

Abstract
The mechanistic (or mammalian) target of rapamycin (mTOR) and the adenosine monophosphate-activated protein kinase (AMPK) regulate cell survival and metabolism in response to diverse stimuli such as variations in amino acid content, changes in cellular bioenergetics, oxygen levels, neurotrophic factors and xenobiotics. This Opinion paper aims to discuss the current state of knowledge regarding how mTOR and AMPK regulate the metabolism and survival of brain cells and the close interrelationship between both signaling cascades. It is now clear that both mTOR and AMPK pathways regulate cellular homeostasis at multiple levels. Studies so far demonstrate that dysregulation in these two pathways is associated with neuronal injury, degeneration and neurotoxicity, but the mechanisms involved remain unclear. Most of the work so far has been focused on their antagonistic regulation of autophagy, but recent findings highlight that changes in protein synthesis, metabolism and mitochondrial function are likely to play a role in the regulatory effects of both mTOR and AMPK on neuronal health. Understanding the role and relationship between these two master regulators of cell metabolism is crucial for future therapeutic approaches to counteract alterations in cell metabolism and survival in brain injury and disease.
Keywords: mammalian target of rapamycin, adenosine monophosphate-activated protein kinase, autophagy, mitochondria, glycolysis, cell death, nutrient deprivation, energy failure
Graphical abstract
1. Introduction
The mechanistic (or mammalian) target of rapamycin (mTOR) and the adenosine monophosphate-activated protein kinase (AMPK) regulate cell growth and metabolism incorporating signals triggered by different stimuli such as variations in the amino acid content, changes in cellular bioenergetics, activation of hormone and growth factor signaling and stress such as oxidative stress, hypoxia or DNA damage. In the brain, AMPK and mTOR signaling are regulated by neurotransmitters and neurotrophin signals. mTOR and AMPK are considered master regulators of cell metabolism. Their activation is directly linked to the regulation of cellular metabolism (mitochondria homeostasis and central carbon metabolism), growth (protein synthesis) and survival (autophagy and cell death pathways). This Opinion paper aims to discuss the current state of knowledge regarding how mTOR and AMPK regulate brain metabolism and survival upon stress and more importantly, the close interrelationship between both signaling molecules (Figure 1 and Supplementary Table 1).
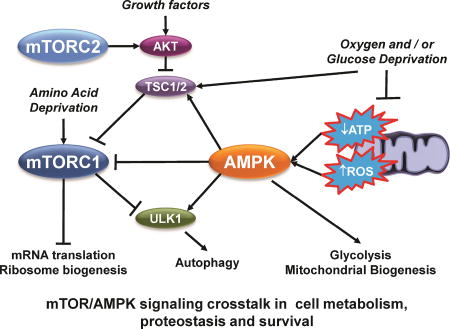
Figure 1. mTOR/AMPK signaling crosstalk.

Signaling by mTOR and AMPK integrates changes in the cellular environment linked to growth factors, nutrient, oxygen and energy availability, excitotoxicity (Ca2+) and stress (oxidative stress). Five major points of convergence include: 1) the transcriptional regulation of genes linked to cell survival/death, proteostasis, redox balance and bioenergetics; 2) metabolism (central carbon and bioenergetics); 3) autophagy; 4) mRNA translation and protein synthesis; and 5) cell survival and growth (AKT). Arrows indicate activation. Stop lines indicate inhibition. Phosphorylation events (P) are color coded according to the kinase involved in phosphorylating the corresponding targeted protein. White background/colored “P” highlights an inhibitory effect; Colored background/white P highlights an stimulatory (activation) effect. A detailed explanation of the phosphorylation events is included in the Supplementary Table 1. Additional crosstalk might involve indirect pathways not highlighted in this figure.
2. mTOR signaling in brief and its role regulating cell metabolism
mTOR is a serine (Ser)/threonine (Thr) protein kinase in the PI3K-related kinase (PIKK) family that forms the catalytic subunit of two distinct protein complexes, known as mTOR Complex 1 (mTORC1) and 2 (mTORC2) (Figure 1). Together with mTOR, the mammalian lethal with sec13 protein 8 (mLST8 or GβL), and the inhibitory DEP domain containing mTOR-interacting protein (DEPTOR) are common to both mTORC1 and mTORC2. Phosphorylation of DEPTOR by mTOR and other kinases promotes its degradation and activation of mTORCs. mLST8 seems to stabilize the kinase activation loop of mTOR but is dispensable for its activity. mTORC1 also includes the regulator-associated protein of the mammalian target of rapamycin (Raptor or RPTOR) and the proline-rich AKT substrate of 40 kDa (PRAS40). Because the interaction between Raptor and mTOR is essential for the stability of the mTORC1 and the recruitment of substrates, post-translational modifications in Raptor (phosphorylation, cleavage or ubiquitination) regulate mTORC1 activity. PRAS40 inhibits the substrate recruitment sites of mTORC1 and its phosphorylation by AKT and mTOR promotes its dissociation from the complex. On the other hand, the mTORC2 includes the rapamycin-insensitive companion of mTOR (Rictor), the mammalian stress-activated MAP kinase-interacting protein 1 (mSIN1), and the proteins observed with rictor 1 and 2 (PROTOR 1 and 2) [1].
The GTP binding protein Ras homolog enriched in brain (Rheb) is an obligate upstream activator of mTORC1 by growth factors and amino acids (Figure 1). Activation of the phosphoinositide-3 kinase (PI3K)/protein kinase B (PKB/AKT) pathway by growth factor receptors inactivates the tuberous sclerosis complex (TSC), a key negative regulator of mTORC1 (Figure 1). TSC is a heterotrimeric complex comprising TSC1, TSC2, and the Tre- 2/BUB2/cdc 1 domain family member TBC1D7. TSC2 acts as a GTPase-activating protein (GAP) to inactivate Rheb (Rheb-GDP). mTORC1 activation requires its localization in the lysosomal surface which is regulated by amino acid and nutrient sensing via Rag GTPases (Figure 1). Induction of protein synthesis by mTORC1 involves the phosphorylation of the ribosomal protein S6 kinase beta-1 (S6K) and the eukaryotic translation initiation factor 4 (eIF4E)-Binding Protein (4E-BP) (Figure 1) [1]. Glucose metabolism has been reported to be regulated by translational regulation of the hypoxia-inducible factor 1-alpha (HIF1α) via 4E-BP and mTORC1 [2]. Via 4E-BP, mTORC1 signaling regulates mitochondrial biogenesis as well [3]. mTORC1 also regulates lipid and nucleotide synthesis through the sterol responsive element binding protein (SREBP) transcription factors and the activation of the transcription factor 4 (ATF4), respectively [4,5].
mTORC2 primarily controls proliferation and survival via the activation of members of the AGC family of protein kinases such as PKC that regulate cytoskeletal remodeling and cell migration. AKT phosphorylation by mTORC2 is required for AKT-mediated phosphorylation of specific substrates such as FOXO1/3a. Reciprocally, AKT has been shown to phosphorylate mSIN1 enhancing mTORC2 kinase activity (Figure 1) [1]. mTORC2 has also been demonstrated to regulate glucose and amino acid metabolism [6–9]. Interestingly, a recent report also demonstrates that mTORC2 has the ability to act as a tyrosine kinase [10].
3. AMPK signaling in brief and its role regulating cell metabolism
AMPK is a Ser/Thr kinase that exists as a heterotrimer composed of catalytic α and regulatory β and γ subunits. In the absence of AMP, the autoinhibitory domain of AMPKα maintains the kinase in an inactive conformation. Upon ATP consumption, AMP/ADP directly bind to AMPKγ promoting the phosphorylation of AMPKα. The liver kinase B1 (LKB1) is the primary kinase that phosphorylates AMPKα at Thr172 (Figure 1). LKB1 is found constitutively active as a complex with Ste20-related adaptor (STRAD) and mouse protein 25 (MO25). A second binding event of AMP/ADP to AMPKγ induces a conformational change that protects AMPKα from dephosphorylation. AMPKα phosphorylation has also been reported to be mediated by the Ca2+/calmodulin (CaM)-dependent protein kinase kinase 2 or β (CAMKK2 or β), independent from changes in the AMP/ADP:ATP ratio (Figure 1), or by the transforming growth factor β-activated kinase 1 (TAK-1). Finally, AMP upregulates allosterically the activation of AMPK [11]. A new study shows that AMPK can also be activated by sensing the absence of fructose-1,6-bisphosphate (FBP), via aldolase even if there are not changes in the AMP/ADP:ATP ratio [12].
AMPK activation inhibits anabolic processes, stimulates catabolism, and restores ATP levels. Several conditions that either interfere with ATP synthesis or promote ATP consumption can lead to AMPK activation. At least 60 targets have been identified as substrates of AMPK [11], but we will focus on those involved in the regulation of cell metabolism (Figure 1). AMPK inhibits the de novo synthesis of fatty acids and activates lipid catabolism via phosphorylation and inactivation of acetyl-CoA carboxylase (ACC). AMPK also stimulates glucose uptake via the phosphorylation of the TBC domain family members TBC1D1 and TBC1D4, and the thioredoxin-interacting protein (TXNIP) that results in an increased translocation of glucose transporters (GLUT) (Figure 1). Phosphorylation of the bifunctional 6-phosphofructo-2-kinase (PFKFB) by AMPK increases fructose-2,6-bisphosphate (F2,6P2), which activates glycolysis via phosphofructokinase 1 (PFK1) (Figure 1). In contrast, glycogen synthesis and hexosamine biosynthesis are inhibited by the phosphorylation and inhibition of glycogen synthase (GS) (Figure 1)and glutamine-fructose-6-phosphate aminotransferase-1 (GFPT1), respectively [13]. AMPK also promotes mitochondrial biogenesis by phosphorylation of the peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α) increasing its transcriptional activity (Figure 1) [14,15], as well as mitochondrial fission and autophagy (mitophagy) [16]. Remarkably, it was recently shown that ROS activate AMPK triggering a PGC-1α-dependent antioxidant response that limits mitochondrial ROS production [17]. Thus, AMPK is a central regulator of mitochondrial homeostasis.
4. mTOR-AMPK signaling crosstalk
While both mTOR and AMPK seem to regulate different pathways, there are key hubs of crosstalk where both converge to regulate homeostatic processes such as autophagy and cell metabolism (Figure 1 and Supplementary Table 1). AMPK utilizes different targets to effectively suppress mTORC1. AMPK inhibits mTORC1 both directly, through the phosphorylation of Raptor [18], as well as indirectly through the phosphorylation and activation of TSC2 [19]. Reciprocally, mTORC1 signaling has also been proposed to regulate AMPK as S6K has been found to phosphorylate and inhibit AMPK (Figure 1) [20].
Autophagy is a proteostatic quality control mechanism that requires the formation of double membraned autophagosomes that deliver cargo for degradation within the lysosome (Figure 1). The unc-51-like autophagy activating kinase 1 (ULK1 or autophagy related protein1 [Atg1]) drives autophagosome formation and mTORC1 suppresses autophagy by phosphorylating ULK1. In contrast, phosphorylation of ULK1 by AMPK promotes autophagy (Figure 1). Therefore, the relative activity of mTORC1 and AMPK in different cellular contexts largely determines the extent of autophagy induction [21].
Another point of convergence is that of protein synthesis. Within the polypeptide translation elongation cycle, translocation of the ribosome is mediated by the elongation factor eEF2. eEF2 is inhibited by phosphorylation catalyzed by the eEF2 kinase (EEF2K). EEF2K is phosphorylated and inactivated by S6K [22] and mTORC1 [23], while its phosphorylation by AMPK promotes its activation (Figure 1) [24].
The forkhead box O3a protein (FOXO3a) is a transcription factor involved in cell survival whose phosphorylation by AKT is well known to inhibit its activity [25], suggesting a possible feedback loop via mTORC2 [26]. FOXO3a has been reported to inhibit mTORC1 activity via transcriptional control of TSC1 [27]. In contrast, phosphorylation by AMPK activates FOXO3a (Figure 1) [28].
5. mTOR/AMPK signaling in the brain
mTOR signaling is involved in a myriad of processes regulating neurodevelopment (proliferation and differentiation) and brain function (neuronal plasticity). Thus, dysregulation in mTOR signaling has been linked to a number of brain disorders associated with neuronal dysfunction and cell death such as neurodegeneration, epilepsy, autism and neurobehavioral alterations. Previous reviews have focused on the role of mTOR in brain function and development [29,30]. In the brain, energy-sensing neurons in the hypothalamus are involved in the regulation of organismal energy balance via AMPK signaling. AMPK activation in the hypothalamus is modulated by energy-related signals associated with food intake (fasting and feeding), energy expenditure (ATP levels), and body weight (hormones and adipokines) [31]. We will next focus on the role of mTOR and AMPK on the survival and cell death of neuro-glial cells and their link with metabolic stress in brain disorders
Brain metabolism is characterized by a complex interplay between different cell types. Glucose is the primary energy substrate of the brain and it has been proposed to be metabolized through two complementary metabolic pathways in neurons and astrocytes. Neurons seem to predominantly metabolize glucose through the pentose phosphate pathway (PPP) to produce the NADPH necessary for antioxidant defense. On the other hand, astrocytes primarily metabolize glucose through glycolysis to produce lactate, where lactate is shuttled as an energy substrate for oxidative phosphorylation in neurons [32–34]. While this astrocyte-neuron lactate shuttle seems to act at rest, recent studies demonstrate that during energy-demanding conditions neurons have the capacity to upregulate glycolysis [35].
Very little is known regarding the role for both AMPK and mTOR signaling in normal brain metabolism. Deletion of Raptor (mTORC1) or Rictor (mTORC2) in the brain leads to neurodevelopmental alterations [36–39], but whether these effects are related to alterations in cell metabolism is unclear. AMPKα2 has been reported to be the predominant catalytic subunit in neurons and astrocytes in the brain and spinal cord [40]. In contrast to mTOR, AMPK is not necessary for neuronal development or survival, but its hyperactivation impairs axon growth via inhibition of mTORC1 [41]. Indeed, AMPK antagonizes mTOR activation by neurotrophic factors in neurons [42]. We have observed that mTOR signaling is important for astrocytes’ basal glycolytic rate (unpublished results). A recent report demonstrates that in astrocytes and neural stem cells, the G-protein-coupled receptor kinase-interacting protein 1 (GIT1) is a novel mTOR-binding protein within a unique mTOR complex regulated by AKT that lacks both Raptor and Rictor [43]. In glioblastoma cells, mTORC2 upregulates glycolysis via acetylation of FOXO1 and FOXO3a transcription factors that result in an increase in c-Myc levels [9].
6. mTOR/AMPK in neuronal cell death and survival
Because mTOR and AMPK are essential for the regulation of metabolism, their dysregulation can generate negative effects leading to cell death and disease progression. There are different examples where the activation of these two kinases has been found to be protective against pathological processes, but it has also been shown that when dysregulated, their increased activity can be detrimental.
6.1. Brain Ischemia and injury
In ischemic brain injury both mTOR and AMPK have been involved in cell death progression. While this topic has been studied extensively [44,45], there are still contradictory results depending on the experimental conditions used. mTOR protects against neuronal cell death induced by ischemic insults [46,47]. Similarly, mTOR signaling via S6K protects astrocytes from oxygen-glucose deprivation (OGD) and mice from middle artery occlusion (MCAO) [48]. Pharmacological inhibition of AMPK also protects against MCAO and OGD [49], but the opposite is found using a global cerebral ischemia model [50]. AMPKα2, but not α1 knockout in mice has been shown to protect against MCAO [51]. Interestingly, a dual role of AMPK in neuronal cell death in neonatal hypoxia has been reported, where inhibition of AMPK prior to OGD increases cell death, while its CAMKK2-dependent activation upon OGD mediates neuronal cell loss [52]. In traumatic brain injury (TBI), pharmacological stimulation of AMPK ameliorates cognitive dysfunction [53].
Accumulation of extracellular excitatory amino acids (glutamate) is a common event associated with neuronal cell death during ischemia and TBI. Neuronal cell death induced by glutamate excitotoxicity has been reported to involve AMPK activation and an increase in the levels of the pro-apoptotic Bim protein [54]. In contrast, AMPK-dependent translocation of GLUT3 and glucose availability have been reported to exert a protective effect against glutamate excitotoxicity [55]. mTOR signaling has also been proposed to contribute to the toxicity of glutamate receptor agonists (N-methyl-D-aspartate [NMDA]) [56]. Finally, mTOR is well known to mediate axon regeneration and myelination after injury [57–59].
6.2. Neurodegeneration
Both mTOR and AMPK signaling modulate the progression of different neurodegenerative disorders, but whether their activation is beneficial or detrimental remains unclear. One of the main mechanisms related to neurodegeneration where mTOR and AMPK converge is autophagy, which can exert protective effects via the degradation of misfolded, damaged or toxic proteins, as well as damaged organelles [60]. Parkinson’s (PD), and Huntington’s disease (HD), are characterized by the accumulation of intracellular misfolded protein aggregates. Because mTORC1 represses autophagy, its inhibition may represent a plausible therapeutic approach [61]. Accordingly, rapamycin has been reported to protect neurons by promoting the clearance of protein aggregates of α-synuclein and mutant huntingtin which are found in PD and HD, respectively [62,63]. A decrease in mTOR activity also reduces Alzheimer’s disease-like amyloid-β (Aβ) deposits in vivo via induction of autophagy [64]. Interestingly, while mTOR activation is usually considered to inhibit autophagy, autophagy can be induced by mTOR stimulation as well. Accordingly, activation of mTORC1 signaling in the striatum stimulates autophagy, ameliorates mitochondrial dysfunction and protects against HD [65]. In contrast, hyperactivation of mTORC1 via striatum-specific deletion of TSC1 has been reported to accelerate motor dysfunction and mortality in HD mouse models [66].
Most of the studies regarding mTOR signaling and neurodegeneration have been focused on autophagy. A recent report demonstrates that inhibition of mTOR with rapamycin preserves neuronal ATP levels upon inhibition of mitochondrial function, and this effect was associated with a decrease in energy consumption by protein synthesis [67]. Rapamycin-induced activation of 4E-BP reduces dopaminergic degeneration and mitochondrial dysfunction linked to mutations in the PD-related genes PTEN-induced putative kinase 1 (PINK1) and E3 ubiquitin-protein ligase Parkin [68]. Conversely, the PD-related protein leucine-rich repeat kinase 2 (LRRK2) has been shown to phosphorylate and inactivate 4E-BP [69]. Together these results suggest that alterations in protein translation via the mTORC1-4E-BP signaling pathway might be involved in PD. Contradicting results have been reported regarding the role of AMPK in dopaminergic cell death associated with PD [70–75]. A protective role for AMPK against mitochondrial dysfunction and toxicity induced by Parkin-, LRRK2-mutations, α-synuclein and mitochondrial toxins has been previously reported [70–72]. In contrast, other reports have shown that AMPK mediates dopaminergic cell death induced by 6-hydroxydopamine (6-OHDA) and mitochondrial toxins as well [73,74]. We have demonstrated that AMPK exerts a protective effect against cell death induced by paraquat (PQ), a pesticide recognized as an important PD risk factor, in combination with the PD-related gene, α-synuclein [76]. Importantly, while autophagy protects against PQ toxicity [77], the modulatory effects of AMPK seem to be primarily related to metabolic dysfunction [76].
The intracellular aggregation of abnormally and hyperphosphorylated tau proteins generates neurofibrillary tangles (NFTs), the pathological hallmark of neurodegenerative diseases commonly known as tauopathies. Aβ oligomers activate AMPK via CAMKK2, which mediates tau phosphorylation and neurotoxicity [78,79]. In contrast, short-term exposure to Aβ oligomers transiently decrease AMPK activity and result in a reduction in glucose transport [80]. In HD, activation of AMPK mediates neuronal atrophy downstream of oxidative stress [81]. However, another study suggests that AMPK activation at early stages of HD pathology protects against neuronal cell death [82].
6.3. Environmental neurotoxicity
Exposure to environmental toxicants is a risk factor for neudegenerative, cognitive and neurodevelopmental disorders. Arsenic, a natural occurring neurotoxicant, impairs neurite outgrowth by inhibition of AMPK signaling [83]. We have observed that AMPK signaling and autophagy promote apoptotic cell death in cortical astrocytes exposed to inorganic arsenic (unpublished results). Cadmium (Cd) neurotoxicity has been shown to be mediated via mTOR signaling [84]. Bisphenol-A (BPA), an endocrine disruptor released from the polycarbonate containers, activates AMPK while it downregulates mTOR signaling, resulting in enhanced autophagy. AMPK signaling and autophagy were reported to exert a protective effect against BPA-induced neurotoxicity in hippocampal neurons [85]. In contrast, the neurotoxicity of tributyltin, a water organotin contaminant, has been linked to the activation of AMPK and excitotoxic glutamate release [84].
7. Conclusions and perspectives
It is now clear that both mTOR and AMPK pathways regulate cellular homeostasis at multiple levels. By continuously sensing the cellular energy and nutrient state mTOR and AMPK have the capacity to facilitate cellular adaptation to stress. In addition, Ca2+, growth factors and oxidative stress have the ability to activate mTOR and AMPK to regulate protein synthesis, autophagy and metabolism. We have aimed to discuss the current state of knowledge regarding how mTOR and AMPK regulate brain metabolism and survival upon stress and the close interrelationship between both signaling pathways. Studies so far have demonstrated that dysregulation in these two pathways is associated with neuronal cell death, but the mechanisms involved remain unclear. Most of the work has been focused on their convergence regulating proteostasis via autophagy, but recent findings highlight that changes in protein synthesis, metabolism and mitochondrial function are likely to play a role in the regulatory effects of mTOR and AMPK on neuronal health. While there is still controversy regarding whether AMPK and mTOR signaling can exert a neuroprotective or neurotoxic effect, some of these controversies might be addressed when considering: 1) the poor specificity of widely used pharmacological approaches to modulate their activity (rapamycin, torin 1, compound C, 5-Aminoimidazole-4- carboxamide ribonucleotide [AICAR] and others); and 2) the time-dependent effect of modulating these signaling pathways. Understanding their roles and the relationship between these two master regulators of cell metabolism is crucial for future therapeutic approaches to counteract alterations in cell metabolism and survival during neuronal cell death and degeneration.
Supplementary Material
Highlights.
mTOR and AMPK regulate cell survival and metabolism in response to diverse stimuli.
Studies so far demonstrate that dysregulation in these two pathways is associated with neuronal injury, degeneration and neurotoxicity.
Recent findings highlight that in addition to autophagy, changes in protein synthesis, metabolism and mitochondrial function play a role in the regulatory effects of mTOR and AMPK on neuronal health.
Acknowledgments
This work was supported by the National Institutes of Health Grant P20RR17675 Centers of Biomedical Research Excellence (COBRE), the Research Council of the University of Nebraska-Lincoln (R.F.), the National Science Foundation Grant DBI-1461240 (R.F. and A.S.), and the University of Arizona Diversity and Inclusion Award and Career Development Award (EMRR).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Saxton RA, Sabatini DM. mTOR Signaling in Growth, Metabolism, and Disease. Cell. 2017;168:960–976. doi: 10.1016/j.cell.2017.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Duvel K, Yecies JL, Menon S, Raman P, Lipovsky AI, Souza AL, Triantafellow E, Ma Q, Gorski R, Cleaver S, et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol Cell. 2010;39:171–183. doi: 10.1016/j.molcel.2010.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Morita M, Gravel SP, Chenard V, Sikstrom K, Zheng L, Alain T, Gandin V, Avizonis D, Arguello M, Zakaria C, et al. mTORC1 controls mitochondrial activity and biogenesis through 4E-BP-dependent translational regulation. Cell Metab. 2013;18:698–711. doi: 10.1016/j.cmet.2013.10.001. [DOI] [PubMed] [Google Scholar]
- 4.Porstmann T, Santos CR, Griffiths B, Cully M, Wu M, Leevers S, Griffiths JR, Chung YL, Schulze A. SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell Metab. 2008;8:224–236. doi: 10.1016/j.cmet.2008.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ben-Sahra I, Hoxhaj G, Ricoult SJH, Asara JM, Manning BD. mTORC1 induces purine synthesis through control of the mitochondrial tetrahydrofolate cycle. Science. 2016;351:728–733. doi: 10.1126/science.aad0489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Albert V, Svensson K, Shimobayashi M, Colombi M, Munoz S, Jimenez V, Handschin C, Bosch F, Hall MN. mTORC2 sustains thermogenesis via Akt-induced glucose uptake and glycolysis in brown adipose tissue. EMBO Mol Med. 2016;8:232–246. doi: 10.15252/emmm.201505610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7*.Gu Y, Albuquerque CP, Braas D, Zhang W, Villa GR, Bi J, Ikegami S, Masui K, Gini B, Yang H, et al. mTORC2 Regulates Amino Acid Metabolism in Cancer by Phosphorylation of the Cystine-Glutamate Antiporter xCT. Mol Cell. 2017;67:128–138. e127. doi: 10.1016/j.molcel.2017.05.030. Using an unbiased proteomic screen, this work identified mTORC2 as a regulator of amino acid metabolism via phosphorylation and inhibition of the cystine-glutamate antiporter xCT that had a direct effect on glutamate release, cystine uptake, and glutathione homeostasis. This study identified a novel target for mTORC2 involved in cellular redox homeostasis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moloughney JG, Kim PK, Vega-Cotto NM, Wu CC, Zhang S, Adlam M, Lynch T, Chou PC, Rabinowitz JD, Werlen G, et al. mTORC2 Responds to Glutamine Catabolite Levels to Modulate the Hexosamine Biosynthesis Enzyme GFAT1. Mol Cell. 2016;63:811–826. doi: 10.1016/j.molcel.2016.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Masui K, Tanaka K, Akhavan D, Babic I, Gini B, Matsutani T, Iwanami A, Liu F, Villa GR, Gu Y, et al. mTOR complex 2 controls glycolytic metabolism in glioblastoma through FoxO acetylation and upregulation of c-Myc. Cell Metab. 2013;18:726–739. doi: 10.1016/j.cmet.2013.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yin Y, Hua H, Li M, Liu S, Kong Q, Shao T, Wang J, Luo Y, Wang Q, Luo T, et al. mTORC2 promotes type I insulin-like growth factor receptor and insulin receptor activation through the tyrosine kinase activity of mTOR. Cell Res. 2016;26:46–65. doi: 10.1038/cr.2015.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hardie DG, Lin SC. AMP-activated protein kinase - not just an energy sensor. F1000Res. 2017;6:1724. doi: 10.12688/f1000research.11960.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12**.Zhang CS, Hawley SA, Zong Y, Li M, Wang Z, Gray A, Ma T, Cui J, Feng JW, Zhu M, et al. Fructose-1,6-bisphosphate and aldolase mediate glucose sensing by AMPK. Nature. 2017;548:112–116. doi: 10.1038/nature23275. Activation of AMPK has been largely ascribed to changes in AMP or ADP:ATP ratios. This study describes an AMP/ADP-independent mechanism for AMPK activation that involves the sensing of fructose-1,6-bisphosphate (FBP) levels via aldolases. This mechanism seems to depend on the lysosomal complex as FBP disrupts its assembly. These results establish a novel “moonlight” role for aldolase in metabolic signaling. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jeon SM. Regulation and function of AMPK in physiology and diseases. Exp Mol Med. 2016;48:e245. doi: 10.1038/emm.2016.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mihaylova MM, Shaw RJ. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol. 2011;13:1016–1023. doi: 10.1038/ncb2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zong H, Ren JM, Young LH, Pypaert M, Mu J, Birnbaum MJ, Shulman GI. AMP kinase is required for mitochondrial biogenesis in skeletal muscle in response to chronic energy deprivation. Proc Natl Acad Sci U S A. 2002;99:15983–15987. doi: 10.1073/pnas.252625599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16*.Toyama EQ, Herzig S, Courchet J, Lewis TL, Jr, Loson OC, Hellberg K, Young NP, Chen H, Polleux F, Chan DC, et al. Metabolism. AMP-activated protein kinase mediates mitochondrial fission in response to energy stress. Science. 2016;351:275–281. doi: 10.1126/science.aab4138. AMPK has been previously reported to regulate mitochondrial dynamics via mitochondrial biogenesis. Now, in this study, a novel role for AMPK mediating mitochondrial fragmentation via the phosphorylation of the mitochondrial fission factor (MFF) is described. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17*.Rabinovitch RC, Samborska B, Faubert B, Ma EH, Gravel SP, Andrzejewski S, Raissi TC, Pause A, St-Pierre J, Jones RG. AMPK Maintains Cellular Metabolic Homeostasis through Regulation of Mitochondrial Reactive Oxygen Species. Cell Rep. 2017;21:1–9. doi: 10.1016/j.celrep.2017.09.026. This study reports that mitochondrial ROS activate AMPK, which reciprocally limits mitochondrial ROS production via a PGC-1α-dependent antioxidant response. Cells lacking AMPK increased mitochondrial ROS, undergo premature senescence, and HIF-1α-dependent Warburg effect, highlighting a key function for AMPK in stress resistance and metabolic balance by controlling mitochondrial ROS. [DOI] [PubMed] [Google Scholar]
- 18.Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, Shaw RJ. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. 2008;30:214–226. doi: 10.1016/j.molcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115:577–590. doi: 10.1016/s0092-8674(03)00929-2. [DOI] [PubMed] [Google Scholar]
- 20.Dagon Y, Hur E, Zheng B, Wellenstein K, Cantley LC, Kahn BB. p70S6 kinase phosphorylates AMPK on serine 491 to mediate leptin's effect on food intake. Cell Metab. 2012;16:104–112. doi: 10.1016/j.cmet.2012.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13:132–141. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang X, Li W, Williams M, Terada N, Alessi DR, Proud CG. Regulation of elongation factor 2 kinase by p90(RSK1) and p70 S6 kinase. EMBO J. 2001;20:4370–4379. doi: 10.1093/emboj/20.16.4370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang X, Regufe da Mota S, Liu R, Moore CE, Xie J, Lanucara F, Agarwala U, Pyr Dit Ruys S, Vertommen D, Rider MH, et al. Eukaryotic elongation factor 2 kinase activity is controlled by multiple inputs from oncogenic signaling. Mol Cell Biol. 2014;34:4088–4103. doi: 10.1128/MCB.01035-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Browne GJ, Finn SG, Proud CG. Stimulation of the AMP-activated protein kinase leads to activation of eukaryotic elongation factor 2 kinase and to its phosphorylation at a novel site, serine 398. J Biol Chem. 2004;279:12220–12231. doi: 10.1074/jbc.M309773200. [DOI] [PubMed] [Google Scholar]
- 25.Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96:857–868. doi: 10.1016/s0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- 26.Feehan RP, Shantz LM. Negative regulation of the FOXO3a transcription factor by mTORC2 induces a pro-survival response following exposure to ultraviolet-B irradiation. Cell Signal. 2016;28:798–809. doi: 10.1016/j.cellsig.2016.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Khatri S, Yepiskoposyan H, Gallo CA, Tandon P, Plas DR. FOXO3a regulates glycolysis via transcriptional control of tumor suppressor TSC1. J Biol Chem. 2010;285:15960–15965. doi: 10.1074/jbc.M110.121871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Greer EL, Oskoui PR, Banko MR, Maniar JM, Gygi MP, Gygi SP, Brunet A. The energy sensor AMP-activated protein kinase directly regulates the mammalian FOXO3 transcription factor. J Biol Chem. 2007;282:30107–30119. doi: 10.1074/jbc.M705325200. [DOI] [PubMed] [Google Scholar]
- 29.Garza-Lombo C, Gonsebatt ME. Mammalian Target of Rapamycin: Its Role in Early Neural Development and in Adult and Aged Brain Function. Front Cell Neurosci. 2016;10:157. doi: 10.3389/fncel.2016.00157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Crino PB. The mTOR signalling cascade: paving new roads to cure neurological disease. Nat Rev Neurol. 2016;12:379–392. doi: 10.1038/nrneurol.2016.81. [DOI] [PubMed] [Google Scholar]
- 31.Ronnett GV, Ramamurthy S, Kleman AM, Landree LE, Aja S. AMPK in the brain: its roles in energy balance and neuroprotection. J Neurochem. 2009;109(Suppl 1):17–23. doi: 10.1111/j.1471-4159.2009.05916.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Magistretti PJ, Allaman I. A cellular perspective on brain energy metabolism and functional imaging. Neuron. 2015;86:883–901. doi: 10.1016/j.neuron.2015.03.035. [DOI] [PubMed] [Google Scholar]
- 33*.Machler P, Wyss MT, Elsayed M, Stobart J, Gutierrez R, von Faber-Castell A, Kaelin V, Zuend M, San Martin A, Romero-Gomez I, et al. In Vivo Evidence for a Lactate Gradient from Astrocytes to Neurons. Cell Metab. 2016;23:94–102. doi: 10.1016/j.cmet.2015.10.010. Using in vivo fluorescecne imaging, this study demonstrates the existence of a lactate gradient from astrocytes to neurons, a prerequisite for the astrocyte-neuron lactate shuttle that substantietes its existance. See also reference 35. [DOI] [PubMed] [Google Scholar]
- 34**.Supplie LM, Duking T, Campbell G, Diaz F, Moraes CT, Gotz M, Hamprecht B, Boretius S, Mahad D, Nave KA. Respiration-Deficient Astrocytes Survive As Glycolytic Cells In Vivo. J Neurosci. 2017;37:4231–4242. doi: 10.1523/JNEUROSCI.0756-16.2017. This is the first report in vivo demonstrating that astrocytes do not require mitochondrial respiration for survival, at least under non-stressed conditions. This observation is key to our understanding regarding the energetic requirements for astrocytes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35**.Diaz-Garcia CM, Mongeon R, Lahmann C, Koveal D, Zucker H, Yellen G. Neuronal Stimulation Triggers Neuronal Glycolysis and Not Lactate Uptake. Cell Metab. 2017;26:361–374. e364. doi: 10.1016/j.cmet.2017.06.021. The exact contribution of glucose vs lactate to neuronal bioenergetics at “rest” or during “stimulation” is still under debate. In this study, using metabolic biosensors, Dr Yellen et al. reported that metabolic responses to neuronal stimulation do not seem to depend on lactate uptake, but instead they require increased glycolysis that provides a rapid response to energy demands. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thomanetz V, Angliker N, Cloetta D, Lustenberger RM, Schweighauser M, Oliveri F, Suzuki N, Ruegg MA. Ablation of the mTORC2 component rictor in brain or Purkinje cells affects size and neuron morphology. J Cell Biol. 2013;201:293–308. doi: 10.1083/jcb.201205030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cloetta D, Thomanetz V, Baranek C, Lustenberger RM, Lin S, Oliveri F, Atanasoski S, Ruegg MA. Inactivation of mTORC1 in the developing brain causes microcephaly and affects gliogenesis. J Neurosci. 2013;33:7799–7810. doi: 10.1523/JNEUROSCI.3294-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang Y, Xu S, Liang KY, Li K, Zou ZP, Yang CL, Tan K, Cao X, Jiang Y, Gao TM, et al. Neuronal mTORC1 Is Required for Maintaining the Nonreactive State of Astrocytes. J Biol Chem. 2017;292:100–111. doi: 10.1074/jbc.M116.744482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Carson RP, Fu C, Winzenburger P, Ess KC. Deletion of Rictor in neural progenitor cells reveals contributions of mTORC2 signaling to tuberous sclerosis complex. Hum Mol Genet. 2013;22:140–152. doi: 10.1093/hmg/dds414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Turnley AM, Stapleton D, Mann RJ, Witters LA, Kemp BE, Bartlett PF. Cellular distribution and developmental expression of AMP-activated protein kinase isoforms in mouse central nervous system. J Neurochem. 1999;72:1707–1716. doi: 10.1046/j.1471-4159.1999.721707.x. [DOI] [PubMed] [Google Scholar]
- 41.Williams T, Courchet J, Viollet B, Brenman JE, Polleux F. AMP-activated protein kinase (AMPK) activity is not required for neuronal development but regulates axogenesis during metabolic stress. Proc Natl Acad Sci U S A. 2011;108:5849–5854. doi: 10.1073/pnas.1013660108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ishizuka Y, Kakiya N, Witters LA, Oshiro N, Shirao T, Nawa H, Takei N. AMP-activated protein kinase counteracts brain-derived neurotrophic factor-induced mammalian target of rapamycin complex 1 signaling in neurons. J Neurochem. 2013;127:66–77. doi: 10.1111/jnc.12362. [DOI] [PubMed] [Google Scholar]
- 43**.Smithson LJ, Gutmann DH. Proteomic analysis reveals GIT1 as a novel mTOR complex component critical for mediating astrocyte survival. Genes Dev. 2016;30:1383–1388. doi: 10.1101/gad.279661.116. This is a very provocative study that suggests that we are still far away from understanding the complexity and tissure/cell type specificity of mTOR signaling. Here, the authors demonstrated that in astrocytes and neural stem cells, the G-protein-coupled receptor kinase-interacting protein 1 (GIT1) acts as as a novel mTOR-binding protein, creating a unique mTOR complex lacking Raptor and Rictor. GIT1 binding to mTOR was found to be regulated by AKT and to be essential for mTOR-mediated astrocyte survival. The indentification of a novel third mTOR complex requires confirmation by other studies. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chong ZZ, Yao Q, Li HH. The rationale of targeting mammalian target of rapamycin for ischemic stroke. Cell Signal. 2013;25:1598–1607. doi: 10.1016/j.cellsig.2013.03.017. [DOI] [PubMed] [Google Scholar]
- 45.Li J, McCullough LD. Effects of AMP-activated protein kinase in cerebral ischemia. J Cereb Blood Flow Metab. 2010;30:480–492. doi: 10.1038/jcbfm.2009.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hwang JY, Gertner M, Pontarelli F, Court-Vazquez B, Bennett MV, Ofengeim D, Zukin RS. Global ischemia induces lysosomal-mediated degradation of mTOR and activation of autophagy in hippocampal neurons destined to die. Cell Death Differ. 2017;24:317–329. doi: 10.1038/cdd.2016.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xie R, Cheng M, Li M, Xiong X, Daadi M, Sapolsky RM, Zhao H. Akt isoforms differentially protect against stroke-induced neuronal injury by regulating mTOR activities. J Cereb Blood Flow Metab. 2013;33:1875–1885. doi: 10.1038/jcbfm.2013.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pastor MD, Garcia-Yebenes I, Fradejas N, Perez-Ortiz JM, Mora-Lee S, Tranque P, Moro MA, Pende M, Calvo S. mTOR/S6 kinase pathway contributes to astrocyte survival during ischemia. J Biol Chem. 2009;284:22067–22078. doi: 10.1074/jbc.M109.033100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McCullough LD, Zeng Z, Li H, Landree LE, McFadden J, Ronnett GV. Pharmacological inhibition of AMP-activated protein kinase provides neuroprotection in stroke. J Biol Chem. 2005;280:20493–20502. doi: 10.1074/jbc.M409985200. [DOI] [PubMed] [Google Scholar]
- 50.Ashabi G, Khodagholi F, Khalaj L, Goudarzvand M, Nasiri M. Activation of AMP-activated protein kinase by metformin protects against global cerebral ischemia in male rats: interference of AMPK/PGC-1alpha pathway. Metab Brain Dis. 2014;29:47–58. doi: 10.1007/s11011-013-9475-2. [DOI] [PubMed] [Google Scholar]
- 51.Li J, Zeng Z, Viollet B, Ronnett GV, McCullough LD. Neuroprotective effects of adenosine monophosphate-activated protein kinase inhibition and gene deletion in stroke. Stroke. 2007;38:2992–2999. doi: 10.1161/STROKEAHA.107.490904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rousset CI, Leiper FC, Kichev A, Gressens P, Carling D, Hagberg H, Thornton C. A dual role for AMP-activated protein kinase (AMPK) during neonatal hypoxic-ischaemic brain injury in mice. J Neurochem. 2015;133:242–252. doi: 10.1111/jnc.13034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hill JL, Kobori N, Zhao J, Rozas NS, Hylin MJ, Moore AN, Dash PK. Traumatic brain injury decreases AMP-activated protein kinase activity and pharmacological enhancement of its activity improves cognitive outcome. J Neurochem. 2016;139:106–119. doi: 10.1111/jnc.13726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Concannon CG, Tuffy LP, Weisova P, Bonner HP, Davila D, Bonner C, Devocelle MC, Strasser A, Ward MW, Prehn JH. AMP kinase-mediated activation of the BH3-only protein Bim couples energy depletion to stress-induced apoptosis. J Cell Biol. 2010;189:83–94. doi: 10.1083/jcb.200909166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Weisova P, Concannon CG, Devocelle M, Prehn JH, Ward MW. Regulation of glucose transporter 3 surface expression by the AMP-activated protein kinase mediates tolerance to glutamate excitation in neurons. J Neurosci. 2009;29:2997–3008. doi: 10.1523/JNEUROSCI.0354-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Swiatkowski P, Nikolaeva I, Kumar G, Zucco A, Akum BF, Patel MV, D'Arcangelo G, Firestein BL. Role of Akt-independent mTORC1 and GSK3beta signaling in sublethal NMDA-induced injury and the recovery of neuronal electrophysiology and survival. Sci Rep. 2017;7:1539. doi: 10.1038/s41598-017-01826-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Park KK, Liu K, Hu Y, Smith PD, Wang C, Cai B, Xu B, Connolly L, Kramvis I, Sahin M, et al. Promoting axon regeneration in the adult CNS by modulation of the PTEN/mTOR pathway. Science. 2008;322:963–966. doi: 10.1126/science.1161566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Figlia G, Gerber D, Suter U. Myelination and mTOR. Glia. 2017 doi: 10.1002/glia.23273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Berry M, Ahmed Z, Morgan-Warren P, Fulton D, Logan A. Prospects for mTOR-mediated functional repair after central nervous system trauma. Neurobiol Dis. 2016;85:99–110. doi: 10.1016/j.nbd.2015.10.002. [DOI] [PubMed] [Google Scholar]
- 60.Menzies FM, Fleming A, Rubinsztein DC. Compromised autophagy and neurodegenerative diseases. Nat Rev Neurosci. 2015;16:345–357. doi: 10.1038/nrn3961. [DOI] [PubMed] [Google Scholar]
- 61.Wong M. Mammalian target of rapamycin (mTOR) pathways in neurological diseases. Biomed J. 2013;36:40–50. doi: 10.4103/2319-4170.110365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG, Scaravilli F, Easton DF, Duden R, O'Kane CJ, et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. 2004;36:585–595. doi: 10.1038/ng1362. [DOI] [PubMed] [Google Scholar]
- 63.Webb JL, Ravikumar B, Atkins J, Skepper JN, Rubinsztein DC. Alpha-Synuclein is degraded by both autophagy and the proteasome. J Biol Chem. 2003;278:25009–25013. doi: 10.1074/jbc.M300227200. [DOI] [PubMed] [Google Scholar]
- 64.Caccamo A, De Pinto V, Messina A, Branca C, Oddo S. Genetic reduction of mammalian target of rapamycin ameliorates Alzheimer's disease-like cognitive and pathological deficits by restoring hippocampal gene expression signature. J Neurosci. 2014;34:7988–7998. doi: 10.1523/JNEUROSCI.0777-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lee JH, Tecedor L, Chen YH, Monteys AM, Sowada MJ, Thompson LM, Davidson BL. Reinstating aberrant mTORC1 activity in Huntington's disease mice improves disease phenotypes. Neuron. 2015;85:303–315. doi: 10.1016/j.neuron.2014.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pryor WM, Biagioli M, Shahani N, Swarnkar S, Huang WC, Page DT, MacDonald ME, Subramaniam S. Huntingtin promotes mTORC1 signaling in the pathogenesis of Huntington's disease. Sci Signal. 2014;7:ra103. doi: 10.1126/scisignal.2005633. [DOI] [PubMed] [Google Scholar]
- 67**.Zheng X, Boyer L, Jin M, Kim Y, Fan W, Bardy C, Berggren T, Evans RM, Gage FH, Hunter T. Alleviation of neuronal energy deficiency by mTOR inhibition as a treatment for mitochondria-related neurodegeneration. Elife. 2016;5 doi: 10.7554/eLife.13378. The protective effects of inhibition of mTOR signaling with rapamycin against neurodegeneration have been largely attributed to autophagy. However, in this work, the authors demonstrate that inhibition of mTOR with rapamycin significantly preserves neuronal ATP levels via a decrease in protein synthesis, a major energy-consuming process, when oxidative phosphorylation is impaired. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tain LS, Mortiboys H, Tao RN, Ziviani E, Bandmann O, Whitworth AJ. Rapamycin activation of 4E-BP prevents parkinsonian dopaminergic neuron loss. Nat Neurosci. 2009;12:1129–1135. doi: 10.1038/nn.2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Imai Y, Gehrke S, Wang HQ, Takahashi R, Hasegawa K, Oota E, Lu B. Phosphorylation of 4E-BP by LRRK2 affects the maintenance of dopaminergic neurons in Drosophila. EMBO J. 2008;27:2432–2443. doi: 10.1038/emboj.2008.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ng CH, Guan MS, Koh C, Ouyang X, Yu F, Tan EK, O'Neill SP, Zhang X, Chung J, Lim KL. AMP kinase activation mitigates dopaminergic dysfunction and mitochondrial abnormalities in Drosophila models of Parkinson's disease. J Neurosci. 2012;32:14311–14317. doi: 10.1523/JNEUROSCI.0499-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Choi JS, Park C, Jeong JW. AMP-activated protein kinase is activated in Parkinson's disease models mediated by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Biochem Biophys Res Commun. 2010;391:147–151. doi: 10.1016/j.bbrc.2009.11.022. [DOI] [PubMed] [Google Scholar]
- 72.Dulovic M, Jovanovic M, Xilouri M, Stefanis L, Harhaji-Trajkovic L, Kravic-Stevovic T, Paunovic V, Ardah MT, El-Agnaf OM, Kostic V, et al. The protective role of AMPactivated protein kinase in alpha-synuclein neurotoxicity in vitro. Neurobiol Dis. 2014;63:1–11. doi: 10.1016/j.nbd.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 73.Xu Y, Liu C, Chen S, Ye Y, Guo M, Ren Q, Liu L, Zhang H, Xu C, Zhou Q, et al. Activation of AMPK and inactivation of Akt result in suppression of mTOR-mediated S6K1 and 4E-BP1 pathways leading to neuronal cell death in in vitro models of Parkinson's disease. Cell Signal. 2014;26:1680–1689. doi: 10.1016/j.cellsig.2014.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kim TW, Cho HM, Choi SY, Suguira Y, Hayasaka T, Setou M, Koh HC, Hwang EM, Park JY, Kang SJ, et al. (ADP-ribose) polymerase 1 and AMP-activated protein kinase mediate progressive dopaminergic neuronal degeneration in a mouse model of Parkinson's disease. Cell Death Dis. 2013;4:e919. doi: 10.1038/cddis.2013.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jiang P, Gan M, Ebrahim AS, Castanedes-Casey M, Dickson DW, Yen SH. Adenosine monophosphate-activated protein kinase overactivation leads to accumulation of alpha-synuclein oligomers and decrease of neurites. Neurobiol Aging. 2013;34:1504–1515. doi: 10.1016/j.neurobiolaging.2012.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76**.Anandhan A, Lei S, Levytskyy R, Pappa A, Panayiotidis MI, Cerny RL, Khalimonchuk O, Powers R, Franco R. Glucose Metabolism and AMPK Signaling Regulate Dopaminergic Cell Death Induced by Gene (alpha-Synuclein)-Environment (Paraquat) Interactions. Mol Neurobiol. 2017;54:3825–3842. doi: 10.1007/s12035-016-9906-2. In this work, our research group demonstrated that while autophagy is indeed a protective mechanism against the toxicity of gene-environment interactions linked to Parkinson’s disease, the protective role of AMPK is associated with the regulation of cell metabolism (glucose) but not autophagy. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Garcia-Garcia A, Anandhan A, Burns M, Chen H, Zhou Y, Franco R. Impairment of Atg5-dependent autophagic flux promotes paraquat- and MPP(+)-induced apoptosis but not rotenone or 6-hydroxydopamine toxicity. Toxicol Sci. 2013;136:166–182. doi: 10.1093/toxsci/kft188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Domise M, Didier S, Marinangeli C, Zhao H, Chandakkar P, Buee L, Viollet B, Davies P, Marambaud P, Vingtdeux V. AMP-activated protein kinase modulates tau phosphorylation and tau pathology in vivo. Sci Rep. 2016;6:26758. doi: 10.1038/srep26758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mairet-Coello G, Courchet J, Pieraut S, Courchet V, Maximov A, Polleux F. The CAMKK2-AMPK kinase pathway mediates the synaptotoxic effects of Abeta oligomers through Tau phosphorylation. Neuron. 2013;78:94–108. doi: 10.1016/j.neuron.2013.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80**.Seixas da Silva GS, Melo HM, Lourenco MV, Lyra ESNM, de Carvalho MB, Alves-Leon SV, de Souza JM, Klein WL, da-Silva WS, Ferreira ST, et al. Amyloid-beta oligomers transiently inhibit AMP-activated kinase and cause metabolic defects in hippocampal neurons. J Biol Chem. 2017;292:7395–7406. doi: 10.1074/jbc.M116.753525. This study links an impairment in AMPK function induced by Aβ oligomers with alterations in neuronal metabolism and survival. Downregulation of AMPK activity led to a decrease in glucose transporters (GLUTs) in dendritic processes, suggesting an impairment in glucose metabolism. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ju TC, Chen HM, Lin JT, Chang CP, Chang WC, Kang JJ, Sun CP, Tao MH, Tu PH, Chang C, et al. Nuclear translocation of AMPK-alpha1 potentiates striatal neurodegeneration in Huntington's disease. J Cell Biol. 2011;194:209–227. doi: 10.1083/jcb.201105010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Vazquez-Manrique RP, Farina F, Cambon K, Dolores Sequedo M, Parker AJ, Millan JM, Weiss A, Deglon N, Neri C. AMPK activation protects from neuronal dysfunction and vulnerability across nematode, cellular and mouse models of Huntington's disease. Hum Mol Genet. 2016;25:1043–1058. doi: 10.1093/hmg/ddv513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wang X, Meng D, Chang Q, Pan J, Zhang Z, Chen G, Ke Z, Luo J, Shi X. Arsenic inhibits neurite outgrowth by inhibiting the LKB1-AMPK signaling pathway. Environ Health Perspect. 2010;118:627–634. doi: 10.1289/ehp.0901510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chen L, Xu B, Liu L, Luo Y, Zhou H, Chen W, Shen T, Han X, Kontos CD, Huang S. Cadmium induction of reactive oxygen species activates the mTOR pathway, leading to neuronal cell death. Free Radic Biol Med. 2011;50:624–632. doi: 10.1016/j.freeradbiomed.2010.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Agarwal S, Tiwari SK, Seth B, Yadav A, Singh A, Mudawal A, Chauhan LK, Gupta SK, Choubey V, Tripathi A, et al. Activation of Autophagic Flux against Xenoestrogen Bisphenol-A-induced Hippocampal Neurodegeneration via AMP kinase (AMPK)/Mammalian Target of Rapamycin (mTOR) Pathways. J Biol Chem. 2015;290:21163–21184. doi: 10.1074/jbc.M115.648998. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.