

Abstract
Motivation
Bivalent ligands are increasingly important such as for targeting G protein-coupled receptor (GPCR) dimers or proteolysis targeting chimeras (PROTACs). They contain two pharmacophoric units that simultaneously bind in their corresponding binding sites, connected with a spacer chain. Here, we report a molecular modelling tool that links the pharmacophore units via the shortest pathway along the receptors van der Waals surface and then scores the solutions providing prioritization for the design of new bivalent ligands.
Results
Bivalent ligands of known dimers of GPCRs, PROTACs and a model bivalent antibody/antigen system were analysed. The tool could rapidly assess the preferred linker length for the different systems and recapitulated the best reported results. In the case of GPCR dimers the results suggest that in some cases these ligands might bind to a secondary binding site at the extracellular entrance (vestibule or allosteric site) instead of the orthosteric binding site.
Availability and implementation
Freely accessible from the Molecular Operating Environment svl exchange server (https://svl.chemcomp.com/).
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Bivalent ligands have emerged to interrogate dimer function or chemically induce biological proximity (Stanton et al., 2018). A bivalent ligand is a single chemical entity composed of two covalently linked, by a spacer chain, pharmacophores with potential to interact simultaneously with two protomers (Lane et al., 2013; Valant et al., 2012). Homobivalent ligands contain the same two pharmacophore units, whereas heterobivalent ligands link two different pharmacophores. Bivalent ligands are proving valuable in multiple research areas such as in the field of G protein-coupled receptors (GPCRs) (Hiller et al., 2013; Shonberg et al., 2011), proteolysis targeting chimeras (PROTACs) (Corson et al., 2008), or bivalent antibody/antigen interactions (Mack et al., 2012), among others.
GPCRs are classically described as monomeric transmembrane (TM) receptors; however, they can also form higher-order complexes of the same (homo) or different (hetero) protomers (Ferre et al., 2014). The study and understanding of these complexes prompt a new dimension for GPCR research (Maurice et al., 2011). The spacer length of GPCR bivalent ligands is a key factor and depends on the dimer interface, the structure of the pharmacophores, and the position and direction of the attachment points (Hiller et al., 2013; Shonberg et al., 2011). Many studies have reported spacers between 15 and 25 atoms such as ligands targeting dimers of the δ- and μ-opioid (Fig. 1, molecule 1) (Daniels et al., 2005), serotonin (2) (Soulier et al., 2005), dopamine (3 and 4) (Kuhhorn et al., 2011; McRobb et al., 2012), cannabinoid and μ opioid/cannabinoid (5 and 6) (Le Naour et al., 2013; Zhang et al., 2010), A2A-D2 (8) (Soriano et al., 2009) and A1-β2 (9) (Barlow et al., 2013) (homo/hetero)dimers to name just a few. Recently, highly active ligands with spacers as large as 44 and 66 atoms (7) (Hubner et al., 2016) were reported.
Fig. 1.

Examples of selected bivalent ligands from the literature. The reported most active molecules for μ-δ (1, MDAN-21), 5HT4-5HT4 (2), D2-D2 (3 and 4), CB1-CB1 (5), μ-CB1 (6), D2-NTS1 (7), A2A-D2 (8), A1-β2 (9), BRD4-VHL (10) and CA-CA (11) dimers
A bivalent PROTAC ligand also covalently links two pharmacophores with a spacer chain (Corson et al., 2008; Stanton et al., 2018). The aim of these ligands is to selectively induce degradation of a target protein, by linking a high affinity binder of the target protein to a strong binder of an E3 ubiquitin ligase. This brings the target and ligase in proximity, triggering polyubiquitination and proteasome-dependent degradation of the target. This post-translational approach offers many benefits to silence proteins in a cell. The very first crystal structure of a bivalent PROTAC ligand (Fig. 1, 10) bound at the bromodomain target protein BRD4 and E3 ligase von Hippel-Lindau protein (VHL) was recently reported (Gadd et al., 2017). The discovery of this ligand involved the exploration of different length polyethylene glycol (PEG) spacers with compound 10 being preferred (Zengerle et al., 2015).
A further application of bivalent ligands has involved the study of bivalent antibody/antigen interactions. The group of Whitesides used a synthetic dimer of carbonic anhydrase (CA) as a model system to replicate the binding of bivalent antibodies to antigens (Mack et al., 2012). A series of bivalent benzenesulfonamides connected by different oligosarcosine spacers were synthesized. The preferred ligand contains four sarcosine units (Fig. 1, 11).
Here we present a MOE-based molecular modelling tool that can define and prioritize the preferred spacer length for simultaneous binding at two protomers within an (homo/hetero)dimer. The tool links the pharmacophore units via the shortest pathway along the proteins’ van der Waals surface, identifying extracellular grooves and plausible conformations for a flexible spacer. We have studied with this tool the spacer size of previously reported bivalent ligands of known dimers of CA, GPCRs and BRD4-VHL. This computational tool can speed-up the design and synthesis of future bivalent ligands.
2 Materials and methods
2.1 Computational models of (homo/hetero)dimers
Crystal structures of the BRD4-VHL heterodimer (PDB id 5T35) (Gadd et al., 2017) and CA homodimer (3PJJ) (Mack et al., 2012) were used, as well as computational models of GPCR (homo/hetero)dimers built based on experimentally reported TM1/2, TM4/5 and TM5/6 interfaces (Cordomi et al., 2015). Crystal structures of GPCR protomers were available for all the cases except dopamine D2 receptor, which was modelled based on the D3 receptor crystal structure. Further details are in supporting information.
2.2 Molecular dynamics simulations
The μ-δ opioid GPCR heterodimer was studied with several bivalent ligands. After system construction and equilibration, several replicas of unrestrained MD trajectories were produced for 1 µs. All simulations used GROMACS v5.01 (Pronk et al., 2013), with force fields described by Cordomi etal (Cordomi et al., 2012). Details for MD simulations are available in supporting information.
2.3 A computational tool to design the size of the spacer
The tool was developed in the Molecular Operating Environment (MOE) software (Chemical computing group Inc., Montreal, QC, Canada). An interaction surface is calculated from a 0-potential contour of the van der Waals potential of a probe atom on a lattice around the receptor atoms. The OPLSA-AA van der Waals parameters for a TIP3P water oxygen were used. The vertices for the triangles making up the interaction surface are used as nodes in a graph, connected where within 1.05 Å. On each of the pharmacophore units an attachment point (a specified atom) to the linker is defined, and the closest vertices on the interaction surface are selected. The shortest path between these points is identified through the graph of the vertices (ignoring duplicate vertices) (Fig. 2). A set of pre-built spacer molecules is read as an input, each with two defined attachment points used to make the connection to the corresponding pharmacophore units. The spacer atoms along the shortest path between the attachment points are identified and transformed to positions on the interaction surface using linear interpolation between the interaction surface vertices. Spacers were ignored if the path across the interaction surface was too long when compared with the sum of the van der Waals radii for the atoms from the shortest path between the linker attachment points. Spacers are defined without hydrogens, allowing the bond to be formed with the pharmacophore attachment point prior to energy minimization A stepped energy minimization protocol is used. Initially, only the spacer atoms are free, while all atoms are included in the later stages of the energy minimization. The strength of the van der Waals and electrostatic repulsion terms are initially scaled by a factor of 10−5; a factor that was increased by 10-fold in each iteration until they are applied at normal strengths. The total force field energy (Etot) and the interaction energy between the spacer atoms and the rest of the system are reported for each of the spacers.
Fig. 2.
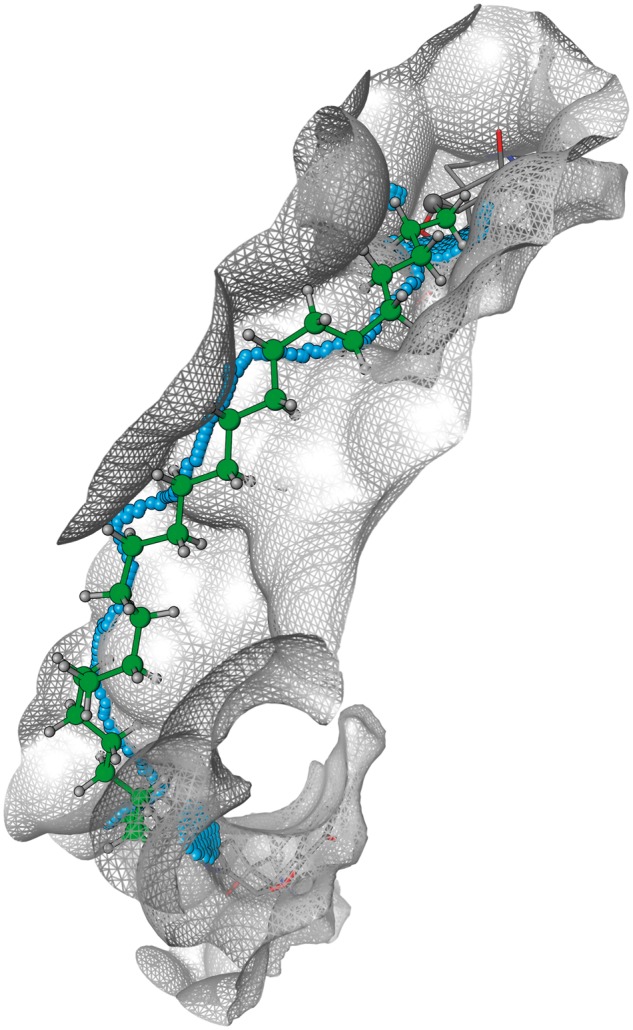
Graphical representation of how the tool generates the shortest path through the graph of surface vertices (blue spheres) and uses this to define the final minimized ‘shortest path’ spacer (green atoms). Multiple close surface vertices to each connection atom point are used as start and end points
3 Results
3.1 Design of PROTAC ligands
The crystal structure of the BRD4-VHL heterodimer in complex with the PROTAC bivalent ligand 10 MZ1 is known (Gadd et al., 2017). MZ1 links the BRD4 inhibitor JQ1 to the VHL ligand VH032 via a three-unit PEG spacer. We used this example to validate the accuracy of the computational tool in predicting the conformation of the spacer as the structure of MZ1 is known. Clearly, the experimental and predicted conformations of the spacer are in very good agreement (Fig. 3). As the PROTAC field expands, more structures will reveal the range of distances between pharmacophore binding sites, and this tool can assist design of bivalent ligands (Chessum et al., 2018; Crew et al., 2018).
Fig. 3.
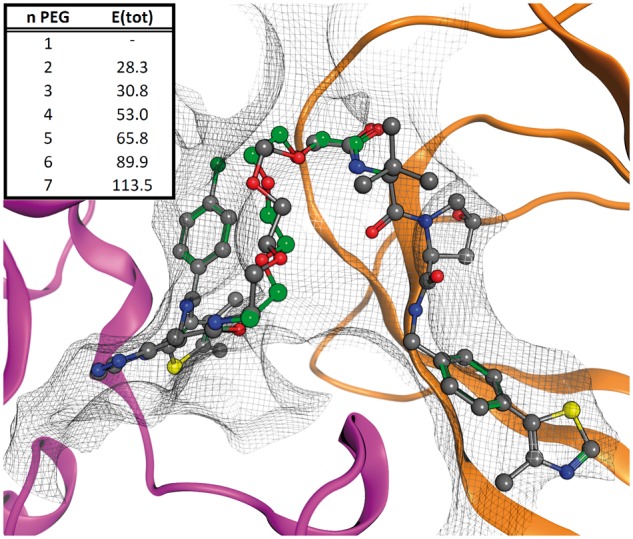
Computational assessment of spacer length for BRD4-VHL bivalent ligand. The tool was used to test a database 1- to 7-unit (n) PEG spacers. The energy of the system (E(tot), kcal/mol) shows the best results use two and three PEG repeats. Both correspond to experimentally valid linkers. The computationally derived three-unit PEG structure from the tool (green) superposes excellently to the X-ray structure (grey)
3.2 Design of CA bivalent ligands
Mack et al. (2012) constructed a synthetic dimer of CA, and a series of bivalent ligands with different lengths, as a model system for the binding of bivalent antibodies to antigens. They obtained the structure of the CA homodimer (although without bivalent ligand) and synthesized a series of benzenesulfonamides connected by oligosarcosine spacers with n = 0, 2, 4, 6, 8 and 10 sarcosine units (LSarnL). The total energy of the systems is reported in Supplementary Figure S1. In agreement with the experimental results (Mack et al., 2012), our MOE-based tool predicts the LSar4L bivalent ligand (11) as the most favorable.
3.3 Design of bivalent ligands of GPCR (homo/hetero)dimers
Despite enormous effort devoted to their design, rational development has been difficult due to the large variety of GPCR members with different extracellular domains, different ligand-binding modes, and the lack of knowledge concerning their dimeric forms.
3.3.1 Factors influencing the spacer size of bivalent ligands
When considering the design of bivalent ligands, the first step understands the structure of the protein dimer complex. In the case of GPCRs, computational models are usually built (Navarro et al., 2016, 2018a, b; Vinals et al., 2015) from crystal structures that contain a variety of symmetric dimerization interfaces (summarized in Cordomi et al., 2015). This mainly includes the TM1/2, TM4/5, TM5 or TM5/6 interfaces (Supplementary Fig. S2). Notably, the TM5/6 dimerization interface reproduces the shortest distance of 27 Å between orthosteric sites, the TM1/2 interface the longer distance of 42 Å, whereas the distances of the TM5 or TM4/5 interfaces are in between and similar to each other (Supplementary Fig. S2). Thus, the optimal length of the spacer of the bivalent ligand depends on the TM interface to target.
The second factor is the influence of ligand orientation. Orthosteric ligand binding typically occurs in a cavity between the extracellular segments of TMs 3, 5–7 or in a minor binding cavity located between the extracellular segments of TMs 1–3, 7 (Rosenkilde et al., 2010). Ligands can penetrate to different depths within these pockets (Venkatakrishnan et al., 2013) and leave atoms exposed available for linking at variable locations as shown in Supplementary Figure S3 using crystal structures of the adenosine A2A receptor. Hence, the variation in orientation and buriedness within the same receptor influences spacer length for bivalent ligands.
Clearly protein surfaces are non-uniform and approximating spacer lengths based only on inter-binding site distances is unsatisfactory. This is especially relevant in GPCRs where the extracellular domain is highly variable in sequence, length, and structure (Gonzalez et al., 2012). For instance, in GPCRs for lipid mediators the extracellular N-terminus and extracellular loop (ECL) 2-folds over the ligand-binding pocket (e.g. CB1R in Supplementary Fig. S4). In contrast, ECL2 of peptide receptors, formed by two β-strands, fully exposes the binding site to the extracellular environment (e.g. opioid receptors in Supplementary Fig. S4). On the other hand, a helical segment forms ECL2 of adrenergic receptors (e.g. β2-adrenergic in Supplementary Fig. S4). Again, this is a further consideration that influences the spacer length of bivalent ligands and prompts the need for a structure based approach.
3.3.2 Design of the optimal spacer size for the μOR–δOR heterodimer
To demonstrate the tool in the more complex GPCR systems, we first study the μ and δ opioid receptor (μ–δ) heterodimer because (i) crystal structures of μ-OR (Manglik et al., 2012) and δ-OR (Granier et al., 2012) are known, (ii) the structure of μOR revealed putative dimer interfaces (Manglik et al., 2012) and (iii) seminal work from the Portoghese group reported a series of μ-δ heterodimer bivalent ligands (Daniels et al., 2005; Yekkirala et al., 2013). The structure of μ-OR shows receptor protomers associated into dimers through two different interfaces: via TM1/2 or TM5/6. The bivalent ligand MDAN-21 (1) for μ-δ contains the μ-OR agonist oxymorphone (OXY) and the δ-OR antagonist naltrindole (NTI) (Daniels et al., 2005) (Fig. 1). As the name indicates, MDAN-21 contains 21-atoms between pharmacophores, including spacer (7 atoms) and linker (2 × 7 atoms). Regarding nomenclature (Shonberg et al., 2011), linker refers to the chemical group that attaches the spacer chain to each pharmacophoric ligand, and hence the spacer is the chain between. Figure 4A shows the μ-δ dimer interacting via the TM5/6 interface (the TM1/2 interface was not considered because the longer distance between binding pockets cannot accommodate ligands such as MDAN-21) bound to MDAN-21. A database of spacers ranging from 2 heavy atom units (16-atom spacer and linker combined) to 14 units (28-atom spacer and linker) was constructed. The tool constructs the linked bivalent ligands and assesses their fit across the dimer surface. Figure 4B shows the best solution obtained from the linking script after energy minimization, which contains 12 heavy-atom units (26-atom spacer and linker, we name it BL-26).
Fig. 4.
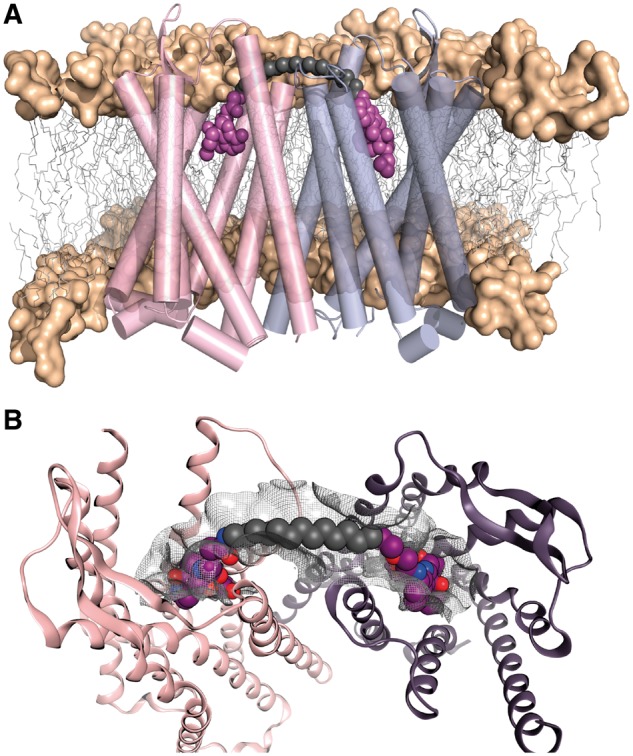
A computational tool to design the spacer size of bivalent ligands. (A) Computational model of the μ–δ heterodimer bound to MDAN-21, the two pharmacophore units (OXY and NTI, respectively) are shown in magenta spheres, carbon spacer is grey. (B) The preferred solution from the computational tool showing the spacer optimally placed across the protein van der Waals surface and connected to each ligand
3.3.3 Computational validation of the calculated spacer size
The tool generates energetically favorable spacers that can be used directly as recommendations for synthesis or for further computational study. In the latter regard we used unbiased molecular dynamics (MDs) simulations in the μs time-scale to evaluate the stability of MDAN-21, the designed BL-26, and an intermediate sized BL-23 ligand at the μ-δ heterodimer interacting via the TM5/6 interface. The stability of the ligands was monitored by the root mean-square deviation (rmsd) of the heavy atoms along the MD trajectories (Supplementary Fig. S5A), whereas the binding of the pharmacophoric units to the orthosteric binding site was monitored by the salt bridge distance between the protonated amine of NTI or OXY and Asp3.32 of δ-OR (Supplementary Fig. S5B) or μ-OR (Supplementary Fig. S5C), respectively. Clearly, the shortest MDAN-21 and BL-23 ligands cannot fulfil the simultaneous binding of the pharmacophoric units to the orthosteric binding site as revealed by both rmsd values and salt bridge distances. In contrast, the pharmacophoric units of the designed BL-26 ligand remain stable at the orthosteric binding cavities through the unbiased 1 μs MD simulation.
3.3.4 The extracellular vestibule
During the MD simulation of MDAN-21 and BL-23, the NTI pharmacophoric element remained bound to the orthosteric site of δ-OR, whereas the OXY pharmacophore unit left the orthosteric site of μ-OR However, before reaching solvent the OXY pharmacophore ligand was retained by electrostatic (Glu2315.35 and Lys2355.39 in TM5) and van der Waals (Trp2285.32, Trp3207.35) interactions in the extracellular domain of μ-OR, as shown by the plateau of the rmsd values (Supplementary Fig. S5A). The presence of a small cavity at the entrance of the orthosteric binding site [named extracellular (Dror et al., 2011) or membrane (Stanley et al., 2016) vestibule or secondary (Gonzalez et al., 2011) or metastable (Fronik et al., 2017) binding site] has been described. To further evaluate the proposed binding of MDAN-21 (NTI in the orthosteric binding site of δ-OR and OXY in the extracellular vestibule or secondary binding site of μ-OR), we performed two additional MD replicas starting at this conformation. The stability of this binding mode was monitored and confirmed by the low rmsd values of ligand heavy atoms (Supplementary Fig. S6). Clearly, MDAN-21 can simultaneously bind the orthosteric site of δ-OR and the vestibule site of μ-OR, providing a plausible alternative explanation for MDAN-21’s interesting pharmacology (Portoghese et al., 2017).
3.3.5 Calculation of the optimal spacer size in reported GPCR bivalent ligands
Having shown how our MOE-based tool can help define preferred spacer lengths of bivalent ligands for the μ-δ heterodimer, we have further applied the tool to the D2–D2 and CB1–CB1 homodimers and μ–CB1, D2–NTS1 and A1–β2 heterodimers (Table 1) for which bivalent ligands are also reported in the literature (Fig. 1). As described above, the TM1/2, TM4/5 and TM5/6 interfaces were considered, in each case permitting different possible linker and spacer lengths. Images of the preferred calculated result for each case are provided in supporting information Supplementary Table S1. The differences between experimentally reported linker lengths versus the corresponding calculated values are immediately apparent. A fundamental result of our systematic approach is the proposition that many reported ligands are too short to link orthosteric sites of adjacent monomers via a receptor-surface bound spacer. Most experimental reports show spacers between 15 and 25 atoms, but even in the shortest possible path such as in the TM5/6 interface for opioid receptors, with little extracellular hindrance (Supplementary Fig. S4), the spacer still requires 26 heavy atoms. Looking in more detail, several trends can be observed. TM1/2 dimers typically prefer a heavy atom linker plus spacer of around 36–40 heavy atoms, the TM4/5 interface is in the range of 50–55, while the TM5/6 interface presents the shortest number of atoms. However, there are exceptions. The TM1/2 interface of the CB1 and D2 homodimers prefer fewer heavy atoms than the other interfaces, because the linker is directed towards TM1 (these differences can be seen by inspecting the images in Supplementary Table S1). Hence, as we have discussed above, specific details of each system impact the spacer length, meaning it is recommended to model each new case in order to optimally apply the approach.
Table 1.
Comparing the preferred experimentally reported linker plus spacer size versus the preferred calculated size for the GPCR (homo/hetero)dimer formed via the TM1/2, TM4/5 or TM5/6 interfaces
| Experimental results from literature |
Calculated results, preferred number of heavy atoms for each respective dimera |
||||
|---|---|---|---|---|---|
| Systemsb | Bivalent ligandc | Number of heavy atomsd | TM1/2 | TM4/5 | TM5/6 |
| μ–δ | 1 | 21 | 40 | 42 | 26 |
| D2–D2 | 4 | 16 | 36 | 56 | 47 |
| CB1–CB1 | 5 | 15 | 33 | 54 | 53 |
| μ–CB1 | 6 | 20 | 41 | 55 | 42 |
| D2–NTS1 | 7 | 44 (66)e | 36 | 52 | 27 |
| A1–β2 | 9 | 25 | 40 | 55 | 33 |
Note: The comparison is performed in terms of number of heavy atoms.
Models were built according to experimental details, the script was applied in each case to determine the preferred linker size.
For references see Section 1 of main text.
Ligand number refers to Figure 1, and represent the most active ligand identified in each associated experimental study.
Based on the preferred ligand.
This study reported that molecule 7 (m = 2, Fig. 1) and another example with m = 3 (66 atom spacer) were both preferred bivalent ligands.
4 Discussion
We have presented a computational tool to assist the design of the optimal spacer and linker size connecting two pharmacophore units bound in a dimer. It provides a scoring based on energy minimization of the resulting ligand receptor complex. Our results suggest inconsistencies between claimed bivalent ligands of GPCRs and what seems plausible assuming an extracellular surface binding mode for the spacer, as has also been pointed by others (Glass et al., 2016; Lane et al., 2013; Shonberg et al., 2011). The alternative is for the spacer to bind between TM helices and disrupt interactions of several helical turns, which would incur a high energetic cost. Thus, these ligands with short spacers may adopt alternative binding modes (schematized in Fig. 5). Some ligands with very short spacers might bind the orthosteric site as well as a secondary binding site at the extracellular entrance (vestibule or allosteric site) of the same protomer (Fig. 5A). However, this mode of binding to a single protomer can also allosterically modulate the binding of a ligand to the orthosteric site of a second protomer (Lane et al., 2014). Second, ligands with larger spacers might bind the orthosteric site of Protomer A and the secondary binding site at the extracellular entrance of Protomer B (Fig. 5B). It has been suggested that the conformations of orthosteric and secondary binding sites of the same protomer are coupled such as that the presence of a ligand in one affects the shape of the other (Bokoch et al., 2010; Dror et al., 2013). And finally, actual bivalent ligands, with the optimal linker size, can simultaneously bind the orthosteric binding sites of Protomers A and B (Fig. 5C).
Fig. 5.

Binding modes of bivalent ligands to GPCR (homo/hetero)dimers. (A) Ligands can bind the orthosteric site as well as a secondary binding site at the extracellular entrance of the same protomer. (B) Ligands with larger spacers might bind the orthosteric site of protomer A and the secondary binding site at the extracellular entrance of Protomer B. (C) Bivalent ligands, with the optimal spacer size, can simultaneously bind the orthosteric binding sites of Protomers A and B
Theoretically, the affinity of a bivalent ligand could approach the sum of the binding free energies for the individual components along with entropic corrections (Foreman, 2017; Mammen et al., 1998). With linkers that are sufficiently long, impressive potency gains have been seen (Mack et al., 2012) but various aspects contribute to a less than expected binding affinity. Negative cooperativity between two protomers can result in the binding of a ligand to the first protomer decreasing the affinity of the ligand for the second protomer (Ferre et al., 2014). Secondly, as mentioned above, the spacer may not permit the two pharmacophores to bind simultaneously in their respective sites. Meanwhile, entropy, is expected to be beneficial because the second pharmacophore unit is present at a much higher local concentration once tethered in a close radius above the dimer (Numata et al., 2012). Nevertheless, binding of the spacer and linker will have an entropic cost once fixed on the receptor surface and recent work to address this has prompted the study of less flexible spacers (Tanaka et al., 2010). Kramer and Karpen (1998) and Mack et al (2012) have shown that flexible spacers much longer than the distance between sites still bind tightly. In many cases however, GPCR bivalent ligands lack the potency gain and our results suggest they are too short to span the high affinity binding sites. Therefore, does the size matter? It becomes clear that a minimum size is required and evaluating this using the presented tool is expected to be a pragmatic and valuable approach. Future reports will show the prospective use of this tool for the design of highly potent bivalent ligands of GPCR homodimers.
Supplementary Material
Acknowledgement
We thank Dr James Edwards for helpful comments on the study.
Funding
This work has been supported by grants from the Spanish Ministerio de Economía y Competitividad [SAF2015-74627-JIN, SAF2016-77830-R].
Conflict of Interest: none declared.
References
- Barlow N. et al. (2013) Effect of linker length and composition on heterobivalent ligand-mediated receptor cross-talk between the A1 adenosine and beta2 adrenergic receptors. ChemMedChem, 8, 2036–2046. [DOI] [PubMed] [Google Scholar]
- Bokoch M.P. et al. (2010) Ligand-specific regulation of the extracellular surface of a G-protein-coupled receptor. Nature, 463, 108–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chessum N.E.A. et al. (2018) Demonstrating in-cell target engagement using a pirin protein degradation probe (CCT367766). J. Med. Chem., 61, 918–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordomi A. et al. (2012) Membrane protein simulations using AMBER force field and berger lipid parameters. J. Chem. Theory Comput., 8, 948–958. [DOI] [PubMed] [Google Scholar]
- Cordomi A. et al. (2015) Structures for G-Protein-Coupled Receptor Tetramers in Complex with G Proteins. Trends Biochem. Sci, 40, 548–551. [DOI] [PubMed] [Google Scholar]
- Corson T.W. et al. (2008) Design and applications of bifunctional small molecules: why two heads are better than one. ACS Chem. Biol., 3, 677–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crew A.P. et al. (2018) Identification and characterization of Von Hippel-Lindau-recruiting proteolysis targeting chimeras (PROTACs) of TANK-binding kinase 1. J. Med. Chem., 61, 583–598. [DOI] [PubMed] [Google Scholar]
- Daniels D.J. et al. (2005) A bivalent ligand (KDAN-18) containing delta-antagonist and kappa-agonist pharmacophores bridges delta2 and kappa1 opioid receptor phenotypes. J. Med. Chem., 48, 1713–1716. [DOI] [PubMed] [Google Scholar]
- Dror R.O. et al. (2011) Pathway and mechanism of drug binding to G-protein-coupled receptors. Proc. Natl. Acad. Sci. USA, 108, 13118–13123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dror R.O. et al. (2013) Structural basis for modulation of a G-protein-coupled receptor by allosteric drugs. Nature, 503, 295–299. [DOI] [PubMed] [Google Scholar]
- Ferre S. et al. (2014) G protein-coupled receptor oligomerization revisited: functional and pharmacological perspectives. Pharmacol. Rev., 66, 413–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foreman K.W. (2017) A general model for predicting the binding affinity of reversibly and irreversibly dimerized ligands. PLoS One, 12, e0188134.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fronik P. et al. (2017) Bitopic ligands and metastable binding sites: opportunities for G protein-coupled receptor (GPCR) medicinal chemistry. J. Med. Chem., 60, 4126–4134. [DOI] [PubMed] [Google Scholar]
- Gadd M.S. et al. (2017) Structural basis of PROTAC cooperative recognition for selective protein degradation. Nat. Chem. Biol., 13, 514–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass M. et al. (2016) One for the price of two…are bivalent ligands targeting cannabinoid receptor dimers capable of simultaneously binding to both receptors? Trends Pharmacol. Sci., 37, 353–363. [DOI] [PubMed] [Google Scholar]
- Gonzalez A. et al. (2011) Molecular basis of ligand dissociation in beta-adrenergic receptors. PLoS One, 6, e23815.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez A. et al. (2012) Impact of helix irregularities on sequence alignment and homology modelling of G protein-coupled receptors. Chembiochem, 13, 1393–1399. [DOI] [PubMed] [Google Scholar]
- Granier S. et al. (2012) Structure of the delta-opioid receptor bound to naltrindole. Nature, 485, 400–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiller C. et al. (2013) Class A G-protein-coupled receptor (GPCR) dimers and bivalent ligands. J. Med. Chem., 56, 6542–6559. [DOI] [PubMed] [Google Scholar]
- Hubner H. et al. (2016) Structure-guided development of heterodimer-selective GPCR ligands. Nat. Commun., 7, 12298.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer R.H., Karpen J.W. (1998) Spanning binding sites on allosteric proteins with polymer-linked ligand dimers. Nature, 395, 710–713. [DOI] [PubMed] [Google Scholar]
- Kuhhorn J. et al. (2011) Development of a bivalent dopamine D(2) receptor agonist. J. Med. Chem., 54, 7911–7919. [DOI] [PubMed] [Google Scholar]
- Lane J.R. et al. (2013) Bridging the gap: bitopic ligands of G-protein-coupled receptors. Trends Pharmacol. Sci., 34, 59–66. [DOI] [PubMed] [Google Scholar]
- Lane J.R. et al. (2014) A new mechanism of allostery in a G protein-coupled receptor dimer. Nat. Chem. Biol., 10, 745–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Naour M. et al. (2013) Bivalent ligands that target mu opioid (MOP) and cannabinoid1 (CB1) receptors are potent analgesics devoid of tolerance. J. Med. Chem., 56, 5505–5513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mack E.T. et al. (2012) Dependence of avidity on linker length for a bivalent ligand-bivalent receptor model system. J. Am. Chem. Soc., 134, 333–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mammen M. et al. (1998) Polyvalent interactions in biological systems: implications for design and use of multivalent ligands and inhibitors. Angew. Chem. Int. Ed. Engl., 37, 2754–2794. [DOI] [PubMed] [Google Scholar]
- Manglik A. et al. (2012) Crystal structure of the micro-opioid receptor bound to a morphinan antagonist. Nature, 485, 321–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurice P. et al. (2011) Asymmetry of GPCR oligomers supports their functional relevance. Trends Pharmacol. Sci., 32, 514–520. [DOI] [PubMed] [Google Scholar]
- McRobb F.M. et al. (2012) Homobivalent ligands of the atypical antipsychotic clozapine: design, synthesis, and pharmacological evaluation. J. Med. Chem., 55, 1622–1634. [DOI] [PubMed] [Google Scholar]
- Navarro G. et al. (2016) Quaternary structure of a G-protein-coupled receptor heterotetramer in complex with Gi and Gs. BMC Biol., 14, 26.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro G. et al. (2018a) Cross-communication between Gi and Gs in a G-protein-coupled receptor heterotetramer guided by a receptor C-terminal domain. BMC Biol., 16, 24.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro G. et al. (2018b) Evidence for functional pre-coupled complexes of receptor heteromers and adenylyl cyclase. Nat. Commun., 9, 1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Numata J. et al. (2012) Influence of spacer-receptor interactions on the stability of bivalent ligand-receptor complexes. J. Phys. Chem. B, 116, 2595–2604. [DOI] [PubMed] [Google Scholar]
- Portoghese P.S. et al. (2017) Heteromer induction: an approach to unique pharmacology? ACS Chem. Neurosci., 8, 426–428. [DOI] [PubMed] [Google Scholar]
- Pronk S. et al. (2013) GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics, 29, 845–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenkilde M.M. et al. (2010) The minor binding pocket: a major player in 7TM receptor activation. Trends Pharmacol. Sci., 31, 567–574. [DOI] [PubMed] [Google Scholar]
- Shonberg J. et al. (2011) Design strategies for bivalent ligands targeting GPCRs. ChemMedChem, 6, 963–974. [DOI] [PubMed] [Google Scholar]
- Soriano A. et al. (2009) Adenosine A2A receptor-antagonist/dopamine D2 receptor-agonist bivalent ligands as pharmacological tools to detect A2A-D2 receptor heteromers. J. Med. Chem., 52, 5590–5602. [DOI] [PubMed] [Google Scholar]
- Soulier J.L. et al. (2005) Design and synthesis of specific probes for human 5-HT4 receptor dimerization studies. J. Med. Chem., 48, 6220–6228. [DOI] [PubMed] [Google Scholar]
- Stanley N. et al. (2016) The pathway of ligand entry from the membrane bilayer to a lipid G protein-coupled receptor. Sci. Rep., 6, 22639.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanton B.Z. et al. (2018) Chemically induced proximity in biology and medicine. Science, 359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T. et al. (2010) Bivalent ligands of CXCR4 with rigid linkers for elucidation of the dimerization state in cells. J. Am. Chem. Soc., 132, 15899–15901. [DOI] [PubMed] [Google Scholar]
- Valant C. et al. (2012) The best of both worlds? Bitopic orthosteric/allosteric ligands of g protein-coupled receptors. Annu. Rev. Pharmacol. Toxicol., 52, 153–178. [DOI] [PubMed] [Google Scholar]
- Venkatakrishnan A.J. et al. (2013) Molecular signatures of G-protein-coupled receptors. Nature, 494, 185–194. [DOI] [PubMed] [Google Scholar]
- Vinals X. et al. (2015) Cognitive impairment induced by Delta9-tetrahydrocannabinol occurs through heteromers between cannabinoid CB1 and serotonin 5-HT2A receptors. PLoS Biol., 13, e1002194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yekkirala A.S. et al. (2013) An immunocytochemical-derived correlate for evaluating the bridging of heteromeric mu-delta opioid protomers by bivalent ligands. ACS Chem. Biol., 8, 1412–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zengerle M. et al. (2015) Selective Small Molecule Induced Degradation of the BET Bromodomain Protein BRD4. ACS Chem. Biol., 10, 1770–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y. et al. (2010) Synthesis and biological evaluation of bivalent ligands for the cannabinoid 1 receptor. J. Med. Chem., 53, 7048–7060. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.