

Abstract
ω3‐polyunsaturated free fatty acids (ω3‐PUFAs), particularly docosahexaenoic (DHA) and eicosapentaenoic acid (EPA), are thought to exert health promoting effects in metabolic and in inflammatory diseases. The molecular mechanisms of these beneficial effects are only partially understood. DHA and EPA activate Free Fatty Acid receptor 4 (GPR120/FFA4). Recently, the first orally available, synthetic ligand of FFA4, 3‐[2‐chloro‐5‐(trifluoromethoxy)phenyl]‐3‐azaspiro[5.5]undecane‐9‐acetic acid (“compound A”; cpd A) has been developed. Cpd A exhibits distinctly higher potency, efficiency, and selectivity at FFA4 than ω3‐PUFAs and ameliorates insulin resistance and adipose tissue inflammation in the mouse. With GPR120/FFA4 activation believed to also attenuate tissue inflammation in autoimmune diseases, cpd A may also have a beneficial effect in these diseases. We have therefore addressed the therapeutic potential of cpd A in mouse models of three prototypical autoimmune diseases, specifically psoriasis, rheumatoid arthritis, and bullous pemphigoid. The effect of cpd A on the course of Aldara™‐induced psoriasis‐like dermatitis, K/BxN serum transfer arthritis, and antibody transfer pemphigoid disease‐like dermatitis was scrutinized. Cpd A did not alter the course of Aldara‐induced psoriasis‐like dermatitis, K/BxN serum transfer arthritis, or antibody transfer pemphigoid disease‐like dermatitis. Our results suggest that therapeutic regimens solely relying on FFA4 activation do not bear the potential to treat inflammatory diseases. With cpd A distinctly more potent in activating GPR120/FFA4 than ω3‐PUFAs, this also suggests that GPR120/FFA4 activation by ω3‐PUFAs does not significantly contribute to the health‐promoting effects of ω3‐PUFAs in autoimmune diseases.
Keywords: autoimmune disease, compound A, GPR120/FFAR4, pemphigoid disease, psoriasis, rheumatoid arthritis
Abbreviations
- ω3‐PUFAs
ω3‐polyunsaturated free fatty acids
- DHA
docosahexaenoic
- EPA
eicosapentaenoic acid
- FFA4
Free Fatty Acid receptor 4
- GPCR
G protein‐coupled receptor
1. INTRODUCTION
ω3‐polyunsaturated free fatty acids (ω3‐PUFAs), particularly docosahexaenoic acid (DHA) and eicosapentaenoic acid (EPA), are abundant in fish oil and considered to exert health promoting effects.1 Thus, high intake of fish oil has been associated with protection from metabolic diseases, such as cardiovascular diseases and type II diabetes, as well as from chronic inflammatory diseases, including psoriasis and rheumatoid arthritis.1, 2, 3, 4 In line with this, the increase in the incidence of chronic inflammatory diseases in Western societies coincided with a shift in ω3‐ and ω6‐PUFA intake from a ratio ω6‐ to ω3‐PUFAs of 1:1 to approximately 15:1.1
These associations have prompted numerous clinical trials investigating the therapeutic potential of fish oil or purified esters of DHA and EPA in different medical conditions. Among chronic inflammatory diseases, their effect has most frequently been studied in psoriasis and in rheumatoid arthritis. The setup of these trials was quite heterogeneous in respect to the ω3‐PUFAs preparations, doses, and routes of administration used as well as in respect to the features of the patient cohorts and the clinical parameters used to benchmark therapeutic effects. The results of these trials have recently been exhaustively summarized and analyzed in two systematic reviews.4, 5 Collectively, most trials investigating the effect in rheumatoid arthritis demonstrated moderate, beneficial effects.5 The results on the therapeutic effects of ω3‐PUFAs on psoriasis have been more contradictory with some trials reporting beneficial effects, others finding no effect.4 The potential reasons for these conflicting results are manifold. An obvious explanation is that the therapeutic potential of ω3‐PUFAs is too moderate, thus, eliciting either no or only little suppression of disease activity in clinical trials including only small numbers of patients. Further, ω3‐PUFAs may be only effective in certain patient subgroups of the respective diseases. This situation precipitates the idea that compounds metabolically more stable than ω3‐PUFAs and pharmacologically engaging the pathways downstream of ω3‐PUFAs more potently than ω3‐PUFAs themselves may be therapeutically more effective.
Mechanistically, the health‐promoting effects of ω3‐PUFAs are thought to be implemented by multiple and diverse modes of action, including the conversion of ω3‐PUFAs to proresolving lipid mediators and PPARγ agonists, substrate competition with arachidonic acid, thus, inhibiting the biosynthesis of proinflammatory eicosanoids, including that of leukotriene B4 (LTB4), as well as changes in the biophysical properties of cell membranes.2, 6
This concept of the molecular mode of action of ω3‐PUFAs has been extended by another putatively most important mechanism since DHA and EPA were identified as cognate ligands of the G protein‐coupled receptor (GPCR) 120. GPR120 was deorphanized in 2005 to bind the ω3‐PUFAs docosahexaenoic acid (DHA), and eicosapentaenoic acid (EPA), and α‐linolenic acid as well as palmitoleic, myristic, and linoleic acid as agonistic, cognate ligands.7, 8 It was subsequently renamed Free Fatty Acid receptor 4 (FFA4) by the International Union of Basic and Clinical Pharmacology.9 Recently, the group of natural GPR120/FFA4 agonists has been extended by a class of newly discovered lipid mediators: the branched fatty acid esters of hydroxy fatty acids (FAHFAs) with the FAHFAs palmitic‐acid‐9‐hydroxy‐stearic‐acid (9‐PAHSA) and palmitic‐acid‐5‐hydroxy‐stearic‐acid (5‐PAHSA) activate FFA4.10 In contrast to ω3‐PUFAs, mammals biosynthesize PAHSAs. Thus, their identification as GPR120/FFA4 ligands marks the discovery of endogenous ligands of FFA4, whose abundance is independent of nutritional intake.
GPR120/FFA4 is widely expressed and is present on parenchymal and epithelial cells of the gut, liver, adipose tissue, pancreas, and taste buds. To date, most extensively investigated physiological role of GPR120/FFA4 is its positive effect on insulin sensitivity, which has sparked broad interest into the development of GPR120/FFA4 agonists as novel antidiabetics.11 In recent years, it has been shown that FFA4 is also expressed on immune cells, including monocytes/macrophages, dendritic cells, and eosinophils,8, 10, 12 suggesting that GPR120/FFA4 may mediate immunomodulatory actions of ω3‐PUFAs. In line with this notion, in vitro activation of GPR120/FFA4 in macrophages curbs their immune response to NLPR3 inflammasome, TLR4, or TNF‐α activation,13, 14, 15 and in vivo GPR120 is required for ω3‐PUFAs to counteract adipose tissue inflammation induced by high‐fat diet in mice by limiting the recruitment of monocyte/macrophages into adipose and reducing the release of proinflammatory M1 macrophage‐derived immunomediators, such as IL‐1β, TNF‐α, MCP‐1, and IL‐6, while in parallel promoting the expression of antiinflammatory M2 macrophages, such as IL‐10, arginase 1, Ym‐1, and Clec7a.14 Notably, these same effects are replicated upon activation of GPR120/FFA4 by the synthetic compound 3‐[2‐chloro‐5‐(trifluoromethoxy)phenyl]‐3‐azaspiro[5.5]undecane‐9‐acetic acid, also designated as compound A (cpd A).16 Cpd A is the first fully selective, high‐affinity, and orally available GPR120/FFA4 agonist and vastly surpasses the pharmacological potency of DHA in GPR120/FFA4 activation in both man and mice,16 thus constituting the first research tool allowing to delineate GPR120/FFA4 ‐mediated actions in a highly specific manner. In mice, cpd A (30 mg/kg body weight/day) improved glucose and insulin tolerance and inhibited monocyte/macrophage migration toward adipose tissue‐derived chemoattractants. Like ω3‐PUFAs, it promoted the establishment of a M2 macrophage microenvironment in adipose tissue. Here, it also enhanced the numbers of antiinflammatory Treg and Breg cells and decreased the levels of LTB4.16
These findings have not only highlighted GPR120/FFA4 agonists as potential, novel class of antidiabetics, but also as possible new therapeutic concept for the treatment of classical inflammatory diseases.8 Studies on the effect of specific GPR120/FFA4 agonists on the course of classical inflammatory diseases are currently, however, not available yet. If effective, such a therapeutic strategy would be a particularly attractive therapeutic strategy in chronic inflammatory diseases often concurrent with type II diabetes, such as rheumatoid arthritis, psoriasis, and bullous pemphigoid.17, 18, 19
In this study, we have addressed the therapeutic potential of cpd A in mouse models of these three common, prototypical, autoimmune diseases using the Aldar™‐induced psoriasis‐like dermatitis model, the K/BxN serum transfer model, and the antibody transfer bullous pemphigoid‐like epidermolysis bullosa acquisita mouse model, respectively.20, 21, 22 In all three models, cpd A did not exhibit therapeutic efficiency, indicating that the antiinflammatory effects, induced by GPR120/FFA4 activation, do not reach the level to ameliorate clinically symptomatic tissue inflammation. Our results therefore discourage the use of GPR120/FFA4 agonists in monotherapeutic regimens in the treatment of inflammatory diseases.
2. MATERIALS AND METHODS
2.1. Mice
C57BL/6J wild‐type were purchased from JANVIER LABS (Saint‐Berthevin Cedex, France). They were housed in a 12‐hour light‐dark cycle in the animal facility of the University of Lübeck (Lübeck, Germany) and fed with standard chow diet. All experiments were performed in 8‐ to 12‐week‐old age‐ and sex‐matched mice by certified personnel. All experiments had been permitted by the state government of Schleswig‐Holstein.
2.2. Preparation and administration of the FF4/GPR120 agonist compound A
The FFA4/GPR120 agonist 3‐[2‐chloro‐5‐(trifluoromethoxy)phenyl]‐3‐azaspiro[5.5]undecane‐9‐acetic acid (compound A; cpd A), previously described,16 was purchased from Biomol GmbH (Hamburg, Germany). Cpd A was solved in 10% DMSO in PBS. In all experiments, 50 mg/kg body weight cpd A was administered by oral gavage daily. In the control group, the mice instead received only the vehicle, 10% DMSO in PBS. Both, cpd A and its vehicle control were well tolerated throughout the experiments. In all three mouse models used in this study, cpd A treatment was started 2 days before the first induction of disease (day −2). This pretreatment regimen was chosen to facilitate detecting possible beneficial effects of cpd A with administration regimens aiming at suppressing emerging tissue inflammation often exhibiting more pronounced effects than therapeutic regimens aiming at reversing established tissue inflammation.
2.3. Induction and clinical evaluation of psoriasis‐like dermatitis
To induce psoriasis‐like dermatitis, the Aldara™‐induced psoriasis‐like dermatitis (AIPD) mouse model, in literature often also referred to as “imiquimod‐induced psoriasis‐like dermatitis mouse model” was conducted, as previously described.23, 24 Briefly, a 2 × 3 cm large area was depilated on the back 2 days before the first application of Aldara™ cream (Meda, Solnau, Sweden). Starting from day 0, 50 mg Aldara™ cream was daily applied on the back, and 5 mg on the dorsal surface of the ears for five consecutive days. On the back, the severity of psoriasis‐like dermatitis was determined using the Psoriasis Activity and Severity Index (PASI). Thus, erythema, infiltration, and desquamation were individually scored on a scale from 0 to 4 with 0, none; 1, mild; 2, moderate; 3, marked; 4; severe. Finally, the scores of these individual aspects of dermatitis were summed up to calculate the cumulative disease score. At the ear, the severity of psoriasis‐like dermatitis was assessed by measuring the dorsal‐ventral distance of the ear daily before the application of Aldara™cream using a micrometer (Mitutoyo Europe, Neuss, Germany) and subsequently calculating ear swelling, i.e., the change of the dorsal‐ventral distance compared to day 0.
2.4. Induction and evaluation of arthritis
To induce arthritis, the K/BxN serum transfer model of rheumatoid arthritis was used, as previously described.25, 26 Thus, K/BxN serum was harvested from 8‐week‐old arthritic K/BxN mice at the Semmelweis University (Budapest, Hungary) and stored at −80°C. For induction of arthritis, 150 μL of serum was injected i.p. into recipient mice on days 0 and 2 of the experiment. Disease severity was assessed using the following clinical score for each paw: 0, no signs of arthritis; 1, localized edema/erythema on one surface of the paw; 2, edema/erythema on the entirety of one surface of the paw; 3, edema/erythema on both surfaces of the paw. Scores were summed up for all four paws to obtain the composite score. Ankle thickness was determined by micrometer (Mitutoyo Europe, Neuss, Germany). Ankle thickening (ankle thickness compared to baseline on day 0) was calculated as the mean difference between the current ankle thickness and the ankle thickness on day 0. For histopathology, ankles were dissected and fixed in 4% neutral buffered paraformaldehyde, demineralized in modified Kristensen's solution, and H&E stained. Stainings were visualized and photographed using the BZ‐9000E series Keyence microscope (Keyence GmbH, Neu‐Isenburg, Germany).
2.5. Induction and clinical scoring of pemphigoid disease‐like dermatitis
To generate pemphigoid disease‐like dermatitis in mice, the antibody transfer bullous pemphigoid‐like epidermolysis bullosa acquisita (BP‐like EBA) mouse model was performed, as previously described.27 Thus, in a first step, to generate IgG antibodies directed to type VII collagen (anti‐Col7 IgG), rabbits were immunized against three epitopes of type VII collagen, and IgG directed to the epitope C (anti‐Col7c IgG) was subsequently isolated from rabbit serum, as previously described.21 Purified anti‐Col7c IgG was filter‐sterilized (pore size: 0.2 μm), and its concentration was assessed by NanoDrop™ (ThermoFisher Scientific, Waltham, MA, USA). The reactivity of each batch of anti‐Col7c IgG was checked on murine tail skin sections by indirect immunofluorescence analysis. To induce pemphigoid disease‐like dermatitis, mice were injected s.c. with 50 μg of the affinity‐purified anti‐Col7c IgG on days 0, 2, and 4 of the experiment. To score the severity of pemphigoid disease‐like dermatitis, skin areas exhibiting erythema, blisters, erosions, crusts, or alopecia were categorized as “affected”, and, subsequently, the percentage of the total body surface affected by skin lesions (ABSA) was calculated.
2.6. Histopathology of the skin
For histopathology, biopsies of the skin were fixed in 4% Histofix® solution (Carl Roth, Karlsruhe, Germany), embedded in paraffin, cut into 6 μm sections, and subsequently H&E stained. Stainings were visualized and photographed using the BZ‐9000E series Keyence microscope (Keyence GmbH, Neu‐Isenburg, Germany). BZ‐II Analyzer software (Keyence, Neu‐Isenburg, Germany) was used to measure epidermal thickness, i.e., the distance between the dermal‐epidermal junction and the epidermal in psoriasis‐like dermatitis, as previously described.23
2.7. Immunofluorescence stainings
For IF stainings, skin biopsies were embedded in Tissue‐Tek® Cryomold® (VWR, Darmstadt, Germany) before 6 μm sections were cut and stored at −20°C until usage. To quantify epidermal cell proliferation in psoriasis‐like skin lesions, sections were stained for Ki‐67 using rat antimurine Ki‐67, biotinylated goat anti‐rat IgG (Biolegend, San Diego, CA, USA), and DyLight 488‐streptavidin (Thermoscientific). The number of Ki‐67+ keratinocytes per μm2 in the epidermis was quantified by BZ‐II Analyzer software, as previously described.28 Alexa Fluor® 594 AffiniPure donkey anti‐rabbit IgG (Jackson Immunoresearch, Suffolk, UK) and FITC‐conjugated goat anti‐mouse complement C3 IgG (MP Biomedicals, Illkirch, France) were used to detect IgG and C3 depositions, respectively, in perilesional skin of pemphigoid disease‐like dermatitis. Biotinylated rat anti‐mouse Ly6G Ab (Biolegend, San Diego, CA, USA) was used in combination with DyLight 488‐conjugated streptavidin (ThermoFisher Scientific, Waltham, MA, USA) to detect Ly6G+ cells. The BZ‐9000E series Keyence microscope and BZ‐II Analyzer software (Keyence GmbH, Neu‐Isenburg, Germany) were used to evaluate IF stainings throughout the study.
2.8. Statistical analysis
All data are presented as mean ± SEM. Results were tested for statistical differences using GraphPad Prism 7.0 (GraphPad, San Diego, CA, USA). P < 0.05 was considered statistically significant. Clinical scores were compared by two‐way ANOVA with Sidak's multiple comparisons test. All data presented are representatives of at least two independent experiments.
3. RESULTS
3.1. CpdA does not modulate the course of psoriasis‐like dermatitis
To evaluate the potential of cpd A as therapeutic in psoriasis, we induced psoriasis‐like skin inflammation in C57Bl/6 wild‐type mice, as described in Methods, and treated one group of mice daily with cpd A by gavage starting 2 days before the first application of Aldara™cream. In parallel, the control group received only the vehicle (10% DMSO in PBS). As expected, application of Aldara™induced skin inflammation clinically and histopathologically resembling in major aspects human plaque psoriasis. Thus, in both groups back skin exhibited signs of skin inflammation, including erythema, infiltration, and desquamation, starting on day 2 of the experiment and continuously rising until the end of the experiment on day 6 (Figure 1A‐D). The severity of these signs of inflammation was scored according to the Psoriasis Activity and Severity Index (PASI), as described in Methods. Vehicle‐ and cpd A‐treated mice did not differ in any of these individual aspects of psoriasis‐like skin inflammation (Figure 1A‐E). We also measured ear swelling after application of Aldara™to the ear in both groups, as an additional parameter for the severity of skin inflammation (Figure 1F). Vehicle‐ and cpd A‐treated mice did not differ in it either. Histopathological analysis confirmed that in both groups psoriasis‐like histopathological alterations had occurred, including infiltration of the dermis with leukocytes and lymphocytes, epidermal hyperproliferation as well as dermal neoangiogenesis (Figure 1G). Staining for Ki‐67 and measuring of the epidermal thickness revealed pronounced keratinocyte proliferation in both groups (Figure 1H/I). Vehicle‐ and cpd A‐treated mice did not differ in any of the listed hallmarks of psoriasis‐like skin inflammation, demonstrating that cpd A treatment is not effective in ameliorating AIPD.
Figure 1.
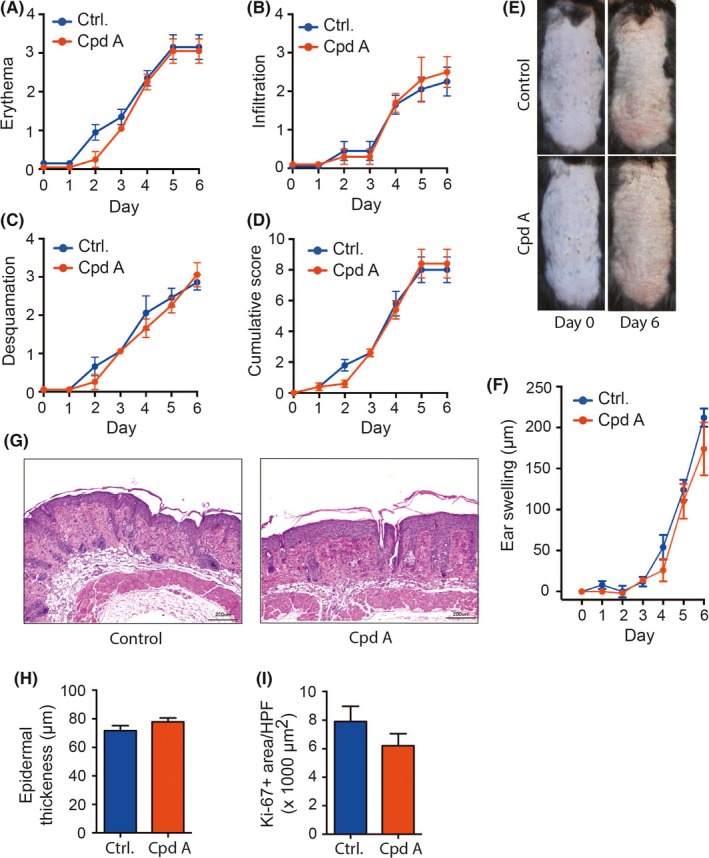
Cpd A in psoriasis‐like dermatitis. Psoriasis‐like dermatitis was induced by daily application of Aldara™ on the shaved back skin and the dorsal ears. 50 mg/kg body weight cpd A or vehicle control was daily administered p.o. by gavage starting 2 days prior to the first application of Aldara™ (day −2). The course of disease severity is shown with the scores for (A) erythema, (B) infiltration, and (C) desquamation, followed by (D) the resulting cumulative score of (A)‐(C). (E) Representative pictures of the clinical presentation of psoriasis‐like dermatitis on back skin on days 0 and 6. (F) Time course of ear swelling in response to local Aldara™ treatment. (G) H&E stainings of back skin harvested day 6. (H) Epidermal thickness of back skin on day 6. (I) Ki‐67 staining in back skin on day 6, and (J) expression level of Ki‐67 in the skin expressed as Ki‐67+ area HPF. One representative of two independent experiments is shown (n = 5 mice/group). All results are presented as mean ± SEM. Results in (A)‐(E) were tested for statistical significance by two‐way ANOVA. Results in (G) and (I) were analyzed by two‐sided, unpaired Student's t test. Scale bars represent 100 μm
3.2. Cpd A does not ameliorate antibody‐induced arthritis
We next tested the effect of cpdA on antibody‐induced arthritis in the K/BxN serum transfer model of rheumatoid arthritis. For this purpose, cpd A or vehicle control was administered to C57BL/6 wild‐type mice by gavage daily starting 2 days prior to the first application of K/BxN serum. In both groups, K/BxN serum precipitated signs of arthritis in fore‐ and hindpaws starting already 2 days after the first injection of K/BxN serum (Figure 2A‐C). Clinical severity of arthritis steadily increased in both groups until the end of the experiment on day 10 (Figure 2A). Likewise, arthritis differed between vehicle‐ and cpdA‐treated mice when severity was assessed by ankle thickening (Figure 2B). For both parameters, the clinical score and ankle thickening, the apparent difference between the two groups, however, did not reach statistical significance probably primarily due to the low magnitude of the therapeutic effect. We also compared the signs of arthritis in the two groups on the histopathological level. Both groups exhibited clear signs of arthritis, including leukocyte infiltration of synovial tissues and into the synovial fluid, synovial fibroblast hyperproliferation, as well cartilage and bone erosion (Figure 2D). There were no detectable differences between the two groups.
Figure 2.
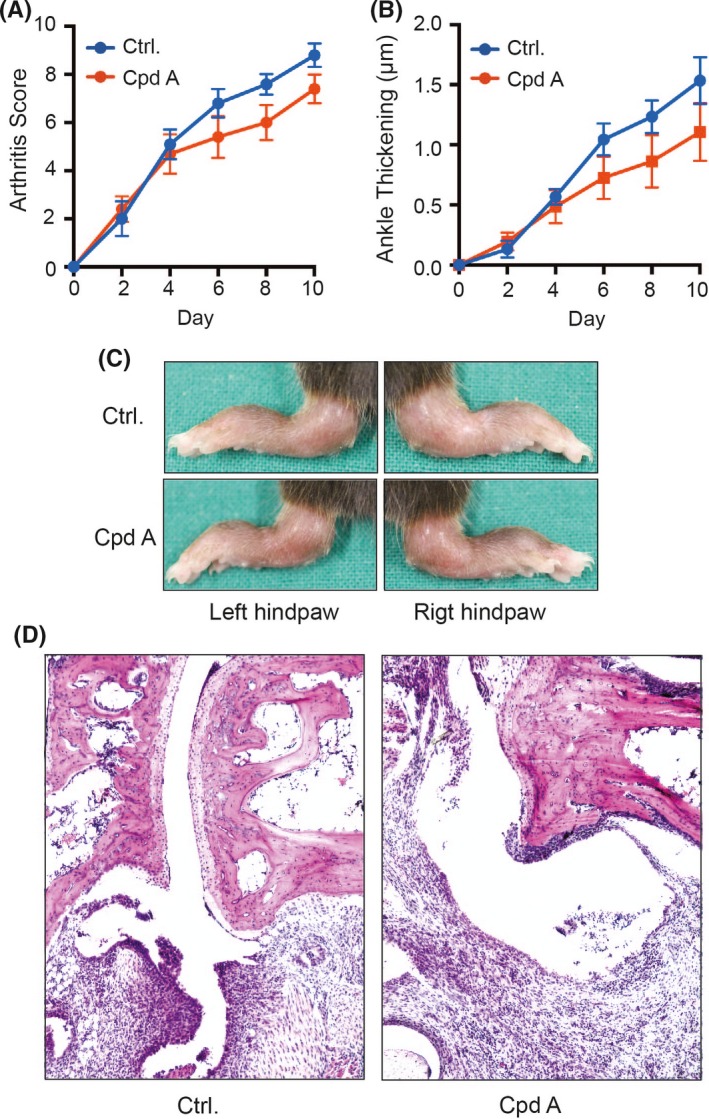
Cpd A in K/BxN serum transfer arthritis. Arthritis was induced by i.p. injection of 150 μL K/BxN serum on days 0 and 2 of the experiment. 50 mg/kg body weight cpd A or vehicle control was daily administered p.o. by gavage starting 2 days prior to the first application of K/BxN serum (day −2). The course of arthritis was evaluated by determining (A) the clinical arthritis score and (B) ankle thickening at the hindpaws. (C) Typical clinical presentation at the hindpaws on day 10 of the experiment. (D) Representative histopathologies of the hindpaws on day 10 presenting with leukocyte infiltrates, synovial fibroblast hyperplasia, and bone and cartilage erosions. The presented are merged from two independent experiments with 10 mice per group in total. All results are presented as mean ± SEM. Results in (A) and (B) were tested for statistical significance by two‐way
3.3. Cpd A does not alter the course of pemphigoid disease‐like dermatitis
We applied the antibody transfer BP‐like EBA mouse model as a third prototypical example for a chronic inflammatory disease to further explore the therapeutic potential of cpd A. We started the administration of cpd A 2 days prior to the first application of anti‐Col7 IgG. In both vehicle‐ and cpd A‐treated mice, pemphigoid disease‐like dermatitis became apparent by day 4 of the experiment and plateaued on day 8 without a significant difference between the two groups (Figure 3A/B). Likewise, on the histopathological level, both groups showed typical pemphigoid disease‐like alterations with a pronounced infiltration of the dermis with granulocytes and subepidermal clefting with no difference between the two groups (Figure 3C).
Figure 3.
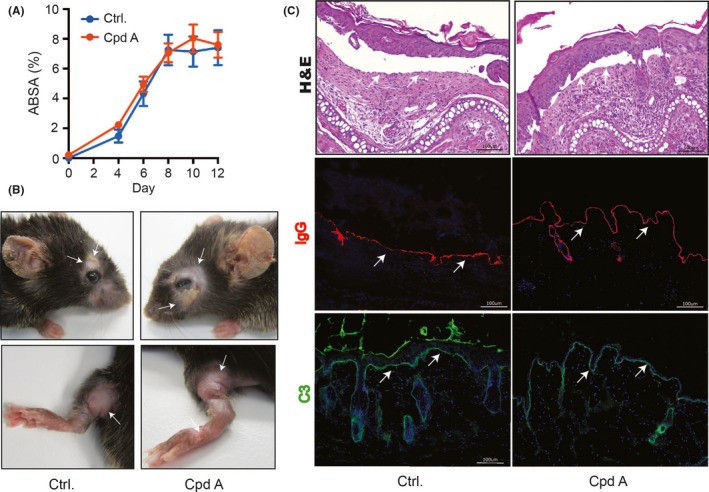
Cpd A in pemphigoid disease‐like dermatitis. Pemphigoid‐like dermatitis was induced by s.c. injection of 50 μg anti‐Col7c IgG on days 0, 2, and 4 of the experiment. 50 mg/kg body weight cpd A or vehicle control was daily administered p.o. by gavage starting 2 days prior to the first application of anti‐Col7c IgG (Day −2). (A) Clinical severity of dermatitis evaluated by the percentage of the total body surface affected by pemphigoid disease‐like skin lesions. (B) Typical clinical presentation of pemphigoid disease‐like dermatitis with erythema, erosions, crusts, and alopecia on day 12. (C) H&E stainings of lesional skin on day 12. Arrows indicate subepidermal clefts, the signature lesion of pemphigoid diseases (1st panel). IgG and C3 deposition at the dermal‐epidermal junction, indicated by white arrows, in perilesional skin on day 12 (second and third panel, respectively). One representative of three independent experiments is shown (n = 10 mice/group). All results are presented as mean ± SEM and were analyzed for statistical significance by two‐way ANOVA. Scale bars represent 100 μm
As in all our disease models cpd A did not exhibit any effect on the course of disease, we scrutinized the general pharmacological activity of the cpd A distributed from our manufacturer by examining the effect of this cpd A on ROS release from Raw 264.7 macrophages. Similar to what had previously been described for other GPR120/FFA4 agonists,29 Cpd A dose‐dependently inhibited PMA‐induced ROS release from Raw 264.7 macrophages (Figure S1).
4. DISCUSSION
High intake of ω3‐PUFAs is associated with protection from a wide spectrum of inflammatory diseases, including rheumatoid arthritis and psoriasis. Accordingly, the therapeutic potential of ω3‐PUFAs has been evaluated in several clinical studies, which, in sum, suggest that the therapeutic administration of ω3‐PUFAs, if at all, only exerts moderate therapeutic effects.4, 30 Thus, to exploit the molecular mechanisms, applied by ω3‐PUFAs, small molecules, which more potently activate these mechanisms and are pharmacologically better controllable and predictable in their effects than ω3‐PUFAs, are required. In pursuit of this goal, we have scrutinized here the therapeutic potential of the first orally available GPR120/FFA4 agonist, cpd A, in three mouse models of prototypic inflammatory diseases.
Although cpd A is in its agonistic, pharmacological properties toward GPR120/FFA4 far superior to ω3‐PUFAs,16 it did not modulate the severity of disease in any of the three mouse models, indicating that activation of GPR120/FFA4 alone is not sufficient to attenuate tissue inflammation, but may require the multimodal pharmacodynamic actions of ω3‐PUFAs. This notion is supported by the attenuation of tissue inflammation in the AIPD and the K/BxN serum transfer model in fat‐1 mice.31, 32 fat‐1 mice transgenically express the enzyme n‐3 desaturase, derived from the roundworm Caenorhabditis elegans, and, consequently, continuously biosynthesize large quantities of ω3‐PUFAs from ω6‐PUFAs in all their tissues.33 One contributory, molecular mechanism may be the biosynthesis of proresolving lipid mediators. Accordingly, deficiency in 12/15‐lipoxygenase, a key enzyme in the biosynthesis of ω3‐PUFA‐derived proresolving lipid mediators, aggravates K/BxN serum transfer arthritis,34 thus hinting at the in vivo relevance of counterregulatory effects of proresolving lipid mediators to contain tissue inflammation in vivo.
In our experiments, we started the administration of cpd A 2 days before the induction of disease. Oh et al, in contrast, treated with cpd A for 5 weeks before detecting a statistically significant improvement in insulin resistance. Such a long‐term administration of GPR120/FFA4 agonists may be required to gradually induce changes in the tissue, implementing an antiinflammatory tissue microenvironment preserving tissue homeostasis and counteracting emerging inflammation. Hence, it is conceivable that continuous long‐term administration of cpd A well in advance to the induction of tissue inflammation does exert more effects than acute application of cpd A. Higher efficiency of cpd A under long‐term application may render cpd A effective preventive drug in these diseases, which would be of interest because all three prototypical diseases included in this study typically proceed in cycles of acute flares and partial remissions. Similarly, although we already applied a higher dose of cpd A than Oh et al, it still cannot be excluded that an even higher dose might have been more effective attenuating disease in one or the other of the three mouse models applied here. A caveat of studies is that cpd A plasma levels were not assessed, thus, complicating the comparison of the results of the two studies with each other and with possible future studies.
Notably, the in vivo significance of GPR120/FFA4 to suppress chronic low‐grade inflammation also has lately been contended. While GPR120/FFA4 activation by ω3‐PUFAs was initially reported to be critical to reduce high‐fat diet‐induced adipose tissue inflammation,14 more recent studies found GPR120/FFA4 to be dispensable for the therapeutic effects of ω3‐PUFAs in mouse models of both high‐fat diet‐induced adipose tissue inflammation, and of atherosclerosis.35, 36 The reason for these discrepant findings is unknown and numerous explanations are conceivable. Inflammation is a most complex process and ω3‐PUFAs can exert diverse pharmacological actions independent of GPR120/FFA4. It would not be surprising if the relative contribution of GPR120 to regulate tissue inflammation varies depending on the detailed conditions of the organism. The conditions under which GPR120/FFA4 activation elicits antiinflammatory effects in vivo therefore must be exactly defined.
In conclusion, our results suggest that targeting GPR120/FFA4 alone is not a promising concept for the development of new treatments for diseases featuring acute tissue inflammation. Future studies should, therefore rather, address whether small molecules individually engaging other molecular mechanisms contributing to the putative beneficial effects of ω3‐PUFAs, such as the receptors for proresolving lipid mediators, are more effective than GPR120/FFA4 engagement in ameliorating tissue inflammation. Alternatively, multipronged therapeutic strategies, in parallel engaging several of these molecular mechanisms to elicit relevant therapeutic effects, may be examined.
ACKNOWLEDGEMENTS
We thank the Deutsche Forschungsgemeinschaft for funding this research through grants to C.D.S., including funding for the Clinical Research Unit (CRU) 303 Pemphigoid Diseases – Molecular Pathways and their therapeutic Potential (Sa1960/5‐1) and for the Excellence Cluster (EXC) 306 Inflammation‐at‐Interfaces.
DISCLOSURES
The authors have no conflict of interest to report.
Supporting information
Wannick M, Bezdek S, Guillen N, et al. Oral administration of the selective GPR120/FFA4 agonist compound A is not effective in alleviating tissue inflammation in mouse models of prototypical autoimmune diseases. Pharmacol Res Perspect. 2018;e00438 10.1002/prp2.438
REFERENCES
- 1. Simopoulos AP. Evolutionary aspects of diet, the omega‐6/omega‐3 ratio and genetic variation: nutritional implications for chronic diseases. Biomed Pharmacother. 2006;60:502‐507. [DOI] [PubMed] [Google Scholar]
- 2. Calder PC. Fatty acids and inflammation: the cutting edge between food and pharma. Eur J Pharmacol. 2011;668(Suppl 1):S50‐S58. [DOI] [PubMed] [Google Scholar]
- 3. Gan RW, Bemis EA, Demoruelle MK, et al. The association between omega‐3 fatty acid biomarkers and inflammatory arthritis in an anti‐citrullinated protein antibody positive population. Rheumatology. 2017;56:2229‐2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Upala S, Yong WC, Theparee T, Sanguankeo A. Effect of omega‐3 fatty acids on disease severity in patients with psoriasis: a systematic review. Int J Rheum Dis. 2017;20:442‐450. [DOI] [PubMed] [Google Scholar]
- 5. Navarini L, Afeltra A, Gallo Afflitto G, Margiotta DPE. Polyunsaturated fatty acids: any role in rheumatoid arthritis? Lipids Health Dis. 2017;16:197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Buckley CD, Gilroy DW, Serhan CN. Proresolving lipid mediators and mechanisms in the resolution of acute inflammation. Immunity. 2014;40:315‐327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hirasawa A, Tsumaya K, Awaji T, et al. Free fatty acids regulate gut incretin glucagon‐like peptide‐1 secretion through GPR120. Nat Med. 2005;11:90‐94. [DOI] [PubMed] [Google Scholar]
- 8. Moniri NH. Free‐fatty acid receptor‐4 (GPR120): cellular and molecular function and its role in metabolic disorders. Biochem Pharmacol. 2016;110–111:1‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Davenport AP, Alexander SP, Sharman JL, et al. International Union of Basic and Clinical Pharmacology. LVIII. G protein‐coupled receptor list: recommendations for new pairings with cognate ligands. Pharmacol Rev. 2013;65:967‐986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yore MM, Syed I, Moraes‐Vieira PM, et al. Discovery of a class of endogenous mammalian lipids with anti‐diabetic and anti‐inflammatory effects. Cell. 2014;159:318‐332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Milligan G, Alvarez‐Curto E, Watterson KR, Ulven T, Hudson BD. Characterizing pharmacological ligands to study the long‐chain fatty acid receptors GPR40/FFA1 and GPR120/FFA4. Br J Pharmacol. 2015;172:3254‐3265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Konno Y, Ueki S, Takeda M, et al. Functional analysis of free fatty acid receptor GPR120 in human eosinophils: implications in metabolic homeostasis. PLoS ONE. 2015;10:e0120386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Li X, Yu Y, Funk CD. Cyclooxygenase‐2 induction in macrophages is modulated by docosahexaenoic acid via interactions with free fatty acid receptor 4 (FFA4). FASEB J. 2013;27:4987‐4997. [DOI] [PubMed] [Google Scholar]
- 14. Oh DY, Talukdar S, Bae EJ, et al. GPR120 is an omega‐3 fatty acid receptor mediating potent anti‐inflammatory and insulin‐sensitizing effects. Cell. 2010;142:687‐698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yan Y, Jiang W, Spinetti T, et al. Omega‐3 fatty acids prevent inflammation and metabolic disorder through inhibition of NLRP3 inflammasome activation. Immunity. 2013;38:1154‐1163. [DOI] [PubMed] [Google Scholar]
- 16. da Oh Y, Walenta E, Akiyama TE, et al. A Gpr120‐selective agonist improves insulin resistance and chronic inflammation in obese mice. Nat Med. 2014;20:942‐947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Innala L, Sjoberg C, Moller B, et al. Co‐morbidity in patients with early rheumatoid arthritis ‐ inflammation matters. Arthritis Res Ther. 2016;18:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kibsgaard L, Bay B, Deleuran M, Vestergaard C. A retrospective consecutive case‐series study on the effect of systemic treatment, length of admission time, and co‐morbidities in 98 bullous pemphigoid patients admitted to a tertiary centre. Acta Derm Venereol. 2015;95:307‐311. [DOI] [PubMed] [Google Scholar]
- 19. Schwandt A, Bergis D, Dapp A, et al. Psoriasis and diabetes: a multicenter study in 222078 type 2 diabetes patients reveals high levels of depression. J Diabetes Res. 2015;2015:792968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kouskoff V, Korganow AS, Duchatelle V, Degott C, Benoist C, Mathis D. A new mouse model of rheumatoid arthritis: organ‐specific disease provoked by systemic autoimmunity. Ryumachi. 1997;37:147. [PubMed] [Google Scholar]
- 21. Sitaru C, Mihai S, Otto C, et al. Induction of dermal‐epidermal separation in mice by passive transfer of antibodies specific to type VII collagen. J Clin Invest. 2005;115:870‐878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. van der Fits L, Mourits S, Voerman JS, et al. Imiquimod‐induced psoriasis‐like skin inflammation in mice is mediated via the IL‐23/IL‐17 axis. J Immunol. 2009;182:5836‐5845. [DOI] [PubMed] [Google Scholar]
- 23. Bezdek S, Hdnah A, Sezin T, et al. The genetic difference between C57Bl/6J and C57Bl/6N mice significantly impacts Aldara‐induced psoriasiform dermatitis. Exp Dermatol. 2016;26:349‐351. [DOI] [PubMed] [Google Scholar]
- 24. Sezin T, Zillikens D, Sadik CD. Leukotrienes do not modulate the course of Aldara‐induced psoriasiform dermatitis in mice. Acta Derm Venereol. 2015;95:341‐342. [DOI] [PubMed] [Google Scholar]
- 25. Sadik CD, Kim ND, Alekseeva E, Luster AD. IL‐17RA signaling amplifies antibody‐induced arthritis. PLoS ONE. 2011;6:e26342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sadik CD, Kim ND, Iwakura Y, Luster AD. Neutrophils orchestrate their own recruitment in murine arthritis through C5aR and FcgammaR signaling. Proc Natl Acad Sci USA. 2012;109:E3177‐E3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sezin T, Krajewski M, Wutkowski A, et al. The leukotriene B4 and its receptor BLT1 act as critical drivers of neutrophil recruitment in murine bullous pemphigoid‐like epidermolysis bullosa acquisita. J Invest Dermatol. 2017;137:1104‐1113. [DOI] [PubMed] [Google Scholar]
- 28. Onken MD, Worley LA, Harbour JW. Association between gene expression profile, proliferation and metastasis in uveal melanoma. Curr Eye Res. 2010;35:857‐863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cheshmehkani A, Senatorov IS, Dhuguru J, Ghoneim O, Moniri NH. Free‐fatty acid receptor‐4 (FFA4) modulates ROS generation and COX‐2 expression via the C‐terminal beta‐arrestin phosphosensor in Raw 264.7 macrophages. Biochem Pharmacol. 2017;146:139‐150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Talukdar S, Olefsky JM, Osborn O. Targeting GPR120 and other fatty acid‐sensing GPCRs ameliorates insulin resistance and inflammatory diseases. Trends Pharmacol Sci. 2011;32:543‐550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Qin S, Wen J, Bai XC, et al. Endogenous n‐3 polyunsaturated fatty acids protect against imiquimod‐induced psoriasis‐like inflammation via the IL‐17/IL‐23 axis. Mol Med Rep. 2014;9:2097‐2104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Woo SJ, Lim K, Park SY, et al. Endogenous conversion of n‐6 to n‐3 polyunsaturated fatty acids attenuates K/BxN serum‐transfer arthritis in fat‐1 mice. J Nutr Biochem. 2015;26:713‐720. [DOI] [PubMed] [Google Scholar]
- 33. Kang JX, Wang J, Wu L, Kang ZB. Transgenic mice: fat‐1 mice convert n‐6 to n‐3 fatty acids. Nature. 2004;427:504. [DOI] [PubMed] [Google Scholar]
- 34. Kronke G, Katzenbeisser J, Uderhardt S, et al. 12/15‐lipoxygenase counteracts inflammation and tissue damage in arthritis. J Immunol. 2009;183:3383‐3389. [DOI] [PubMed] [Google Scholar]
- 35. Paerregaard SI, Agerholm M, Serup AK, et al. FFAR4 (GPR120) signaling is not required for anti‐inflammatory and insulin‐sensitizing effects of omega‐3 fatty acids. Mediators Inflamm. 2016;2016:1536047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Shewale SV, Brown AL, Bi X, et al. In vivo activation of leukocyte GPR120/FFAR4 by PUFAs has minimal impact on atherosclerosis in LDL receptor knockout mice. J Lipid Res. 2017;58:236‐246. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials