

Abstract
In this study we performed a systematic search to identify breast cancer-specific small non-coding RNAs, which we have collectively termed orphan non-coding RNAs (oncRNAs). We subsequently discovered that one of these oncRNAs, which originates from the 3’ end of TERC, acts as a regulator of gene expression and is a robust promoter of breast cancer metastasis. This oncRNA, which we have named T3p, exerts its pro-metastatic effects by acting as an inhibitor of RISC complex activity and increasing the expression of the pro-metastatic genes NUPR1 and PANX2. Furthermore, we have shown that oncRNAs are present in cancer cell-derived extracellular vesicles, raising the possibility that these circulating oncRNAs may also play a role in non-cell autonomous disease pathogenesis. Additionally, these circulating oncRNAs present a novel avenue for cancer fingerprinting using liquid biopsies.
Main
The widespread reprogramming of the gene expression landscape is a hallmark of cancer development. Thus, the systematic identification of regulatory pathways that drive pathologic gene expression patterns is a crucial step towards understanding and treating cancer. Many regulatory mechanisms have been implicated in the oncogenic expression of genes involved in tumor progression. In addition to the transcriptional networks that underlie metastasis, post-transcriptional regulatory pathways have also emerged as major regulators of this process. MicroRNAs (miRNAs), a subclass of small RNAs involved in gene silencing, were among the first post-transcriptional regulators to be functionally implicated in breast cancer progression1. RNA-binding proteins (RBPs) are also critical regulators of gene expression, and several specific RBPs have been shown to affect oncogenesis and cancer progression2–5. Recently, we demonstrated that tRNAs6 and tRNA fragments7, two other classes of small non-coding RNAs, also play important roles in breast cancer metastasis.
Despite the diversity of known regulatory mechanisms involved in cancers, they share the characteristic of deregulating existing cellular pathway. To activate oncogenic processes and down-regulate tumor suppressive pathways, cancer cells adopt many strategies, including somatic mutations (e.g. KRAS8), genetic amplifications/deletions (e.g. EGFR9), gene fusions (e.g. BCR-ABL10), and epigenetic modifications (e.g. promoter hypermethylation11). While these oncogenic strategies rely on the genetic or epigenetic modulation of existing regulatory programs, there is an unexplored possibility that cancer cells may be capable of engineering regulatory pathways that function at the RNA or protein level to drive tumorigenesis by enforcing pro-oncogenic gene expression patterns. This idea is further reinforced by the current understanding of cancer progression as an evolutionary and ecological process12. In this study, we set out to ask whether tumors can evolve this type of novel regulatory program that drives cancer progression. We envisioned that new regulatory pathways could emerge through a two-step evolutionary process: the appearance of a pool of sufficiently abundant and diverse macromolecules with regulatory potential and the subsequent adoption of these molecules as functional neo-regulators of gene expression patterns. Since non-coding RNAs rely on their base-pairing capacity and interactions with RNA-binding proteins to carry out their regulatory functions, it follows that novel cancer cell-specific RNA species have this same potential. Based on this broad regulatory potential, we focused on cancer cell-specific small non-coding RNAs as a possible source of tumor-evolved regulators capable of modulating disease-relevant pathways and processes.
To search for small RNAs that are expressed in breast cancer cells and are undetectable in normal breast tissue, we implemented an unbiased approach, combining small RNA sequencing (smRNA-seq) of cancer cell lines and patient-derived xenograft models, as well as integrating analysis of existing clinical breast cancer datasets. We discovered and annotated 201 previously unknown small RNAs that are expressed in breast cancer cells and not in mammary epithelial cells. We have named these RNAs ‘orphan’ non-coding RNAs (oncRNAs) to highlight their cancer-specific biogenesis. To assess whether any members of this class play a direct role in breast cancer progression, we compared the expression of oncRNAs in poorly and highly metastatic cells. We successfully identified, characterized, and validated the cancer-relevant function of one such oncRNA that is generated from the 3’-end of TERC (the RNA component of telomerase). This oncRNA, which we have named T3p, promotes breast cancer metastasis by acting as a decoy for the RISC complex in breast cancer cells. Furthermore, we demonstrated that a number of oncRNAs, including T3p, can be detected in extracellular vesicles originating from cancer cells, raising the possibility that they may play an emergent role in “educating” non-tumoral cells. Clinically, given their absence in normal cells, extracellular oncRNAs could serve as a specific digital fingerprint of the underlying cancer cells.
Results
A systematic search for orphan small non-coding RNAs in breast cancer
We first sought to determine if a set of small RNAs exists that is only expressed in cancer cells and could provide a pool of potential regulators. We reasoned that such oncRNAs would only be detectable in cancer cell lines and not in normal cells. To test this hypothesis, we performed smRNA-seq on eight breast cancer cell lines (representing all major breast cancer subtypes), as well as human mammary epithelial cells (HMEC) as a non-transformed reference sample. We identified 437 unannotated small RNAs that were detected above a significant threshold across all the breast cancer lines and were undetected in HMEC samples (Fig. 1a).
Fig 1: Discovery, annotation, and validation of cancer-specific orphan non-coding RNAs in breast cancer.
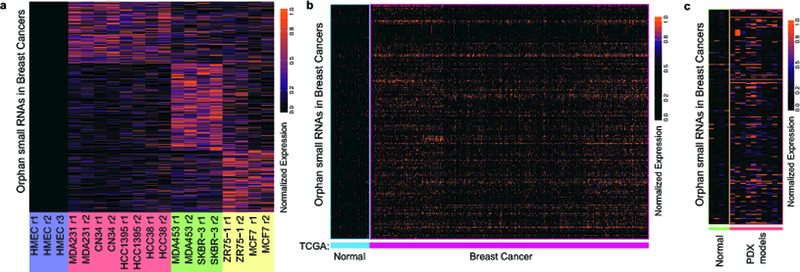
a, Heatmap representing the relative abundance of 437 small non-coding RNAs that are significantly expressed in breast cancer lines but not in normal HMECs. HMEC samples were prepared in biological triplicate while all other cell lines were prepared in duplicate. Cell lines color coded by sub-type: HMEC (purple), triple negative breast cancer (TNBC; red), HER2 positive (green), and luminal (yellow). b, Of the 437 small RNAs identified in (a), 201 were significantly expressed in breast tumor biopsy small RNA gene expression profiles collected as part of The Cancer Genome Atlas (TCGA-BRCA), and these 201 were also largely absent from the adjacent normal tissue collected from the ~200 individuals in this dataset. c, These 201 cancer-specific small RNAs were classified as oncRNAs and were independently validated in a third dataset comprised of small RNA profiles from four normal epithelial samples and 10 patient-derived breast cancer xenograft models (PDX models).
To further narrow our search we next performed a similar analysis on smRNA-seq data obtained from The Cancer Genome Atlas, which consisted of small RNA expression profiles across roughly 200 normal tissue samples and 1000 breast cancer biopsies. The highly significant overlap between these two independent analyses revealed a high-confidence set of 201 oncRNAs (Fig. 1b and Supplementary Fig. 1a). To independently validate this oncRNA set, we generated a dataset of small RNA profiles from 10 breast cancer patient-derived xenograft (PDX) models and four normal epithelial samples (unmatched). As shown in Fig. 1c, these oncRNAs are largely absent in normal samples yet are frequently detected in this set of PDX models. By summing the expression of all 201 oncRNAs across every sample, we derived a simple classification rule that perfectly assigns the normal and PDX profiles to their correct group (Supplementary Fig. 1b). Together, these findings establish the existence of a pool of orphan ncRNAs whose expression is strongly associated with breast cancer and is largely undetected in normal tissue.
Identification of T3p, an oncRNA associated with breast cancer progression
Orphan non-coding RNAs provide a cancer cell-specific pool of RNA species. However, the potential oncogenic function of these RNAs remained unknown. To address this question, we performed a series of analyses to identify those oncRNAs that are strongly associated with breast cancer progression. First, in addition to the cell lines assayed in Fig. 1, we also profiled two highly metastatic breast cancer cell lines that were previously in vivo selected in immunocompromised mice for higher metastatic capacity to the lung1,13. Upon comparing the expression of oncRNAs in these highly metastatic cells relative to their poorly metastatic parental lines, we noted one oncRNA with significantly increased levels in highly metastatic cells (Fig. 2a). This 45-nucleotide oncRNA mapped to the 3’-end of the TERC gene, which codes for the RNA component of telomerase (Fig. 2b). As such, we have named this previously unknown small RNA T3p (for TERC 3’ RNA). Analysis of our previously published smRNA-seq dataset from the same poorly and highly metastatic pairs of cell lines7 corroborated the higher expression of T3p in highly metastatic cells (Fig. 2c). We validated this upregulation of T3p expression in metastatic cells using quantitative RT-PCR (qRT-PCR) (Fig. 2c). In our smRNA-seq data we also observed T3p expression across our panel of breast cancer cell lines but not in HMECs (Supplementary Fig. 2a).
Fig 2: The oncRNA T3p is associated with breast cancer progression.
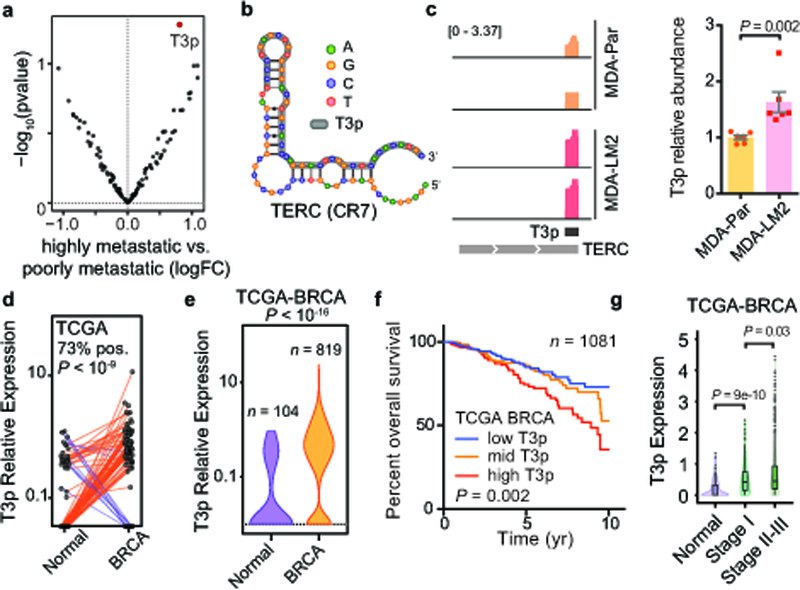
a, A volcano plot comparing the expression of oncRNAs in poorly metastatic breast cancer cells relative to their highly metastatic derivatives. T3p is highlighted in red. n = 4 biologically independent experimental comparisons. P values and log fold change were calculated using the DE-seq2 package (two-sided, no correction for multiple testing). b, T3p (in grey) maps to the 3’ end (CR7 domain) of TERC, the RNA component of telomerase. c, Normalized (per million reads mapped) coverage plots for reads mapped to TERC in smRNA-seq data from highly metastatic MDA-LM2 cells and poorly metastatic MDA-MB-231 parental cells in our previously published data18 (identical y-axes), and validation of this T3p upregulation by qRT-PCR (n = 6, comprised of two biologically independent sets of biologically independent triplicates, using a two-tailed Mann-Whitney test). Shown are mean ± s.e.m. d, Expression of T3p (count-per-million; cpm) in breast tumor biopsies and their matched normal tissue in the TCGA-BRCA dataset. n = 96 biologically independent paired samples. A paired two-way Wilcoxon test was used to calculate the associated P-value. e, Violin plots of T3p expression across the TCGA-BRCA dataset. n = 923 biologically independent samples. A two-way Mann-Whitney test was used to calculate the P value. Violin plots show the distribution along the minima and maxima for each cohort (median = 0 for normal and 0.34 for BRCA). f, Survival analysis in the TCGA-BRCA dataset for patients stratified into tertiles based on the expression of T3p in their tumors. All tertiles are shown. P value calculated from a two-sided log-rank test. Hazard-ratio (HR) equals 0.5 for the low vs. high tertiles. g, Expression of T3p across normal, stage I, and stage II and III samples in the TCGA-BRCA dataset shown as violin and boxplots (quartiles). Normal: n = 104 (median = 0), Stage I: n = 169 (median = 0.42), Stage II-III: n = 799 (median = 0.46). The violin plots show the distribution along the minima and maxima. The boxplots show the quartiles. The whiskers indicate quartile ± IQR (inter-quartile range). Outliers are also shown as points. P value calculated using a two-way Mann-Whitney test.
We then asked whether increased expression of T3p is associated with breast cancer pathogenesis. We first analyzed ~200 matched normal and breast cancer tumor tissue samples from TCGA-BRCA (The Cancer Genome Atlas, Breast Cancer) and noted that consistent with its classification as an oncRNA, the expression of T3p was highly cancer specific (Fig. 2d). We then included the entire TCGA-BRCA dataset (~1,000 tumor samples) in this analysis, and observed that T3p, which was not detected in the majority of normal samples, was present at relatively high levels in tumor biopsies (Fig. 2e and Supplementary Fig. 2b). Importantly, consistent with the higher expression of T3p in highly metastatic cells, we observed a significant association between patient survival and T3p expression, but not with TERC expression (Fig. 2f and Supplementary Fig. 2c). Higher expression of T3p in clinical breast cancer samples was also significantly correlated with advanced breast cancer (Fig. 2g). Interestingly, stratification of these tumor samples by hormone receptor and HER2 status showed no strong association between T3p levels and expression of a specific receptor (Supplementary Fig. 2d). Finally, we also noted increased expression of T3p in PDX breast cancer models relative to normal epithelial tissue, as well as in metastatic PDX models relative to non-metastatic (Supplementary Fig. 2e,f). Together, these results establish the oncRNA T3p as a cancer-specific biomarker with robust prognostic value.
T3p acts as a broad regulator of gene expression in breast cancer cells
The strong association between T3p expression and breast cancer progression from multiple independent datasets raised the possibility that T3p may be playing a direct and functional role in breast cancer progression. To elucidate its molecular function, we first asked whether modulating T3p expression levels had any regulatory consequences. To answer this question, we silenced T3p by transfecting highly metastatic MDA-LM2 breast cancer cells with antisense locked nucleic acid oligonucleotides (LNAs) targeting T3p or with control LNAs. We then performed gene expression profiling to measure the genome-wide regulatory impact of silencing T3p. Surprisingly, we observed a highly significant change in the gene expression landscape of the cell upon T3p inhibition, affecting thousands of genes (Fig. 3a). This is on par with the impact of many well-established post-transcriptional regulators such as miRNAs1,14. However, the full length TERC transcript remained a potential confounding factor, as the T3p-targeting LNA may also impact TERC function, which in turn could be responsible for the observed gene expression changes. To distinguish between these two possibilities, we used two independent approaches. First, in addition to using a non-targeting control LNA, we also used an LNA complementary to a sequence upstream of T3p, thereby only targeting full length TERC. Gene expression changes induced by the anti-T3p LNA were similar regardless of whether the scrambled control or anti-full length TERC LNA was used as the reference (Supplementary Fig. 3a), indicating that LNA interaction with full length TERC does not induce the same magnitude of gene expression changes generated by T3p inhibition. To further strengthen these findings we performed a gain-of-function experiment by transfecting MDA-MB-231 breast cancer cells with either a synthetic T3p mimetic or a control scrambled oligonucleotide and then performed gene expression profiling. We again observed a significant change in the gene expression landscape in the mimetic compared to control transfected cells, similar to that seen with LNA transfection. Importantly, these gene expression changes were generally anti-correlated with those observed in the loss-of-function LNA experiment (Fig. 3b,c). This is consistent with our expectation that anti-T3p LNAs and T3p mimetics should elicit opposite gene expression changes. Together, these observations establish T3p as a broad regulator of gene expression in breast cancer cells.
Fig 3: T3p regulates gene expression and drives metastatic progression.
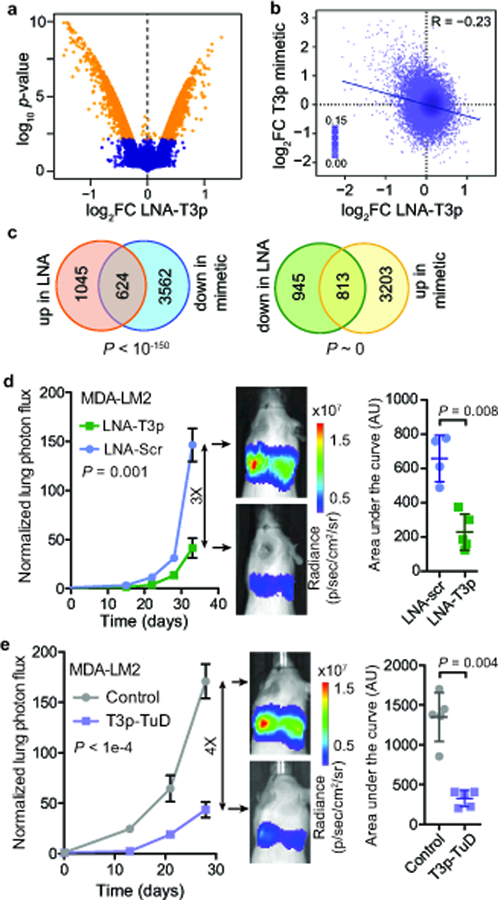
a, A volcano plot of gene expression changes following transfection of anti-T3p LNA relative to scrambled LNA in highly metastatic MDA-LM2 cells. n = 2 biologically independent experiments. P and log fold change values were calculated using the Lumi package (significance threshold was set at two-tailed adjusted P = 0.01). b, Scatter plot comparing gene expression changes induced by anti-T3p LNA in MDA-LM2 cells versus T3p mimetic in MDA-MB-231 parental cells. Reported is the associated Pearson correlation (R = −0.234, P ~ 0). c, Venn diagrams showing overlap of genes upregulated in LNA-transfected and downregulated in T3p mimetic-transfected MDA-LM2 cells, as well as overlap of genes downregulated in LNA-transfected and upregulated in T3p mimetic-transfected MDA-LM2 cells. P-values from hypergeometric distribution. d, Bioluminescence imaging plot of lung colonization by MDA-LM2 cells transfected with anti-T3p LNA (LNA-T3p) or scrambled LNA (LNA-Scr); n = 4 (LNA-Scr) or n = 5 (LNA-T3p) biologically independent animals per cohort; mean ± s.e.m. are reported for each day; two-way ANOVA. Area under the curve is presented as mean ± s.d.; P was calculated using a one-tailed Mann-Whitney test. e, Bioluminescence imaging plot of lung colonization by MDA-LM2 cells stably expressing T3p-TuD or a control hairpin; n = 5 biologically independent animals per cohort; mean ± s.e.m. are reported for each day; two-way ANOVA. Area under the curve is presented as mean ± s.d.; P was calculated using a one-tailed Mann-Whitney test.
T3p promotes breast cancer metastasis
Given the broad regulatory effect of T3p on gene expression and its association with metastasis and with poor survival in breast cancer, we next asked whether this oncRNA could affect metastasis in vivo. To test this, we transfected highly metastatic MDA-LM2 cells with anti-T3p LNAs and injected these cells into the venous circulation of immunocompromised mice and followed their metastatic colonization over time using in vivo imaging. We observed that cells transfected with anti-T3p LNAs had significantly diminished lung colonization capacity (Fig. 3d). Gross histology of lungs from each cohort also revealed a significantly lower number of visible metastatic nodules in the lungs of mice injected with T3p-LNA transfected cells (Supplementary Fig. 3b). This observation indicates that even transient inhibition of T3p can lead to lower metastatic capacity. Given that T3p and TERC share a common sequence, RNAi-mediated inhibition of T3p is not a suitable strategy for isolating its function. Thus, to achieve stable and sustained inhibition of T3p we took advantage of Tough Decoy (TuD) sponges—hairpin-shaped bulged RNAs containing two recognition sites that bind and sequester the small RNA of interest15. We generated cells stably expressing TuD RNAs targeting T3p and performed additional lung colonization assays. TuD-mediated inhibition of T3p resulted in a similarly substantial reduction in the metastatic capacity of MDA-LM2 cells (Fig. 3e and Supplementary Fig. 3c). Interestingly, T3p inhibition did not significantly impact in vitro proliferation rates, cell cycle length, or in vivo tumor growth rates of MDA-LM2 cells (Supplementary Fig. 3d-f). To ensure these observations were not limited to the MDA-LM2 background, we also performed in vivo lung colonization assays with an independent breast cancer cell line, HCC1395. Consistent with our previous results, T3p inhibition in HCC1395 cells also resulted in decreased metastatic capacity, and also did not affect in vitro proliferation rates (Supplementary Fig. 3g,h). Together, these observations strongly support a functional role for T3p, a previously unknown ncRNA, in driving breast cancer metastasis.
Biogenesis of T3p in breast cancer cells
We next sought to identify the T3p biogenesis pathway to explain its cancer specific expression. We considered two possibilities for the biogenesis of T3p: (i) transcription from a cryptic promoter, and (ii) nucleolytic digestion of the TERC RNA. Ectopic transcription has been implicated in a variety of diseases16; to assess this possibility, we examined RNA-seq and global run-on sequencing (GRO-seq) datasets from MDA-MB-231 cells and did not find any evidence for increased active transcription at the T3p locus. Consistent with this, RNA PolII ChIP-seq datasets also show no increased RNA PolII binding at the 5’ end of T3p, suggesting that T3p is not a product of ectopic transcription (Supplementary Fig. 4a). These findings suggested that T3p may be a byproduct of TERC RNA digestion or cleavage. Supporting this, we noted a significant positive correlation between TERC and T3p expression across the TCGA-BRCA dataset (Supplementary Fig. 4b). To identify RNA binding proteins (RBPs) and nucleases that may be involved in the biogenesis of T3p we first performed a co-expression network analysis17 centered on T3p expression by analyzing the TCGA-BRCA dataset to identify those genes whose expression is significantly correlated with that of T3p. We then further limited our candidates to RBPs and nucleases in order to focus on factors that could have a direct role in T3p biogenesis. We also required candidates to be (i) overexpressed in breast tumors relative to normal tissue, and (ii) upregulated in highly metastatic MDA-LM2 cells compared to poorly metastatic MDA-MB-231 cells. This analysis yielded two candidates: the double-stranded RBP TARBP2 and the double-stranded RNA-specific endoribonuclease DROSHA (Fig. 4a). The expression of each of these genes is also correlated with T3p expression (Supplementary Fig. 4c). Interestingly, our published TARBP2 HITS-CLIP data18 showed a direct interaction between TARBP2 and the 3’-end of TERC (Fig. 4b). We also analyzed recently published data from DROSHA fCLIP-seq19, and observed evidence of DROSHA binding to the 3’-end of TERC (Fig. 4c). Together, these analyses suggested TARBP2 and DROSHA as candidates directly involved in the nucleolytic biogenesis of T3p from TERC.
Fig 4: T3p biogenesis and function.
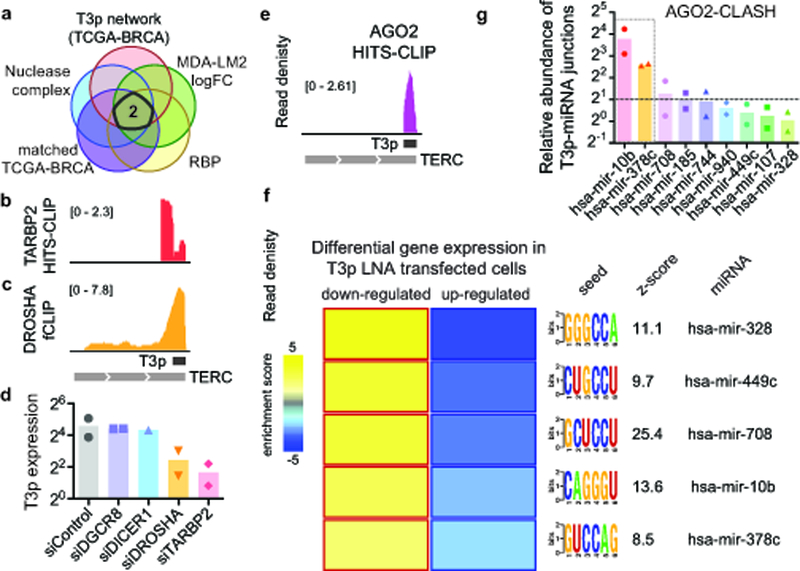
a, A network analytical approach combined with a computational gene prioritization step (see Methods) was used to identify two candidate proteins that may be involved in T3p biogenesis: TARBP2 and DROSHA. RNA-seq experiments were performed in biological replicates. b, c Read densities from previously published CLIP datasets showing direct interactions between TARBP2 (b) and DROSHA (c) proteins with the 3’ end of TERC RNA18,19. d, Normalized T3p levels in smRNA-seq data from MDA-LM2 cells transfected with each of the indicated siRNAs. n = 2 biologically independent samples. Normalized expression was calculated using the DE-Seq2 package (center = mean). e, Genome browser view of AGO2 CLIP24 read density at the T3p locus. f, A heatmap depicting the activity of miRNAs predicted to be targeted by T3p in vivo. The miRNA seed sequences were used to evaluate gene expression changes in their targets induced by an anti-T3p LNA. For each listed miRNA, we observed a significant enrichment of the miRNA target genes among those downregulated in T3p silenced cells. Also shown are the mutual information values (MI, measured in bits) and the associated z-score for each association26. Yellow boxes with red borders indicate significant enrichment and blue boxes with dark blue borders indicate significant depletion. n = 2 biologically independent experimental comparisons. g, AGO2 CLASH-qPCR was performed on candidate and control miRNAs (hsa-mir-940 and hsa-mir-107) to test for in vivo interactions with T3p. Assays performed in biological duplicate.
To directly test the impact of TARBP2 and DROSHA on T3p levels in MDA-MB-231 cells we used siRNAs to knock down each factor, as well as DGCR8 and DICER1 as controls. We then performed smRNA-seq to measure changes in T3p levels. RNAi-mediated knockdown of either TARBP2 or DROSHA resulted in a substantial reduction in T3p levels, while silencing DICER and DGCR8 did not affect T3p levels (Fig. 4d). We confirmed this observation using qRT-PCR (Supplementary Fig. 4d). Consistently, in TCGA-BRCA, T3p levels are higher in samples that have higher expression of both DROSHA and TARBP2 (Supplementary Fig. 4e). Together, these results implicate TARBP2 and DROSHA in the biogenesis of T3p from TERC, and also provide an explanation for the presence of T3p in cancer cells and its absence in normal cells. Increased TERC expression and telomerase activity are hallmarks of tumorigenesis. This increase in TERC, combined with higher expression of TARBP2 and DROSHA in cancer cells, provides an explanation for the cancer-specific generation of T3p. As we will demonstrate below, the production of T3p as an oncRNA provides an opportunity for cancer cells to modulate the expression of key promoters of metastatic progression.
T3p exerts its regulatory effects through interaction with the RISC complex
After assessing the regulatory and phenotypic consequences of T3p expression in breast cancer cells we sought to determine the regulatory mechanism(s) through which T3p drives deregulated and pathologic gene expression. To address this we searched a large dataset of binding preferences for RBPs20 to find potential T3p binding proteins, and similarly searched a curated database of CLIP experiments that provides in vivo binding sites for roughly 100 RBPs2,7,21–24. The most robust of these observed and inferred interactions was a highly significant and precise interaction between T3p and Argonaute 2 (AGO2) in a previously published CLIP-seq dataset24. Unlike TARBP2 and DROSHA, which show binding to regions of TERC outside of T3p, AGO2 binding signal is confined to the T3p boundaries (Fig. 4e). This clear demarcation suggests that AGO2 binds T3p but not TERC. We confirmed the interaction between T3p and AGO2 using UV crosslinking and AGO2 immunoprecipitation followed by qRT-PCR in MDA-MB-231 cells (Supplementary Fig. 4f).
The interaction between T3p and AGO2 raised the possibility that the broad regulatory effects of T3p could be carried out either by T3p acting as a miRNA, leading to targeted silencing of downstream transcripts, or as inhibitor of AGO2-miRNA complex activity. To test the first possibility, we performed an unbiased search for all 7-mers across T3p that could act as seed sequences. For each 7-mer, we asked whether the levels of the mRNAs that contain its complementary sequence change when T3p levels are modulated using transfected LNAs or exogenous mimetics. The absence of a seed sequence that meets this criterion, and the fact that T3p is longer than conventional miRNAs, largely rules out the former model. We next hypothesized that T3p may interfere with the activity of the RISC complex by competing with endogenous miRNA targets for binding. To assess this possibility, we used three criteria to identify candidate miRNAs that may be targeted by T3p: (i) the ability of the miRNA to form a stable duplex with T3p (e.g. Supplementary Fig. 4g), (ii) downregulation of predicted mRNA targets of the miRNA when T3p is silenced, and (iii) the miRNA is expressed at a level similar to or lower than that of T3p (i.e. favorable stoichiometry). We reasoned that decreased T3p expression should result in increased miRNA activity, which in turn would decrease levels of the mRNA targets of the miRNA. We identified a set of five miRNAs that satisfy these criteria (Fig. 4f and Supplementary Fig. 4h,i). To test whether any of these miRNAs directly interact with T3p in the RISC complex, we used a qRT-PCR derivative of the CLASH method25 (see Methods for details). As shown in Fig. 4g, this assay provided evidence that RISC-associated miR-10b-5p (miR-10b) and miR-378c-5p (miR-378c) directly interact with T3p.
As a consequence of this T3p-mediated interaction, the targets of these miRNAs also respond to LNA-mediated inhibition of T3p activity. Consistent with this observation, the miR-10b and miR-378c regulons were upregulated in the presence of T3p mimetic oligos as well as in MDA-LM2 cells, whose endogenous T3p levels are higher than those in MDA-MB-231 cells (Fig. 5a). Together, these results provide further evidence that T3p is a modulator of miRNA activity in breast cancer cells.
Fig 5: T3p-mediated inhibition of miR-10b and miR-378c results in overexpression of metastasis promoters NUPR1 and PANX2.
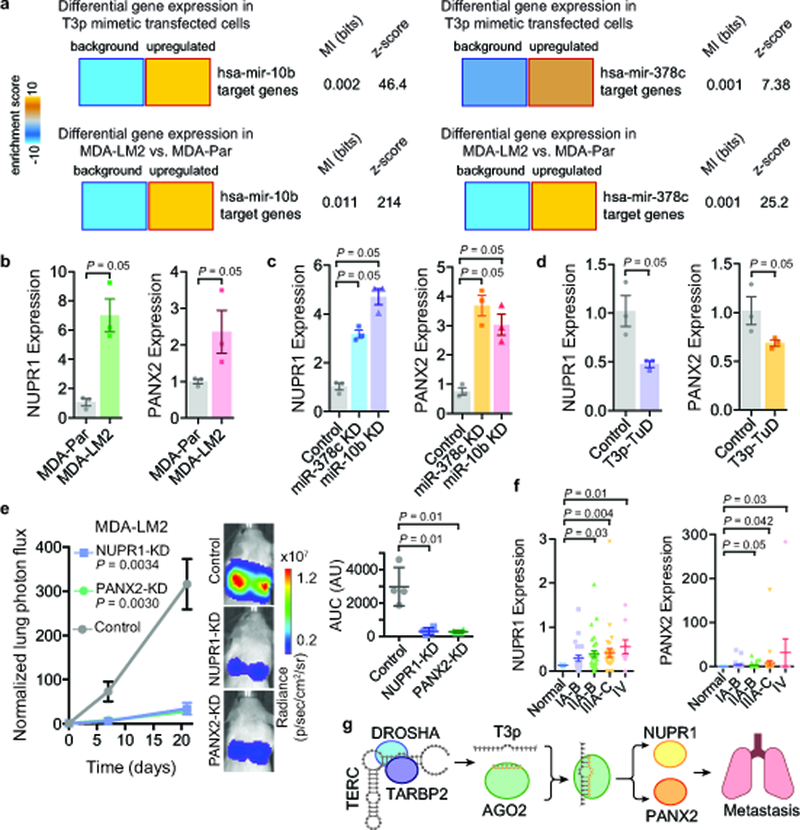
a, Heatmaps showing significant upregulation of miR-10b and miR-378c targets upon transfection of MDA-MB-231 cells with T3p mimetic. Significant upregulation of these targets was also observed in highly metastatic MDA-LM2 cells compared to poorly metastatic MDA-MB-231 cells. Mutual information values (MI, measured in bits) and the associated z-scores are also shown. n = 2 biologically independent experimental comparisons. b, qRT-PCR for NUPR1 and PANX2 in MDA-LM2 and the parental MDA-MB-231; n = 3 biologically independent experiments. c, qRT-PCR for NUPR1 and PANX2 in control, miR-378c, and miR-10b knockdown cells; n = 3 biologically independent experiments. d, NUPR1 and PANX2 expression levels in cells expressing a Tough Decoy against T3p (T3p-TuD) relative to control cells; n = 3 biologically independent experiments. A one-tailed Mann-Whitney test was used to calculate P for (b-d). e, Bioluminescence imaging plot of lung colonization by MDA-LM2 cells stably expressing CRISPRi guide RNAs against NUPR1, PANX2, or a control guide; n = 4 biologically independent animals per cohort. Statistical significance of knockdown versus control cohorts was measured using two-way ANOVA. The area under the curve was also calculated for each mouse; mean ± s.d. shown (change in normalized lung photon flux times days elapsed); P was calculated using a one-tailed Mann-Whitney test. f, NUPR1 and PANX2 expression levels were measured using qRT-PCR across 96 clinical samples composed of normal tissue and tissue from the indicated breast cancer stages. P was calculated using a one-tailed Mann-Whitney test (without adjustment). g, A schematic of model for T3p-mediated control of target genes in highly metastatic cells.
In order to identify the downstream genes that drive the phenotypic consequences of T3p expression, we performed a search for the most robust targets of this pathway. To do this we first identified genes predicted to be targeted by miR-10b or miR-378c26, and then searched for those also (i) up-regulated in large breast cancer datasets (TCGA-BRCA), (ii) down-regulated upon LNA-mediated inhibition of T3p, and (iii) up-regulated in MDA-LM2 compared to MDA-MB-231 cells. We calculated the sum of all ranks across these datasets, and identified NUPR1 and PANX2 as the two top candidate genes. These genes, which are predicted targets of miR-10b and miR-378c, have increased levels in highly metastatic MDA-LM2 cells, in which T3p expression is also elevated (Fig. 5b and Supplementary Fig. 5a). We also observed that T3p, NUPR1 and PANX2 are all up-regulated in bone, but not brain metastatic cells lines derived from the MDA-MB-231 cell line13,27 (Supplementary Fig. 5b). Moreover, NUPR1 and PANX2 expression was positively correlated with T3p expression and negatively correlated with miR-10b and miR-378c expression across a set of 10 breast cancer PDX models as well as a panel of nine breast cancer cell lines (Supplementary Fig. 5c). Furthermore, expression of NUPR1 and PANX2 were reduced upon transfection of the anti-T3p LNA in both MDA-LM2 and HCC38 cells (Supplementary Fig. 5d,e). We observed that inhibition of miR-10b and miR-378c using antisense inhibitors targeting these miRNAs resulted in an increase in the expression of NUPR1 and PANX2 mRNA (Fig. 5c). We also observed a decrease in the expression of NUPR1 and PANX2 in the presence of the T3p Tough Decoy (T3p-TuD; Fig. 5d). To assess if these observed changes in expression were due to post-transcriptional changes in RNA stability rather than transcriptional modulation, we also measured NUPR1 and PANX2 pre-mRNA levels upon T3p, miR-10b and miR-378c inhibition, and noted little change in the pre-mRNA levels of these genes, further implicating RNA stability as the underlying cause of the observed gene expression changes (Supplementary Fig. 5f,g). In support of these findings, we observed that NUPR1 and PANX2 mRNA co-immunoprecipitated with AGO2, and that inhibition of miR-10b and miR378c resulted in the loss of this association (Supplementary Fig. 5h). To test if NUPR1 and PANX2 are downstream of T3p as well as miR-10b and miR-378c, we used qRT-PCR to measure NUPR1 and PANX2 levels in cells transfected with inhibitors of T3p and these miRNAs. This showed that PANX2 and NUPR1 expression decreases upon T3p downregulation only when the miRNA levels are not perturbed (Supplementary Fig. 5i). Taken together, these observations establish NUPR1 and PANX2 as downstream targets of this T3p-mediated regulatory pathway.
Consistent with increased expression of these genes in highly metastatic cells, analysis of multiple breast cancer gene expression datasets showed a negative association between the expression of these genes in breast tumors and patient survival, as well as increased expression of these genes in advanced breast cancers (Fig. 5f and Supplementary Fig. 5j,k). We did not observe any significant association between NUPR1 or PANX2 expression and breast cancer subtype (Supplementary Fig. 5l). Together, these clinical associations support a role for NUPR1 and PANX2 in breast cancer progression. To experimentally test this possibility, we used CRISPRi to knock down these genes in the highly metastatic MDA-LM2 cells. As shown in Fig. 5e, decreased expression of these genes significantly reduced metastatic colonization of the lungs in xenografted mice. Additionally, we stably expressed T3p-TuD or the control decoy in these NUPR1 and PANX2 knockdown cells. We then performed in vivo metastatic lung colonization assays with these lines and, consistent with the premise that T3p impacts metastasis through these genes, we observed no further reduction in metastatic lung colonization when T3p was inhibited (Supplementary Fig. 5m). In vitro assays showed little change in the proliferation rates of these NUPR1 and PANX2 knockdown cells (data not shown), indicating that the observed effect on metastasis is not due to a general reduction in cell proliferation. These results establish T3p, a previously unknown non-coding RNA, as a promoter of breast cancer progression (Fig. 5g).
Specific oncRNAs are present in the extracellular compartment
We next asked if oncRNAs could be detected in the extracellular space, a beneficial characteristic for clinical markers. To explore this possibility, we isolated RNA from media conditioned by eight breast cancer cell lines as well as HMECs. SmRNA-seq of this material revealed that of our 201 annotated oncRNAs, close to half were detected in the conditioned media (CM) from one or more of these breast cancer lines but not in HMECs (Fig. 6a), demonstrating that oncRNAs are present in the extracellular space. Given that extracellular vesicles (EVs) have been previously reported as a biologically functional destination for small RNAs28, we asked whether oncRNAs are also present in EVs. To do this, we sequenced RNA purified from EVs isolated from the same conditioned media we used for smRNA-seq in Fig. 6a (Supplementary Fig. 6a), and found that of the 201 annotated oncRNAs, a third were observed in the EVs from one or more of these breast cancer lines but not in HMECs. There is a small positive correlation between oncRNA levels in cells and conditioned media, as well as oncRNA levels in cells and EVs; suggesting that there are elements of both passive and active secretion of oncRNAs into the extracellular space (Supplementary Fig. 6b). Additionally, analysis of publicly available exosomal small RNA-seq data from MDA-MB-231 cells29 showed that 20% of oncRNAs, including T3p, were present in this independent dataset (Supplementary Fig. 6c). Given that extracellular oncRNAs are specifically generated and secreted by cancer cells, they may potentially serve as a fingerprint for the underlying tumor and would be accessible through liquid biopsies. To assess this possibility, we employed a machine learning approach by training a gradient boosted classifier (GBC) on the TCGA-BRCA dataset to distinguish normal tissue from breast cancer biopsies using oncRNAs as features. We first asked if this classifier could correctly identify serum samples collected from healthy individuals as ‘normal’. To do this, we processed raw smRNA-seq data from 35 healthy volunteers (11 samples from from exoRNA atlas and 24 samples from30) to measure circulating oncRNAs and used our previously trained GBC to classify this data. We found that our GBC successfully labeled 34/35 samples as normal. We then tested the same classifier on a dataset of circulating small RNAs from 40 breast cancer patients31 and successfully classified 37/40 patient samples. The strong overall performance of our classifier in distinguishing sera collected from healthy individuals and breast cancer patients (Area Under the ROC curve, AUROC=0.97; Supplemental Fig. 6d) further highlights the cancer-specificity and clinical utility of oncRNAs. For example, T3p alone showed markedly different expression levels in the serum of 40 breast cancer patients and healthy volunteers (Fig. 6b and Supplementary Fig. 6e). We also used qRT-PCR to measure T3p levels in EV RNA from an independent set of serum samples from breast cancer patients and healthy volunteers, and observed significantly increased T3p levels in Stage I and II samples compared to the unaffected controls (Supplementary Fig. 6f). These data demonstrate that T3p is present in circulating EVs in human cancer patients, and may be useful in detecting stage I breast cancers and well as more advanced cancer stages. It important to note that although this classifier is trained on oncRNA levels in tumor biopsies, it can correctly classify serum samples, highlighting the fact that circulating oncRNAs can directly reflect the predictive oncRNA content of the underlying tumor. To test the possibility that any random classifier can perform similarly well, we also trained a GBC on features (i.e. small RNAs) and labels (i.e. normal vs. tumor) randomly selected from the TCGA dataset. As expected, this random classifier performed poorly on the 35 healthy and 40 cancer samples (AUROC: 0.422).
Fig 6: Systematic profiling of oncRNAs in the extracellular compartment.
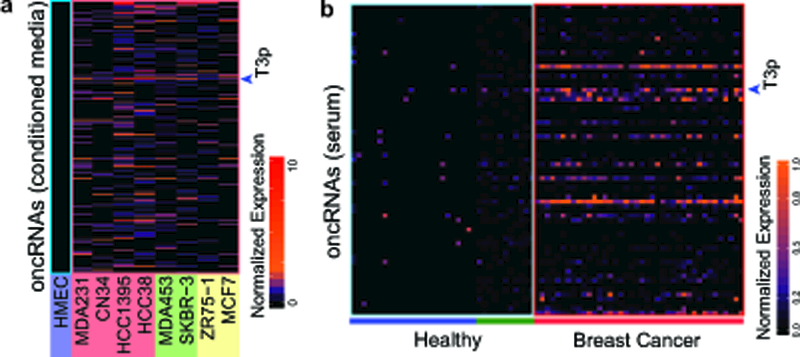
a, Small RNA sequencing of RNA collected from conditioned media (CM) from breast cancer cell lines and normal HMECs. CM from each cell line was prepared in biologically independent duplicate, which were combined prior to count-per-million calculations. Heatmap shows the detection of oncRNAs in the extracellular compartment. T3p is indicated with an arrow. b, The detection of oncRNAs in serum samples collected from breast cancer patients with stage II and III disease36. As a point of reference, we have also included data from 35 healthy individuals from two independent studies (green: 11 samples from the exoRNA atlas; blue: 24 samples from30). T3p is indicated with an arrow.
We then sought to compare the performance of circulating oncRNAs to that of previously reported breast cancer serum miRNA signatures in classifying these clinical samples. To do so, we compiled a set of 36 miRNA signatures associated with breast cancer tumorigenesis and progression from four independent studies32–35. These independent studies did not identify any overlapping miRNAs, highlighting the high degree of variation involved in circulating miRNA quantification. Taking the same approach, we used these previously published miRNAs to train a GBC on the TCGA-BRCA dataset. Testing this miRNA-based classifier on the serum small RNA dataset showed a relatively poor performance (AUROC=0.69; Supplementary Fig. 6g). These results imply that serum miRNA signatures do not reflect changes in miRNA expression levels of the underlying tumor and, unlike oncRNAs, may not directly originate from cancer cells. Consistent with this notion, a 5-fold cross-validation of a miRNA-based classifier on the serum small RNA dataset showed 89% (±11%) accuracy, which is substantially superior to its performance when trained on the TCGA-BRCA dataset. However, oncRNAs performed better in this sample type as well: a five-fold cross-validation of an oncRNA-based classifier achieved 98% (±8%) accuracy in serum small RNA data. Based on these results, we surmise that detection of circulating oncRNAs may potentially act as a more reliable readout of the underlying cancer.
Discussion
Through the application of a systematic and unbiased discovery platform that includes breast cancer cell lines, patient-derived xenograft models, and clinical breast cancer data, we have identified a set of 201 RNA species that are specifically expressed in breast cancer cells and are largely undetected in normal cells and tissue. These RNA molecules, which we have collectively named orphan non-coding RNAs, provide a pool of novel potential regulators that breast cancer cells may use to engineer new regulatory circuits. We compared poorly and highly metastatic cells to ask whether any of these oncRNAs are adopted by cancer cells to promote disease progression. We discovered that T3p, an oncRNA generated from the TERC RNA, is strongly associated with breast cancer progression both in cell line models and clinical datasets. We then used computational and experimental methodologies to (i) establish its role as a regulator of gene expression and a promoter of breast cancer metastasis, (ii) characterize its regulatory function, and (iii) uncover its biogenesis pathway. Finally, we have discovered that oncRNAs can be detected in circulating and EV compartments originating from cancer cells, and therefore have the potential to play functional roles in non-cell autonomous pathways.
Our findings introduce a new paradigm of cancer progression in which tumors evolve new regulatory pathways en route to metastatic spread. Furthermore, these cancer-specific post-transcriptional regulatory pathways comprise a set of potential novel therapeutic targets that are exclusive to cancer cells. Moreover, as circulating oncRNAs provide a snapshot of the cancer cells’ regulatory state, oncRNA profiling presents an opportunity to obtain highly tumor-specific data from liquid biopsies, and may complement current diagnostic methods. Therefore, oncRNA profiling in serum samples could act as an informative digital fingerprint—i.e., a profile of the presence/absence of each oncRNAs— of the underlying tumor.
Although our study has focused on the role of T3p in breast cancer metastasis, the approaches and concepts presented here are generalizable and can be applied to other cancer types. Taken together, our findings raise the possibility that further examination of the cancer-specific RNA landscape may yield novel strategies in developing diagnostic and therapeutic methods in breast cancer.
Methods:
Tissue culture
All cells were cultured in a 37°C, 5% CO2 humidified incubator. Cell lines MDA-MB-231, MDA-LM2, CN34-par, CN34-Lm1a, MCF7 and MDA-MB-453 were propagated in DMEM base media supplemented with 4.5 g/L glucose, 10% FBS, 4mM L-glutamine, 1mM sodium pyruvate, penicillin (100 units/mL), streptomycin (100 μg/mL) and amphotericin (1μg/mL). Cell lines HCC1395, ZR-75–1 and HCC38 were propagated in RPMI 1640 base media supplemented with 10% FBS, 2mM L-glutamine, penicillin (100 units/mL), streptomycin (100 μg/mL) and amphotericin (1μg/mL). SK-BR-3 cell line was propagated in McCoy’s 5a modified media supplemented with 10% FBS, penicillin (100 units/mL), streptomycin (100 μg/mL) and amphotericin (1μg/mL). HMECs were obtained from Thermo Fisher Scientific and propagated in HuMEC ready media (Thermo Fisher Scientific).
Small and extracellular vesicle RNA extraction and sequencing
Preparation of conditioned media for isolation of RNA from extracellular vesicle and total conditioned media was carried out by seeding cells at 7×105. 24 hours later, cells were washed twice with PBS and 10 mL exosome-depleted media was added. 48 hours later, media was harvested by spinning at 200 x g for 15 minutes and taking the supernatant. Exosome-depleted media was prepared by substituting exosome-depleted FBS (Thermo Fisher Scientific) for FBS. Exosome-depleted HMEC media was prepared by centrifuging the bovine pituitary extract media component at 100,000 x g at 4°C for 16 hours.
Extracellular vesicle RNA was isolated from 5 mL conditioned media, prepared as outlined above, using the Cell Culture Media Exosome Purification and RNA Isolation kit (Norgen Biotek). RNA from conditioned media was isolated from 400 μL total conditioned media using the miRNeasy serum/plasma kit (Qiagen). Total cellular small RNA samples were extracted using Norgen Biotek small RNA purification kit according to the manufacturer’s protocol. RNA samples were subsequently prepared for high-throughput sequencing with the NEXTflex Small RNA Sequencing Kit v3 using the manufacturer’s protocol (Bioo Scientific). The resulting libraries were then sequenced and processed as recommended by the manufacturer. Briefly, cutadapt (v1.4) was used to remove the adapter sequences and trim the degenerate sequences at the beginning and end of each read. We then used bowtie2 (v2.3.3) to align the resulting sequences to the human genome (build hg38). The resulting BAM files were then sorted and converted to BED for further analysis.
Extracellular vesicle RNA was isolated from serum samples using the Plasma/serum Exosome Purification and RNA isolation kit (Norgen Biotek) according to the manufacturer’s instructions.
TCGA-BRCA small RNA sequencing data and identification of oncRNAs
Reads from the TCGA-BRCA project were downloaded from the Genomic Data Commons (GDC) in BAM format (hg38) and the samples were annotated using the GDC API. Upon conversion to the BED format, the Piranha package7 was used to identify the expressed small RNA loci. The resulting loci were merged across all samples using mergeBed to create a comprehensive list of small RNA loci expressed in breast tissue and breast cancer.
By enumerating the small RNA sequences obtained from breast cancer cell lines and HMECs, we generated a count table for each small RNA locus. We then normalized the resulting table by library size and retained only those loci with no observed reads across the three HMEC replicates. We used two independent statistical tests to compare either all cancer cell lines or each subtype individually (TNBC, HER2+, and Luminal): (i) we used the DESeq package in R to calculate an adjusted P value, and (ii) we used Fisher’s exact test to compare presence and absence of each small RNA. We selected those loci with either an adjusted P <0.05 in the former or P <0.1 in the latter test across all comparisons. There were 437 loci that satisfied these criteria (shown in Fig. 1a). For visualization, we max-normalized each row and performed a k-means clustering (k = 3).
For the TCGA-BRCA database, we generated a similar count table across all subtype annotated samples (based on PAM50 classification) and all small RNA loci and normalized the resulting table to generate a count-per-million reads (cpm) table. In order to identify ‘orphan’ small RNAs, i.e. small RNAs that are largely absent in normal cells, we retained only the loci with their 90th percentile expression in normal samples below 0.5 cpm. Of the 437 loci above, 268 passed this step. We then performed Fisher’s exact tests to compare the presence of all small RNAs across the tumor samples and normal biopsies. We performed similar comparisons between normal samples and each of the breast cancer subtypes. We then retained those loci that were significant in at least one of these tests with an adjusted P-value of <0.05. 201 of the small RNAs satisfied this final step and were thus classified as orphan non-coding RNAs. We confirmed that none of these small RNAs were previously annotated as miRNAs, snoRNA, or tRNAs.
Small RNA sequencing of PDX models and normal epithelial samples
All human samples used to generate PDX tumors, as well as the human non-tumor samples, were previously described37. Small RNA profiling and data pre-processing was carried out by Q2Solutions. The abundance of oncRNAs in these samples was determined as described above.
Comparing oncRNA expression between poorly and highly metastatic cells
We used the R package DESeq2 to compare expression of oncRNAs between parental cell lines in Figure 1 (MDA231 and CN34) and their highly metastatic in vivo selected derivatives to identify those oncRNAs that were significantly upregulated in highly metastatic cells. We identified T3p in this analysis, which we also confirmed in a small RNA dataset we had previously generated for these lines7. In addition, we also performed quantitative RT-PCR assays. For this, we extracted small RNAs from MDA-MB-231 parental cells and their highly metastatic MDA-LM2 derivative cell line (microRNA Purification Kit; Norgen) and performed stem-loop qPCR. T3p primers: 5’-CCAGTGCAGGGTCCGAGGTA and 5’-CCCAGGACTCGGCTCACAC. 18S (endogenous control) primers: 5’-GTAACCCGTTGAACCCCATT and 5’-CCATCCAATCGGTAGTAGCG.
T3p expression and clinical association in the TCGA-BRCA dataset
We used the metadata accompanying the TCGA-BRCA dataset to perform survival analysis based on T3p expression in tumor samples. We stratified the patients based on T3p levels and generated Kaplan-Meier curves using all tertiles and performed log-rank (Mantel-Cox) test to calculate the associated P-value. We similarly used the clinical data to compare T3p expression across early and late stage tumors (one-tailed Mann-Whitney U-test).
T3p modulation and gene expression profiling
We used miRCURY LNA inhibitors (Exiqon) against the following sequences: T3p: CAGGACTCGGCTCACACATGC; TERC: TTGTCTAACCCTAACTGAGAAGG; Scrambled: AGACGACAGCTGGATCACACG. Similarly, we used T3p mimetics (IDT): 5’-rU*rCrCrCrUrGrArGrCrUrGrUrGrGrGrArCrGrUrGrCrArCrCrCrArGrGrArCrUrCrGrGrCrUrCrArCrArCrArUrG*rC-3′ (T3p mimetic) and rA*rGrArCrGrArCrArGrCrUrGrGrArUrCrArCrArC*rG (control). We then transfected the LNAs in the highly metastatic MDA-LM2 cells and the mimetics in the parental MDA-MB-231 cells and performed gene expression profiling and differential gene expression analysis as previously described7.
Tough Decoys and in vivo lung colonization and tumor growth assays
MDA-LM2 cells were transfected with anti-T3p or scrambled LNA (same as above) and after 48 hours, cells were injected via tail-vein into the vasculature of immunocompromised NOD SCID gamma (NSG) mice (2.5×104 per mouse; n = 5 per cohort). In vivo imaging and comparison of curves was performed as previously described18. Lungs from at least three mice per cohort (middle signal) were then extracted, fixed, sectioned, stained with hematoxylin and eosin, and quantified as previously described18.
To generate stable inhibition of T3p, we designed Tough Decoys (TuDs) against this small RNA in a lentiviral backbone under a RNA PolIII promoter (pLKO.1). We then stably transduced MDA-LM2 cells and performed lung colonization assays, injecting 5×104 cells per NSG mouse. HCC1395 cells were similarly transduced and injected at 2×105 cells per NSG mouse.
Orthotopic tumor growth assays were performed by injecting 2.5×105 cells resuspended in 50 μL PBS mixed with 50 μL matrigel into the mammary glands of age-matched 6–8 week old female NOD/SCID gamma mice using a 28 gauge needle. Tumor volume was determined by using calipers to measure the tumor length (L) and width (W) every two days and calculated using the formula πLW2/6. The experiment endpoint was reached once tumors reached a volume of 800mm3.
Cell proliferation
In vitro cancer cell proliferation assays are performed by seeding 5×104 cells at day 0 and then counting them in triplicate on day 3 and day 5. The slope of the best fitted line between log of cell counts and days is the reported proliferation rate (logNt = logN0 + rt; t: time (days); r: proliferation rate (days−1)).
For cell-cycle analysis, cells were grown to 80% confluency in 6-cm plates, harvested and fixed in 70% ethanol. Cells were then pelleted and resuspended in 50 μg/mL propidium iodide (Thermo Fisher Scientific) and 1 mg/mL RNase A (Thermo Fisher Scientific) and allowed to incubate 1 hour at 37°C. BD Aria2 flow cytometer was then used for FACS analysis; post-FACS analysis and cell cycle quantification was performed using the python package ‘fcsparser’. FCSparser: https://github.com/eyurtsev/fcsparser/tree/master/fcsparser
Co-expression analysis for finding T3p biogenesis factors
In order to identify the regulators of T3p biogenesis, we listed the genes with known nuclease activity (GO:0004540 and GO:0004525) and further added RNA-binding proteins that are known to interact with these nucleases38. We then performed co-expression analysis between T3p levels and those of the genes on this list across the TCGA-BRCA dataset. We overlapped the genes with strong associations with those that are upregulated in highly metastatic MDA-LM2 cells and are also higher in breast cancer samples relative to normal biopsies in the TCGA-BRCA dataset. Based on these criteria, we identified seven candidates, two of which had known double-stranded binding activity. Since the CR7 domain of TERC is structured (i.e. forms double-stranded regions), we reasoned that these proteins, namely DROSHA and TARBP2, were the best candidates for follow-up. We used siRNAs (IDT) to knockdown DROSHA and TARBP2, as well as DGCR8 and DICER1, which are known to interact with DROSHA and TARBP2, respectively. We used the following target sequences: TARBP2: 5’-ACCTGGGATTCTCTACGAAATTCAGT, DROSHA: 5’-CCTTGATTGAGGTATAGTTCTTGTCT, DICER1: 5’-TGGTGCTTAGTAAACTCTTGGTTCCA, and DGCR8: 5’-CTGCAGGAGTAAGGACAGGAAGGTGC. Following siRNA transfection and knockdown verification, we performed small RNA sequencing as described above.
Identifying and validating miRNA targets of T3p
For each miRNA, we asked whether their target regulon, predicted based on matches to their seed sequences, was significantly downregulated when T3p was silenced. For this, we used the motif discovery platform FIRE26 to analyze the T3p-LNA differential gene expression data against the list of miRNA seed sequences. For those miRNAs that passed this initial test, we used RNAHybrid39 to further select those miRNAs that can form duplexes with T3p (maximum MFE of −14.0). We also required these miRNAs to be expressed at a similar or lower level than T3p in our small RNA profiling dataset. Finally, we performed AGO2 CLASH qPCR assays, based on the AGO2-CLASH method, to confirm direct interactions in vivo between candidate miRNAs and T3p.We used an AGO2 antibody (Proteintech 10686–1-AP) and used a preparation method similar to AGO2 HITS-CLIP40. Briefly, samples were CIP and PNK treated on bead after immunoprecipitation, and then T4 RNA ligase 1 (in the absence of the 3’ linker commonly used in CLIP experiments) was used to achieve proximity ligation. Samples were then treated with Proteinase K and the RNA was extracted and cleaned up (Zymogen RNA Clean & Concentrator kit). The T3p matching primer 5’-TGTGAGCCGAGTCCTGGGTG, along with primers based on each of the candidate miRNAs, were used to perform qRT-PCR (SYBR Green). Among the tested candidates, miR-10b and miR-378c showed strong evidence of physical interaction with T3p in vivo. We obtained predicted targets of these two miRNAs (StarBase) and confirmed their upregulation in the presence of T3p based on our gene expression profiles. AGO2 CLIP-qPCR was performed using the AGO2 HITS-CLIP immunoprecipitation method, without any nuclease digestion or other enzymatic treatment of the crosslinked RNA. Protein-RNA complexes were immunoprecipitated using an antibody to AGO2 (Proteintech 10686–1-AP) or rabbit IgG. After washing, RNA was isolated by treating beads with Proteinase K followed by phenol chloroform extraction. The relative abundance of NUPR1 and PANX2 in the resulting RNA was assessed by qRT-PCR. To assess T3p and miR-378c copy number, we used synthetic RNA versions of these molecules (IDT) to create a standard curve with known concentrations of synthetic T3p and miR-378c. We then used qRT-PCR to measure T3p and miR-378c concentration in small RNA extracted from 8×105 MDA-MB-231 cells by comparing to the standard curve.
T3p downstream target selection and functional validation
In order to identify downstream targets that are mediated by T3p expression in breast cancer cells, we used the following criteria: (i) targets of miR-10b and miR-378c (StarBase), (ii) upregulated in breast tumors (TCGA-BRCA dataset), (iii) upregulated in highly metastatic MDA-LM2 cells where T3p is highly expressed, and (iv) down-regulated when T3p inhibited. Based on these criteria, we identified NUPR1 and PANX2 as targets of T3p. We used qRT-PCR to verify changes in the levels of these genes in response to modulations in components of this pathway. We used miRNA inhibitors (IDT) and Tough Decoys for this assay. Primer pairs used for qRT-PCR measurement were: 5’-AAGCAGAGACAGACAAAGCG and 5’-TGGGCATAGGCATGATGAGA were for NUPR1 mRNA; 5’-AACCCAGATCTCTGTCCCTTT and 5’-CCGCAGTCCCGTCTCTATT for NUPR1 pre-mRNA. Similarly, 5’-CCAAGAACTTCGCAGAGGAAC and 5’-GGGCAGGAACTTGTGCTCA were used for for PANX2 mRNA, and 5’-AGCCCGTGTCTCCTCTT and 5’-AGCTCCGTCCAGCAGTA for PANX2 pre-mRNA. We used HPRT1 as the endogenous control in these experiments: 5’-GACCAGTCAACAGGGGACAT and 5’-CCTGACCAAGGAAAGCAAAG.
To examine the association between NUPR1 and PANX2 expression with breast cancer progression, we acquired 96 cDNA samples from breast cancer clinical specimens including five normal epithelial cell samples, 23 stage I, 30 stage II, 29 stage III, and nine stage IV metastatic biopsies (Origene). We then used SYBR Green qRT-PCR to measure NUPR1 and PANX2 expression levels, using ACTB as the endogenous control.
We used CRISPRi guides to generate NUPR1 and PANX2 knockdown cells in the highly metastatic MDA-LM2 cells. The choice of CRISPRi was based on its minimal off-target effects. We achieved 4–5 fold knockdown for each gene using the following guides: 5’-TTGGGGATCCCGGCCCGGAGCGCGTTTAAGAGC for PANX2 and 5’-TTGGGCCTTATAAGCTGAGGGAGGTTTAAGAGC for NUPR1. We then performed lung colonization assays as described above.
Training and testing of oncRNA-based classifiers
Of the 201 oncRNAs, 100 were detected in at least one serum sample. We used these 100 oncRNAs to train a GBC on subtype-annotated TCGA-BRCA samples (sklearn module). We then bootstrapped our compendium of serum samples from 35 healthy and 40 cancer patients 100 times to calculate the performance parameters of the classifier, namely average AUROC, precision, and accuracy scores. We also performed an independent assessment of oncRNAs by performing training and testing on the serum data as opposed to TCGA-BRCA (5-fold cross-validation).
We used the following miRNAs to perform similar analyses as above: miR-10b-5p, miR-10b-3p, miR-148b-3p, miR-148b-5p, miR-155–3p, miR-155–5p, miR-34a-3p, miR-376a-3p, miR-652–3p, miR-133a-3p, miR-139–3p, miR-143–3p, miR-145–3p, miR-15a-3p, miR-18a-3p, miR-425–3p, miR-34a-5p, miR-376a-5p, miR-652–5p, miR-133a-5p, miR-139–5p, miR-143–5p, miR-145–5p, miR-15a-5p, miR-18a-5p, miR-425–5p, miR-127–3p, miR-194–5p, miR-205–5p, miR-21–5p, miR-375, miR-376c-3p, miR-382–5p, miR-409–3p, and miR-411–5p.
Supplementary Material
Acknowledgments:
We are grateful to Davide Ruggero, David Erle, Felix Feng, Sohail Tavazoie, and Saeed Tavazoie for reading earlier versions of this manuscript. We acknowledge the UCSF Center for Advanced Technology (CAT) for high-throughput sequencing and other genomic analyses. We also acknowledge David Erle and Paula Godoy for providing early access to their serum small RNA profiles. We thank Byron Hann and the Preclinical Therapeutics core as well as the Laboratory Animal Resource Center (LARC). We thank Seda Kilinc for her assistance with extracellular vesicle size analysis. We thank Faranak Fattahi for helpful discussion. We are also grateful for the genomic data contributed by the TCGA Research Network, including donors and researchers. We acknowledge support from our colleagues at the Breast Oncology Program (Helen Diller Family Comprehensive Cancer Center). This work was supported by the NIH (R01GM123977 and R00CA194077), Friends for an Earlier Breast Cancer Test, the American Cancer Society (130920-RSG-17–114-01-RMC), and the Goldberg-Benioff Fund in Translational Research. S.Z. was supported through the HHMI Medical Research Fellowship. J.Y. is an NSF GRFP fellow. A.G. was supported by the CDMRP Breast Cancer Research Program W81XWH-12–1-0272 and W81XWH-16–1-0603.
Footnotes
Competing interests: The authors declare no competing interests.
Animal studies
All animal studies were completed according to University of California San Francisco IACUC guidelines.
Statistical methods
Statistical tests used to assess data significance are described in the legends.
Reporting summary
Additional information on experimental design is available in the Nature Research Reporting Summary linked to this article.
Data Availability
All sequencing data generated for this study has been deposited in the Gene Expression Omnibus under the accession number GSE114366.
References
- 1.Tavazoie SF et al. Endogenous human microRNAs that suppress breast cancer metastasis. Nature 451, 147–152 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fish L et al. Muscleblind-like 1 suppresses breast cancer metastatic colonization and stabilizes metastasis suppressor transcripts. Genes Dev 30, 386–398 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vanharanta S et al. Loss of the multifunctional RNA-binding protein RBM47 as a source of selectable metastatic traits in breast cancer. eLife 3, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.David CJ, Chen M, Assanah M, Canoll P & Manley JL HnRNP proteins controlled by c-Myc deregulate pyruvate kinase mRNA splicing in cancer. Nature 463, 364–368 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen L-Y & Lingner J AUF1/HnRNP D RNA binding protein functions in telomere maintenance. Mol. Cell 47, 1–2 (2012). [DOI] [PubMed] [Google Scholar]
- 6.Goodarzi H et al. Modulated Expression of Specific tRNAs Drives Gene Expression and Cancer Progression. Cell 165, 1416–1427 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goodarzi H et al. Endogenous tRNA-Derived Fragments Suppress Breast Cancer Progression via YBX1 Displacement. Cell 161, 790–802 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Simanshu DK, Nissley DV & McCormick F RAS Proteins and Their Regulators in Human Disease. Cell 170, 17–33 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bhargava R et al. EGFR gene amplification in breast cancer: correlation with epidermal growth factor receptor mRNA and protein expression and HER-2 status and absence of EGFR-activating mutations. Mod. Pathol. Off. J. U. S. Can. Acad. Pathol. Inc 18, 1027–1033 (2005). [DOI] [PubMed] [Google Scholar]
- 10.Ren R Mechanisms of BCR-ABL in the pathogenesis of chronic myelogenous leukaemia. Nat. Rev. Cancer 5, 172–183 (2005). [DOI] [PubMed] [Google Scholar]
- 11.Lin R-K & Wang Y-C Dysregulated transcriptional and post-translational control of DNA methyltransferases in cancer. Cell Biosci 4, 46 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu C-I, Wang H-Y, Ling S & Lu X The Ecology and Evolution of Cancer: The Ultra-Microevolutionary Process. Annu. Rev. Genet 50, 347–369 (2016). [DOI] [PubMed] [Google Scholar]
- 13.Minn AJ et al. Distinct organ-specific metastatic potential of individual breast cancer cells and primary tumors. J. Clin. Invest 115, 44–55 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Loo JM et al. Extracellular metabolic energetics can promote cancer progression. Cell 160, 393–406 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bak RO, Hollensen AK, Primo MN, Sørensen CD & Mikkelsen JG Potent microRNA suppression by RNA Pol II-transcribed ‘Tough Decoy’ inhibitors. RNA N. Y. N 19, 280–293 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cooper DN, Berg LP, Kakkar VV & Reiss J Ectopic (illegitimate) transcription: new possibilities for the analysis and diagnosis of human genetic disease. Ann. Med 26, 9–14 (1994). [DOI] [PubMed] [Google Scholar]
- 17.Margolin AA et al. ARACNE: an algorithm for the reconstruction of gene regulatory networks in a mammalian cellular context. BMC Bioinformatics 7 Suppl 1, S7 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goodarzi H et al. Metastasis-suppressor transcript destabilization through TARBP2 binding of mRNA hairpins. Nature 513, 256–260 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim B, Jeong K & Kim VN Genome-wide Mapping of DROSHA Cleavage Sites on Primary MicroRNAs and Noncanonical Substrates. Mol. Cell 66, 258–269.e5 (2017). [DOI] [PubMed] [Google Scholar]
- 20.Ray D et al. A compendium of RNA-binding motifs for decoding gene regulation. Nature 499, 172–177 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang Y-CT et al. CLIPdb: a CLIP-seq database for protein-RNA interactions. BMC Genomics 16, 51 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Van Nostrand EL et al. Robust transcriptome-wide discovery of RNA-binding protein binding sites with enhanced CLIP (eCLIP). Nat. Methods 13, 508–514 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goodarzi H et al. Systematic discovery of structural elements governing stability of mammalian messenger RNAs. Nature 485, 264–268 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kishore S et al. A quantitative analysis of CLIP methods for identifying binding sites of RNA-binding proteins. Nat. Methods 8, 559–564 (2011). [DOI] [PubMed] [Google Scholar]
- 25.Helwak A, Kudla G, Dudnakova T & Tollervey D Mapping the human miRNA interactome by CLASH reveals frequent noncanonical binding. Cell 153, 654–665 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Elemento O, Slonim N & Tavazoie S A universal framework for regulatory element discovery across all genomes and data types. Mol. Cell 28, 337–350 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bos PD et al. Genes that mediate breast cancer metastasis to the brain. Nature 459, 1005–1009 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fiskaa T et al. Distinct Small RNA Signatures in Extracellular Vesicles Derived from Breast Cancer Cell Lines. PloS One 11, e0161824 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhou W et al. Cancer-secreted miR-105 destroys vascular endothelial barriers to promote metastasis. Cancer Cell 25, 501–515 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Noren Hooten N et al. Age-related changes in microRNA levels in serum. Aging 5, 725–740 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu X et al. De novo sequencing of circulating miRNAs identifies novel markers predicting clinical outcome of locally advanced breast cancer. J. Transl. Med 10, 42–42 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Roth C et al. Circulating microRNAs as blood-based markers for patients with primary and metastatic breast cancer. Breast Cancer Res. BCR 12, R90 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huo D, Clayton WM, Yoshimatsu TF, Chen J & Olopade OI Identification of a circulating microRNA signature to distinguish recurrence in breast cancer patients. Oncotarget 7, 55231–55248 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cuk K et al. Plasma microRNA panel for minimally invasive detection of breast cancer. PloS One 8, e76729 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kodahl AR et al. Novel circulating microRNA signature as a potential non-invasive multi-marker test in ER-positive early-stage breast cancer: a case control study. Mol. Oncol 8, 874–883 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wu X et al. De novo sequencing of circulating miRNAs identifies novel markers predicting clinical outcome of locally advanced breast cancer. J. Transl. Med 10, 42 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.DeRose YS et al. Tumor grafts derived from women with breast cancer authentically reflect tumor pathology, growth, metastasis and disease outcomes. Nat. Med 17, 1514–1520 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fabregat A et al. The Reactome Pathway Knowledgebase. Nucleic Acids Res 46, D649–D655 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rehmsmeier M, Steffen P, Hochsmann M & Giegerich R Fast and effective prediction of microRNA/target duplexes. RNA N. Y. N 10, 1507–1517 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moore MJ et al. Mapping Argonaute and conventional RNA-binding protein interactions with RNA at single-nucleotide resolution using HITS-CLIP and CIMS analysis. Nat. Protoc 9, 263–293 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.