChE and BChE activity was monitored in the whole blood and brain 0.5, 1, 3, and 6 hours after sarin or VX exposure. Although all mice survived the poisoning, symptoms and their intensity differed in regard to OP. Mice exposed to VX showed only mild symptoms of poisoning (i.e., the mice were calm), whereas mice exposed to sarin exhibited immediate moderate to severe tremors that lasted up to 2 hours after exposure. Later the mice were very calm, but they exhibited continued tachycardia. Symptoms were attenuated if mice were treated with oxime RS194B. This difference in symptoms between VX and sarin can be assigned to different physical and chemical properties of sarin and VX (John et al., 2015) and their tissue dispositions, as reported by Shih et al. (2005, 2010). Indeed, blood AChE and BChE activity was significantly inhibited by both OPs, whereas AChE in the brain was more inhibited by sarin than by VX.
The effect of the RS194B oxime in terms of AChE/BChE activity in the whole blood and brain is shown in Fig. 4. Despite large variability, it seems that recovery of brain activity by RS194B in mice exposed to sarin or even VX was evidence for its fast distribution to tissues and efficient blood-brain barrier penetration. The efficacy of antidotal therapy was most evident in the brain of mice exposed to sarin, with the peak in the 1-hour group. The obtained results are in accordance with an RS194B half-life of 1 hour or less in plasma, as well as brain levels exceeding those in plasma after 40 minutes (Radić et al., 2012), staying elevated for longer time periods. RS194B reactivation was not evident in other peripheral tissues studied (data not shown). A similar phenomenon was reported in a study on tabun-exposed rats, in which significant AChE reactivation with a pyridinium oxime was observed only in whole blood (Katalinić et al., 2015).
Fig. 4.

The effects of oxime RS194B (administered intramuscularly) on AChE/BChE activities in whole blood and brain of sarin- or VX-exposed mice at 0.5, 1, 3, and 6 hours after treatment. Activities are expressed as the median and interquartile range of four values obtained for each mouse and sample and compared with the control group (100% value). The control group received 2 ml/kg saline i.m. Open squares indicate mice that were exposed to 0.75 LD50 of sarin or VX subcutaneously. Solid circles indicate mice that were exposed to the same dose of sarin or VX and 1 minute after exposure to OP received RS194B (125 mg/kg with atropine 10 mg/kg, i.m.). Statistical significance between mice that received RS194B and atropine therapy was determined using t tests (two-sided, with 95% confidence interval). Differences were considered significant when *P < 0.05; **P < 0.005; ***P < 0.001.
Discussion
These studies with RS194B and related hydroxyiminoacetamido alkylamines provide justification for further investigations of zwitterionic oxime antidotes in the treatment of OP exposure. The superior efficacy of these hydroxyiminoacetamido alkyl amines may arise from multiple pharmacodynamic and pharmacokinetic considerations that are applicable to antidotes, the principles of which have been shown for candidate therapeutic agents. First, we note that a comparison of antidote indices (Fig. 1) shows that when reactivation rates are scaled to toxicity of the respective antidotes, the values for RS194B exceed those for the 2-PAM standard in mice. The compounds have roughly comparable reactivation rates for hAChE with the various OP conjugates, but the toxicity of 2-PAM in the mouse is 104 mg/kg, whereas it is greater than 500 mg/kg for RS194B (Radić et al., 2012).
When antidote efficacy is analyzed in terms of intrinsic activity of the antidote at the target site, several factors are likely to come into play. First, the quaternary ligands, such as 2-PAM, may be favored in terms of their affinity for the active center over a tertiary amine that exists in association with H2O as a hydronium ion or in hydrogen bonding. Hence, binding or association within the active center gorge is likely reflected in the parameter Kox, an apparent dissociation constant. However, in the case of a nucleophilic reaction at the phosphorus atom in the conjugated alkyl phosphonate or phosphorate, orientation of the bound ligand positioning the oximate nucleophile may become the critical factor. Among the aldoximes, the attached methylpyridinium ring is far larger than the proximal carbamoyl group in our zwitterionic oximes. Hence, within the spatially impacted active center gorge, a smaller molecular moiety proximal to the aldoxime that has electron withdrawing characteristics may be preferred. An extensive analysis of various oximes (in particular, those of the hydroxyiminoacetamido alkylamine series) shows a wide variation in the deconstructed reactivation parameters, Kox and k2, when examined for alkylphosphonates and phosphorates of different structures (Radić et al., 2012). Among the enantiomeric Rp and Sp methylphosphonates, substantial differences exist between enantiomers for reactivation (Wong et al., 2000; Kovarik et al., 2003, 2004). Thus, the more reactive OP enantiomer is also the one with the highest propensity for reactivation, implying a similarity in transition states associated with OP conjugation and oxime- or general base-catalyzed hydrolysis of the cholinesterase conjugate.
A second parameter of efficacy that arises relates to achieving pharmacokinetics and tissue disposition that favor the zwitterionic oximes (e.g., RS194B and RS2-138B) over the quaternary oximes (2-PAM) and their congeners such as MMB4 (oxo-[[1-[[4-(oxoazaniumylmethylidene)pyridin-1-yl]methyl]pyridin-4-ylidene]methyl]azanium;dibromide) and HI-6 ([(E)-[1-[(4-carbamoylpyridin-1-ium-1-yl)methoxymethyl]pyridin-2-ylidene]methyl]-oxoazanium). Here, several factors (both known or predictable and unknown) come into play. The ability of OPs to compromise respiratory and cardiovascular parameters arises from both central and peripheral inhibition of AChE activity and enhanced acetylcholine concentrations at synapses (Edery et al., 1986; Buccafusco, 1996). Moreover, oximes can reverse the actions of locally applied OPs in the ventral (rostral) medulla oblongata (Edery et al., 1986). OPs are hydrophobic compounds; some, such as sarin, have high vapor pressure and would be expected to cross the blood-brain barrier rapidly, exhibiting acute toxicity. They may also become sequestered at or partition into hydrophobic sites, where over time, they may redistribute from these storage sites back into the circulation. Accordingly, an antidote with the potential to cross the blood-brain barrier rapidly would not only reactivate the immediately exposed and inhibited OP-sensitive sites in the CNS but should also prevent secondary inhibition. Hence, there may be a justification for longer-term reactivation potential of an oxime antidote that is retained in the brain. Multiple ionization states, a capacity to be retained in tissue, a larger volume of distribution, and prolonged oral absorption may favor less frequent repeat dosing of the antidote.
Several pharmacokinetic studies have been reported for 2-PAM and the more extended pyridinium structures such as MMB4 and HI-6 (Sidell and Groff, 1971; Baggot et al., 1993; Hong et al., 2013). Typically, small volumes of distribution (0.3–0.5 l/kg) and short half-lives (approximately 30 minutes) are evident. This likely arises from appreciable glomerular filtration and renal tubular secretion of the quaternary amine (Swartz and Sidell, 1974). Thus, antidotes retained within tissues with larger volumes of distribution may carry a pharmacokinetic preference.
A final question relates to the fractional distribution of species at physiologic pH. In the case of RS194B, its pKa values for the protonation of the amine and dissociation of a proton from the oxime forming the oximate are nearly equivalent at 8.8 and 8.9 (Radić et al., 2012). Approximately equal fractions of neutral species and zwitterions exist at physiologic pH values, and one can ask whether the nonionized species population at pH 7.4 is sufficient to meet the pharmacokinetic and pharmacodynamic criteria. In terms of transfer across the blood-brain barrier, the enlarged potential surface area of the blood-brain barrier should allow crossing of the neutral species at a rate sufficient to account for the kinetics of CNS accumulation seen in Fig. 2. RS194B shows transfer into and retention in the brain that is superior to RS41A despite its difference only in ring size of the pyrrole and azepine rings. Although it has yet to be measured, RS2-138B, with its additional methylene groups, may even show greater brain retention. Further analysis should also determine whether carrier-mediated transport processes, efflux from the brain, and/or biliary or renal transport are critical factors governing the plasma pharmacokinetics and tissue disposition.
Identifying efficacious antidotes capable of reactivating OP-inhibited AChE in the central and peripheral nervous systems has proven elusive, despite many years of intense effort. RS194B, compared with several pyridinium aldoximes, did not show high antidote activity when OPs were administered subcutaneously in the guinea pig (Wilhelm et al., 2014). Recent studies have demonstrated that a lethal dose of inhaled sarin vapor and paraoxon aerosol in macaques resulted in a sequelae of symptoms very similar to humans exposed to insecticide poisoning (either by inhalation or ingestion), and these symptoms could be rapidly reversed by intramuscular administration of a centrally acting oxime RS194B (Rosenberg et al., 2017a,b). Herein, our studies in mice and macaques show that RS194B has pharmacokinetic and reactivating capabilities, enabling potentially effective antidotal actions for acute exposure to a volatile OP. Subsequent oral dosing offers the potential of sustained antidotal action for redistributed OP or other thioate OP pesticides that are slowly converted to the active oxons.
Abbreviations
- 2-PAM
2-pyridinealdoxime methiodide
- ACh
acetylcholine
- AChE
acetylcholinesterase
- BChE
butyrylcholinesterase
- BW284C51
4,4′-(3-oxo-1,5-pentanediyl)bis(N-allyl-N,N-dimethylanilinium)
- CNS
central nervous system
- DAM
diacetylmonooxime
- hAChE
human acetylcholinesterase
- HI-6
[(E)-[1-[(4-carbamoylpyridin-1-ium-1-yl)methoxymethyl]pyridin-2-ylidene]methyl]-oxoazanium
- MDP
maximal dose of organophosphate poison
- MINA
monoisonitrosoacetone
- MMB4
oxo-[[1-[[4-(oxoazaniumylmethylidene)pyridin-1-yl]methyl]pyridin-4-ylidene]methyl]azanium
- OP
organophosphate
- PI
protective index
- RA41A
N-(2-(pyrrolidinyl)ethyl)-2-(hydroxyimino)acetamide
- RS2-90C
N-(2-(azepan-1-yl)-2-oxoethyl)-2-(hydroxyimino)acetamide
- RS2-138B
N-(2-(4-azatricyclo[4.3.1.13,8]undecan-4-yl)ethyl)-2-(hydroxyimino)acetamide
- RS2-140B
N-(2-(4,4-difluoroazepan-1-yl)ethyl)-2-(hydroxyimino)acetamide
- RS2-148B
N-(2-(1,4-thiazepan-4-yl)ethyl)-2-(hydroxyimino)acetamide
- RS2-150C
N-(3-(4-azatricyclo[4.3.1.13,8]undecan-4-yl)propyl)-2-(hydroxyimino)acetamide
- RS194B
N-(2-(azepan-1-yl)ethyl)-2-(hydroxyimino)acetamide
- RS2-234D
N-(2-(7-azabicyclo[2.2.1]heptan-7-yl)ethyl)-2-(hydroxyimino)acetamide
- RS2-237D
N-(2-(8-azabicyclo[3.2.1]octan-8-yl)ethyl)-2-(hydroxyimino)acetamide
- RS2-245C
(S)-N-(2-(azepan-1-yl)propyl)-2-(hydroxyimino)acetamide
- RS2-292A
N-(2-(azepan-1-yl)ethyl)-2-(hydroxyamino)-2-iminoacetamide
- RS3-36D
N-(2-(2-azaadamantan-2-yl)ethyl)-2-(hydroxyimino)acetamide
- RS3-43D
N-(2-(3-azabicyclo[3.2.1]octan-3-yl)ethyl)-2-(hydroxyimino)acetamide
- VX
O-ethyl S-[2-(disopropylamino)ethyl]methylphosphonothioat
Authorship Contributions
Participated in research design: Sit, Kovarik, Green, Fokin, Sharpless, Radić, Taylor.
Conducted experiments: Sit, Kovarik, Maček Hrvat, Žunec, Green, Fokin, Radić.
Performed data analysis: Sit, Kovarik, Maček Hrvat, Žunec, Green, Fokin, Radić, Sharpless, Radić, Taylor.
Wrote or contributed to writing of the manuscript: Sit, Kovarik, Maček Hrvat, Sharpless, Radić, Taylor.
Footnotes
This work was supported by the National Institutes of Health Countermeasures Against Chemical Threats (CounterACT) program, the National Institutes of Health Office of the Director, and the National Institutes of Health National Institute of Neurological Disorders and Stroke [Grants U01-NS 5058048 (to P.T.) and 1R21-NS084904] (to Z.R.)], and the Croatian Science Foundation [Grant 4307 (to Z.K.)].
References
- Baggot JD, Buckpitt A, Johnson D, Brennan P, Chung H. (1993) Bioavailability and disposition kinetics of HI-6 in Beagle dogs. Biopharm Drug Dispos 14:93–105. [DOI] [PubMed] [Google Scholar]
- Bhonsle JB, Causey R, Oyler BL, Bartolucci C, Lamba D, Pesaresi A, Bhamare NK, Soojhawon I, Garcia GE. (2013) Evaluation and computational characterization of the facilitated transport of Glc carbon C-1 oxime reactivators across a blood brain barrier model. Chem Biol Interact 203:129–134. [DOI] [PubMed] [Google Scholar]
- Bodor N, Shek E, Higuchi T. (1975) Delivery of a quaternary pyridinium salt across the blood-brain barrier by its dihydropyridine derivative. Science 190:155–156. [DOI] [PubMed] [Google Scholar]
- Buccafusco JJ. (1996) The role of central cholinergic neurons in the regulation of blood pressure and in experimental hypertension. Pharmacol Rev 48:179–211. [PubMed] [Google Scholar]
- Chambers JE, Chambers HW, Funck KE, Meek EC, Pringle RB, Ross MK. (2016) Efficacy of novel phenoxyalkyl pyridinium oximes as brain-penetrating reactivators of cholinesterase inhibited by surrogates of sarin and VX. Chem Biol Interact 259:154–159. [DOI] [PubMed] [Google Scholar]
- de Koning MC, van Grol M, Noort D. (2011) Peripheral site ligand conjugation to a non-quaternary oxime enhances reactivation of nerve agent-inhibited human acetylcholinesterase. Toxicol Lett 206:54–59. [DOI] [PubMed] [Google Scholar]
- Dolgin E. (2013) Syrian gas attack reinforces need for better anti-sarin drugs. Nat Med 19:1194–1195. [DOI] [PubMed] [Google Scholar]
- Edery H, Geyer MA, Taylor P, Berman HA. (1986) Target sites for anticholinesterases on the ventral surface of the medulla oblongata: hypotension elicited by organophosphorus agents. J Auton Pharmacol 6:195–205. [DOI] [PubMed] [Google Scholar]
- Ellman GL, Courtney KD, Andres V, Jr, Feather-Stone RM. (1961) A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem Pharmacol 7:88–95. [DOI] [PubMed] [Google Scholar]
- Garcia GE, Campbell AJ, Olson J, Moorad-Doctor D, Morthole VI. (2010) Novel oximes as blood-brain barrier penetrating cholinesterase reactivators. Chem Biol Interact 187:199–206. [DOI] [PubMed] [Google Scholar]
- Harvilchuck JA, Hong SP, Richey JS, Osheroff MR, Johnson JD. (2013) In vivo acetylcholinesterase reactivation in male guinea pigs and rhesus macaques following cyclosarin exposure and treatment with 1,1′-methylenebis4-[(hydroxyimino)methyl] pyridinium dimethanesulfonate. Int J Toxicol 32 (Suppl):99S–107S. [DOI] [PubMed] [Google Scholar]
- Hong SP, Gibbs ST, Kobs DJ, Osheroff MR, Johnson JD, Burback BL. (2013) Pharmacokinetics of MMB4 DMS in rats, rabbits, and dogs following a single IV administration. Int J Toxicol 32 (Suppl):30S–37S. [DOI] [PubMed] [Google Scholar]
- John H, Balszuweit F, Kehe K, Worek F, Thiermann H. (2015) Toxicokinetic aspects of nerve agents and vesicants, in Handbook of Toxicology of Chemical Warfare Agents, 2nd ed, (Gupta RC. ed) pp 817–856, Academic Press, Boston. [Google Scholar]
- Kalisiak J, Ralph EC, Zhang J, Cashman JR. (2011) Amidine-oximes: reactivators for organophosphate exposure. J Med Chem 54:3319–3330. [DOI] [PubMed] [Google Scholar]
- Katalinić M, Maček Hrvat N, Žďárová Karasová J, Misik J, Kovarik Z. (2015) Translation of in vitro to in vivo pyridinium oxime potential in tabun poisoning. Arh Hig Rada Toksikol 66:291–298. [DOI] [PubMed] [Google Scholar]
- Kliachyna M, Santoni G, Nussbaum V, Renou J, Sanson B, Colletier JP, Arboléas M, Loiodice M, Weik M, Jean L, et al. (2014) Design, synthesis and biological evaluation of novel tetrahydroacridine pyridine- aldoxime and -amidoxime hybrids as efficient uncharged reactivators of nerve agent-inhibited human acetylcholinesterase. Eur J Med Chem 78:455–467. [DOI] [PubMed] [Google Scholar]
- Kovarik Z, Maček N, Sit RK, Radić Z, Fokin VV, Sharpless KB, Taylor P. (2013) Centrally acting oximes in reactivation of tabun-phosphoramidated AChE. Chem Biol Interact 203:77–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovarik Z, Radić Z, Berman HA, Simeon-Rudolf V, Reiner E, Taylor P. (2003) Acetylcholinesterase active centre and gorge conformations analysed by combinatorial mutations and enantiomeric phosphonates. Biochem J 373:33–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovarik Z, Radić Z, Berman HA, Simeon-Rudolf V, Reiner E, Taylor P. (2004) Mutant cholinesterases possessing enhanced capacity for reactivation of their phosphonylated conjugates. Biochemistry 43:3222–3229. [DOI] [PubMed] [Google Scholar]
- Mercey G, Verdelet T, Saint-André G, Gillon E, Wagner A, Baati R, Jean L, Nachon F, Renard PY. (2011) First efficient uncharged reactivators for the dephosphylation of poisoned human acetylcholinesterase. Chem Commun (Camb) 47:5295–5297. [DOI] [PubMed] [Google Scholar]
- Radić Z, Sit RK, Kovarik Z, Berend S, Garcia E, Zhang L, Amitai G, Green C, Radić B, Fokin VV, et al. (2012) Refinement of structural leads for centrally acting oxime reactivators of phosphylated cholinesterases. J Biol Chem 287:11798–11809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajapurkar MV, Koelle GB. (1958) Reactivation of DFP-inactivated acetylcholinesterase by monoisonitrosoacetone (MINA) and diacetylmonoxime (DAM) in vivo. J Pharmacol Exp Ther 123:247–253. [PubMed] [Google Scholar]
- Renou J, Mercey G, Verdelet T, Păunescu E, Gillon E, Arboléas M, Loiodice M, Kliachyna M, Baati R, Nachon F, et al. (2013) Syntheses and in vitro evaluations of uncharged reactivators for human acetylcholinesterase inhibited by organophosphorus nerve agents. Chem Biol Interact 203:81–84. [DOI] [PubMed] [Google Scholar]
- Rosenberg YJ, Mao L, Jiang X, Lees J, Zhang L, Radić Z, Taylor P. (2017a) Post-exposure treatment with the oxime RS194B rapidly reverses early and advanced symptoms in macaques exposed to sarin vapor. Chem Biol Interact 274:50–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg YJ, Wang J, Ooms T, Rajendran N, Mao L, Jiang X, Lees J, Urban L, Momper JD, Sepulveda Y, et al. (2017b) Post-exposure treatment with the oxime RS194B rapidly reactivates and reverses advanced symptoms of lethal inhaled paraoxon in macaques. Toxicol Lett 293:229–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih T-M, Kan RK, McDonough JH. (2005) In vivo cholinesterase inhibitory specificity of organophosphorus nerve agents. Chem Biol Interact 157–158:293–303. [DOI] [PubMed] [Google Scholar]
- Shih T-M, Koplovitz I, Kan RK, McDonough JH. (2012) In search of an effective in vivo reactivator for organophosphorus nerve agent −inhibited acetylcholinesterase in the central nervous system. Adv Stud Biol 10:451–478. [Google Scholar]
- Shih T-M, Skovira JW, O’Donnell JC, McDonough JH. (2010) In vivo reactivation by oximes of inhibited blood, brain and peripheral tissue cholinesterase activity following exposure to nerve agents in guinea pigs. Chem Biol Interact 187:207–214. [DOI] [PubMed] [Google Scholar]
- Sidell FR, Groff WA. (1971) Intramuscular and intravenous administration of small doses of 2-pyridinium aldoxime methochloride to man. J Pharm Sci 60:1224–1228. [DOI] [PubMed] [Google Scholar]
- Sit RK, Fokin VV, Amitai G, Sharpless KB, Taylor P, Radić Z. (2014) Imidazole aldoximes effective in assisting catalysis of organophosphate detoxification. J Med Chem 57:1378–1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sit RK, Radić Z, Gerardi V, Zhang L, Garcia E, Katalinić M, Amitai G, Kovarik Z, Fokin VV, Sharpless KB, et al. (2011) New structural scaffolds for centrally acting oxime reactivators of phosphylated cholinesterases. J Biol Chem 286:19422–19430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somani SM, Solana RP, Dube SN. (1992) Toxicodynamics of nerve agents, in Chemical Warfare Agents, pp 67–123, Academic Press, New York. [Google Scholar]
- Swartz RD, Sidell FR. (1974) Renal tubular secretion of pralidoxime in man. Proc Soc Exp Biol Med 146:419–424. [DOI] [PubMed] [Google Scholar]
- Taylor P. (2018) Anticholinesterase agents, in Goodman & Gilman’s The Pharmacological Basis of Therapeutics, 13th ed (Brunton LL. ed) pp 163–176, McGraw-Hill, New York. [Google Scholar]
- Thiermann H, Steinritz D, Worek F, Radtke M, Eyer P, Eyer F, Felgenhauer N, Zilker T. (2011) Atropine maintenance dosage in patients with severe organophosphate pesticide poisoning. Toxicol Lett 206:77–83. [DOI] [PubMed] [Google Scholar]
- Thompson WR. (1947) Use of moving averages and interpolation to estimate median-effective dose. Bacteriol Rev 11:115–145. [PubMed] [Google Scholar]
- Weil CS. (1952) Tables for convenient calculation of median-effective dose (LD50 or ED50) and instruction in their use. Biometrics 8:249–263. [Google Scholar]
- Wilhelm CM, Snider TH, Babin MC, Jett DA, Platoff GE, Jr, Yeung DT. (2014) A comprehensive evaluation of the efficacy of leading oxime therapies in guinea pigs exposed to organophosphorus chemical warfare agents or pesticides. Toxicol Appl Pharmacol 281:254–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson IB. (1951) Acetylcholinesterase XI: reversibility of tetraethylpyrophosphate inhibition. J Biol Chem 190:110–117. [PubMed] [Google Scholar]
- Wilson IB. (1959) Molecular complementarity and antidotes for alkylphosphate poisoning. Fed Proc 18:752–758. [PubMed] [Google Scholar]
- Wilson IB, Ginsburg B. (1955) A powerful reactivator of alkylphosphate-inhibited acetylcholinesterase. Biochim Biophys Acta 18:168–170. [DOI] [PubMed] [Google Scholar]
- Wong L, Radić Z, Brüggemann RJM, Hosea N, Berman HA, Taylor P. (2000) Mechanism of oxime reactivation of acetylcholinesterase analyzed by chirality and mutagenesis. Biochemistry 39:5750–5757. [DOI] [PubMed] [Google Scholar]
- Worek F, Thiermann H. (2013) The value of novel oximes for treatment of poisoning by organophosphorus compounds. Pharmacol Ther 139:249–259. [DOI] [PubMed] [Google Scholar]